

Abstract
β-Defensin 3 (BD3) was identified as a ligand for the melanocortin receptors (MCRs) in 2007, although the pharmacology activity of BD3 has not been clearly elucidated. Herein, it is demonstrated that human and mouse BD3 are full micromolar agonists at the MCRs. Furthermore, mouse and human β-defensin 1 (BD1) are also MCR micromolar agonists. This work identifies BD1 as an endogenous MCR ligand and clarifies the controversial role of BD3 as a micromolar agonist.
Keywords: Melanocortin receptors, obesity, β-defensin-3, β-defensin-1, endogenous agonist, GPCR, feeding, pigmentation, MC1R, MC3R, MC4R, anti-inflammatory, innate immunity
Graphical Abstract
Introduction
The five melanocortin receptors (MC1-5R) are postulated to be involved in numerous physiological processes, including pigmentation1–2 and energy homeostasis.3–6 Endogenous agonists for the MCRs include the α-, β-, and γ-melanocyte-stimulating hormones (MSH) and adrenocorticotropic hormone. The melanocortin system also possesses the only currently known endogenous antagonists to GPCRs, agouti-signaling protein (ASP) and agouti-related protein (AGRP). Many ligands from the naturally occurring agonists and antagonists have been developed and advanced to clinical trials, including afamelanotide, bremelanotide, and setmelanotide (as previously reviewed).7 Novel ligands for the MCRs with new scaffolds may generate probes and therapeutic leads with unique pharmacological profiles.
In most mammals, coat coloration is regulated by the epithelial pigments eumelanin and pheomelanin (producing brown/black and red/yellow coloration, respectively). Agonist stimulation of the MC1R increases intracellular cAMP levels, resulting in eumelanin production and darker pigmentation.8–9 Antagonism of the MC1R by ASP decreases intracellular cAMP concentrations, resulting in pheomelanin production.10 While interactions between the endogenous agonists, ASP, and the MC1R control the majority of mammal pigmentation, an interesting case of dominantly-inherited dark coloration not involving these genes is observed in canines.11–12 Originally described as the KB locus,13 a seminal paper by Candille et al. identified the N-terminal Gly deletion from the 45-residue canine β-defensin (cBD103) peptide to be responsible for this black coat coloration.14 Wild type and deletion cBD103 peptides possessed sub-micromolar affinities for the canine MC1R (221 nM and 37 nM, respectively), and for the mouse and human MC1R (9.7 to 35.5 nM affinities), indicating that cBD103 interacted with the MC1R to influence coat coloration.14 The human ortholog of cBD103, β-defensin 3 (hBD3), and human β-defensin 1 (hBD1) were also shown to possess binding affinity at the hMC1R (13.8 and 30.0 nM affinities, respectively), and the deletion cBD103 peptide possessed 105 nM binding affinity at the hMC4R,14 an indication additional β-defensins may interact with multiple MCRs.
In the same publication, generation of transgenic mice expressing either the native or deletion form of cBD103 resulted in mice with a black coat color despite being on an Agouti background, suggesting MC1R agonism.14 These transgenic mice maintained a lower body weight and size compared to non-cBD103 expressing littermates,14 a possible sign of MC4R agonism.3 When the hBD3 was centrally administered in male Wistar rats following a fast, treated rats consumed less food and maintained a lower body weight than vehicle,15 additional indications of MC4R agonist activity. Transgenic MC1R-null mice were generated that expressed the deletion form of cBD103 and did not have a dark coat coloration, supporting the hypothesis that cBD103 interacts with the MC1R to generate the inheritable pigmentation observed in canines.15 As the dark coloration suggests eumelanin production, cBD103 could be directly stimulating the MC1R as an agonist, or could be preventing ASP-induced MC1R antagonism. Despite cBD103 possessing nM binding affinities at the canine, mouse, and human MC1R, a cAMP functional assay in mouse Melan-a cells (presumably expressing the mMC1R) indicated that cBD103 was unable to stimulate the mMC1R at concentrations up to 1 μM (or 1.6 μM in a follow-up study).14–15 No evidence of direct cBD103-ASP interactions was observed, indicating cBD103 was not sequestering ASP to modulate MC1R signaling.14 Thus, while cBD103 was identified to be involved with pigmentation and a dominant inheritance of dark coat coloration (most likely through MC1R signaling), the pharmacological activity of the cBD103 at the MC1R was not elucidated.
Additional studies have not unambiguously clarified how β-defensins interact with the MCRs. Studies in HEK293 cells using the hBD3 peptide (84% identical shared identity with cBD103 with seven conservative substitutions) resulted in a dose dependent increase in cAMP levels (100 and 300 nM concentrations of hBD3).16 The cAMP response at the highest dose of hBD3 assayed (300 nM) was lower than the maximal response of 1 nM NDP-MSH, with the authors concluding hBD3 may be a partial agonist at HEK293 cells expressing the MC1R.17 While no cAMP signal was observed when up to 100 nM concentrations of hBD3 were assayed in B16 cells16–17 or in primary melanocytes,18 100 nM hBD3 did lower the agonist induction of cAMP by α-MSH (in primary melanocytes18) and NDP-MSH (in B16 cells16–17 and HEK293 cells16 expressing the MC1R), exhibiting suspected antagonist activity (in the absence of a Schild analysis,19 antagonist activity cannot be confirmed). In a DNA repair model system that did not directly measure cAMP levels, hBD3 exhibited antagonist-like activity following DNA damage mediated through UV light20 or cisplatin,21 similar to the effect of ASP treatment. Due to the binding affinity of hBD3 in transiently transfected HEK293 cells expressing the hMC1R or hMC4R (42 or 110 nM, respectively) without apparent agonist activity (but resulting in an agonist-like coat coloration inheritance pattern), the hBD3 has also been labeled a neutral antagonist.15 Thus, the hBD3 has been described as a partial agonist, antagonist, and neutral antagonist.
As the functional effects of hBD3 have been variable, further work has been performed to identify the structural requirements for hBD3 binding affinity. In HEK293 cells transiently expressing the hMC1R or hMC4R, truncated hBD3 analogues did not possess binding affinity, while linear and single residue substitution variants of hBD3 maintained similar binding to the native sequence.15 To disrupt the affinity of hBD3 at the hMC1R or hMC4R, large patches of positively charged residues were replaced with neutral amino acids.15 The authors concluded that hBD3 interacts with the MCRs through electrostatic patches between the cationic hBD3 and the anionic MCRs.15 A follow-up study examining electrostatic potential of multiple β-defensins and binding affinity at the MC1R suggested β-defensins with similar charge distributions to hBD3 had greater affinity at the MC1R.22 This work also demonstrated that numerous β-defensins have affinity to the MC1R, though additional MCRs were not examined. Additionally, the hBD1 was not examined.
The β-defensins are a peptide family hypothesized to be involved in the innate immune system that are classified by a conserved 6 Cys residues that form 3 disulfide bonds in a 1–5, 2–4, 3–6 pattern, as previously reviewed.23 Outside of the Cys amino acids, there is extensive sequence diversity, although NMR and X-ray crystal structures of three human β-defensins (hBD1, hBD2, and hBD3) indicate a similar over fold.24–28 Due to the similar purported structures of the β-defensins, and the ability of the hBD3 to bind to the hMC4R, it is hypothesized that additional β-defensins may interact with additional MCRs beyond the MC1R. Since hBD1 is expressed in many tissues including the brain,29 it was hypothesized that hBD1 may interact with the MC3R and MC4R, MCRs involved in energy homeostasis that are also centrally expressed.3–6, 30
Therefore, the following experiments were performed to probe the interactions between BD3 and the MCRs and to determine if BD1 is a functional ligand at the MCRs. To clarify how BD3 interacts with the MCRs, oxidized and reduced forms of hBD3 and the mouse ortholog (the accepted mouse ortholog for hBD3 is mBD14,31 but will be referred to as mBD3* in this manuscript for simplicity) were synthesized and characterized at both the human and mouse MCRs. Notably, dose-response curves were generated using concentrations up to 100 μM, approximately 100-fold higher concentrations than previously utilized to determine if BD3 possesses agonist activity at micromolar concentrations. Additionally, oxidized and reduced forms of hBD1 and mBD1 were synthesized and characterized for activity at the human and mouse MCRs. Both human and mouse β-defensins were utilized in this study as differential agonist responses have been previously reported between human and mouse MCR subtypes.32 Due to the central expression of mBD1 and the MC3R/MC4R responsible for energy homeostasis, mBD1 was centrally administered in mice while monitoring food intake and body weight to examine if this β-defensin has potential physiological effects on appetite.
Results and Discussion
Details about peptide characterization (SI Table 1), the amplified luminescent proximity homogeneous cAMP assay (PerkinElmer), and the competitive displacement assay utilized in this study can be found in the supporting information.
β-defensin 3 Pharmacology
Due to the previous conflicting reports regarding the activity of hBD3 (the ability of hBD3 to stimulate MCRs and generate cAMP production), these studied herein were performed using the competitive displacement cAMP assay. The oxidized, folded form of hBD3 was a full agonist at the mMC1R and mMC5R (EC50 = 2.2 and 4.7 μM, respectively) and was able to stimulate some cAMP response at both the mMC3R and mMC4R at 100 μM concentrations (35% and 40% of maximal NDP-MSH signal, respectively; Table 1 and Figure 1). At the human MCRs, hBD3 was observed to be a full agonist at the hMC1R and hMC4R (EC50 = 0.40 and 2.6 μM, respectively) and partially stimulated the hMC3R and hMC5R at 100 μM concentrations (35% and 45% relative to the maximal stimulatory signal of NDP-MSH; Table 1 and Figure 1). The linear, reduced form (hBD3red) was unable to stimulate the mMC1R, mMC5R, and hMC3R at up to 100 μM concentrations, maintained similar activity at the mMC3R, mMC4R, and hMC5R (30%, 35%, and 35% of maximal NDP-MSH signal at 100 μM concentrations) to hBD3, and partially activated the hMC1R and hMC4R at 100 μM concentrations (45% maximal NDP-MSH stimulation at both receptors; Table 1). Competitive displacement binding assays with [125I]-NDP-MSH indicated hBD3 possessed micromolar binding affinities at the mMC4R and hMC1R (17 and 7 μM, respectively) and displaced 50% of the radiolabeled NDP-MSH at 100 μM concentrations at the hMC4R (SI Table 2, SI Figure 1).
Table 1.
Agonist Pharmacology of Human and Mouse β-Defensin 1 and 3 at the Mouse and Human Melanocortin Receptors.a
| Peptide | Mouse Melanocortin Receptors Agonist EC50 (nM)
|
Human Melanocortin Receptors Agonist EC50 (nM)
|
||||||
|---|---|---|---|---|---|---|---|---|
| mMC1R | mMC3R | mMC4R | mMC5R | hMC1R | hMC3R | hMC4R | hMC5R | |
| NDP-MSH | 0.040 ± 0.008 | 0.33 ± 0.05 | 0.87 ± 0.38 | 0.41 ± 0.09 | 0.070 ± 0.008 | 0.30 ±0.07 | 0.13 ± 0.02 | 0.40 ± 0.03 |
| mBD1red | 55% @ 100 μM | >100,000 | >100,000 | 55% @ 100 μM | 4,500 ± 400 | >100,000 | 70% @ 100 μM | >100,000 |
| mBD1 | 11,000 ± 6,000 | 14,000 ± 2,000 | 75% @ 100 μM | 5,200 ± 400 | 1,400 ± 200 | 22,000 ± 4,000 | 3,800 ± 600 | >100,000 |
| mBD3*red | 40% @ 100 μM | 35% @ 100 μM | 40% @ 100 μM | 35% @ 100 μM | 45% @ 100 μM | >100,000 | 50% @ 100 μM | 40% @ 100 μM |
| mBD3* | 45% @ 100 μM | 35% @ 100 μM | 30% @ 100 μM | 35% @ 100 μM | 55% @ 100 μM | >100,000 | 60% @ 100 μM | 40% @ 100 μM |
| hBD1red | >100,000 | >100,000 | >100,000 | >100,000 | 60% @ 100 μM | >100,000 | 35% @ 100 μM | >100,000 |
| hBD1 | 65% @ 100 μM | 35% @ 100 μM | 30% @ 100 μM | 55% @ 100 μM | 7,400 ± 3,400 | >100,000 | 21,000 ± 8,000 | >100,000 |
| hBD3red | >100,000 | 30% @ 100 μM | 35% @ 100 μM | >100,000 | 45% @ 100 μM | >100,000 | 45% @ 100 μM | 35% @ 100 μM |
| hBD3 | 2,200 ± 1,700 | 35% @ 100 μM | 40% @ 100 μM | 4,700 ± 900 | 400 ± 30 | 35% @ 100 μM | 2,600 ± 600 | 45% @ 100 μM |
The indicated errors represent the standard error of the mean determined from at least three independent experiments.
>100,000 indicates that the compound was examined but lacked agonist activity at up to 100 μM concentrations. A % indicates the maximal stimulatory response observed at 100 μM concentrations but not enough stimulation was observed to determine an EC50 value. The accepted mouse ortholog nomenclature for hBD3 is mBD14,31 but will be referred to as mBD3* in this manuscript for simplicity.
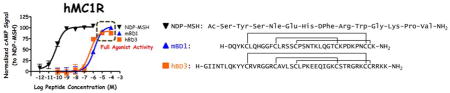
Figure 1.

Illustrations of the in vitro agonist pharmacology of NDP-MSH, mBD1, mBD3*, hBD1, and hBD3 at the hMC1R, hMC4R, mMC1R, and mMC4R. The cAMP signal was normalized as previously described.33 The accepted mouse ortholog nomenclature for hBD3 is mBD14,31 but will be referred to as mBD3* in this manuscript for simplicity.
Evaluation of the mouse ortholog of hBD3 demonstrated that mBD3* (reported in the literature as mBD14)31 generated an increased cAMP agonist response at the mMCRs at 100 μM concentrations compared to maximal signal of NDP-MSH (45%, 35%, 30%, and 35% at the mMC1R, mMC3R, mMC4R, and mMC5R, respectively; Table 1 and Figure 1). An increased CAMP agonist response was also observed at the hMC1R, hMC4R, and hMC5R (55%, 60%, and 40% relative to NDP-MSH control), while no activity was found at the hMC3R at concentrations up to 100 μM (Table 1 and Figure 1). Surprisingly, the reduced form (mBD3*red) possessed similar activity to the folded, oxidized mBD3* peptide at all receptor subtypes (Table 1). [This mBD3* reduced form was intended as a structural control (i.e. no disulfide bridges, no secondary and tertiary structure) and hypothesized to result in a lack of stimulatory activity.] Similar binding affinities were observed for mBD3* at the mMC4R and hMC1R (5 and 6 μM, respectively) compared to the hBD3, while mBD3* also displaced the radiolabeled NDP-MSH at the hMC4R (18 μM).
Previous studies indicated that hBD3 did not possess full agonist pharmacology at the MC1R using concentrations up to 2 μM, with varying results between cell types.14–18 This contrasts to the in vivo effects, including a dark coat coloration,14–15 decreased body length and weight in transgenic mice expressing the BD3,14 and central administration of hBD3 resulting in decreased food intake and decreased weight gain following a fast in rats,15 effects that may indicate melanocortin agonist activity. Therefore, in the present study, higher concentrations (up to 100 μM) were used to generate 7-point dose-response curves to fully characterize potential cAMP based agonist activity of the hBD3 and mBD3*. Full agonist activity was observed by using increased concentrations of hBD3 at the mMC1R, mMC5R, hMC1R and hMC4R utilizing higher concentrations. Notably, at 10−7 M concentrations (100 nM), the dose-response curves have not reached the sigmoidal increase (Figure 1), indicating that single dose- response points at these concentrations would appear to be inactive or possess minimal activity. Thus, the current data with hBD3 possessing full agonist efficacy is likely from extending the dose-activity curve to higher concentrations and correlates to previous reports of no activity at sub-micromolar concentrations. The present study also utilized HEK293 cells expressing the MCRs, which have been reported to possess partial agonist activity at 300 nM concentrations when expressing the MC1R.16 Other prior results using primary mouse (Melan-a, B16)14–17 and human (melanocytes)18 cells expressing the MC1R have not reported agonist activity, which may suggest that differing levels of receptor expression and receptor density at the cell surface influence the reported hBD3 pharmacology.
A notably difference in hBD3 binding affinities was also observed, which have previously been reported to be 13.8 nM14 and 42 nM15 at the hMC1R and 110 nM15 at the hMC4R. The present study observed binding affinities of 7 μM at the hMC1R and 50% displacement at 100 μM concentrations at the hMC4R, differences of approximately 500-fold. Extensive substitution work on hBD3 by Nix et al. support the sub-micromolar binding affinity, with numerous substitutions required for significant loss of affinity.15 While all binding experiments were performed using HEK293 cells, difference in protocols may explain the relatively large change in binding affinities. The current study used stably expressing MCR constructs, [125I]-NDP-MSH label, and a 1 h incubation time versus transiently transfected cells, Eu-NDP-MSH label, and a 2 h incubation time in the prior report.15 Further experiments examining both protocols may clarify the observed differences in hBD3 binding affinity. The micromolar binding affinity of hBD3 to the mMC4R, hMC1R, and hMC4R correlate with the functional agonist EC50 values reported herein.
β-defensin 1 Pharmacology
In addition to the BD3 analogs, BD1 peptides were also examined [both the human and mouse orthologs (hBD1 and mBD1)]. The hBD1 was selected as PCR suggested that this defensin is ubiquitously expressed in many tissues, including the heart, pancreas, and brain.29 The expression of hBD1 in the brain suggests that this β-defensin may be a ligand for the centrally expressed hMC3R and hMC4R that modulate energy homeostasis. Derivatives from this scaffold may therefore result in novel probes and therapeutic leads for altered energy states, including obesity. At the mouse MCRs, hBD1 partially stimulated cAMP accumulation at 100 μM concentrations compared to the full response of NDP-MSH (65%, 35%, 30%, and 55% at the mMC1R, mMC3R, mMC4R, and mMC5R, respectively; Table 1 and Figure 1). Full agonist efficacy was observed at the hMC1R and hMC4R (7.4 and 21 μM, respectively) while no activity was observed at up to 100 μM at the hMC3R and hMC5R (Table 1 and Figure 1). The linear peptide (hBD1red) also possessed partial agonist stimulation at the hMC1R and hMC4R (60% and 35% of NDP-MSH maximal response) and no activity at the remaining receptor subtypes (Table 1). Competitive binding data indicated hBD1 possessed 26 μM binding affinities at both the mMC4R and hMC1R, while there was not sufficient displacement of the radiolabel to generate an IC50 values at the hMC4R (SI Table 2 and SI Figure 1).
The mouse homolog of hBD1 (mBD1) possessed full agonist efficacy at the mMC1R, mMC3R, and mMC5R (EC50 = 11, 14, and 5.2 μM) and stimulated the mMC4R to 75% the maximal signal of NDP-MSH at 100 μM concentrations (Table 1 and Figure 1). Full agonist efficacy was also observed at the hMC1R, hMC3R and hMC4R (EC50 = 1.4, 22, and 3.8 μM), while no activity was observed at the hMC5R at up to 100 μM concentrations (Table 1 and Figure 1). The linear mBD1red did not possess agonist activity at the mMC3R and mMC4R, and partially activated the mMC1R and mMC5R at 100 μM concentrations (55% maximal NDP-MSH signal at both receptor subtypes; Table 1). The reduced peptide was also unable to stimulate the hMC3R and hMC5R, partially stimulated the hMC4R (70% NDP-MSH maximal signal), and was a full agonist at the hMC1R albeit with a 3-fold lower potency compared to the cyclic mBD1 (Table 1). Despite possessing full agonist efficacy at the mMC4R, hMC1R, and hMC4R, mBD1 was unable to displace I125-NDP-MSH at these three receptors (SI Table 2 and SI Figure 1).
While the hBD3 has been utilized in several studies, hBD1 has not been as well characterized, with a single reported binding affinity of 30 nM.14 Similar to the observed micromolar agonist EC50 values for hBD3 reported herein, hBD1 also possessed full agonist efficacy at the hMC1R and hMC4R at micromolar concentrations, demonstrating that at least two peptides from the β-defensins family are full agonists at select MCRs. The mBD1 also possessed full agonist efficacy with micromolar potency at the mMC1R, mMC3R, mMC5R hMC1R, hMC3R, and hMC4R. These data suggest that the BD1 and BD3 are novel melanocortin agonist scaffolds that may be optimized to act as novel probes and therapeutic lead molecules for the MCRs.
Analogous to the hBD3, the binding affinity of hBD1 in the present study (26 μM at the hMC1R) has 1,000-fold decreased potency compared to a prior study.14 The differences in binding assays were detailed above, and may explain this discrepancy. In support of the micromolar affinities is the micromolar agonist potencies observed herein.
In Vivo Intracerebroventricular Administration Studies
The original paper describing β-defensins as MCR ligands noted that transgenic mice overexpressing cBD3 maintained a reduced body weight and length,14 perhaps indicative of MC3R/MC4R agonism. Central administration of hBD3 in rats has also been shown to decrease food intake and slower return to pre-fast weight following a 24 h fast,15 suggestive of MC3R/MC4R agonist activity. Since BD1 has been demonstrated to be expressed in the brain,29 the mBD1 peptide was selected to be centrally administered in cannulated male mice to determine potential in vivo responses to mBD1 on food intake. It is well documented that central administration of melanocortin agonists result in decreased food intake while antagonists increase food intake.4, 34 Due to the melanocortin agonist pharmacology observed in the present study, and the past demonstration that hBD3 decreases food intake in rats following a 24 h fast,15 it was hypothesized that mBD1 would result in decreased food intake when administered in mice.
In an ad libitum feeding experiment, mBD1 was administered 2 h before light outs in 10, 20, and 30 nmol doses to mice given free access to normal food and drink. No differences in food intake or body weight were observed at the lower doses (10 and 20 nmol; data not shown). While a significant decrease was observed starting at the 2 h time point for the 30 nmol dose in male mice (data not shown), this was likely due to toxicity of mBD1 as one of four male mice did not survive this dosing. The toxicity is hypothesized to result from residual trifluoroacetic acid remaining in the sample from the purification process35 and not due to inherent toxicity of mBD1. Previous reports have suggested that this feeding paradigm does not result in a statistical change in food intake or body weight change in adult male Wistar rats when administer up to 2 nmol hBD3.15 Further studies were done using 20.9 nmol (100 μg) as no adverse effects were noted with this dose.
Since there was no significant change in feeding parameters during the ad libitum experiment, a fasting feeding paradigm was explored where food was removed 22 h before being re-introduced. This experimental design was utilized by Nix et. al. in rats and resulted in modest, but significant decreases in cumulative food intake at 8 and 24 h and resulted in rats maintaining a greater weight loss at 24 h compared to vehicle treated rats following the fast.15 Male mice were administered saline or 100 μg mBD1 following a Latin square fasting feeding paradigm at the start of a 22 h fast. Fasting was ended 2 hours before lights out (t = 0 h) by reintroduction of a pre-massed amount of food placed in the cage. Cumulative food intake and body weight were measured for 48 h. Administration of mBD1 showed a significant main effect of mBD1 treatment on food intake (seen in the trend of decreased food intake in SI Figure 2 for mBD1 treated animals), but follow-up analysis did not result in significance at a specific time point. A similar trend of lower body weight following the fast was also observed (SI Figure 2), with a significant main effect of mBD1 treatment on body weight, but no significance was observed at specific time points.
Nix et al. used a 7 μg (1.2 nmol) hBD3 dose in male rats and observed significant decreases in food intake and body weight after an intracerebroventricular (ICV) injection at the onset of a 24 h fast.15 A 100 μg dose of mBD1 was used in the current study as the highest dose that could be utilized without side effects, and due to the relative weak (micromolar) agonist potency observed in the in vitro cell assays. A similar trend of decreased food intake and slower return to pre-fast body weight was observed in both experiments. This represents the first time the centrally expressed BD1 peptide has been utilized in vivo and resulted in a significant treatment effect in mice, demonstrating that the BD1 peptide may signal through the MC3R/MC4R in mice.
The trend of decreased food intake and body weight is similar to the effects observed when hBD3 is administered in rats.15 This data supports the hypothesis put forth by Nix et al. that β-defensins may interact with the MCRs as neutral antagonists and block the activity of endogenous AGRP.15 However, in vitro results presented herein demonstrated that β-defensins also act as micromolar, full agonists at the MCRs. It is possible that mBD1 may be acting through two different mechanisms in vivo. First, mBD1 may antagonize the effects of endogenous AGRP, resulting in decreased feeding and decreased body weight. Second, mBD1 may decrease food intake and body weight directly by melanocortin agonist signaling. This dual effect is supported by both functional data and binding data presented herein, as well as the differences between ad libitum experiments and fasting experiments. The number of ARC neurons expressing AGRP mRNA and the level of AGRP mRNA expression is increased in fasted mice.36 Therefore in a fasting paradigm, it may be hypothesized that AGRP levels are increased, generating a system that is more sensitive to the functional antagonist effects of the micromolar mBD1 agonist. The level of AGRP in free feeding mice would be expected to be much lower, and therefore no significant effect may be observed. In such a paradigm, the higher affinity endogenous agonists may negate the effects of mBD1 and result in non-significant results. Considering the micromolar agonist activity of mBD1, the lack of significance for the ad libitum study may not be surprising. Administration of higher levels of mBD1 without residual TFA or converting mBD1 to a more potent agonist may help clarify these results.
Conclusions
While previous studes have not clearly elucidated the functional pharmacology of BD3 orthologs at the MCRs, data presented herein demonstrates that β-defensins act as full cAMP based agonists at micromolar concentrations. Lower concentrations (<10−6 M) result in no observable increase in cAMP levels, in agreement with previously published studies. The centrally expressed mBD1 was further investigated for feeding effects in mice. Central administration of mBD1 was shown to have an effect on fasted male mice, with trends of decreased food intake and body weight following the fast suggestive of potential melanocortin agonist activity. The in vitro cellular assays and in vivo functional data suggest that the β-defensins are micromolar full agonists at the MCRs. Further work examining additional β-defensins as well as determining the functionally signficant parts of BD1 and BD3 may lead to the development of novel melanocortin ligands and probes with unique pharmacological profiles due to their novel scaffolds as melanocortin ligands.
Experimental
Peptides were synthesized using standard Fmoc chemical techniques, the details of which can be found in the supporting information. Oxidative folding was achieved by dissolving the purified linear peptides in a redox buffer (1.0 M guanidine hydrochloride, 0.1 M Tris buffer, 1.0 mM reduced glutathione, 0.1m M oxidized glutathione, pH ~8) for 48 h at a peptide concentration of 0.1 mg/mL. The folded peptides were then purified by preparative RP-HPLC. Peptide purity (determined to be > 95%) was assessed by analytical RP-HPLC in two different solvent systems before functional characterization in the AlphaScreen bioassay.
Detailed methodology for the AlphaScreen bioassay (PerkinElmer), competitive displacement assay, and feeding studies can be found in the supporting information.
Supplementary Material
Acknowledgments
Funding Sources
This work has been supported in part by NIH Grants R01DK097838, RO1DK064250, R01DK091906, and R01DK108893 (C.H.-L.) and an NIH Postdoctoral Fellowship F32DK108402 (M.D.E.). The Haskell-Luevano laboratory is the recipient of a 2017 Wallin Neuroscience Discovery Fund Award through the University of Minnesota.
ABBREVIATIONS
- AGRP
agouti-related protein
- ASP
agouti-signaling protein
- MCR
melanocortin receptor
- BD
β-defensin
- MSH
melanocyte stimulating hormone
Footnotes
Supporting Information. Details about the peptide synthesis, in vitro pharmacological characterization, animal studies are available in the supporting information. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Allen BM. The results of extirpation of the anterior lobe of the hypophysis and of the thyroid of rana pipiens larvae. Science. 1916;44(1143):755–758. doi: 10.1126/science.44.1143.755. [DOI] [PubMed] [Google Scholar]
- 2.Smith PE. Experimental ablation of the hypophysis in the frog embryo. Science. 1916;44(1130):280–282. doi: 10.1126/science.44.1130.280. [DOI] [PubMed] [Google Scholar]
- 3.Huszar D, Lynch CA, FairchildHuntress V, Dunmore JH, Fang Q, Berkemeier LR, Gu W, Kesterson RA, Boston BA, Cone RD, Smith FJ, Campfield LA, Burn P, Lee F. Targeted disruption of the melanocortin-4 receptor results in obesity in mice. Cell. 1997;88(1):131–141. doi: 10.1016/s0092-8674(00)81865-6. [DOI] [PubMed] [Google Scholar]
- 4.Irani BG, Xiang ZM, Yarandi HN, Holder JR, Moore MC, Bauzo RM, Proneth B, Shaw AM, Millard WJ, Chambers JB, Benoit SC, Clegg DJ, Haskell-Luevano C. Implication of the melanocortin-3 receptor in the regulation of food intake. Eur J Pharmacol. 2011;660(1):80–87. doi: 10.1016/j.ejphar.2010.10.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Butler AA, Kesterson RA, Khong K, Cullen MJ, Pelleymounter MA, Dekoning J, Baetscher M, Cone RD. A unique metabolic syndrome causes obesity in the melanocortin-3 receptor-deficient mouse. Endocrinology. 2000;141(9):3518–3521. doi: 10.1210/endo.141.9.7791. [DOI] [PubMed] [Google Scholar]
- 6.Chen AS, Marsh DJ, Trumbauer ME, Frazier EG, Guan XM, Yu H, Rosenblum CI, Vongs A, Feng Y, Cao LH, Metzger JM, Strack AM, Camacho RE, Mellin TN, Nunes CN, Min W, Fisher J, Gopal-Truter S, MacIntyre DE, Chen HY, Van der Ploeg LHT. Inactivation of the mouse melanocortin-3 receptor results in increased fat mass and reduced lean body mass. Nat Genet. 2000;26(1):97–102. doi: 10.1038/79254. [DOI] [PubMed] [Google Scholar]
- 7.Ericson MD, Lensing CJ, Fleming KA, Schlasner KN, Doering SR, Haskell-Luevano C. Bench-top to clinical therapies: A review of melanocortin ligands from 1954 to 2016. Biochim Biophys Acta, Mol Basis Dis. 2017;1863(10):2414–2435. doi: 10.1016/j.bbadis.2017.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bitensky MW, Burstein SR. Effects of cyclic adenosine monophosphate and melanocyte-stimulating hormone on frog skin in vitro. Nature. 1965;208(5017):1282–1284. doi: 10.1038/2081282a0. [DOI] [PubMed] [Google Scholar]
- 9.Novales RR, Davis WJ. Melanin-dispersing effect of adenosine 3′,5′-monophosphate on amphibian melanophores. Endocrinology. 1967;81(2):283–290. doi: 10.1210/endo-81-2-283. [DOI] [PubMed] [Google Scholar]
- 10.Lu D, Willard D, Patel IR, Kadwell S, Overton L, Kost T, Luther M, Chen W, Woychik RP, Wilkison WO, Cone RD. Agouti protein is an antagonist of the melanocyte-stimulating-hormone receptor. Nature. 1994;371(6500):799–802. doi: 10.1038/371799a0. [DOI] [PubMed] [Google Scholar]
- 11.Little CC. The Inheritance of Coat Colour in Dogs. Comstock Publishing Associates; Ithaca, New York: 1957. pp. 1–194. [Google Scholar]
- 12.Kerns JA, Olivier M, Lust G, Barsh GS. Exclusion of melanocortin-1 receptor (MC1R) and agouti as candidates for dominant black in dogs. J Hered. 2003;94(1):75–79. [PubMed] [Google Scholar]
- 13.Kerns JA, Cargill EJ, Clark LA, Candille SI, Berryere TG, Olivier M, Lust G, Todhunter RJ, Schmutz SM, Murphy KE, Barsh GS. Linkage and segregation analysis of black and brindle coat color in domestic dogs. Genetics. 2007;176(3):1679–1689. doi: 10.1534/genetics.107.074237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Candille SI, Kaelin CB, Cattanach BM, Bin Y, Thompson DA, Nix MA, Kerns JA, Schmutz SM, Millhauser GL, Barsh GS. A β-defensin mutation causes black coat color in domestic dogs. Science. 2007;318(5855):1418–1423. doi: 10.1126/science.1147880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nix MA, Kaelin CB, Ta T, Weis A, Morton GJ, Barsh GS, Millhauser GL. Molecular and functional analysis of human β-defensin 3 action at melanocortin receptors. Chem Biol. 2013;20(6):784–795. doi: 10.1016/j.chembiol.2013.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Beaumont KA, Smit DJ, Liu YY, Chai E, Patel MP, Millhauser GL, Smith JJ, Alewood PF, Sturm RA. Melanocortin-1 receptor-mediated signalling pathways activated by NDP-MSH and HBD3 ligands. Pigm Cell Melanoma Res. 2012;25(3):370–374. doi: 10.1111/j.1755-148X.2012.00990.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Beaumont KA, Wong SS, Ainger SA, Liu YY, Patel MP, Millhauser GL, Smith JJ, Alewood PF, Leonard JH, Sturm RA. Melanocortin MC1 receptor in human genetics and model systems. Eur J Pharmacol. 2011;660(1):103–110. doi: 10.1016/j.ejphar.2010.11.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Swope VB, Jameson JA, McFarland KL, Supp DM, Miller WE, McGraw DW, Patel MA, Nix MA, Millhauser GL, Babcock GF, Abdel-Malek ZA. Defining MC1R regulation in human melanocytes by its agonist α-melanocortin and antagonists agouti signaling protein and β-defensin 3. J Invest Dermatol. 2012;132(9):2255–2262. doi: 10.1038/jid.2012.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schild HO. pAx and competitive drug antagonism. Br J Pharmacol Chemother. 1949;4(3):277–280. doi: 10.1111/j.1476-5381.1949.tb00548.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jarrett SG, Horrell EMW, Boulanger MC, D’Orazio JA. Defining the contribution of MC1R physiological ligands to ATR phosphorylation at Ser435, a predictor of DNA repair in melanocytes. J Invest Dermatol. 2015;135(12):3086–3095. doi: 10.1038/jid.2015.280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jarrett SG, Carter KM, Shelton BJ, D’Orazio JA. The melanocortin signaling cAMP axis accelerates repair and reduces mutagenesis of platinum-induced DNA damage. Sci Rep. 2017;7(1):11708. doi: 10.1038/s41598-017-12056-5. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 22.Nix MA, Kaelin CB, Palomino R, Miller JL, Barsh GS, Millhauser GL. Electrostatic similarity analysis of human β-defensin binding in the melanocortin system. Biophys J. 2015;109(9):1946–1958. doi: 10.1016/j.bpj.2015.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pazgier M, Hoover DM, Yang D, Lu W, Lubkowski J. Human β-defensins. Cell Mol Life Sci. 2006;63(11):1294–1313. doi: 10.1007/s00018-005-5540-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bauer F, Schweimer K, Kluver E, Conejo-Garcia JR, Forssmann WG, Rosch P, Adermann K, Sticht H. Structure determination of human and murine β-defensins reveals structural conservation in the absence of significant sequence similarity. Protein Sci. 2001;10(12):2470–2479. doi: 10.1110/ps.24401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hoover DM, Chertov O, Lubkowski J. The structure of human β-defensin-1 - New insights into structural properties of β-defensins. J Biol Chem. 2001;276(42):39021–39026. doi: 10.1074/jbc.M103830200. [DOI] [PubMed] [Google Scholar]
- 26.Hoover DM, Rajashankar KR, Blumenthal R, Puri A, Oppenheim JJ, Chertov O, Lubkowski J. The structure of human β-defensin-2 shows evidence of higher order oligomerization. J Biol Chem. 2000;275(42):32911–32918. doi: 10.1074/jbc.M006098200. [DOI] [PubMed] [Google Scholar]
- 27.Sawai MV, Jia HP, Liu L, Aseyev V, Wiencek JM, McCray PB, Jr, Ganz T, Kearney WR, Tack BF. The NMR structure of human β-defensin-2 reveals a novel α-helical segment. Biochemistry. 2001;40(13):3810–3816. doi: 10.1021/bi002519d. [DOI] [PubMed] [Google Scholar]
- 28.Schibli DJ, Hunter HN, Aseyev V, Starner TD, Wiencek JM, McCray PB, Tack BF, Vogel HJ. The solution structures of the human β-defensins lead to a better understanding of the potent bactericidal activity of HBD3 against. Staphylococcus aureus J Biol Chem. 2002;277(10):8279–8289. doi: 10.1074/jbc.M108830200. [DOI] [PubMed] [Google Scholar]
- 29.Kao CY, Chen Y, Zhao YH, Wu R. ORFeome-based search of airway epithelial cell-specific novel human β-defensin genes. Am J Respir Cell Mol Biol. 2003;29(1):71–80. doi: 10.1165/rcmb.2002-0205OC. [DOI] [PubMed] [Google Scholar]
- 30.Mountjoy KG, Mortrud MT, Low MJ, Simerly RB, Cone RD. Localization of the melanocortin-4 receptor (MC4-R) in neuroendocrine and autonomic control circuits in the brain. Mol Endocrinol. 1994;8(10):1298–1308. doi: 10.1210/mend.8.10.7854347. [DOI] [PubMed] [Google Scholar]
- 31.Roehrl J, Yang D, Oppenheim JJ, Hehlgans T. Identification and biological characterization of mouse β-defensin 14, the orthologue of human β-defensin 3. J Biol Chem. 2008;283(9):5414–5419. doi: 10.1074/jbc.M709103200. [DOI] [PubMed] [Google Scholar]
- 32.Joseph CG, Yao H, Scott JW, Sorensen NB, Marnane RN, Mountjoy KG, Haskell-Luevano C. γ2-Melanocyte stimulation hormone (γ2-MSH) truncation studies results in the cautionary note that γ2-MSH is not selective for the mouse MC3R over the mouse MC5R. Peptides. 2010;31(12):2304–2313. doi: 10.1016/j.peptides.2010.08.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lensing CJ, Freeman KT, Schnell SM, Adank DN, Speth RC, Haskell-Luevano C. An in vitro and in vivo investigation of bivalent ligands that display preferential binding and functional activity for different melanocortin receptor homodimers. J Med Chem. 2016;59(7):3112–3128. doi: 10.1021/acs.jmedchem.5b01894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fan W, Boston BA, Kesterson RA, Hruby VJ, Cone RD. Role of melanocortinergic neurons in feeding and the agouti obesity syndrome. Nature. 1997;385(6612):165–168. doi: 10.1038/385165a0. [DOI] [PubMed] [Google Scholar]
- 35.Cornish J, Callon KE, Lin CQX, Xiao CL, Mulvey TB, Cooper GJS, Reid IR. Trifluoroacetate, a contaminant in purified proteins, inhibits proliferation of osteoblasts and chondrocytes. Am J Physiol: Endocrinol Metab. 1999;277(5):E779–E783. doi: 10.1152/ajpendo.1999.277.5.E779. [DOI] [PubMed] [Google Scholar]
- 36.Hahn TM, Breininger JF, Baskin DG, Schwartz MW. Coexpression of AGRP and NPY in fasting-activated hypothalamic neurons. Nat Neurosci. 1998;1(4):271–272. doi: 10.1038/1082. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.