

Abstract
The arenavirus Lassa causes severe hemorrhagic fever and a significant disease burden in West Africa every year. The glycoprotein, GPC, is the sole antigen expressed on the viral surface and the critical target for antibody-mediated neutralization. Here we present the crystal structure of the trimeric, prefusion ectodomain of Lassa GP bound to a neutralizing antibody from a human survivor at 3.2-angstrom resolution. The antibody extensively anchors two monomers together at the base of the trimer, and biochemical analysis suggests that it neutralizes by inhibiting conformational changes required for entry. This work illuminates pH-driven conformational changes in both receptor-binding and fusion subunits of Lassa virus, illustrates the unique assembly of the arenavirus glycoprotein spike, and provides a much-needed template for vaccine design against these threats to global health.
Lassa virus (LASV) is the etiologic agent of Lassa fever, an often-fatal viral hemorrhagic fever that is endemic in West Africa, with an estimated 20 to 70% lethality (1, 2). The virus appears to have extended its geographic spread (3), and outbreaks in 2016 were accompanied by demonstrated human-to-human transmission in Africa and Germany (4). There is no approved Lassa fever vaccine, and the nucleoside analog ribavirin and supportive therapy are the only treatment options currently in use for LASV infection. Attempts to use convalescent plasma against LASV have met with variable results (5). LASV is a member of the arenavirus family, which includes more than 30 known pathogens that exist on all populated continents on Earth. The Old World family of arenaviruses contains LASV; lymphocytic choriomeningitis virus (LCMV), which causes febrile illness, neurological disease, and birth defects with a 2 to 5% seroprevalence in North America and Europe; and the 80% lethal hemorrhagic fever virus Lujo (LUJV), which emerged in Southern Africa in 2008 (6). Arenaviruses in the New World category include Machupo virus (MACV) and Junín virus (JUNV), the causative agents of Bolivian and Argentinian hemorrhagic fever, respectively, as well as numerous other agents such as Sabía and Guanarito.
As the sole antigen on the viral surface, the arenavirus glycoprotein complex (GPC) is the primary target of protective humoral immune responses (7, 8) and a focus for vaccine design efforts. The virion form of GPC is a trimer of heterodimers, each containing the receptor-binding subunit GP1 and the transmembrane, fusion-mediating subunit GP2 (9). GPC also encodes an unusual stable signal peptide (SSP) that has many critical functions and is retained in the virion as part of the complex. Specifically, it is required for proper processing of GPC into the GP1 and GP2 subunits and modulation of the pH of infectivity, and it may have additional chaperone functions during GPC maturation (10–17). The GPC precursor is trafficked from the endoplasmic reticulum to the Golgi, where it is heavily N-glycosylated and processed by cellular proteases [signal peptidase (SPase) SKI1/SP1] into its mature form, which comprises noncovalently linked GP1, GP2, and SSP (18). GPC must also interact with ERGIC-53 in the exocytic pathway in order to form infectious virions (19).
The GPC trimer on the resulting virion must engage several host receptors to mediate entry of target cells. GPC of LASV binds to a xylose–glucaronic acid sugar, called matriglycan (20); on α-dystroglycan (α-DG) (21–23); or to alternative receptors at the cell surface (24). LASV then enters the endocytic pathway, where it binds to lysosome-associated membrane protein 1 (LAMP1) before membrane fusion (9, 25, 26). The GPC of pathogenic New World arenaviruses binds to transferrin receptor 1 (27) as its cellular receptor.
Multiple structures of individual GP1 and GP2 subunits of various arenaviruses have been determined (28–33), along with a GP1-GP2 complex of LCMV (34). That structure, however, did not reveal the organization of GPC on the arenavirus surface that is relevant for antibody neutralization and vaccine design.
A study of more than 100 antibodies from human survivors of Lassa fever found that the majority of the neutralizing response to LASV targeted the quaternary assembly of the prefusion GPC trimer, rather than either subunit alone (7). Further, the majority of the neutralizing response was contained in a single competition group, termed GPC-B. Here, we present the 3.2-Å crystal structure of the prefusion GPC trimer of LASV, in complex with the human neutralizing antibody 37.7H, which is directed against the quaternary GPC-B epitope. This structure reveals the first look at the prefusion arenavirus GP trimer; suggests that conformational changes occur in the GP1 subunit, as well as the GP2 subunit upon exposure to low pH; and illuminates reasons why GPC must be enzymatically processed to oligomerize and bind one of its extracellular receptors. It also illuminates what appears to be the most vulnerable region on the LASV trimer targeted for antibody-mediated neutralization and suggests that such antibodies function by blocking conformational changes required for binding an intracellular receptor and for fusion.
Structure determination
Structure determination of an arenavirus GPC trimer has been previously hindered by metastability of the protein: the propensity of GP1 and GP2 to separate and for GP2 to spring into its postfusion, six-helix bundle conformation. Mindful of the success of cysteine-linkage strategies for HIV and Rous sarcoma virus (35–40), we genetically modified the LASV glycoprotein ectodomain by (i) making the point mutations R207C and G360C (41) to covalently link GP1 and GP2 together, (ii) introduction of a proline via an E329P mutation in the metastable region of HR1 of GP2, and (iii) by replacing the native S1P GP1-GP2 cleavage site with a furin site (RRLL to RRRR) to enable efficient processing of the GP in Drosophila S2 cells (fig. S1A). Among hundreds of modifications screened over 10 years, these three, in combination, provided stable, crystallizable protein. Size-exclusion chromatography coupled with multiangle light scattering (SEC-MALS) and SDS– polyacrylamide gel electrophoresis analysis of the resulting protein (termed GPCysR4) demonstrates that the GP elutes as a monomer and that the protein is efficiently processed into GP1 and GP2 subunits but that the two subunits remain associated (fig. S1, B and C). Further, enzyme-linked immunosorbent assay analysis with a panel of human antibodies demonstrates that GPCysR4 is recognized by neutralizing antibodies that require native association between the GP1 and GP2 subunits (7) and is not recognized by antibodies against post-fusion GP2 (fig. S1D). Together, these results suggest that GPCysR4 is in its native, prefusion state.
Monomeric GPCysR4 was incubated with excess Fab 37.7H and subjected to SEC-MALS analysis. SEC-MALS indicated the formation of trimeric GP-Fab complexes in addition to monomeric GP-Fab complexes (fig. S1B).
Crystals of both the monomeric and trimeric fractions of the GPCysR4-Fab 37.7H complex formed in space group P6122 and diffract to 3.2 Å with a trimer of GP bound to three Fabs in the asymmetric unit (table S1 and fig. S1E). Phases were determined with an iterative approach by using molecular replacement with a related Fab structure and the LCMV GP crystal structure (34).
Architecture of the trimer
In the crystal structure, the soluble LASV GPCysR4 trimer adopts a compact tripod shape that closely matches the tomographic reconstruction of the trimeric GPC spike from authentic Lassa virions (9) (Fig. 1, A and B). Notably, the arenavirus GP lacks a central three-helix fusion subunit core evident in other class I glycoproteins such as Ebola virus GP (42), HIV-1 Env (36, 39), and influenza hemagglutinin (HA) (43) (fig. S2). Instead, the 1775-Å2 surface area buried at the trimeric interface arises from interactions between both the GP1 and GP2 subunits between monomers, particularly α helices 1, 2, and 3; the C-terminal tail of GP1; and heptad repeat 1 (HR1) of GP2 (Fig. 1C and fig. S4). Our recent work demonstrated that mutation of either S138 or L143 (Lassa numbering) to arginine prevented rescue of recombinant LCMV virions (34). In LASV, these residues lie at the trimeric interface on α2 and contact the GP1 C terminus of the neighboring monomer (Fig. 1C and fig. S4). Hence, contacts observed to form the trimer interface in the crystal structure are essential for virus viability.
Fig. 1. Organization and glycosylation of the LASV trimer.
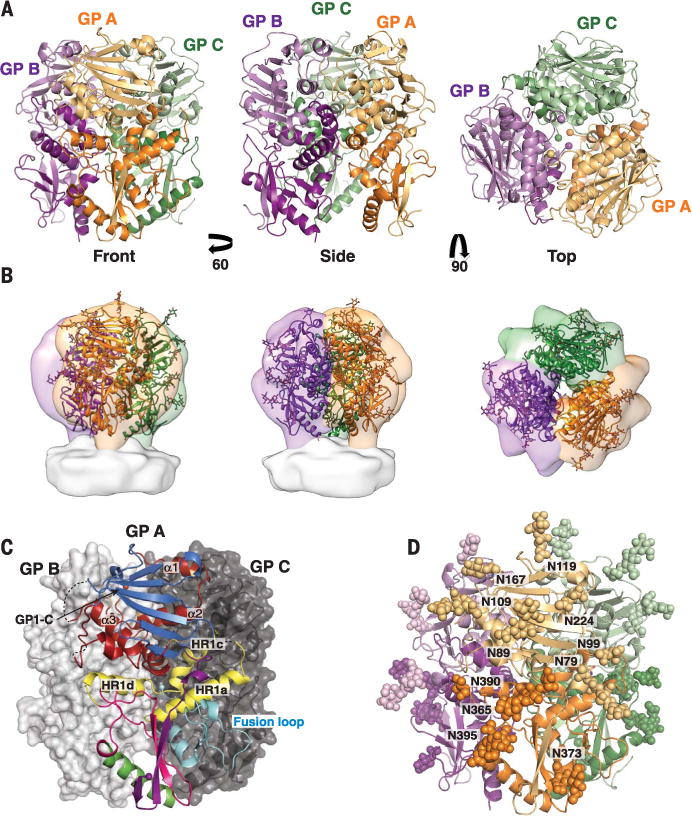
(A) Cartoon representation of the trimer from the front, side, and top. The GP1 subunit of each monomer is in a light shade and the GP2 subunit in a dark shade. In the top view, spheres indicate positions of the C terminus of GP1 and the N terminus of GP2 at the trimeric interface. (B) The crystal structure of the LASV GP trimer (cartoon) docked into the tomographic reconstruction of the LASV GPC spike from fixed virions (surface, EMD-3290), in the same orientations as shown in (A). (C) GP monomer A is shown in cartoon representation and is colored by domain as in fig. S3. Structural elements involved in trimerization are indicated. GP monomers B and C are shown as surfaces. (D) The glycans visible in the crystal structure are illustrated as atomic spheres, with those glycans attached to GP1 illustrated in light shades and those attached to GP2 illustrated in dark shades.
GPs of the arenavirus family are heavily glycosylated. Lassa GP has 11 potential N-linked glycosylation sites on each monomer, which together comprise ~25% of the total mass of the protein. In the trimeric structure presented here, we can now visualize the location of the majority of these glycans (fig. S5). The glycans shield the side and lower portions of the trimer, leaving only a few regions vulnerable to antibody binding—(i) the β-sheet face of GP1 where LAMP-1 and the New World arenavirus receptor transferrin receptor 1 (TfR1) bind (28, 30), (ii) three regions of GP2, the fusion peptide, fusion loop, and HR2, and (iii) the GP1- and GP2-involving trimeric interfaces (Fig. 1D and fig. S6A). Modeling of the larger, oligo-mannose glycans that might be present on the viral surface (44) reinforces the significant effect that glycosylation has on the neutralizing antibody evasion for the arenavirus family (8, 45) (fig. S6A). Further, analysis of glycanprotein interactions in this structure reveals the molecular basis for the specific and strong influences of glycosylation on viral fitness (46). For example, Bonhomme and co-workers found that recombinant LCMV bearing a mutation to remove the N79 glycan site (Lassa numbering) was unable to be rescued (46). We find here that this glycan packs against the fusion peptide (fig. S6B) and may shield and provide stability to this highly hydrophobic region.
Receptor binding in the context of the trimer
The overall structure of LASV GPCysR4 aligns well with the previously determined structure of the LCMV GP monomer with a 2-Å RMSD over the entire structure and a 1-Å RMSD over the core elements (β sheets and α helices of GP1 and all of GP2) (Fig. 2A). Differences between the two structures outside the core can be mapped to three main regions: (i) the flexible loops connecting the upper, β-sheet face and the lower α-helical face of GP1, (ii) the ~20 C-terminal residues of GP1, and (iii) the fusion peptide (Fig. 2A). In LCMV, the C terminus of GP1 lies in close apposition to the N terminus of GP2. In the LASV trimer, the C terminus is translated 30 Å, points away from GP2 into the apex of the trimer, and packs against α1 and α2 of the neighboring monomer (Figs. 1C and 2B). Enzymatic cleavage of GP1 from GP2 is required to achieve this separation of the termini and formation of this trimeric assembly. Last, the fusion peptide adopts a conformation in the LASV trimer different from that in the LCMV dimer, where it packs into a dimer interface (Fig. 2A) (34).
Fig. 2. Receptor binding in the context of the LASV GP trimer.
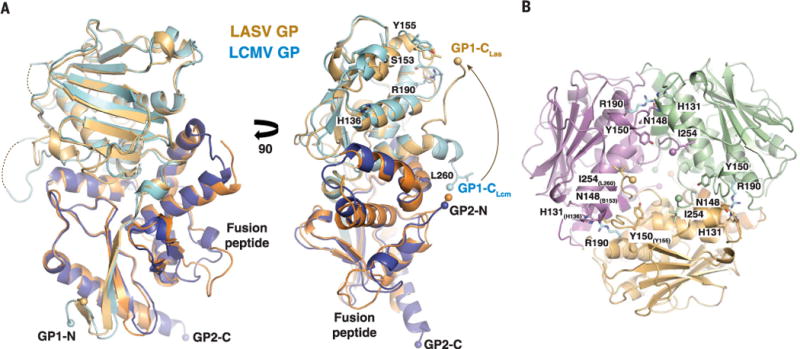
(A) The crystal structure of one monomer of the LASV GP trimer (GP1 and GP2 colored light and dark orange, respectively) is superimposed on one monomer of the LCMV GP dimer (GP1 and GP2 colored light and dark blue). Residues known to confer high-affinity binding to α-DG by LCMV are shown with LCMV numbering. (B) View of the LASV GP trimer from the top. The locations of the residues important for α-DG binding are shown and numbered according to LASV, with the equivalent residues in LCMV noted as subscript. Note: R190 in LCMV is required for α-DG binding. This residue is G186 in LASV but is modeled here as arginine (in cyan).
Lassa virus uses extracellular and intracellular receptors for efficient entry into host cells. On the cell surface, the virus engages matriglycan on α-DG and other receptors (21–24). Residues involved in binding to α-DG (34, 47–49) can now be mapped in the context of the trimer. Note that all of these residues are found at the trimeric interfaces (Fig. 2B), suggesting that LASV GP may need to be a trimer to interact with α-DG. Indeed, we previously observed that α-DG does not bind to the monomeric LCMV GP1 alone (34).
Once inside the cell, LASV binds to the lysosomal receptor LAMP1 (10–12). A histidine triad (H92, H93, and H230) has been identified as important for pH sensing and LAMP1 binding (30, 50) and is located on the β-sheet face of GP1 (Fig. 3A). Comparison of this structure of LASV GP1 in this prefusion assembly solved at pH 8 to the structure of LASV GP1 solved as an isolated subunit at pH 5 shows differences in the positions and interactions made by the three histidines. In the neutral-pH, prefusion complex, H92 packs against N89, another glycan identified by Bonhomme and coauthors (46) as essential for LCMV rescue (Fig. 3B and fig. S6B). In the low-pH structure of GP1 alone, H92 is in essentially the same location, but its interaction partner, N89, is shifted ~10 Å away. Similarly, in the neutral-pH, prefusion complex, H93 is located on the underside of a β sheet and makes a hydrogen bond with the main-chain oxygen of N90. In the low-pH structure of GP1 alone, H93 is instead oriented 180 degrees away from its location in the GP trimer and is unable to make the same hydrogen bond. Presumably, the local environment experienced by these residues at neutral and low pH may alter the structural stability of the entire GP1 subunit to release its hold on GP2.
Fig. 3. Conformational changes and receptor binding by LASV GP.
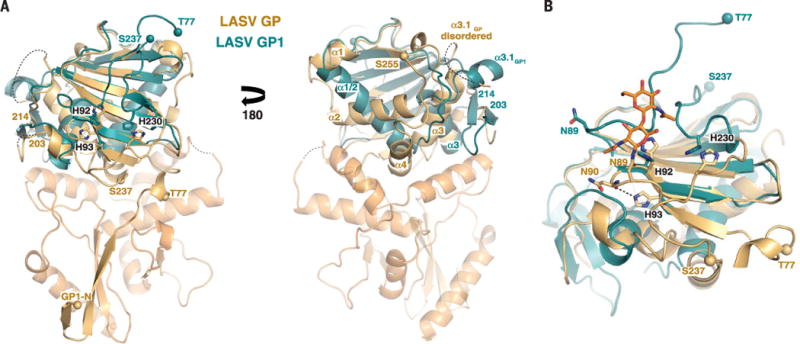
(A) Comparison of the crystal structure of the isolated GP1 subunit of LASV, at pH 5, to the GP1 subunit of the LASV GP trimer, at neutral pH. Positions of the histidine triad involved in LAMP1 binding are shown. Structural elements of LASV GP1 that vary between the low- and neutral-pH forms are indicated. Spheres indicate the N- and C-terminal residues visible in the low-pH GP1 structure and the location of the equivalent residues in the LASV GP trimer structure. (B) Detailed view of the histidine triad. Glycan N89 is shown as orange ball-and-stick. The hydrogen bond between H92 and the backbone oxygen of N90 is shown as a dotted line.
Other significant structural differences between the neutral-pH complex and the low-pH GP1 include elongation and rotation of α1 and α2 from two antiparallel helices in GP1 of the neutral-pH prefusion complex to a single helix in the low-pH GP1 monomer, as well as rearrangement of residues 200 to 214 from a helix-loop structure in the prefusion complex to a two-stranded β sheet in the low-pH monomer (Fig. 3A). Further, in the low-pH GP1, the visible termini are T77 and S237, and both are oriented upward, away from where GP2 would be. In the prefusion complex, residues 59 to 75 in the N terminus form an extended βstrand, which assembles with strand β11 of the GP2 T loop; residue S237 continues on to form α4 in the GP1 C terminus. Hence, the helices, loops, and C terminus of GP1 are flexible and adopt alternate positions when not bound by GP2.
Conformational changes in GP2 are well known during fusion. Based on these structural data, it is tempting to speculate that important conformational changes also occur in GP1 during low-pH–mediated LASV entry.
Old and New World arenaviruses share a similar organization of the GP1 core and are ~60% identical in GP2 by sequence, which allows modeling of other arenavirus receptor and antibody interactions in the context of a GPC trimer. Arenaviruses in the New World category bind TfR1 as their cellular receptor, and a structure of MACV GP1 in complex with a TfR1 monomer is available (28). MACV GP1 aligns to the prefusion LASV GP1 with a 2.5-Å RMSD for the core, and modeling illustrates the relative positions of the TfR1-binding sites relative to each other in the arenavirus trimer (fig. S7A). Neutralizing antibodies are known against this TfR1-binding site for JUNV (32), and modeling of the JUNV-Fab complex into the LASV trimer assembly suggests that three such Fab fragments could bind in the context of the JUNV GPC trimer (fig. S7B).
Structural definition of the anti-LASV 37.7H epitope
The antibody 37.7H against LASV was isolated from a Sierra Leonean survivor of Lassa fever. This antibody neutralizes viruses representing all four known lineages of LASV in vitro (7) and offers protection from lethal LASV challenge in guinea pig (51) and potentially nonhuman primates. The antibody simultaneously binds two GP monomers at the base of the GP trimer, where it engages four discontinuous regions of LASV GP, two in “site A” and two in “site B” (Fig. 4, A and B). Site A contains residues 62 to 63 of the N-terminal loop of GP1 and residues 387 to 408 in the T-loop and HR2 of GP2. Site B contains residues 269 to 275 of the fusion peptide and residues 324 to 325 of HR1 of GP2 (Fig. 4B). In total, 37.7H buries 1620 Å2 of GP: ~1000 Å2 of GP at site A and ~620 Å2 of GP at site B. Although nearly the entire surface buried on GP belongs to GP2, the presence of both GP1 and GP2 is critical for 37.7H recognition (7), likely because GP1 is required to maintain the proper prefusion conformation of GP2 for 37.7H binding.
Fig. 4. Structural definition of the 37.7H epitope.
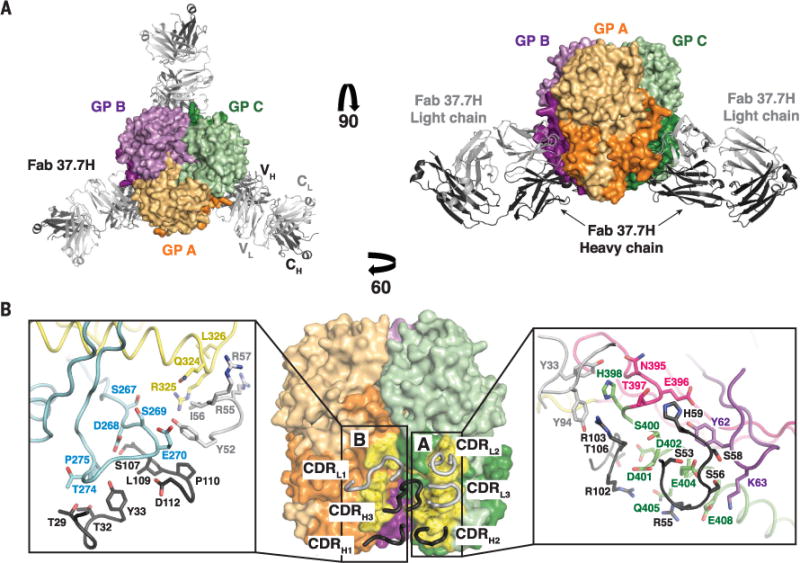
(A) The GP trimer bound to three 37.7H Fabs from the top (left) and side (right). Cartoon representations of the 37.7H Fab heavy and light chains are shown in dark and light gray, respectively. Each Fab binds to two GP monomers, shown as surface representations, near the base of the trimer. (B) Monomers A and C are shown as surface representations, with the heavy- and light-chain CDRs shown as dark and light gray tubes, respectively. The footprint of the antibody is colored yellow, with the binding site indicated. Side-chain interactions at the GP-37.7H interface are magnified in the inset boxes. GP elements are colored as indicated in fig. S3. Note that only selected residues are shown for clarity.
The antibody 37.7H also recognizes the GPC of LCMV (7) but does not recognize the GPC of the more distantly related Old World arenavirus LUJV nor the GPC of New World arenaviruses (7). Sequence comparison among these arenaviruses demonstrates nearly complete sequence conservation throughout the 37.7H epitope for all LASV lineages and LCMV (fig. S8). However, the sequences of LUJO, JUNV, and MACV GPCs are far more divergent, particularly in HR2 of GP2, which is heavily involved in binding to 37.7H.
The 37.7H antibody neutralizes by stabilizing the prefusion GP
The quaternary nature and the involvement of the fusion peptide in the 37.7H epitope suggest that this antibody neutralizes the virus by stabilizing GPC in the prefusion conformation, thereby preventing the conformational changes required for infection. To test this hypothesis, we analyzed the ability of LASV GP–pseudotyped recombinant vesicular stomatitis virus (rVSV-LASV GP) to mediate fusion with cell membranes. We first confirmed the ability of 37.7H to neutralize rVSV-LASV GP. Similar to previously published results, 37.7H effectively prevented cellular infection by rVSV-LASV GP, as did the antibody 12.1F, which binds to the upper, β-sheet face of LASV GP1 and is presumed to block cell attachment (7). In contrast, antibodies 13.4E, which binds a linear epitope in the T-loop, and 9.7A, which is a non-neutralizing GPC-B antibody, did not prevent viral infection (Fig. 5, A and B). Next, we determined whether 37.7H could prevent fusion of rVSV-LASV GP with cell membranes when exposed to low pH. Unlike the non-neutralizing antibodies 9.7A and 13.4E, which were not effective in preventing fusion, 37.7H reduced fusion by nearly 80% compared with rVSV-LASV GP alone (Fig. 5C). In contrast, the neutralizing antibody against GP1 (anti-GP1) 12.1F showed only a slight reduction in infectivity, suggesting that the effect of 37.7H was strictly due to disruption in fusogenicity of the GPC and not attachment to cells.
Fig. 5. Effect of antibodies on rVSV-LASV GP infection and fusion.
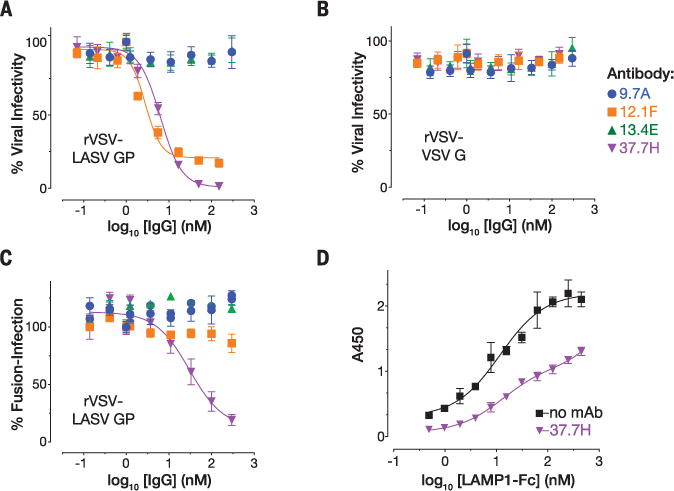
Antibody-mediated neutralization of (A) rVSV-LASV GP or (B) rVSV-VSV-G. The antibody 9.7A is non-neutralizing and in the same competition group as 37.7H (GPC-B); 13.4E binds to a linear epitope in the T-loop of GP2; 12.1F binds to the GP1 subunit of LASV. Error bars indicate the standard deviation of at least six (two biological replicates, each having three or more technical replicates). (C) Antibody-mediated inhibition of rVSV-LASV GP fusion at the cell surface. Error bars indicate the standard error of the mean of six (except 37.7H, where N = 9). (D) Fab 37.7H reduces binding of a LAMP1-Fc fusion protein to LASV GPCysR4. Error bars indicate the standard deviation of six and three technical replicates.
Before exposure of the GP2 fusion peptide and loop and subsequent fusion of the viral and host-cell membranes, LASV GP1 engages LAMP1. Engagement of this receptor is thought to require conformational changes in GP1 that are triggered by exposure to the low pH in the endosome. Tomography of LASV spikes in the presence of low pH and LAMP1 indeed shows an opening of the trimer compared with its neutral pH conformation (9). To determine whether 37.7H could prevent these conformational changes, we analyzed the ability of GPCysR4 to bind to a soluble LAMP1-Fc fusion alone and when bound to Fab 37.7H. In the absence of Fab 37.7H, GPCysR4 effectively bound to LAMP1 when exposed to low pH. In the presence of Fab 37.7H, however, interaction between GPCysR4 and LAMP1 was markedly reduced (Fig. 5D). Note that the footprint of 37.7H and the footprint of LAMP1 are separated by ~50 Å, and the angle adopted by the bound Fab fragments of 37.7H suggests that it is unlikely to sterically interfere with LAMP1 (fig. S9). Thus, there are likely to be conformational changes in GP1 required for LAMP1 binding that are prevented by this human survivor antibody.
Taken together, these results demonstrate that the probable mechanism of action for 37.7H and probably for other antibodies in its potent GPC-B competition group is stabilization of the prefusion GPC trimer and prevention of the conformational changes required for binding of LAMP1 and triggering of the GP2 fusion peptide and fusion loop in the endosome.
Discussion
This structure provides the first high-resolution image of the trimeric, prefusion arrangement of an arenavirus GP. The lack of a three-helix core, the tripod-like arrangement of HR2, and strong involvement of the GP1 subunit in trimerization are unique among members of the class I viral glycoproteins.
This structure illuminates the quaternary epitope of the most abundant class of LASV human neutralizing antibodies, maps the binding sites on the GPC trimer for α-DG and LAMP1, and indicates that this class of neutralizing antibodies inhibits conformational changes required for both LAMP1 and membrane fusion. Further, this structure provides the initial foundation for understanding the molecular mechanisms of neutralization of LASV. The availability of this structure may facilitate development of epitope-targeted vaccines or broadly reactive antibody-based therapeutics. Protective antibody epitopes like 37.7H may well exist for other disease-causing arenaviruses; this structure now provides the road-map to developing prefusion GPC constructs for these other threats to global health as well.
Supplementary Material
Acknowledgments
The authors acknowledge the Viral Hemorrhagic Fever Research Consortium, the Viral Hemorrhagic Fever Immunotherapeutic Consortium (VIC), and NIH grant 1U19AI109762-01 (E.O.S., K.C., J.E.R., L.M.B., and R.F.G); NIH grant R21 AI116112 (E.O.S.); NIH Training Program grant T32 GM007491 (L.M.K.); NIH contract HHSN272200900049C (J.E.R., E.O.S., R.F.G., and L.M.B.); an Investigators in Pathogenesis of Infectious Diseases award from the Burroughs Wellcome Fund (E.O.S.); and beamline 12-2 of the Stanford Synchrotron Radiation Lightsource (Palo Alto, CA), as well as beamlines 19-ID, 23-ID-B, and 23-ID-D of the Advanced Photon Source (Argonne, IL) for data collection. We are also grateful to S. Whelan, Harvard Medical School, for providing the LAMP1-Fc expression plasmid used in these studies. K.M.H. designed and cloned the constructs, produced recombinant protein, crystallized the complex, built and refined the model, performed the receptor-binding experiments, analyzed the data, and wrote the manuscript; M.A.Z. cloned the constructs and produced recombinant protein; L.M.K. and K.C. performed the VSV neutralization and fusion assays; J.E.R., L.M.B., M.L.H., M.M.R., and R.F.G generated and produced antibodies used throughout the studies; and E.O.S. analyzed the data and wrote the manuscript. All authors commented on the manuscript. Crystallographic structure factors and coordinates are deposited into the Protein Data Bank with accession number 5VK2. Use of the Stanford Synchrotron Radiation Lightsource (SSRL), SLAC National Accelerator Laboratory, is supported by the U.S. Department of Energy (DOE), Office of Science, Office of Basic Energy Sciences, under contract DE-AC02-76SF00515. The SSRL Structural Molecular Biology Program is supported by the DOE Office of Biological and Environmental Research and by the National Institute of General Medical Sciences (NIGMS), NIH (including P41GM103393). This research used resources of the Advanced Photon Source, a DOE Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under contract DE-AC02-06CH11357. The contents of this publication are solely the responsibility of the authors and do not necessarily represent the official views of NIGMS or NIH. Correspondence and requests for materials should be addressed to E.O.S.. This is manuscript 29458 from The Scripps Research Institute.
Footnotes
SUPPLEMENTARY MATERIALS
www.sciencemag.org/content/356/6341/923/suppl/DC1
Materials and Methods
References (52–69)
REFERENCES AND NOTES
- 1.Shaffer JG, et al. PLOS Negl Trop Dis. 2014;8:e2748. doi: 10.1371/journal.pntd.0002748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Asogun DA, et al. PLOS Negl Trop Dis. 2012;6:e1839. doi: 10.1371/journal.pntd.0001839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sogoba N, Feldmann H, Safronetz D. Zoonoses Public Health. 2012;59(suppl 2):43–47. doi: 10.1111/j.1863-2378.2012.01469.x. [DOI] [PubMed] [Google Scholar]
- 4.Wolff S, et al. Genome Announc. 2016;4:e00938–16. [Google Scholar]
- 5.Jahrling PB, Peters CJ. Infect Immun. 1984;44:528–533. doi: 10.1128/iai.44.2.528-533.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Briese T, et al. PLOS Pathog. 2009;5:e1000455. doi: 10.1371/journal.ppat.1000455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Robinson JE, et al. Nat Commun. 2016;7:11544. doi: 10.1038/ncomms11544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sommerstein R, et al. PLOS Pathog. 2015;11:e1005276. doi: 10.1371/journal.ppat.1005276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li S, et al. PLOS Pathog. 2016;12:e1005418. doi: 10.1371/journal.ppat.1005418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Eichler R, Lenz O, Strecker T, Garten W. FEBS Lett. 2003;538:203–206. doi: 10.1016/s0014-5793(03)00160-1. [DOI] [PubMed] [Google Scholar]
- 11.Froeschke M, Basler M, Groettrup M, Dobberstein B. J Biol Chem. 2003;278:41914–41920. doi: 10.1074/jbc.M302343200. [DOI] [PubMed] [Google Scholar]
- 12.Kunz S, Edelmann KH, de la Torre JC, Gorney R, Oldstone MB. Virology. 2003;314:168–178. doi: 10.1016/s0042-6822(03)00421-5. [DOI] [PubMed] [Google Scholar]
- 13.Saunders AA, et al. J Virol. 2007;81:5649–5657. doi: 10.1128/JVI.02759-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schrempf S, Froeschke M, Giroglou T, von Laer D, Dobberstein B. J Virol. 2007;81:12515–12524. doi: 10.1128/JVI.01481-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.York J, Nunberg JH. J Virol. 2006;80:7775–7780. doi: 10.1128/JVI.00642-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.York J, Nunberg JH. Virology. 2007;359:72–81. doi: 10.1016/j.virol.2006.08.048. [DOI] [PubMed] [Google Scholar]
- 17.York J, Romanowski V, Lu M, Nunberg JH. J Virol. 2004;78:10783–10792. doi: 10.1128/JVI.78.19.10783-10792.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Oppliger J, et al. J Virol. 2015;90:705–714. doi: 10.1128/JVI.01751-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Klaus JP, et al. Cell Host Microbe. 2013;14:522–534. doi: 10.1016/j.chom.2013.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yoshida-Moriguchi T, Campbell KP. Glycobiology. 2015;25:702–713. doi: 10.1093/glycob/cwv021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Oppliger J, Torriani G, Herrador A, Kunz S. J Virol. 2016;90:6412–6429. doi: 10.1128/JVI.00257-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kunz S, Borrow P, Oldstone MB. Curr Top Microbiol Immunol. 2002;262:111–137. doi: 10.1007/978-3-642-56029-3_5. [DOI] [PubMed] [Google Scholar]
- 23.Kunz S, et al. J Virol. 2005;79:14282–14296. doi: 10.1128/JVI.79.22.14282-14296.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shimojima M, Ströher U, Ebihara H, Feldmann H, Kawaoka Y. J Virol. 2012;86:2067–2078. doi: 10.1128/JVI.06451-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jae LT, et al. Science. 2014;344:1506–1510. doi: 10.1126/science.1252480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pasqual G, Rojek JM, Masin M, Chatton JY, Kunz S. PLOS Pathog. 2011;7:e1002232. doi: 10.1371/journal.ppat.1002232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Radoshitzky SR, et al. Nature. 2007;446:92–96. doi: 10.1038/nature05539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Abraham J, Corbett KD, Farzan M, Choe H, Harrison SC. Nat Struct Mol Biol. 2010;17:438–444. doi: 10.1038/nsmb.1772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bowden TA, et al. J Virol. 2009;83:8259–8265. doi: 10.1128/JVI.00761-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cohen-Dvashi H, Cohen N, Israeli H, Diskin R. J Virol. 2015;89:7584–7592. doi: 10.1128/JVI.00651-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Igonet S, et al. Proc Natl Acad Sci USA. 2011;108:19967–19972. doi: 10.1073/pnas.1108910108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mahmutovic S, et al. Cell Host Microbe. 2015;18:705–713. doi: 10.1016/j.chom.2015.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Parsy ML, Harlos K, Huiskonen JT, Bowden TA. J Virol. 2013;87:13070–13075. doi: 10.1128/JVI.02298-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hastie KM, et al. Nat Struct Mol Biol. 2016;23:513–521. doi: 10.1038/nsmb.3210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Joyce MG, et al. Nat Struct Mol Biol. 2016;23:811–820. doi: 10.1038/nsmb.3267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Julien JP, et al. Science. 2013;342:1477–1483. doi: 10.1126/science.1245625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Krarup A, et al. Nat Commun. 2015;6:8143. doi: 10.1038/ncomms9143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kwon YD, et al. Nat Struct Mol Biol. 2015;22:522–531. doi: 10.1038/nsmb.3051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lyumkis D, et al. Science. 2013;342:1484–1490. doi: 10.1126/science.1245627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sanders RW, et al. PLOS Pathog. 2013;9:e1003618. doi: 10.1371/journal.ppat.1003618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Single-letter abbreviations for the amino acid residues are as follows:; Ala A, Cys C, Asp D, Glu E, Phe F, Gly G, His H, Ile I, Lys K, Leu L, Met M, Asn N, Pro P, Gln Q, Arg R, Ser S, Thr T, Val V, Trp W, Tyr Y. The R207C mutation is Cys replacing Arg207 [Google Scholar]
- 42.Lee JE, et al. Nature. 2008;454:177–182. doi: 10.1038/nature07082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gamblin SJ, et al. Science. 2004;303:1838–1842. doi: 10.1126/science.1093155. [DOI] [PubMed] [Google Scholar]
- 44.Goncalves AR, et al. J Virol. 2013;87:11504–11515. doi: 10.1128/JVI.01893-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.McCormick JB, Mitchell SW, Kiley MP, Ruo S, Fisher-Hoch SP. J Med Virol. 1992;37:1–7. doi: 10.1002/jmv.1890370102. [DOI] [PubMed] [Google Scholar]
- 46.Bonhomme CJ, Knopp KA, Bederka LH, Angelini MM, Buchmeier MJ. PLOS ONE. 2013;8:e53273. doi: 10.1371/journal.pone.0053273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Smelt SC, et al. J Virol. 2001;75:448–457. doi: 10.1128/JVI.75.1.448-457.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sullivan BM, et al. Proc Natl Acad Sci USA. 2011;108:2969–2974. doi: 10.1073/pnas.1019304108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Teng MN, Borrow P, Oldstone MB, de la Torre JC. J Virol. 1996;70:8438–8443. doi: 10.1128/jvi.70.12.8438-8443.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cohen-Dvashi H, Israeli H, Shani O, Katz A, Diskin R. J Virol. 2016;90:10329–10338. doi: 10.1128/JVI.01624-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cross RW, et al. Antiviral Res. 2016;133:218–222. doi: 10.1016/j.antiviral.2016.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.