

Abstract
Gα13 mediates the ability of the morphogen retinoic acid to promote primitive endoderm formation from mouse P19 embryonal carcinoma stem cells, a process that includes the obligate activation of Jun N-terminal kinase. Expression of the constitutively activated (Q226L) GTPase-deficient form of Gα13 mimics retinoic acid and was used to investigate the signaling upstream of primitive endoderm formation. Jun N-terminal kinase 1 activity, MEK1,2, MKK4, and MEKK1 were constitutively activated in clones stably transfected to express Q226L Gα13. Dominant negative forms of MEKK1 and MEKK4 were expressed stably in the clones harboring Q226L Gα13. Expression of dominant negative versions of either MEKK1 or MEKK4 effectively blocks both the activation of Jun N-terminal kinase as well as the formation of primitive endoderm. Depletion of MEKK1, −2, or −4 by antisense oligodeoxynucleotides suppressed signaling from Q226L Gα13 to JNK1 and primitive endoderm formation. We demonstrate that the signal linkage map from Gα13 activation to primitive endoderm formation in these stem cells requires activation at three levels of the mitogen-activated protein kinase cascade: MEKK1, −2, or −4 for MAP kinase kinase kinase; MKK4 and/or MEK1 for MAP kinase kinase; and JNK1 for MAP kinase.
The mitogen-activated protein (MAP)1 kinase network has emerged as an essential signaling component in development (1, 2). In a variety of model systems, members of the MAP kinase family play central roles. The activation of Erk1,2 is an obligate element in the ability of the morphogen retinoic acid (RA) to promote primitive endoderm (PE) formation in mouse F9 teratocarcinoma stem cells (3) and in the adipogenic conversion of mouse 3T3-L1 fibroblasts to yield mature adipocytes (4). Jun N-terminal kinase (JNK) has been implicated in development in Drosophila (5), Caenorhabditis elegans (6), as well as in mammalian embryonal carcinoma P19 stem cells (7). The P19 stem cells are particularly attractive as a model system of early development, because three germ layers (endoderm, mesoderm, and ectoderm) can be derived from these mouse embryonal carcinoma cells in response to specific inducers and culture conditions (8). RA stimulates the loss of the embryonic marker SSEA-1 and positive staining by the TROMA antibody specific for the cytokeratin endo A, a hallmark for primitive endoderm.
The search to link RA action to activation of JNK revealed an important intermediary, the heterotrimeric G-protein G13. Expression of Gα13 rises sharply in P19 cells in response to RA as well as in early mouse embryos (8). In P19 cells, suppression of this rise in Gα13 by antisense oligodeoxynucleotides blocks the ability of RA to promote formation of PE (8). The concept that heterotrimeric G-proteins are critical elements of development has been bolstered by recent studies of developmental pathways in which the following α-subunits have been implicated: Gs, Go, Gq, Gi2, and G12/G13(2,9). One of the most powerful approaches to the study of G-protein α-subunits takes advantage of specific mutations that suppress the intrinsic GTPase activity, yielding an α-subunit that is constitutively activated. Herein we explored the upstream control of JNK activation in P19 clones that have been stably transfected to express the constitutively active (CA) Q226L mutant form of Gα13. Expression of Q226L Gα13 in P19 cells leads to robust activation of JNK and formation of PE in the absence of RA. At the level of MAP kinase kinase upstream of JNK, MEK1 and MKK4 (also known as JNKK1) are activated. At the level of MAP kinase kinase kinase, expression of Q226L Gα13 results in activation of MEKK1. Expression of the dominant negative form of either MEKK4 or MEKK1 blocks Q226L Gα13-induced differentiation. Treatment with oligodeoxynucleotides antisense to MEKK1, 2, or 4 blocks Q226L Gα13-induced JNK activation and PE formation. Thus, MEKK1, 2, and 4 act downstream of Gα13 and upstream of c-Jun N-terminal kinase in the pathway mediating differentiation of these stem cells to primitive endoderm.
EXPERIMENTAL PROCEDURES
Cell Culture and Differentiation
The P19 embryonal carcinoma cells were purchased from the American Type Culture Collection (Rockville, MD). Both the stable transfectants and the wild-type clones were cultured in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum (Hyclone, Logan, UT) in a humidified atmosphere of 6% CO2. P19 cells cultured as monolayers on tissue culture plates in the Dulbecco’s modified Eagle’s medium with 10% serum were induced to primitive endoderm by the addition of 10 nM all-trans-retinoic acid (Sigma) for 2–4 days.
Plasmids and Transfections
pCDNA3 plasmids harboring no insert (i.e. empty vector), the wild-type, constitutively active, and dominant negative (DN) mutant forms of MEKK1 and MEKK4 were provided generously by Dr. Gary L. Johnson (Dept. of Pharmacology, University of Colorado Health Sciences Center, Denver, CO). The pCMV5 plasmid harboring the Q226L mutant form of Gα13 was obtained from Dr. Alfred Gilman (Pharmacology, University of Texas Southwestern Medical School, Dallas, TX). The P19 cells were transfected with one or more plasmids using LipofectAMINE. Stably transfected P19 clones were selected in the presence of the neomycin analog G418 (400 μg/ml). The wild-type and mutant forms of the MEKK1 and MEKK4 had been epitope-tagged with the hemagglutinin antigen to follow expression of each independently of endogenous MEKKs.
Immunoblotting
Samples (10–50 μg of protein per lane) of total cell lysates were subjected to electrophoresis in SDS on 10% polyacrylamide gels. The resolved proteins were transferred electrophoretically to nitrocellulose blots. The blots were stained with primary antibodies and the immune complexes made visible using a second antibody coupled with horseradish peroxidase and developed using the ECL method. The antibodies were purchased from the following sources: JNK1, MEKK1, MEK2, and MEK7 antibodies from Santa Cruz Biotechnology (Santa Cruz, CA); MEK1 from Zymed Laboratories Inc. (South San Francisco, CA); MKK4 antibodies from Pharmogel Antibodies (San Diego, CA); phosphospecific antibodies to MEK1,2, MEK3,6, and MKK4 from Cell Signaling Technology (Beverly, MA).
Immunoprecipitation and Activity Assays of MEKKs and JNK
The immunoprecipitation reactions and solid-state assay of MEKKs and JNK were performed as detailed earlier (7), using the rGST-JNKK1 and rGST-c-Jun N-terminal fusion protein as substrates for the MEKKs and JNK activity assays, respectively. The vectors expressing the rGST-c-Jun and rGST-JNKK1 were provided generously by Dr. Roger Davis (HHMI, University of Massachusetts Medical Center, Worcester, MA.) In some cases, P19 cells were treated with oligodeoxynucleotides antisense to MEKK1, 2, 3, and 4 as described earlier (8).
Indirect Immunofluorescence Staining of SSEA-1 and TROMA
The staining of the embryonic antigen SSEA-1 with the monoclonal antibody MC-80 and the endoderm-specific marker antigen cytokeratin endo A by the monoclonal antibody TROMA was performed with antibodies purchased from the Developmental Studies Hybridoma Bank (University of Iowa, Iowa City, IO). The P19 cells were cultured, stained, and subjected to analysis by epifluorescence microscopy as described previously. Because the differentiated cells often grow from monolayers to whorls of cells with multiple layers, the indirect immunofluorescence and phase contrast images may not appear to be in focus. This artifact is unavoidable under these conditions of differentiation.
Data Analysis
For all of the experiments reported, the data are compiled from at least three independent replicate experiments performed on separate cultures on separate occasions with highly reproducible results. The indirect immunofluorescence and phase contrast images are of representative fields of interest.
RESULTS
The mouse P19 embryonal carcinoma cells were stably transfected with an expression vector pCDNA3 either lacking an insert (empty vector) or harboring the cDNA for the constitutively active mutant form of Gα13 with the Q226L substitution (Gα13QL). The Q226L mutation reduces the intrinsic α-subunit GTPase activity responsible for the hydrolysis of GTP bound in the activated state of Gα13. Analysis of the levels of Gα13 mRNA in the stably transfected cells was performed by reverse transcription-polymerase chain reaction amplification. Expression of Gα13 mRNA was increased dramatically in the clones harboring the Q226L Gα13-containing expression vector (Fig. 1A). Amplification of the marker mRNA for GAPDH, used to evaluate loading consistency, was equivalent among the clones expressing Q226L Gα13 or empty vector. Crude membranes prepared from P19 clones harboring empty vector or vector expressing the Q226L Gα13 subunit were subjected to SDS-polyacrylamide gel electrophoresis, blotting to nitrocellulose, and staining with antibody against Gα13 (Fig. 1B). Expression of Gα13 Q226L as deduced from the increase in immunoreactive Gα13 in blots of crude membranes of stably transfected clones was about 1.2- to 1.7-fold greater than the expression of the endogenous Gα13 (Fig. 1C).
FIG. 1. Expression of Gα13 mRNAs and immunoreactive Gα13 is increased in P19 embryonal carcinoma clones transfected with the cDNA of Q226L Gα13.
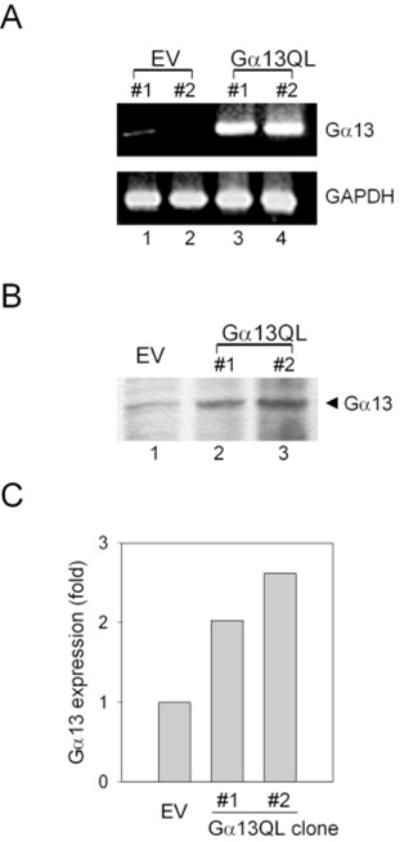
A, reverse transcriptase-PCR amplification of total cellular RNA isolated from stable transfectant clones harboring empty vector (pCDNA3, lanes 1 and 2) or pCDNA3 vector harboring the cDNA for the constitutively active mutant form of Gα13Q226L (lanes 3 and 4). Simultaneous amplification was performed with primers specific for GAPDH, demonstrating equivalent loading for lanes 1–4. B, crude membranes were prepared from P19 clones harboring either the empty vector (pCDNA3) or pCDNA3 expression vector harboring the cDNA encoding the constitutively active mutant form of Gα13 (Q226L). An aliquot (200 μg of protein/lane) of crude membranes was separated on a 10% SDS-polyacrylamide gel, transferred to nitrocellulose membranes, and probed with a primary antibody against Gα13. Immunoblots stained for Gα13 were made visible by use of the chemiluminescence reagent. The data shown are representative of at least three independent experiments performed on separate occasions. C, quantification of the amount of expression of Gα13 (wild-type plus Q226L) immunoreactivity in crude membranes of P19 clones.
Treating P19 cells with RA induces activation of c-Jun N-terminal kinase and PE formation (7). Expression of Q226L Gα13 mimics the activation of c-Jun N-terminal kinase by RA, as measured by the solid-state kinase assay (Fig. 2). Examined in a number of independent stably transfected clones, the increase in JNK activity ranged from 3.0- to 7.6-fold in clones expressing Q226L Gα13, and the average increase in kinase activity over basal from more than five clones was 4.2-fold. The amount of JNK1 expression itself was monitored in the Q226L Gα13-expressing clones and found to be the same as that observed for clones harboring the empty vector (Fig. 2) or for the wild-type cells (not shown).
FIG. 2. Activation of JNK1 in P19 embryonal carcinoma clones expressing the Q226L mutant version of Gα13.
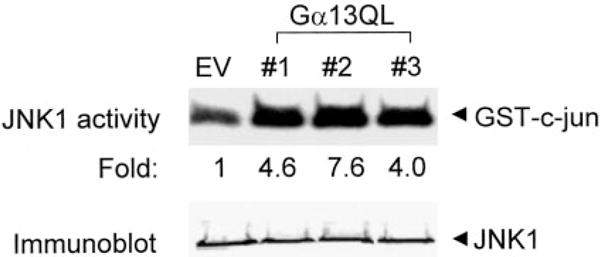
Cell lysates were prepared from cells harboring the empty expression vector or the expression vector harboring the cDNA to Q226L Gα13. JNK1 activity was measured using the solid-phase kinase assay that employs immunoprecipitation of JNK1 with a polyclonal antibody to JNK 1. Bacterially expressed GST-cJun was used as substrate. The phosphorylation reaction products were analyzed on 10% SDS-PAGE. The gel was stained, dried, and subjected to autoradiography. Replicates of the immunoprecipitants used for the solid-phase kinase assay were subjected to immunoblotting and stained with an antibody to JNK1. JNK1 protein was equivalent in all of the clones studied. The data shown are representative of at least three separate experiments.
The ability of Q226L Gα13 to provoke JNK activation focused the analysis on the upstream control of kinase activation, commencing with a measurement of the expression and activity of members of the MAP kinase kinase family (Fig. 3). Expression of MEK1–4, 6, and MKK7 in the clones overex-pressing Q226L Gα13 was probed by immunoblotting. Expression of MEK2 only was modestly greater (10–15%) in the clones expressing the Q226L Gα13 (Fig. 3A). The expression of MKK7 was similarly increased (not shown). The expression levels of MEK1, MEK3, and MKK4 were not changed (Fig. 3, A and B). MEK6 expression in F9 stem cells was not detected (not shown). Activation of MEKs occurs through phosphorylation of serine/threonine residues in MEK1 and 2 (Ser-217 and Ser-221), MEK3 (Ser-189 and Ser-207), and MKK4 (Ser-219 and Thr-261). Solid-state kinase assays of MEK kinase activities were precluded by a lack of suitable antibodies. As an alternative, we employed phosphospecific antibodies that recognize the phosphorylated, activated MEKs to evaluate the activities of MEKs. The activities of MEK1 and MEK2 were found to increase 80 and 40%, respectively, in the clones expressing Q226L Gα13, whereas the activity of MEK3 was unchanged (Fig. 3, A–D). Activity of MKK4, in contrast, was increased in the clones expressing Q226L Gα13. The activity of MKK4 (also known as JNKK1) was increased 60–70% in the P19 cells expressing the mutant Gα13. Thus, expression of constitutively active Q226L Gα13 provokes essentially no change in the expression of MAP kinase kinase family members. MEK1,4 and to a lesser extent MEK2 are constitutively activated similar to JNK1 in the clones expressing the Q226L mutant of Gα13. These levels of MEK activation are similar to reports in mouse macrophage RAW 264.7 cells (10), mouse BaF/3 cells (11), human airway smooth muscle cells (12), rat cerebral neuronal culture cells (13), simian COS-7 cells (14), and human embryonal kidney (HEK293) cells (15, 16).
FIG. 3. Activation of MEK1,2 and MKK4 in P19 embryonal carcinoma clones expressing the Q226L version of Gα13.
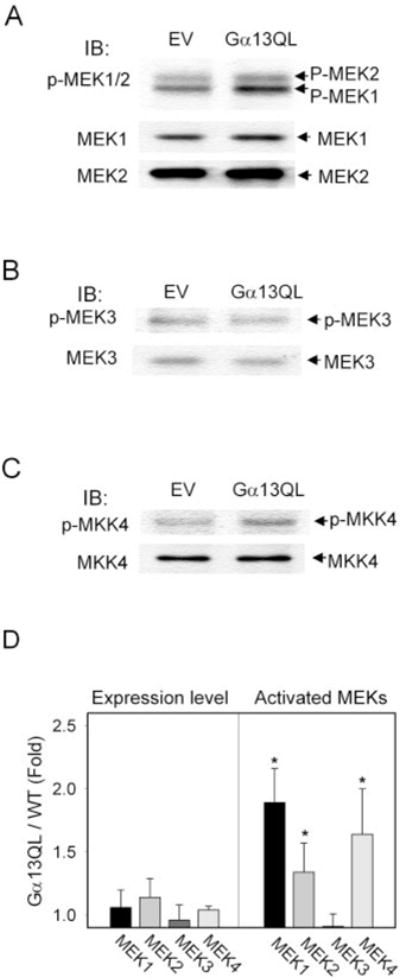
Crude whole-cell lysates were prepared from P19 clones harboring the empty vector (pCDNA3) or expression vector containing the cDNA for Q226L Gα13. Crude whole-cell lysates were subjected to electrophoresis and then immunoblotting. The immunoblots were probed with antibodies specific for MEK1 and MEK2 (A), MEK3 (B), and MKK4 (C) to measure protein expression as well as with antibodies specific for phospho-MEK1/2 (phospho-Ser-217 and -221), phospho-MEK3 (phospho-Ser-189 and -207), and MKK4 (phospho-Ser-219 and phospho-Thr-261). The combined results (mean ± S.E.) from three experiments for MEK expression and activation are shown (D). Asterisks denote values statistically significant from the control with p ≤ 0.05.
Using recombinant MEK1 as a substrate enabled the solid-phase assay of the activity of MEKK1. MEKK1 activity increased 1.5- to 2.0-fold in P19 cells expressing the Gα13 Q226L mutant (Fig. 4). Assays performed using a GST-tagged version of JNKK1 as a substrate reported a 1.9- to 2.2-fold increase in activation in response to expression of the constitutively active versions of either MEKK1 or MEKK4 (17). Thus, expression of Q226L Gα13 yields an activation of MEKK1 not unlike that measured in these same stem cells in response to expression of either constitutively activated MEKK1 or MEKK4. Although the constitutively activated Q226L Gα13 led to chronic activation of MEKK1, the expression of MEKK1 itself was unchanged. The lack of suitable antibodies precluded similar direct measurements of MEKK4 activity.
FIG. 4. Activation of MEKK1 in P19 embryonal carcinoma clones expressing the constitutively active Q226L mutant form of Gα13QL.

Crude cell lysates were prepared from P19 clones harboring empty vector (pCDNA3) or the expression vector containing the cDNA for Q226L Gα13. MEKK1 was immunoprecipitated from the crude whole-cell lysates using a polyclonal antibody to MEKK1. A solid-phase kinase assay was performed using the recombinant MEK 1 protein as substrate. The kinase assay products were separated on a10% SDS-PAGE, stained, dried and exposed to autoradiography. Immunoblots of samples from the whole-cell lysates were prepared and stained with a rabbit polyclonal antibody to MEKK1. The data displayed are representative of three replicate experiments.
To test further the roles of the MAP kinase kinase kinases MEKK1 and MEKK4 in the activation of JNK1 in P19 cells expressing Gα13 Q226L, clones were co-transfected with vectors expressing the dominant negative form of either MEKK1 (DN-MEKK1) or MEKK4 (DN-MEKK4). The expression of DN versions of these two prominent MAP kinase kinase kinases was evaluated for ability to influence the downstream signaling to JNK. Expression of Q226L Gα13 mutant increased JNK1 activity ~3-fold (Figs. 2 and 5), and co-expression of the dominant negative form of MEKK1 abolished this activation of JNK1. Not only did the DN-MEKK1 block the activation by Q226L Gα13, but it also suppressed the level of JNK1 below that observed in clones expressing the empty vector (Fig. 5). Expression of the dominant negative form of MEKK4 likewise abolished the ability of the Q226L Gα13 to activate JNK1 and suppressed the basal activity of JNK1 by 70%. These data demonstrate that activation of JNK by Gα13 Q226L mutant can be blocked by dominant negative forms of either MEKK1 or MEKK4.
FIG. 5. Expression of dominant negative versions of either DN-MEKK1 or DN-MEKK4 blocks the constitutive activation of JNK1 in clones expressing Q226L Gα13.
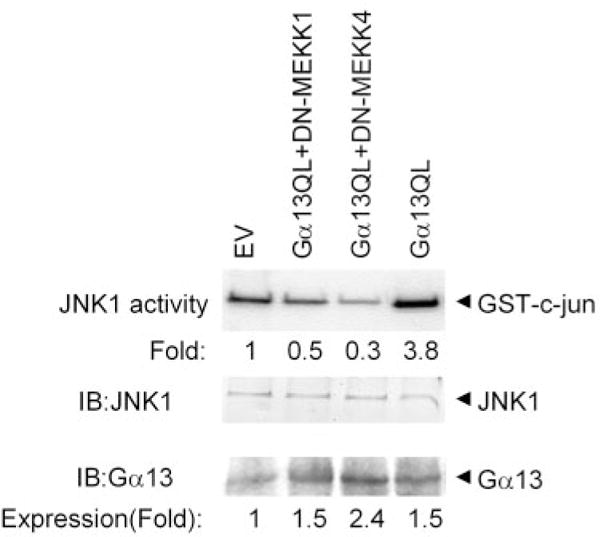
P19 cells were transfected stably with Q226L Gα13 alone as well as in combination with DN-MEKK1 or with DN-MEKK4. Clones were selected based on the expression of both Q226L Gα13 and dominant negative forms of the MEKKs. Cell lysates were prepared from the three types of clones and subjected to immunoprecipitation using an antibody specific for JNK1. The solid-phase kinase assay was performed using the JNK1 immunoprecipitates in combination with bacterially expressed GST-cJun as the substrate. The phosphorylated products were resolved on a 10% SDS-PAGE gel and subjected to autoradiography. Replicates of the immunoprecipitates were subjected to immunoblotting and stained with an antibody specific for JNK1. The level of JNK1 protein expression was equivalent among the various clones. The results displayed are representative of at least three separate experiments.
Would the expression of DN-MEKK1 or DN-MEKK4, which blocks the ability of Q226L Gα13 to activate JNK, also block the ability of Q226L Gα13 to promote PE formation? P19 cells were stably co-transfected with expression vectors harboring Q226L Gα13 in the absence or presence of those harboring either DN-MEKK1 or DN-MEKK4. The clones were selected, propagated, and stained with either antibodies for the embryonic marker SSEA-1 or with TROMA, a monoclonal antibody that recognizes cytokeratin endo A, a PE-specific marker protein in early development (Fig. 6). Expression of Q226L Gα13 provoked not only the activation of JNK but also the loss of expression of the embryonic marker SSEA-1. The clones expressed Q226L Gα13 stain positively by TROMA, in the absence of the morphogen retinoic acid, demonstrating the ability of the constitutively active Gα13 to promote PE formation without RA. Expression of the dominant negative mutant of DN-MEKK1 blocked PE formation as evidenced by the absence of TROMA staining and continued expression of the SSEA-1 embryonic antigen (Fig. 6). Expression of the dominant negative form of DN-MEKK4 in P19 clones co-expressing Gα13Q226L likewise resulted in the retention of the embryonal phenotype and suppression of PE formation established by the presence of SSEA-1 antigen and lack of positive staining by the TROMA antibody. Thus, the expression of dominant negative mutants of either MEKK1 or MEKK4 is capable of blocking the downstream signaling by Gα13 to activation of JNK and differentiation to primitive endoderm.
FIG. 6. Expression of dominant negative versions of either dominant negative MEKK1 or MEKK4 blocks the ability of Q226L Gα13 to induce formation of primitive endoderm.
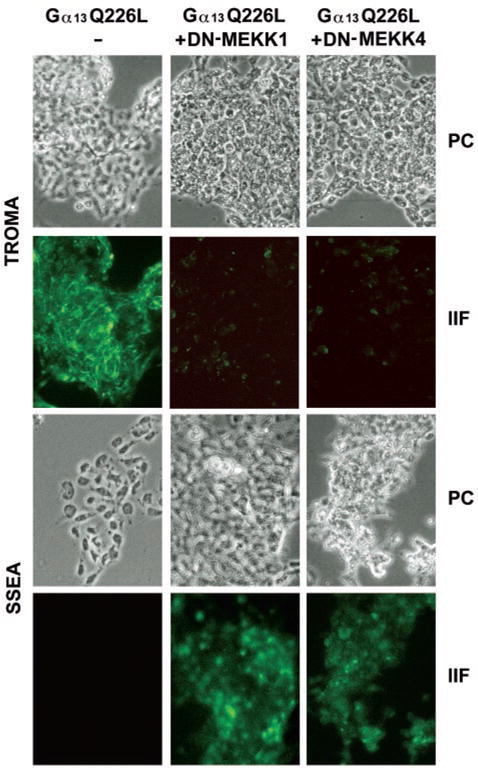
Cells were transfected with empty vector (pCDNA3), expression vector harboring Q226L Gα13 alone, or Q226L Gα13 in combination with the dominant negative form of either DN-MEKK1 or DN-MEKK4. Selected clones were grown in chamber slides, and the cells were processed for immunocytochemistry using either a monoclonal antibody to the embryonic marker SSEA-1 antigen or a monoclonal antibody to the cytokeratin endo A (TROMA 1) marker protein for primitive endoderm. Fluorescein isothiocyanate-conjugated secondary antibodies were employed in tandem with indirect epifluorescence to detect the immune complexes. The phase-contrast images (PC) and the indirect immunofluorescence images (IIF) are shown from an experiment representative of four independent experiments.
To further address the role of MEKK1–4 in JNK activation and PE formation, we examined the effects of oligodeoxynucleotides antisense to MEKK1, −2, −3, or −4 on P19 cells transiently transfected with an expression vector harboring Gα13Q226L (Fig. 7). Transient transfected P19 cells assayed at 48 h posttransfection displayed an increase in both JNK1 activity as well as in the PE marker stained with TROMA antibody (Fig. 7A), although less than that was obtained when the clones were allowed 96 h prior to sampling (which was not possible here). Cells transiently transfected with Gα13Q226L and treated with oligodeoxynucleotides (ODNs) antisense to MEKK1 (as-MEKK1) displayed a loss of JNK1 activity and failed to form PE. Treatment with ODNs antisense to MEKK4 by the same approach also resulted in a reduction in JNK1 activity and failure to form PE. The treatment with antisense ODNs suppressed the expression of MEKK1–4, as determined by immunoblotting (Fig. 7B). Treating cells with ODNs antisense to MEKK2 displayed a suppression of JNK1 activity and a reduction in PE formation, whereas treatment with ODNs antisense to MEKK3 failed to suppress the activation of JNK1 by Gα13Q226L. Despite the inability to block JNK activity, treatment with ODNs antisense to MEKK3 produced a reduction in PE formation in response to Gα13Q226L. These data suggest that MEKK2 is capable of transducing Gα13Q226L signals into activation of JNK1 and PE formation, whereas MEKK3 may be involved in Gα13Q226L-stimulated PE formation in a pathway independent of JNK1 activation.
FIG. 7. Treatment with oligodeoxynucleotides antisense to MEKK1, 2, or 4 blocks the ability of Q226L Gα13 to activate JNK1 activity.
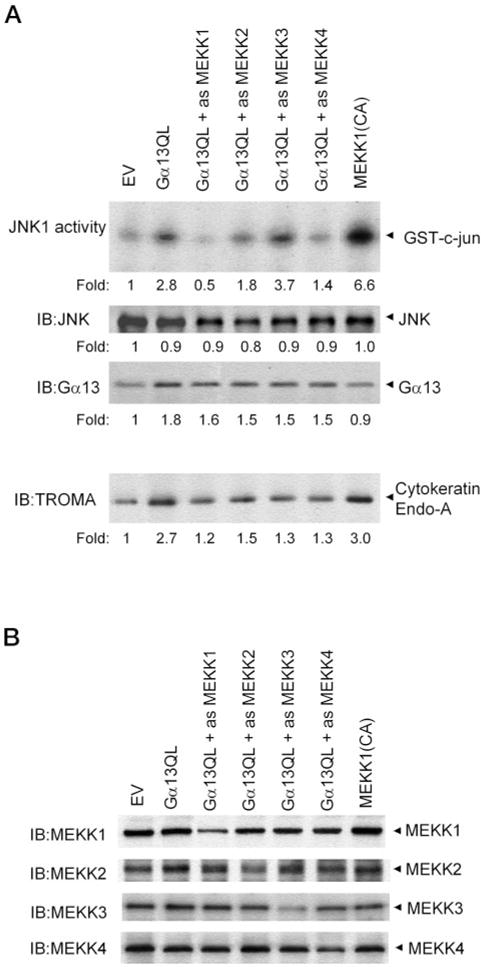
Cells were transiently transfected with empty vector (EV), the expression vector harboring Q226L Gα13 alone, or the empty vector and the Q226L Gα13 vector in combination with the oligodeoxynucleotides antisense to MEKK1–4. Cells were treated for 12 h prior with the antisense ODNs and transiently transfected with the empty vector (pCDNA3) or expression vector harboring Q226L Gα13 for the next 48 h in the presence of the antisense ODNs. The ODNs were replenished every 24 h for the entire 60-h period. At the end of the transient transfection, JNK1 activity, JNK expression, Gα13 immunoreactivity, and PE formation were assayed in the cells (A). The results shown are representative of at least three separate experiments. The amounts of MEKK1, MEKK2, MEKK3, and MEKK4 were determined by immunoblotting for all of the clones treated with antisense ODNs specifically targeting one of the MEKK family (B). A representative experiment is displayed. The suppression of MEKK forms in response to treatment with antisense ODNs under these conditions ranged from 70–85%.
We explored whether the activation of JNK1 was obligate for PE formation in response to Gα13Q226L, as it is in response to PE formation caused by the morphogen retinoic acid. Cells transiently transfected to express Gα13Q226L were transfected concurrently with the dominant negative form of JNK1, JNK1(DN), and PE formation was measured by the staining of the TROMA1 antigen marker protein (Fig. 8). Expression of the FLAG-tagged JNK1(DN) mutant and Gα13 was confirmed by immunoblotting. The expression of Gα13Q226L provoked expression of the PE marker, and this Ga13Q226L-stimulated PE formation was blocked by co-expression of the JNK1(DN). We examined also whether PE formation in response to expression of constitutively activated MEKK1 would be sensitive to the expression of the dominant negative JNK1. Expression of JNK1(DN) and the hemagglutinin antigen-tagged MEKK1(CA) were established by immunoblotting. Expression of the MEKK1(CA) stimulated PE formation, and this PE response to MEKK1(CA) was abolished when the cells were concurrently expressing the dominant negative version of JNK1. These data demonstrate that JNK1 activation is obligate for signaling from Gα13Q226L or MEKK1(CA) to PE formation.
FIG. 8. Co-expression of dominant negative JNK1 (JNK1(DN)) blocks PE formation stimulated by the expression of either Q226L Gα13 or constitutively active MEKK1 (MEKK1(CA)).
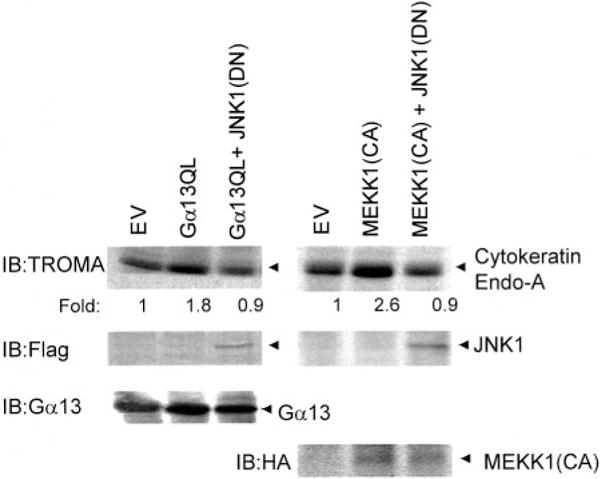
Cells were transiently transfected with empty vector (EV), the expression vector harboring Q226L Gα13 alone, or the empty vector and the Q226L Gα13 vector in combination with expression vector harboring FLAG-tagged JNK1(DN). Cells were transiently transfected for 48 h. At the end of the transient transfection, PE formation was assayed by immunoblotting of the TROMA antigen, which is a hallmark of PE. The expression of JNK1(DN) was assayed by immunoblotting with antibodies to the FLAG epitope. MEKK1 and MEKK1(CA) were hemagglutinin antigen-tagged, and expression was determined by immunoblotting. The expression of Gα13 Q226L was determined by immunoblotting with antibodies that stain endogenous and mutant Gα13 proteins alike. The results shown are representative of at least three separate experiments.
DISCUSSION
The present studies explore the signaling upstream of JNK in a mammalian model of development, the pluripotent mouse P19 embryonal stem cell. A role for JNK in signaling primitive endoderm formation in response to the morphogen RA was first identified in the P19 stem cells (7, 8) (Fig. 9). JNK has been implicated since in several models of development. In Drosophila dorsal closure, the basket gene product is an ortholog of JNK (18, 19). The MAP kinase network has been implicated in C. elegans embryogenesis, although the jnk-1 gene product most homologous to JNK appears to be obligate for coordinated locomotion (20). How the activity of JNK is activated in these systems remains to be fully elucidated. Expression of a constitutively active mutant form of Gα13 (Q226L) obviated the need for the morphogen and has been shown to promote robust PE formation (8). Expression of the Q226L Gα13 provokes persistent activation of JNK as well as formation of PE, enabling this group to detail signaling upstream of JNK in these mammalian stem cells.
FIG. 9. Q226L Gα13 regulates the MAP kinase regulatory network in promoting the formation of primitive endoderm.
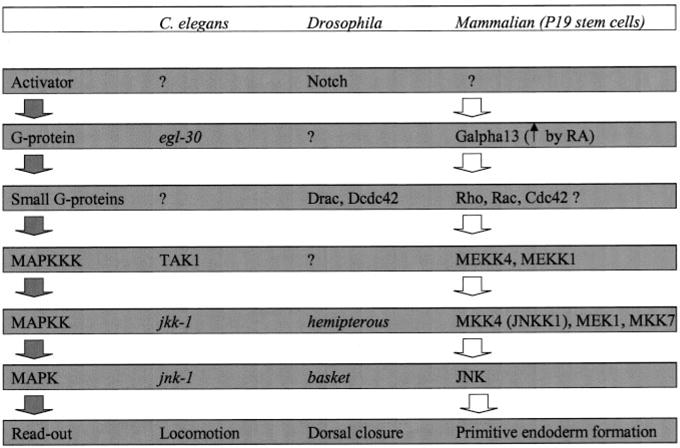
A comparison of emerging signaling patterns in C. elegans, Drosophila, and mammalian stem cells is shown. See the “Discussion” section.
At the level of MAP kinase kinase, it is clear that both MEK1 and MKK4 (which is also known as JNKK1) were persistently activated in clones expressing Q226L Gα13 (Fig. 9). MEK2 also displayed some activation, whereas MEK 3 activity was unaffected by the expression of constitutively active Q226L Gα13. Upstream signaling of JNK in C. elegans requires the jkk-1 gene (20), whereas in Drosophila the hemipterous gene product is obligate (21). There appears to be a high degree of conservation for signaling pathways operating in development at the levels of the MAP kinase kinase and MAP kinase.
Direct analysis at the level of MEKK reveals the activation of MEKK1 in clones expressing Q226L Gα13, establishing a clear linkage between Gα13 and MAP kinase kinase kinases. Although direct analysis of MEKK4 was not possible, we employed the expression of dominant negative mutants of both MEKK1 and MEKK4 in an attempt to test if either of the DN-MEKKs could block constitutive signaling from Q226L Gα13 to JNK and PE formation. The results of the analysis demonstrate that expression of DN-MEKK1 blocks the activation of JNK1 as well as the ability of the Q226L Gα13 to promote PE formation. Expression of DN-MEKK4 also blocks signaling from Gα13 to JNK and PE formation. These observations were extended by studies in which cells were treated with ODNs antisense to MEKK1–4 individually. Treatment with ODNs antisense to MEKK1, 2, or 4 suppressed both JNK1 activation and PE formation in response to expression of Gα13Q226L. Depletion of MEKK3, in contrast, blocked PE formation in response to expression of Gα13Q226L, but failed to suppress JNK1 activation. Thus, signaling via MEKK1, 2, or 4 from Gα13Q226L to PE formation requires JNK1, although MEKK3 appears to play a role in Gα13Q226L-stimulated PE formation independent of JNK1 activation.
These studies reveal a central role for JNK1, and MEKK1/4 in the downstream signaling from Q226L Gα13 to the control of JNK and PE formation (Fig. 7). Orthologs of MKK4 and MEKK1/4 likely play a central role in the control of development in C. elegans (6, 22), Drosophila (23), and other models of development (1). Absence of the JNK and JNKK homologs, basket and hemipterous, in Drosophila leads to an absence in embryonic dorsal closure (5, 23–26). For mammalian P19 stem cells, inhibition of either JNK1 or MEKK4 by dominant negative versions leads to a block in the formation of primitive endoderm in response to either RA (17) or to the expression of Q226L Gα13 (present work). Expression of dominant negative JNK1 was shown to block PE formation in response to either Gα13Q226L or constitutively active MEKK1. Expression of the constitutively active form of JNK, in contrast, failed to stimulate PE formation (7), suggesting that additional signals, perhaps the result of activation of MAP kinase kinase or MAP kinase kinase kinase, are obligate for the PE formation to be initiated. In Drosophila, small molecular weight G-proteins have been implicated upstream of MAP kinase kinase activation in dorsal closure (21, 27). The fact that Gα13 can signal to members of the Rho family of G-proteins (28) provides an important lead for further study on the linkages between Gα13 and MEKKs in early vertebrate development (29).
Footnotes
This work was supported by United States Public Health Service Grant DK30111 from NIDDK, National Institutes of Health.
The abbreviations used are: MAP, mitogen-activated protein; CA, constitutively active; DN, dominant negative, JNK, c-Jun N-terminal kinase; JNKK, JNK kinase; MEK, mitogen and extracellularly activated protein kinase; MEKK, MEK kinase; PE, primitive endoderm; ODN, oligodeoxynucleotides; GST, glutathione S-transferase; RA, retinoic acid; SSEA-1, stem cell surface embryonal antigen-1 marker.
References
- 1.Ip YT, Davis RJ. Curr Opin Cell Biol. 1998;10:205–219. doi: 10.1016/s0955-0674(98)80143-9. [DOI] [PubMed] [Google Scholar]
- 2.Morris AJ, Malbon CC. Physiol Rev. 1999;79:1373–1430. doi: 10.1152/physrev.1999.79.4.1373. [DOI] [PubMed] [Google Scholar]
- 3.Gao P, Malbon CC. J Biol Chem. 1996;271:9002–9008. doi: 10.1074/jbc.271.15.9002. [DOI] [PubMed] [Google Scholar]
- 4.Crespo P, Xu N, Simonds WF, Gutkind JS. Nature. 1994;369:418–420. doi: 10.1038/369418a0. [DOI] [PubMed] [Google Scholar]
- 5.Noselli S. Trends Gen. 1998;14:33–38. doi: 10.1016/S0168-9525(97)01320-6. [DOI] [PubMed] [Google Scholar]
- 6.Garrington TP, Johnson GL. Curr Opin Cell Biol. 1999;11:211–218. doi: 10.1016/s0955-0674(99)80028-3. [DOI] [PubMed] [Google Scholar]
- 7.Jho EH, Davis RJ, Malbon CC. J Biol Chem. 1997;272:24468–24474. doi: 10.1074/jbc.272.39.24468. [DOI] [PubMed] [Google Scholar]
- 8.Jho EH, Malbon CC. J Biol Chem. 1997;272:24461–24467. doi: 10.1074/jbc.272.39.24461. [DOI] [PubMed] [Google Scholar]
- 9.Malbon CC. Biochem Pharmacol. 1997;53:1–4. doi: 10.1016/s0006-2952(96)00662-4. [DOI] [PubMed] [Google Scholar]
- 10.Swantek JL, Cobb MH, Geppert TD. Mol Cell Biol. 1997;17:6274–6282. doi: 10.1128/mcb.17.11.6274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Craddock BL, Hobbs J, Edmead CE, Welham MJ. J Biol Chem. 2001;276:24274–24283. doi: 10.1074/jbc.M009098200. [DOI] [PubMed] [Google Scholar]
- 12.Lee JH, Johnson PR, Roth M, Hunt NH, Black JL. Am J Physiol Lung Cell Mol Physiol. 2001;280:L1019–L1029. doi: 10.1152/ajplung.2001.280.5.L1019. [DOI] [PubMed] [Google Scholar]
- 13.Chandler LJ, Sutton G, Dorairaj NR, Norwood D. J Biol Chem. 2001;276:2627–2636. doi: 10.1074/jbc.M003390200. [DOI] [PubMed] [Google Scholar]
- 14.Voyno-Yasenetskaya TA, Faure MP, Ahn NG, Bourne HR. J Biol Chem. 1996;271:21081–21087. doi: 10.1074/jbc.271.35.21081. [DOI] [PubMed] [Google Scholar]
- 15.Yamauchi J, Hirasawa A, Miyamoto Y, Itoh H, Tsujimoto G. Biochem Biophys Res Commun. 2001;284:1199–1203. doi: 10.1006/bbrc.2001.5103. [DOI] [PubMed] [Google Scholar]
- 16.Yamauchi J, Tsujimoto G, Kaziro Y, Itoh H. J Biol Chem. 2001;276:23362–23372. doi: 10.1074/jbc.M011752200. [DOI] [PubMed] [Google Scholar]
- 17.Kanungo J, Potapova I, Malbon CC, Wang H. J Biol Chem. 2000;275:24032–24039. doi: 10.1074/jbc.M002747200. [DOI] [PubMed] [Google Scholar]
- 18.Riesgo-Escovar JR, Jenni M, Fritz A, Hafen E. Genes Dev. 1996;10:2759–2768. doi: 10.1101/gad.10.21.2759. [DOI] [PubMed] [Google Scholar]
- 19.Sluss HK, Han Z, Barrett T, Davis RJ, Ip YT. Genes Dev. 1996;10:2745–2758. doi: 10.1101/gad.10.21.2745. [DOI] [PubMed] [Google Scholar]
- 20.Kawasaki M, Hisamoto N, Iino Y, Yamamoto M, Ninomiya-Tsuji J, Matsumoto K. EMBO J. 1999;18:3604–3615. doi: 10.1093/emboj/18.13.3604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Glise B, Noselli S. Genes Dev. 1997;11:1738–1747. doi: 10.1101/gad.11.13.1738. [DOI] [PubMed] [Google Scholar]
- 22.Ishitani T, Ninomiya-Tsuji J, Nagai S, Nishita M, Meneghini M, Barker N, Waterman M, Bowerman B, Clevers H, Shibuya H, Matsumoto Nature. 1999;399:798–802. doi: 10.1038/21674. [DOI] [PubMed] [Google Scholar]
- 23.Noselli S, Agnes F. Curr Opin Genet Dev. 1999;9:466–472. doi: 10.1016/S0959-437X(99)80071-9. [DOI] [PubMed] [Google Scholar]
- 24.Boutros M, Paricio N, Strutt DI, Mlodzik M. Cell. 1998;94:109–118. doi: 10.1016/s0092-8674(00)81226-x. [DOI] [PubMed] [Google Scholar]
- 25.Neo SY, Zhang Y, Yaw LP, Li P, Lin SC. Biochem Biophys Res Commun. 2000;272:144–150. doi: 10.1006/bbrc.2000.2751. [DOI] [PubMed] [Google Scholar]
- 26.Najlerahim A. Neurochem Res. 1993;18:291–295. doi: 10.1007/BF00969085. [DOI] [PubMed] [Google Scholar]
- 27.Magie CR, Meyer MR, Gorsuch MS, Parkhurst SM. Development. 1999;126:5353–5364. doi: 10.1242/dev.126.23.5353. [DOI] [PubMed] [Google Scholar]
- 28.Mao J, Yuan H, Xie W, Wu D. Proc Natl Acad Sci U S A. 1998;95:12973–12976. doi: 10.1073/pnas.95.22.12973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hart MJ, Jiang X, Kozasa T, Roscoe W, Singer WD, Gilman AG, Sternweis PC, Bollag G. Science. 1998;280:2112–2114. doi: 10.1126/science.280.5372.2112. [DOI] [PubMed] [Google Scholar]