

Abstract
Drugs do not act solely by canonical ligand-receptor binding interactions. Amphiphilic drugs partition into membranes thereby perturbing bulk lipid bilayer properties and possibly altering the function of membrane proteins. Distinguishing membrane perturbation from more direct protein-ligand interactions is an ongoing challenge in chemical biology. Herein, we present one strategy for doing so, using dimeric 6-bromo-2-mercaptotryptamine (BrMT) and synthetic analogs. BrMT is a chemically unstable marine snail toxin that has unique effects on voltage-gated K+ channel proteins, making it an attractive medicinal chemistry lead. BrMT is amphiphilic and perturbs lipid bilayers, raising the question of whether its action against K+ channels is merely a manifestation of membrane perturbation. To determine whether medicinal chemistry approaches to improve BrMT might be viable, we synthesized BrMT and 11 analogs and determined their activities in parallel assays measuring K+ channel activity and lipid bilayer properties. Structure-activity relationships were determined for modulation of the Kv1.4 channel, bilayer partitioning, and bilayer perturbation. Neither membrane partitioning, nor bilayer perturbation, correlate with K+ channel modulation. We conclude that BrMT’s membrane interactions are not critical for its inhibition of Kv1.4 activation. Further, we found that alkyl or ether linkages can replace the chemically labile disulfide bond in the BrMT pharmacophore, and we identified additional regions of the scaffold that are amenable to chemical modification. Our work demonstrates a strategy for determining if drugs act by specific interactions or bilayer-dependent mechanisms, and chemically stable modulators of Kv1 channels are reported.
INTRODUCTION
Biological membranes are composites of lipid bilayers and embedded proteins. It has long been known that membrane protein function is sensitive to the composition of the host bilayer.(1-4) Commonly, drugs that modulate membrane proteins are presumed to target proteins, while in fact many act by changing the bulk properties of the host bilayer, thereby altering membrane protein conformational equilibria.(5-8) Modulators that act by bilayer perturbation promiscuously modulate a broad spectrum of unrelated membrane proteins.(6, 7, 9-15) When interpreting the mechanisms underlying the physiological actions of a drug, it thus becomes crucial to determine whether the action of an amphiphilic modulator may involve bulk bilayer perturbation, in addition to more specific interactions.
A prominent example of the importance of understanding drug mechanism involves capsaicin, a natural product of chili peppers that stimulates mammalian peripheral neurons to evoke a sensation of burning heat. Capsaicin perturbs bilayers and modulates a wide variety of membrane proteins, including Na+, K+, and TRP channels. Capsaicin modulates voltage gated Na+ and K+ channels via lipid bilayer perturbation,(5, 7) but capsaicin also has a specific receptor site on TRPV1.(16) Medicinal chemistry approaches have been successful in generating analogs of capsaicin that are selective TRPV1 inhibitors.(17) Similar efforts to selectively modulate Na+ or K+ channels with capsaicin analogs would be foolhardy, however, because modulation of membrane proteins via bilayer perturbation is fundamentally promiscuous. Thus, determining if lipid bilayer perturbation underlies modulation of a target is critical for the prediction of undesired effects on other membrane proteins.
Because lipophilic and amphiphilic drugs, by their chemical nature, partition into membranes and perturb the function of transmembrane proteins, it is a significant challenge to determine whether bilayer perturbation is the relevant mechanism underlying modulation of any particular target protein. To identify whether drugs operate by a bilayer mechanism, we previously developed a method of testing modulators for promiscuous activity against multiple unrelated classes of membrane proteins.(7) Although this method is effective, it requires significant resources and expertise with many membrane protein preparations. Herein we report a greatly simplified strategy for using structure-activity relationships (SARs) of a modulator against a single target of interest, in combination with synthetic membrane assays, to dissect the effects of bulk bilayer perturbation from those of direct protein binding.
The medicinal chemistry target in this study is the natural product ion channel modulator dimeric 6-bromo-2-mercaptotryptamine (BrMT, 1a). A component of the defensive mucus of the marine snail Calliostoma canaliculatum, it inhibits voltage-gated K+ channels of the Kv1 and Kv4 subfamilies.(18) BrMT is an allosteric modulator that inhibits channels by slowing the voltage activation steps that precede pore opening, without blocking the central channel pore.(19, 20) Allosteric modulators of Kv channels are valuable not only as research tools but also potentially as therapeutics.(21, 22) BrMT itself has limited utility because it contains a chemically labile disulfide bond that is degraded by light and reducing conditions.(19) BrMT is thus an attractive target for medicinal chemistry efforts to improve its stability.
Several observations suggest that the activity of BrMT against Kv channels may be affected by non-specific membrane partitioning. First, high concentrations of BrMT applied to outside-out membrane patches disrupt the patch-clamp seal.(19) Second, a series of chimeras between the BrMT-sensitive Shaker Kv channel and the insensitive Kv2.1 channel suggest that the region imparting sensitivity is in the S1, S2, and/or S3 transmembrane regions of sensitive channels.(23) Third, the wash-in and wash-out kinetics of BrMT are multiphasic, suggesting slow accumulation of BrMT in the cell membrane during prolonged exposures.(23, 24) Together, these effects are consistent with BrMT partitioning in and out of cell membranes and acting through the membrane to alter channel function. Similar to many other amphiphilic molecules that act by bilayer perturbation, the biological effect of BrMT, with its two aminoethyl groups, depends on the side of the membrane to which it is applied.(25-28) BrMT slows Kv channel voltage activation only when applied from the extracellular side of the membrane,(19) suggesting that its two positive charges may prevent it from crossing the membrane entirely. Certain Kv modulator peptides from animal venoms partition into, but do not cross, the outer leaflet of the plasma membrane bilayer. Many of these peptides bind to the transmembrane voltage sensor domains of the channels.(29-31) However, other closely related venom peptides modulate ion channels via bilayer perturbation.(32) It remains unclear whether BrMT modulates K+ channels by direct channel binding, or in an indirect manner, by perturbing the bilayer, or a combination of both.(33) To elucidate the mode of action of BrMT and potentially improve its properties as a lead compound for future mechanistic or therapeutic studies, we synthesized a series of analogs, including several with stable disulfide replacements. The resulting SARs were assessed separately in membrane partitioning, perturbation, and ion channel assays to test whether specific or non-specific interactions drive K+ channel activities with these bis-indole compounds.
MATERIALS AND METHODS
The Supporting Information contains detailed descriptions of the synthesis of all BrMT analogs, cell culture, electrophysiology, gramicidin-based fluorescence quench assay, and isothermal titration calorimetry.
RESULTS
Synthesis of novel BrMT analogs
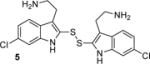
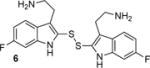
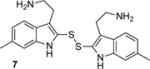
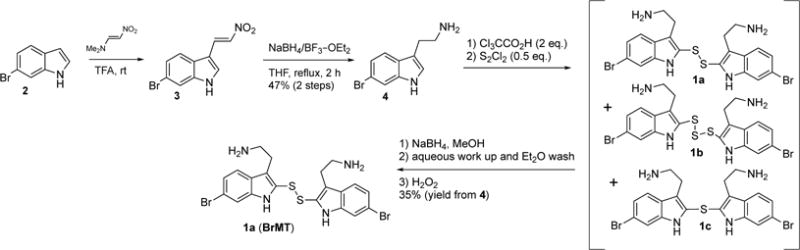
To determine the minimal structural features required for modulation of Kv channels with BrMT (1a), we designed a flexible synthesis that would enable facile modification of the tryptamine scaffold as well as the disulfide linker (Scheme 1). Our synthetic route is similar to that reported by Gallin and Hall.(34) 6-Bromotryptamine (4) was prepared from 6-bromoindole according to the sequence reported by Davidson.(35) Protonation of 4 with trichloroacetic acid, followed by reaction with freshly distilled S2Cl2,(36, 37) yielded a mixture of mono-, di-, and trisulfides 1a–c that was characterized by LC-MS. Using a protocol reported by Showalter for the preparation of bisindole diselenides as tyrosine kinase inhibitors,(38) we increased the yield of the desired disulfide product 1a by treating the mixture with sodium borohydride to reduce the di- and trisulfides. Extraction of the non-polar monosulfide 1c with ether from the basic aqueous solution of the resulting indole-2-thiolate, followed by oxidation of the thiolate with hydrogen peroxide gave the disulfide 1a, which was purified by semi-preparative HPLC and treated with HCl in dioxane to yield the bis-hydrochloride salt. Five different tryptamines were prepared via variations of literature protocols (see Supporting Information for details), and these were transformed into the analogous bistryptamine-disulfides 5–8 (Table 1) according to the sequence of reactions in Scheme 1.
Table 1.
Compilation of properties of BrMT derivatives
| Structure | IC50 (μM)a | Hill coeff.a | ClogPb | KPW→Lc | gA2 (μM)d |
|---|---|---|---|---|---|
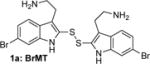
|
1.1 ± 0.1 N = 10 |
1.7 ± 0.3 N = 10 |
5.32 | 560 ± 5 N = 3 |
40 ± 5 N = 3 |
|
0.8 ± 0.2 N = 10 |
1.4 ± 0.3 N = 10 |
4.99 | 1000 ± 200 N = 5 |
13 ± 1 N = 3 |
|
26 ± 9 N = 9 |
1.1 ± 0.5 N = 9 |
4.06 | 800 ± 200 N = 5 |
28 ± 1 N = 3 |
|
2.1 ± 0.2 N = 21 |
1.8 ± 0.2 N = 21 |
4.81 | 450 ± 80 N = 4 |
12 ± 1 N = 3 |
|
2.1 ± 0.2 N = 7 |
1.3 ± 0.2 N = 7 |
5.32 | 1900 ± 600 N = 4 |
inactivee (50 μΜ) N = 3 |
|
|
inactivee (100 μΜ) N = 8 |
– | 5.15 | 600 ± 200 N = 4 |
inactivee (50 μΜ) N = 3 |
|
2.7 ± 0.3 N = 23 |
1.9 ± 0.3 N = 23 |
3.98 | 3100 ± 700 N = 3 |
2.9 ± 0.2 N = 3 |
|
70 ± 30 N = 10 |
0.7 ± 0.2 N = 10 |
3.62 | 3900 ± 500 N = 4 |
1.9 ± 0.1 N = 3 |
|
2.3 ± 0.3 N = 6 |
1.8 ± 0.2 N = 6 |
4.72 | 3300 ± 800 N = 3 |
12.8 ± 0.7 N = 3 |
|
35 ± 7 N = 8 |
1.3 ± 0.3 N = 8 |
5.77 | 670 ± 90 N = 4 |
5.1 ± 0.3 N = 3 |
|
|
inactivee (25 μΜ) N = 6 |
– | 4.82 | 1400 ± 200 N = 4 |
inactivee (50 μΜ) N = 3 |
|
inactivee (25 μΜ) N = 8 |
– | 4.21 | 3500 ± 1100 N = 3 |
32 ± 5 N = 3 |
Calculated as best fit for inhibition of Kv1.4 ± standard deviation of parameter fit.
Calculated using ChemAxon MarvinSketch v. 17.4.3.0.
KPW→L is the equilibrium constant for partitioning from water to lipid. Calculated as the geometric mean ± positive standard error.
Gramicidin A (gA) channel activity, as measured by the rate of quenching of intravesicular ANTS fluorescence by Tl+. gA2 fits shown in Supp. Figure S2.
Indicates IC50 or gA2 was not measured (highest concentration tested in parentheses).
The relative instability of bistryptamine-disulfides, and their potential for disulfide exchange reactions in vivo (39), inspired us to explore the use of alternative linkers between the indole moieties. Several two-carbon indole linkers have been reported in the literature,(40, 41) but we expected that these would be too rigid and/or short to be effective disulfide replacements. Accordingly, we pursued a convergent synthesis of symmetrical bis-indoles by reacting suitable aniline derivatives with bis-alkynes. Dipropargyl ether was selected as our first choice, as it could provide a three-atom, ether-based linker between the two indole rings that would provide a distance between indole moieties comparable to that of the disulfide linker.
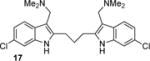
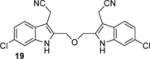
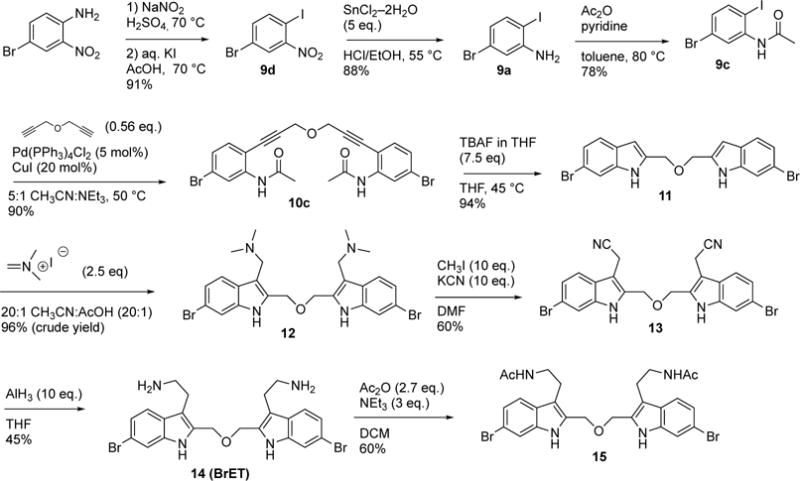
The optimized synthesis of the ether-linked compounds is given in Scheme 2. Commercially available 4-bromo-2-nitroaniline was subjected to a Sandmeyer reaction(42) to yield aryliodide 9d in 94% yield. The nitro group of 9d was reduced to aniline 9a using SnCl2 and conc. HCl, followed by N-acylation with acetic anhydride to provide the amide 9c. Amide 9c was subjected to the double Sonogashira cross coupling conditions (Table S1, entry 7) to yield the bis-alkyne 10c in 90% yield. Treatment of 10c with aqueous TBAF, followed by amide hydrolysis gave 11 (see Supporting Information for details). Alkylation of the bis-indole 11 with Eschenmosher’s salt (43) gave the bis-gramine 12 in excellent yield. Owing to the potential instability of 12, it was converted without purification to the bis-nitrile 13 upon treatment with excess iodomethane and sodium cyanide in DMF. Reduction of the nitrile groups in 13 with LiAlH4 was problematic and led to partial reduction of the aryl bromide, but reduction of 13 with alane(44) proceeded smoothly to give the bis-amine 14 (termed BrET) in 45% yield. Acetylation of 14 with acetic anhydride furnished the bis-amide 15 in 60% yield. The related BrMT analogs 16–19 (Table 1) were prepared by a sequence of reactions similar to those depicted in Scheme 2. Initial attempts to reduce 19 to the corresponding diamine were unsuccessful in generating compound of acceptable purity.
BrMT derivatization alters potency of ion channel modulation
We assessed the modulatory effects of BrMT analogs on currents through voltage-gated potassium channels. To measure the dose–response relations of many compounds in parallel, we developed an automated whole-cell voltage clamp assay against a BrMT-sensitive ion channel. BrMT inhibits Kv1 (Shaker-type) channels from invertebrates and vertebrates.(18) Among members of the Kv1 family, Kv1.4 was chosen because it is a potential target for chronic pain, and lacks high-affinity modulators.(45-48) Moreover, Kv1.4 traffics efficiently and reliably to the plasma membrane in mammalian cell lines, making it optimal for automated electrophysiology.(49, 50) We therefore created a CHO-K1 cell line with tetracycline-inducible expression of Kv1.4 to provide a scalable cell culture with consistent current levels amenable to automated patch clamp methods.
Amphiphilic drugs often have variable potencies in different experimental preparations. This preparation-dependent variability may stem from membrane partitioning that is sensitive to the exact composition of the cell membrane, solution flow, and other factors.(24, 51, 52) Not surprisingly, BrMT has different potencies against Shaker Kv1 channels in patches and whole cell voltage clamp of HEK cells, CHO cells, and Xenopus oocytes.(23) To serially compare the effects of BrMT derivatives with minimal variability, the assays were conducted with a commercially available automated patch clamp system that applies consistent solution flow for different experiments.
When BrMT was applied to voltage-clamped cells expressing Kv1.4, it inhibited the currents by slowing gating kinetics and reducing peak currents (Figure 1A). The effects of BrMT on kinetics were quantified by fitting a double exponential to the rise and decay of current after a depolarizing voltage step (Figure 1B). Increasing concentrations of BrMT progressively slowed the gating kinetics (Figure 1C). These effects are similar to those seen with Shaker Kv1 channels.(19) BrMT has two phenomenological effects on Kv1 channels: a slowing of activation kinetics and a diminishment of peak current amplitude after a voltage step.(19) Peak current amplitudes of Kv1.4 are affected more dramatically than the rate of channel activation by BrMT (Figures 1, 2A). This greater sensitivity of the peak amplitudes may be due to stronger coupling between activation and inactivation in Kv1.4 as compared to Shaker, channels.(53) For comparison among the BrMT analogs, we therefore used the IC50 values determined from the changes in peak current because their fitting was better constrained than the results obtained from the more complex curve fitting procedures needed to analyze the activation kinetics. Modulation of Kv1.4 currents is apparent over a range of concentrations (Figure S1), and IC50 estimates were successfully obtained for most BrMT analogs (Figure 2). The Hill slopes deduced from these fits are between 1 and 2 for most analogs. Hill slopes of greater than 1 could arise from cooperative interactions of the BrMT analog with multiple subunits of Kv channels.(20) One analog, 15, has a lower Hill slope, which could indicate that this weakly modulating analog acts by a different mechanism, but its low potency precludes any significant interpretations.
Figure 1. BrMT inhibits Kv1.4 channel activation.
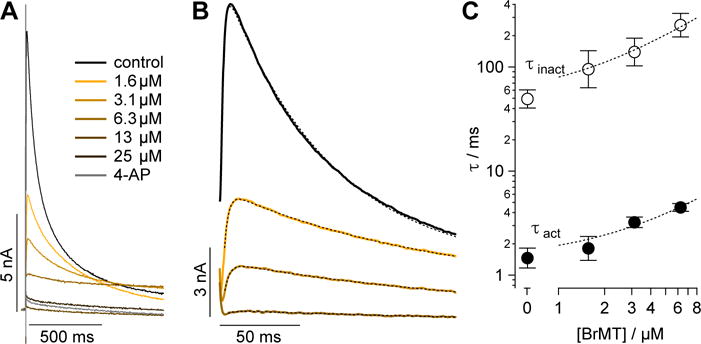
(A) Kv1.4 current responses to indicated BrMT concentrations during voltage steps to from −100 mV to 0 mV. (B) Kv1.4 current responses. Colored lines correspond to same BrMT concentrations as panel A. Stippled lines are fits of Equation 2, in Supporting Information. Voltage step to from −100 mV to 0 mV. Vehicle τact = 1.72 ± 0.02, τinact = 67.3 ± 0.2; 1.5 μM τact = 2.54 ± 0.02, τinact = 136.6 ± 0.6; 3 μM τact = 3.17 ± 0.03, τinact = 181 ± 2; 6 μM τact = 4.2 ± 0.4, τinact = 300 ± 20. (C) Time constants of activation (filled circles) and inactivation (empty circles). Error bars indicate SEM. n = 9. Stippled lines are fits for Kd as in ref. 19. Kd from τact = 2.9 ± 1.0 μM, Kd from τinact = 1.6 ± 0.7 μM.
Figure 2. Structure-activity relationship of Kv1.4 inhibition.
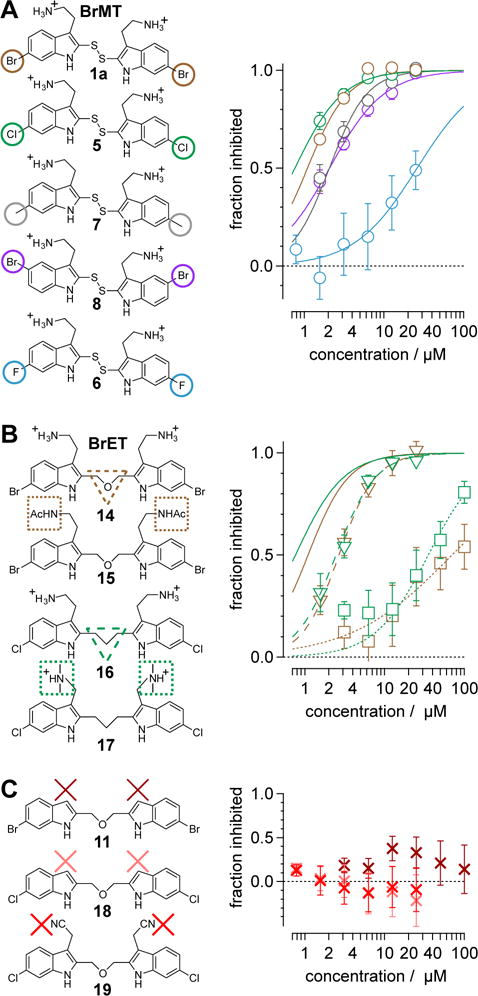
Average inhibition of peak Kv1.4 currents as function of concentration. The shapes and colors of markers correspond to compounds with modifications indicated by shapes in left column. Error bars indicate SEM. Lines are fits of Equation 1, in Supporting Ingormation, with parameter values indicated in Table 1. Color and pattern of lines correspond to compounds with modifications indicated by shapes in left column. (A) BrMT and analogs with modifications at the 6-positions on the indole rings. (B) Analogs with modifications of the disulfide linker and amines. (C) Ineffective analogs without aminoethyl groups.
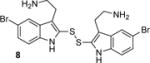
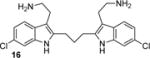
Factors affecting channel inhibition emerge from analysis of SAR data. Replacement of the 6-bromo moiety of BrMT with a chloro or methyl group as in 5 and 7 had little effect on inhibitor potency, whereas substitution with the smaller and more electronegative fluoro group as in 6 led to a decrease in potency by an order of magnitude (IC50 = 26 μM). Prior measurements with a BrMT analog containing only a hydride at position 6 indicated that it was also an order of magnitude less potent than BrMT against Shaker K+ channels.(54) Moving the bromo group to the 5-position on the indole ring as in 8 had only a minor effect on potency. These results suggest that variable indole substituents are tolerated.
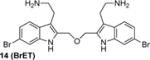
BrMT loses its potency against Kv channels when the disulfide is reduced to form monomeric compounds.(19) On the other hand, the disulfide linkage between the indole groups is remarkably tolerant to replacement. For example, activity is retained when the disulfide moiety in BrMT is replaced with the ether linkage in 14, or when the disulfide in the 6-chloro-substituted analog 5 is replaced with the trimethylene linker in 16. That alterations in the linker between the indoles have only mild effects on Kv potency was also reported by Gallin and Hall,(34) who found that Kv1 channel inhibition was maintained for 1a–c, wherein the number of sulfur atoms bridging the indole rings was varied from one to three.
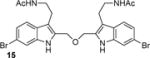
The discovery that Kv inhibition is retained with an ether or alkyl linkage between the indole rings permitted additional SAR studies using these more chemically stable scaffolds. We found that compounds 11, 18, and 19, which each lack the aminoethyl side chain, are inactive at the highest concentrations tested. Moreover, the N-acetyl derivative 15 and the N,N-dimethylaminoethyl analog 17 are less potent than the parent compound BrET (14) by more than an order of magnitude. Collectively, these results indicate that modification at the 2- and 6-positions of the indole rings of BrMT are well-tolerated, but the aminoethyl groups at the 3-positions are critical for channel activity: compounds lacking basic sidechains at the 3-position were inactive (e.g., 11, 18, 19).
BrMT derivatization alters bilayer partitioning, but partitioning does not predict ion channel modulation
We measured the degree to which the BrMT analogs partition into membranes using isothermal titration calorimetry (ITC).(55) These experiments were conducted with suspensions of large unilamellar phospholipid vesicles (LUVs) (see Supporting Information) as a surrogate for the bilayer component of cell membranes. All of the analogs partitioned into the bilayer with water→bilayer partition coefficients (KPW→L) ranging between 450 and 3900 (Table 1); the calculated log ([n-octanol]/[water]) (ClogP) values are also listed for comparison. KPW→L increased when the disulfide linkage was replaced with a 3-atom alkyl or an ether linkage. For example, replacing the disulfide linkage of BrMT (1a) (KPW→L = 560) with an ether linkage as in 14 (KPW→L = 3100) led to an increase in KPW→LP, as did replacing the disulfide bridge of the chloro analog 5 (KPW→L = 1000) with a trimethylene linkage in 16 (KPW→L = 3300). If channel modulation results from compounds partitioning into the membrane bilayer, we would expect that the Kv1.4 IC50 values would correlate with the KPW→L values, but this is not the case (Figure 3). Examination of small structural changes that influence partitioning and/or channel modulation also indicate that the two properties are independent. Replacing the disulfide linkage of BrMT (1a) with the ether moiety in 14, increases KPW→L but has little impact on IC50. The acetylated, non-basic compound 15 has an increased fraction in membranes versus its basic parent 14 (KPW→L = 3900 vs. 3100), but its potency against Kv1.4 drops substantially (IC50 = 70 vs. 2.7 M) (Figure 3, brown arrows). Replacing the disulfide linkage of 5 with a trimethylene linkage in 16 also increases KPW→L with little impact on IC50. Comparison of 16 to the related N,N-dimethylaminomethyl compound 17 is a rare example of a relatively minor structural change that simultaneously decreases both the KPW→L and inhibitory potency (Figure 3, green arrows). Overall, increases in the partition coefficient were not coupled to an increase of inhibitory potency. Fitting a linear regression to KPW→L and IC50 values did not yield significance (r = 0.34, p = 0.37). The fraction of a BrMT analog in the bilayer does not correlate with its potency, which suggests that bilayer partitioning is not sufficient for Kv channel modulation.
Figure 3. Membrane partition coefficients do not predict channel inhibition.
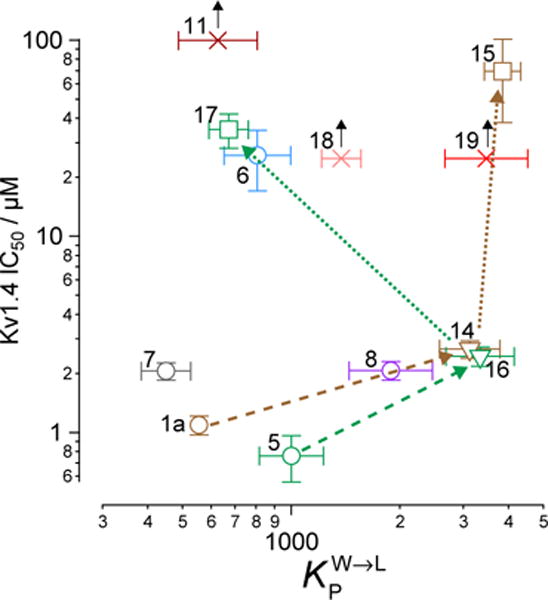
Numbers by each marker denote the analog identity. Shape and color of markers correspond to Figure 2. Values are from Table 1. Black arrows denote analogs with so low activity that we could not measure it (null response), with the marker indicating highest concentration tested. Brown and green arrows with dashed lines indicate modification of disulfide linker. Brown and green arrows with dotted lines indicate modification of aminoethyl groups.
Membrane perturbation by BrMT derivatives is not correlated with bilayer partitioning
Bilayer partitioning itself did not underlie the variable potencies of Kv channel inhibitors, but it remains a possibility that non-specific bilayer perturbation might affect their inhibition of Kv1.4, because small membrane perturbing amphiphiles are known to inhibit many Kv channels(7). We therefore probed for bilayer-modifying effects using a gramicidin A (gA) based fluorescence assay that monitors changes of gA activity in LUVs.(56, 57) This assay exploits the gramicidin channel-permeant thallous ion (Tl+), which quenches the fluorescence of the water-soluble fluorophore 8-aminonaphthalene-1,3,6-trisulfonate (ANTS). gA permits transmembrane Tl+ flux only as a dimer, and the equilibrium between monomeric and dimeric gA is altered by lipid bilayer perturbation. ANTS fluorescence is measured after addition of Tl+ to a suspension of ANTS-loaded gA-containing LUVs, and the time course of fluorescence quenching (Figure 4A) provides a measure of the changes in intravesicular [Tl+]. The initial rate of fluorescence quenching provides a measure of the initial rate of Tl+ influx into the LUVs (58-60), which varies with changes in the number of dimeric gA molecules in the LUV membrane. It is thus possible to determine how a drug, or other amphiphile, shifts the gA monomer/dimer equilibrium, which provides a measure of the bilayer-modifying potency of the molecule of interest. The BrMT analog concentration that elicits a doubling of the rate of gA-dependent quenching is denoted as gA2 and serves as a metric for the bilayer-perturbing potency (Figure S2). Note that a lower gA2 concentration indicates more potent bilayer perturbation. Some noticeable structural trends were observed (Table 1). Analogs 11 and 18 that lack aminoethyl side chains did not have a measurable gA2, because they caused no changes in the quench rate at the highest concentration tested. This result indicates that these analogs have minimal effects on bilayer properties. The linkage between the tryptamine monomers also affected bilayer perturbation. For example, the ether-linked derivatives 14 and 15 had the lowest gA2, and replacing the disulfide linkage of 5 with a trimethylene chain as in 16 did not detectably change gA2. Other changes to the aminoethyl side chains as shown by 14 vs. 15 and 16 vs. 17 increased the bilayer-modifying potency.
Figure 4. Membrane perturbation is distinct from partitioning and does not predict channel inhibition.
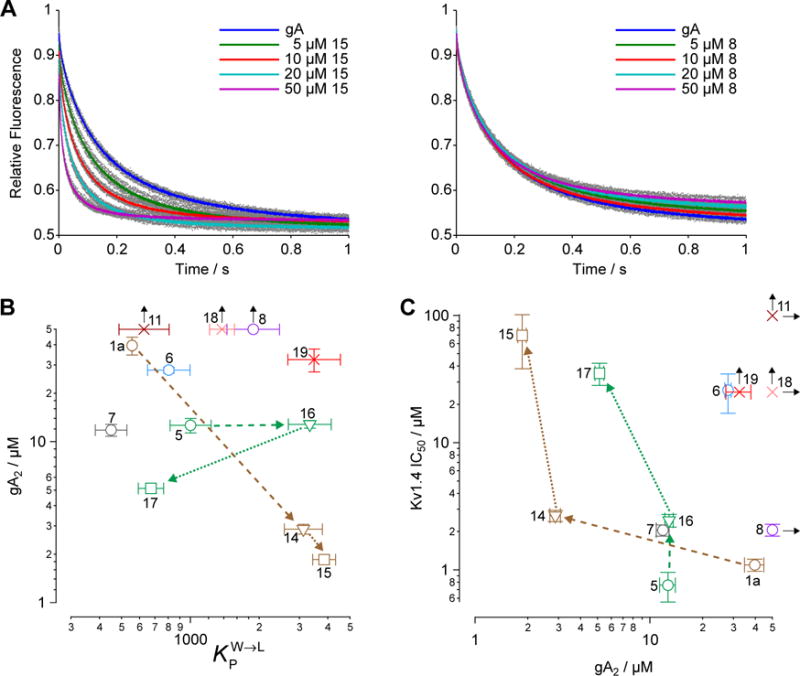
(A) Gramicidin A (gA) channel activity, as measured by the rate of quenching of intravesicular ANTS fluorescence by Tl+. For each trace, the gray dot denote the results obtained in the nine individual repeats, whereas the colored dots denote the average value at each time point. (B) Membrane partition–perturbation relationships. Numbers next to the different markers denote the analog’s identity. Shape and color of markers as in Figure 2. Data from Table 1. Black arrows indicate null response, with marker indicating highest concentration tested. Brown and green arrows with dashed lines indicate modification of disulfide linker. Brown and green arrows with dotted lines indicate modification of aminoethyl groups. (C) Membrane perturbation–inhibition relationship (data from Table 1).
Bilayer partitioning did not predict the bilayer-perturbing potency, as there was no obvious correlation between KPW→L and gA2 (Figure 4B) (r = −0.27, p = 0.47). Divergent effects of structure on KPW→L and gA2 can also be seen when comparing the related analogs 1a vs. 14 and 15, as well as 5 vs. 16 and 17. For compounds 1a, 14, and 15, a decreasing gA2 was consistent with an increasing KPW→L, as would be expected if an increase in partition coefficient decreased the aqueous concentration needed to reach a mole-fraction in the bilayer that caused the perturbation. Substituting an ether linkage into 1a giving 14, for example, increased bilayer partitioning and perturbation. When the aminoethyl groups in 14 are acetylated to give 15, there is a further slight increase of bilayer partitioning and perturbation (Figure 4B, brown arrows). However, in a sequence of modifications at the same positions of the Cl-substituted disulfide in the series 5, 16, and 17, gA2 appeared insensitive to KPW→L (5→16, Figure 4B, stippled green arrows), and then anti-correlated (16→17). A particularly striking example of the divergence between gA2 and KPW→L is when the bromo group at C6 in 1a is moved to C5 in 8 resulting in a moderate increase in KPW→L, yet complete inactivity in the gA assay (Figure 4A, right panel). Overall, BrMT analogs did not show a consistent dependence of bilayer perturbation on partitioning.
Potency of channel modulation is not dictated by bilayer perturbation
Bilayer perturbation was altered by BrMT analogs in a fashion distinct from partitioning, so we then assessed whether perturbation might be the mechanism underlying Kv modulation. When Kv1.4 IC50s are plotted vs. gA2 as a measure of bilayer perturbation, there is no obvious correlation (r = −0.37, p = 0.36) (Figure 4C). In fact, more potent bilayer-perturbing compounds are generally weaker inhibitors of Kv1.4. Substitution of the native disulfide linkage of 1a for the ether linker in 14 led to a 10-fold decrease in gA2 (10-fold increase in bilayer-perturbing potency) with a slight decrease in potency of channel inhibition (IC50 = 2.7 μM for 14 vs 1.1 μM for 1a). In contrast, when the disulfide linkage in the 6-chloro analog 5 is replaced with the trimethylene linker in 16, there is little change in gA2, but a three-fold increase in IC50. Changes to the aminoethyl side chain (e.g., 14→15 or 16→17) decrease gA2 but increase IC50 (Figure 4C). Notably, analog 8, which does not perturb bilayers at the highest concentration tested, is a strong modulator of Kv1.4, whereas the most bilayer-perturbing analog 15 is a weak modulator of Kv1.4 channels. Inspection of the current traces recorded in the presence of 15 reveals that inactivation is accelerated. This action is consistent with prior findings that bilayer-perturbing detergents modulate inactivation of Kv1 channels,(61) and suggests Kv1.4 modulation by 15 may be via a different mechanism than the less bilayer-perturbing BrMT analogs.
DISCUSSION
Overall, these relationships show that Kv1.4 inhibition does not correlate with either bilayer partitioning or bilayer perturbation. We conclude that though BrMT and its analogs that are active against ion channels all partition into bilayer and usually perturb bilayer properties, the partitioning and alterations in bilayer properties do not drive potassium channel modulation. We conclude that specific interactions between Kv1.4 and “strong” modulators such as BrMT and BrET are critical for activity.
Limitations
Our results show that bilayer partitioning and perturbation are not critical determinants of modulation of Kv channels by BrMT or its analogs. These conclusions rely on interpretations that have caveats:
The bilayer partitioning and perturbation experiments were conducted with bilayers formed by synthetic phosphocholine lipids. Results in multi-component bilayers,(62-64) and whole cells,(10) have been found to be similar to those obtained in single-component bilayers. On the basis of these previous validations, we expect the impact of compounds on single-component bilayers will extend to living cells more generally. However, our experiments do not exclude the possibility that BrMT derivatives partition differently into CHO cells and/or interact in a binding pocket with specialized lipids.
Our interpretation assumes that the BrMT derivatives alter Tl+ flux through gA channels by perturbing lipid membranes, not by interacting directly with the gA channel itself. Our experiments cannot completely exclude such direct effects; results with many different structurally diverse amphiphiles show that they have similar effects on right- and left-handed channels, which effectively excludes direct interactions.(6, 8, 65) The achirality of the BrMT derivatives here precludes such validation.
Implications
By using a series of analogs of the snail defensive toxin BrMT, we identified certain molecular features that determine its inhibition of potassium channels as well as its partitioning into and perturbation of membranes. Our findings are consistent with BrMT acting through a drug binding pocket at the lipid bilayer–potassium channel interface.(31, 66, 67) The positive charges of other amphiphilic, membrane partitioning compounds are crucial for their inhibition of Kv1 channels,(68) which is consistent with our observations for BrMT and analogs. Compounds 1a, 5, 7, 8, 14, and 16 each strongly inhibit Kv channels, but of these only the native compound, BrMT (1a) has a minimal impact on membranes. The BrMT analog 7 is the only strong Kv1.4 inhibitor that shows a relative decrease in bilayer partitioning and is a more potent bilayer perturber. On the other hand, 8 is the only strong Kv1.4 inhibitor that is a less potent bilayer perturber, and that has a higher partition coefficient than BrMT. Thus, all of our strongly Kv1.4 inhibiting analogs either partitioned into or perturbed the bilayer more than BrMT. We speculate that membrane partitioning and perturbation may have played a role in the natural selection of BrMT to be the defensive toxin of the sea snail, possibly because of its minimal negative impacts on snail membranes. We also conclude that bilayer interactions are not correlated with inhibition of Kv1.4.
BrMT analogs as research tools
Determining what conformations ion channels adopt is an ongoing challenge of molecular physiology. BrMT is a toxin that selectively binds to resting Kv conformations(19), making BrMT a powerful tool to explore conformational changes of potassium channels. Conformation-selective ligands are of increasing importance for associating functional states of proteins with structurally-defined conformations. For example, conformation-selective toxins were the enabling factors for reconstructing TRPV1 ion channels in an open state(16, 69-71), and co-crystals of ASIC ion channels with peptide toxin gating modifiers led to identifying structures of new channel conformations(72-74). In functional studies, tagged conformation-selective toxins allow imaging of Kv2 channels adopting specific conformations in live cells.(75, 76) BrMT-like compounds similarly can stabilize the resting conformation of Kv1 channel voltage sensors, and they have the potential to be useful tools for studying channel conformations in cells and live tissue. The ether-linked analog BrET (14) identified in this study is comparable in potency to BrMT, but lacks the unstable disulfide moiety of the natural product. With the information that modifications to the indole ring and the linker between them are well-tolerated, a path is revealed for generating additional conformation-selective ligands targeting resting conformations of Kv1 ion channel voltage sensors.
Supplementary Material
Scheme 1.
Total synthesis of BrMT
Scheme 2.
Synthesis of ether-linker analogs 14 (BrET) and 15
Acknowledgments
the staff of the UT-Austin NMR and Mass Spectrometry facilities; Dr. Sheng Cai (Marquette University), for analytical chemistry support; Drs. Ali Yehia and Juliette Johnson (Fluxion) as well as Jeffrey Webber (Molecular Devices) for generous access to and technical assistance with the IonFlux automated patch clamp; Justin Du Bois and Justin Litchfield (Stanford University) for help conceiving the project, preliminary synthesis attempts, and electrophysiology; ACD Labs for NMR processing software; ChemAxon Ltd. for software to calculate ClogP. This work was financially supported by Marquette University, the Texas Institute for Drug and Diagnostics Development (TI-3D), and the University of California, Davis. Support for our research programs was also provided by NIH grants R15 HL127636 (C.D.), T32 GM099608 (I.H.K.), R01 NS096317 (J.T.S), and R01 GM021432 (O.S.A.).
ABBREVIATIONS
- ACN
acetonitrile
- ANTS
8-aminonaphthalene-1,3,6-trisulfonate (di-sodium salt)
- ASIC
acid-sensing ion channel
- 4-AP
4-aminopyridine
- BrET, BrMT analog
2-[2-(<3-(2-aminoethyl)-6-bromo-1>H-indol-2-yl]methoxy}methyl)-6-bromo-1H-indol-3-yl]ethan-1-amine
- BrMT
snail toxin 6-bromo-2-mercaptotryptamine dimer
- ClogP
calculated log of the partition coefficient ([n-octanol]/[water])
- gA
gramicidin A
- gA2
concentration of compound that elicits a doubling of the rate of gA-dependent quenching
- HPW→L
enthalpy of partitioning from water into lipid
- IC50
half-maximal inhibitory concentration
- ITC
isothermal calorimetry
- KPW→L
equilibrium constant for partitioning from water into lipid
- Kv
voltage-gated potassium channel
- LUV
large unilamellar vesicles
- SAR
structure-activity relationship
- TRPV1
transient receptor potential vanilloid 1 channel
Footnotes
SUPPORTING INFORMATION
Contains supplementary figures: Table S1. Optimization of bis-indole synthesis; Figure S1. Representative Kv1.4 current responses to modulators; Figure S2. Gramicidin A assay data; Figure S3. Representative normalization of fluorescence in Gramicidin A assay. Also contains detailed methods: Synthesis of tryptamine building blocks; Protocol for bistryptamine–disulfide formation; Protocols for synthesis of BrET (14) and 15; Electrophysiology methods; Isothermal calorimetry methods; Gramicidin assay methods.
AUTHOR CONTRIBUTIONS
Chemical synthesis: C.D., M.W.D., D.G.
Electrophysiology: K.S.E., I.H.K., J.T.S.
Bilayer assays: O.S.A., H.I.I., R.K., T.P., R.R.
Conceived of project: R.W.A., O.S.A., C.D., S.F.M., J.T.S.
Writing: O.S.A., C.D., I.H.K., J.T.S., S.F.M.
The authors declare no competing financial interests. An earlier version of this manuscript was submitted to the ChemRxiv preprint server: https://doi.org/10.26434/chemrxiv.5908276.v1
References
- 1.Spector AA, Yorek MA. Membrane lipid composition and cellular function. Journal of lipid research. 1985;26:1015–1035. [PubMed] [Google Scholar]
- 2.Bienvenüe A, Marie JS. Chapter 12 - Modulation of Protein Function by Lipids. In: Hoekstra D, editor. Current Topics in Membranes. Academic Press; 1994. pp. 319–354. [Google Scholar]
- 3.Andersen OS, Koeppe RE., 2nd Bilayer thickness and membrane protein function: an energetic perspective. Annu Rev Biophys Biomol Struct. 2007;36:107–130. doi: 10.1146/annurev.biophys.36.040306.132643. [DOI] [PubMed] [Google Scholar]
- 4.Altenbach RJ, Adair RM, Bettencourt BM, Black LA, Fix-Stenzel SR, Gopalakrishnan SM, Hsieh GC, Liu HQ, Marsh KC, McPherson MJ, Milicic I, Miller TR, Vortherms TA, Warrior U, Wetter JM, Wishart N, Witte DG, Honore P, Esbenshade TA, Hancock AA, Brioni JD, Cowart MD. Structure-Activity Studies on a Series of a 2-Aminopyrimidine-Containing Histamine H-4 Receptor Ligands. J Med Chem. 2008;51:6571–6580. doi: 10.1021/jm8005959. [DOI] [PubMed] [Google Scholar]
- 5.Lundbaek JA, Birn P, Tape SE, Toombes GE, Sogaard R, Koeppe RE, 2nd, Gruner SM, Hansen AJ, Andersen OS. Capsaicin regulates voltage-dependent sodium channels by altering lipid bilayer elasticity. Mol Pharmacol. 2005;68:680–689. doi: 10.1124/mol.105.013573. [DOI] [PubMed] [Google Scholar]
- 6.Rusinova R, Herold KF, Sanford RL, Greathouse DV, Hemmings HC, Jr, Andersen OS. Thiazolidinedione insulin sensitizers alter lipid bilayer properties and voltage-dependent sodium channel function: implications for drug discovery. J Gen Physiol. 2011;138:249–270. doi: 10.1085/jgp.201010529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ingolfsson HI, Thakur P, Herold KF, Hobart EA, Ramsey NB, Periole X, de Jong DH, Zwama M, Yilmaz D, Hall K, Maretzky T, Hemmings HC, Jr, Blobel C, Marrink SJ, Kocer A, Sack JT, Andersen OS. Phytochemicals perturb membranes and promiscuously alter protein function. ACS Chem Biol. 2014;9:1788–1798. doi: 10.1021/cb500086e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lundbaek JA, Koeppe RE, 2nd, Andersen OS. Amphiphile regulation of ion channel function by changes in the bilayer spring constant. Proc Natl Acad Sci U S A. 2010;107:15427–15430. doi: 10.1073/pnas.1007455107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lundbaek JA, Birn P, Girshman J, Hansen AJ, Andersen OS. Membrane stiffness and channel function. Biochemistry. 1996;35:3825–3830. doi: 10.1021/bi952250b. [DOI] [PubMed] [Google Scholar]
- 10.Lundbaek JA, Birn P, Hansen AJ, Sogaard R, Nielsen C, Girshman J, Bruno MJ, Tape SE, Egebjerg J, Greathouse DV, Mattice GL, Koeppe RE, 2nd, Andersen OS. Regulation of sodium channel function by bilayer elasticity: the importance of hydrophobic coupling. Effects of Micelle-forming amphiphiles and cholesterol. J Gen Physiol. 2004;123:599–621. doi: 10.1085/jgp.200308996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sogaard R, Werge TM, Bertelsen C, Lundbye C, Madsen KL, Nielsen CH, Lundbaek JA. GABA(A) receptor function is regulated by lipid bilayer elasticity. Biochemistry. 2006;45:13118–13129. doi: 10.1021/bi060734+. [DOI] [PubMed] [Google Scholar]
- 12.Huang CJ, Harootunian A, Maher MP, Quan C, Raj CD, McCormack K, Numann R, Negulescu PA, Gonzalez JE. Characterization of voltage-gated sodium-channel blockers by electrical stimulation and fluorescence detection of membrane potential. Nat Biotechnol. 2006;24:439–446. doi: 10.1038/nbt1194. [DOI] [PubMed] [Google Scholar]
- 13.Morris CE, Juranka PF. Nav channel mechanosensitivity: activation and inactivation accelerate reversibly with stretch. Biophys J. 2007;93:822–833. doi: 10.1529/biophysj.106.101246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Howery AE, Elvington S, Abraham SJ, Choi KH, Dworschak-Simpson S, Phillips S, Ryan CM, Sanford RL, Almqvist J, Tran K, Chew TA, Zachariae U, Andersen OS, Whitelegge J, Matulef K, Du Bois J, Maduke MC. A designed inhibitor of a CLC antiporter blocks function through a unique binding mode. Chem Biol. 2012;19:1460–1470. doi: 10.1016/j.chembiol.2012.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lundbaek JA, Collingwood SA, Ingolfsson HI, Kapoor R, Andersen OS. Lipid bilayer regulation of membrane protein function: gramicidin channels as molecular force probes. Journal of the Royal Society Interface / the Royal Society. 2010;7:373–395. doi: 10.1098/rsif.2009.0443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cao E, Liao M, Cheng Y, Julius D. TRPV1 structures in distinct conformations reveal activation mechanisms. Nature. 2013;504:113–118. doi: 10.1038/nature12823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Walpole CS, Bevan S, Bovermann G, Boelsterli JJ, Breckenridge R, Davies JW, Hughes GA, James I, Oberer L, Winter J, Wrigglesworth R. The discovery of capsazepine, the first competitive antagonist of the sensory neuron excitants capsaicin and resiniferatoxin. J Med Chem. 1994;37:1942–1954. doi: 10.1021/jm00039a006. [DOI] [PubMed] [Google Scholar]
- 18.Kelley WP, Wolters AM, Sack JT, Jockusch RA, Jurchen JC, Williams ER, Sweedler JV, Gilly WF. Characterization of a novel gastropod toxin (6-bromo-2-mercaptotryptamine) that inhibits shaker K channel activity. J Biol Chem. 2003;278:34934–34942. doi: 10.1074/jbc.M301271200. [DOI] [PubMed] [Google Scholar]
- 19.Sack JT, Aldrich RW, Gilly WF. A gastropod toxin selectively slows early transitions in the Shaker K channel’s activation pathway. J Gen Physiol. 2004;123:685–696. doi: 10.1085/jgp.200409047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sack JT, Aldrich RW. Binding of a gating modifier toxin induces intersubunit cooperativity early in the Shaker K channel’s activation pathway. J Gen Physiol. 2006;128:119–132. doi: 10.1085/jgp.200609492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wulff H, Castle NA, Pardo LA. Voltage-gated potassium channels as therapeutic targets. Nature reviews. Drug discovery. 2009;8:982–1001. doi: 10.1038/nrd2983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bagal SK, Brown AD, Cox PJ, Omoto K, Owen RM, Pryde DC, Sidders B, Skerratt SE, Stevens EB, Storer RI, Swain NA. Ion channels as therapeutic targets: a drug discovery perspective. J Med Chem. 2013;56:593–624. doi: 10.1021/jm3011433. [DOI] [PubMed] [Google Scholar]
- 23.Sack JT. Ph D Thesis. Stanford University; Palo Alto, California: 2003. Voltage gating of the Shaker potassium channel is modified by 6-bromo-2-mercaptotryptamine, a novel gastropod toxin. [Google Scholar]
- 24.Gingrich KJ, Burkat PM, Roberts WA. Pentobarbital produces activation and block of {alpha}1{beta}2{gamma}2S GABAA receptors in rapidly perfused whole cells and membrane patches: divergent results can be explained by pharmacokinetics. J Gen Physiol. 2009;133:171–188. doi: 10.1085/jgp.200810081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sheetz MP, Singer SJ. Equilibrium and kinetic effects of drugs on the shapes of human erythrocytes. J Cell Biol. 1976;70:247–251. doi: 10.1083/jcb.70.1.247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sheetz MP, Singer SJ. Biological membranes as bilayer couples. A molecular mechanism of drug-erythrocyte interactions. Proc Natl Acad Sci U S A. 1974;71:4457–4461. doi: 10.1073/pnas.71.11.4457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Martinac B, Adler J, Kung C. Mechanosensitive ion channels of E. coli activated by amphipaths. Nature. 1990;348:261–263. doi: 10.1038/348261a0. [DOI] [PubMed] [Google Scholar]
- 28.Browning JL, Nelson DL. Amphipathic amines affect membrane excitability in paramecium: role for bilayer couple. Proc Natl Acad Sci U S A. 1976;73:452–456. doi: 10.1073/pnas.73.2.452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lee SY, MacKinnon R. A membrane-access mechanism of ion channel inhibition by voltage sensor toxins from spider venom. Nature. 2004;430:232–235. doi: 10.1038/nature02632. [DOI] [PubMed] [Google Scholar]
- 30.Milescu M, Vobecky J, Roh SH, Kim SH, Jung HJ, Kim JI, Swartz KJ. Tarantula toxins interact with voltage sensors within lipid membranes. J Gen Physiol. 2007;130:497–511. doi: 10.1085/jgp.200709869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gupta K, Zamanian M, Bae C, Milescu M, Krepkiy D, Tilley DC, Sack JT, Vladimir YY, Kim JI, Swartz KJ. Tarantula toxins use common surfaces for interacting with Kv and ASIC ion channels. eLife. 2015;4:e06774. doi: 10.7554/eLife.06774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Suchyna TM, Tape SE, Koeppe RE, 2nd, Andersen OS, Sachs F, Gottlieb PA. Bilayer-dependent inhibition of mechanosensitive channels by neuroactive peptide enantiomers. Nature. 2004;430:235–240. doi: 10.1038/nature02743. [DOI] [PubMed] [Google Scholar]
- 33.Andersen OS. Perspectives on how to drug an ion channel. J Gen Physiol. 2008;131:395–397. doi: 10.1085/jgp.200810012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gao D, Sand R, Fu H, Sharmin N, Gallin WJ, Hall DG. Synthesis of the non-peptidic snail toxin 6-bromo-2-mercaptotryptamine dimer (BrMT)(2), its lower and higher thio homologs and their ability to modulate potassium ion channels. Bioorg Med Chem Lett. 2013;23:5503–5506. doi: 10.1016/j.bmcl.2013.08.070. [DOI] [PubMed] [Google Scholar]
- 35.Schumacher RW, Davidson BS. Synthesis of didemnolines A-D, N9-substituted beta-carboline alkaloids from the marine ascidian Didemnum sp. Tetrahedron. 1999;55:935–942. [Google Scholar]
- 36.Wieland T, Weiberg O, Fischer E, Horlein G. *Darstellung Schwefel-Haltiger Indol-Derivate. Liebigs Ann Chem. 1954;587:146–161. [Google Scholar]
- 37.Freter K, Weissbach H, Redfield B, Udenfriend S, Witkop B. Oxindole Analogs of (5-Hydroxy)-Tryptamine and (5-Hydroxy)-Tryptophan, as Inhibitors of the Biosynthesis and Breakdown of Serotonin. J Am Chem Soc. 1958;80:983–987. [Google Scholar]
- 38.Showalter HDH, Sercel AD, Leja BM, Wolfangel CD, Ambroso LA, Elliott WL, Fry DW, Kraker AJ, Howard CT, Lu GH, Moore CW, Nelson JM, Roberts BJ, Vincent PW, Denny WA, Thompson AM. Tyrosine kinase inhibitors .6. Structure-activity relationships among N- and 3-substituted 2,2′-diselenobis(1H-indoles) for inhibition of protein tyrosine kinases and comparative in vitro and in vivo studies against selected sulfur congeners. J Med Chem. 1997;40:413–426. doi: 10.1021/jm960689b. [DOI] [PubMed] [Google Scholar]
- 39.Nagy P. Kinetics and mechanisms of thiol-disulfide exchange covering direct substitution and thiol oxidation-mediated pathways. Antioxid Redox Signal. 2013;18:1623–1641. doi: 10.1089/ars.2012.4973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dann O, Wolff HP, Schlee R, Ruff J. Syntheses of Antileukemic (Indolylvinyl)Indoles. Liebigs Annalen Der Chemie. 1986:2164–2178. [Google Scholar]
- 41.Sercel AD, Showalter HDH. The synthesis of symmetrical (2-indolyl)ethynes and reduced congeners via palladium-catalyzed couplings of 2-bromoindole precursors. J Heterocyclic Chem. 2006;43:701–707. [Google Scholar]
- 42.Altenbach R, Black L, Chang SJ, Cowart M, Faghih R, Gfesser G, Ku YY, Liu H, Lukin K, Nersesian D. WO 2004/043458 Bicyclic-substituted amines as histamine-3 receptor ligands. 2004
- 43.Baran PS, Shenvi RA. Total synthesis of (+/−)-chartelline C. J Am Chem Soc. 2006;128:14028–14029. doi: 10.1021/ja0659673. [DOI] [PubMed] [Google Scholar]
- 44.Yoon NM, Brown HC. Selective Reductions. 12. Explorations in Some Representative Applications of Aluminum Hydride for Selective Reductions. J Am Chem Soc. 1968;90:2927–2938. [Google Scholar]
- 45.Du X, Gamper N. Potassium channels in peripheral pain pathways: expression, function and therapeutic potential. Current Neuropharmacology. 2013;11:621–640. doi: 10.2174/1570159X113119990042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rasband MN, Park EW, Vanderah TW, Lai J, Porreca F, Trimmer JS. Distinct potassium channels on pain-sensing neurons. Proc Natl Acad Sci U S A. 2001;98:13373–13378. doi: 10.1073/pnas.231376298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wells JE, Rose ET, Rowland KC, Hatton JF. Kv1.4 subunit expression is decreased in neurons of painful human pulp. J Endod. 2007;33:827–829. doi: 10.1016/j.joen.2007.03.013. [DOI] [PubMed] [Google Scholar]
- 48.Gutman GA, Chandy KG, Grissmer S, Lazdunski M, McKinnon D, Pardo LA, Robertson GA, Rudy B, Sanguinetti MC, Stuhmer W, Wang X. International Union of Pharmacology. LIII. Nomenclature and molecular relationships of voltage-gated potassium channels. Pharmacol Rev. 2005;57:473–508. doi: 10.1124/pr.57.4.10. [DOI] [PubMed] [Google Scholar]
- 49.Manganas LN, Trimmer JS. Subunit composition determines Kv1 potassium channel surface expression. J Biol Chem. 2000;275:29685–29693. doi: 10.1074/jbc.M005010200. [DOI] [PubMed] [Google Scholar]
- 50.Manganas LN, Wang Q, Scannevin RH, Antonucci DE, Rhodes KJ, Trimmer JS. Identification of a trafficking determinant localized to the Kv1 potassium channel pore. Proc Natl Acad Sci U S A. 2001;98:14055–14059. doi: 10.1073/pnas.241403898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Guo L, Guthrie H. Automated electrophysiology in the preclinical evaluation of drugs for potential QT prolongation. J Pharmacol Toxicol Methods. 2005;52:123–135. doi: 10.1016/j.vascn.2005.04.002. [DOI] [PubMed] [Google Scholar]
- 52.Sorota S, Zhang XS, Margulis M, Tucker K, Priestley T. Characterization of a hERG screen using the IonWorks HT: comparison to a hERG rubidium efflux screen. Assay Drug Dev Technol. 2005;3:47–57. doi: 10.1089/adt.2005.3.47. [DOI] [PubMed] [Google Scholar]
- 53.Rasmusson RL, Morales MJ, Wang S, Liu S, Campbell DL, Brahmajothi MV, Strauss HC. Inactivation of voltage-gated cardiac K+ channels. Circ Res. 1998;82:739–750. doi: 10.1161/01.res.82.7.739. [DOI] [PubMed] [Google Scholar]
- 54.Litchfield JD. Ph D Thesis. Stanford University; Palo Alto, California: 2010. Using synthetic small molecules to probe the structure and function of voltage-gated ion channels. [Google Scholar]
- 55.Heerklotz H, Seelig J. Titration calorimetry of surfactant-membrane partitioning and membrane solubilization. Biochim Biophys Acta. 2000;1508:69–85. doi: 10.1016/s0304-4157(00)00009-5. [DOI] [PubMed] [Google Scholar]
- 56.Ingolfsson HI, Andersen OS. Screening for small molecules’ bilayer-modifying potential using a gramicidin-based fluorescence assay. Assay Drug Dev Technol. 2010;8:427–436. doi: 10.1089/adt.2009.0250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ingolfsson HI, Sanford RL, Kapoor R, Andersen OS. Gramicidin-based fluorescence assay; for determining small molecules potential for modifying lipid bilayer properties. Journal of Visualized Experiments. 2010;44:e2131. doi: 10.3791/2131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Moore HP, Raftery MA. Direct spectroscopic studies of cation translocation by Torpedo acetylcholine receptor on a time scale of physiological relevance. Proc Natl Acad Sci USA. 1980;77:4509–4513. doi: 10.1073/pnas.77.8.4509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ingólfsson HI, Andersen OS. Screening for small molecules’ bilayer-modifying potential using a gramicidin-based fluorescence assay. Assay Drug Dev Technol. 2010;8:427–436. doi: 10.1089/adt.2009.0250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ingólfsson HI, Sanford RL, Kapoor R, Andersen OS. Gramicidin-based fluorescence assay for determining small molecules potential for modifying lipid bilayer properties. J Vis Exp. 2010:e2131. doi: 10.3791/2131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hollerer-Beitz G, Heinemann SH. Influence of detergents on the function of cloned potassium channels. Receptors Channels. 1998;5:61–78. [PubMed] [Google Scholar]
- 62.Bruno MJ, Koeppe RE, 2nd, Andersen OS. Docosahexaenoic acid alters bilayer elastic properties. Proc Natl Acad Sci U S A. 2007;104:9638–9643. doi: 10.1073/pnas.0701015104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Rusinova R, Koeppe RE, 2nd, Andersen OS. A general mechanism for drug promiscuity: Studies with amiodarone and other antiarrhythmics. J Gen Physiol. 2015;146:463–475. doi: 10.1085/jgp.201511470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Herold KF, Sanford RL, Lee W, Andersen OS, Hemmings HC., Jr Clinical concentrations of chemically diverse general anesthetics minimally affect lipid bilayer properties. Proc Natl Acad Sci U S A. 2017;114:3109–3114. doi: 10.1073/pnas.1611717114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hwang TC, Koeppe RE, 2nd, Andersen OS. Genistein can modulate channel function by a phosphorylation-independent mechanism: importance of hydrophobic mismatch and bilayer mechanics. Biochemistry. 2003;42:13646–13658. doi: 10.1021/bi034887y. [DOI] [PubMed] [Google Scholar]
- 66.Ottosson NE, Silvera Ejneby M, Wu X, Yazdi S, Konradsson P, Lindahl E, Elinder F. A drug pocket at the lipid bilayer-potassium channel interface. Sci Adv. 2017;3:e1701099. doi: 10.1126/sciadv.1701099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Milescu M, Bosmans F, Lee S, Alabi AA, Kim JI, Swartz KJ. Interactions between lipids and voltage sensor paddles detected with tarantula toxins. Nat Struct Mol Biol. 2009;16:1080–1085. doi: 10.1038/nsmb.1679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Borjesson SI, Parkkari T, Hammarstrom S, Elinder F. Electrostatic tuning of cellular excitability. Biophys J. 2010;98:396–403. doi: 10.1016/j.bpj.2009.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bae C, Anselmi C, Kalia J, Jara-Oseguera A, Schwieters CD, Krepkiy D, Won Lee C, Kim EH, Kim JI, Faraldo-Gomez JD, Swartz KJ. Structural insights into the mechanism of activation of the TRPV1 channel by a membrane-bound tarantula toxin. eLife. 2016;5 doi: 10.7554/eLife.11273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Gao Y, Cao E, Julius D, Cheng Y. TRPV1 structures in nanodiscs reveal mechanisms of ligand and lipid action. Nature. 2016;534:347–351. doi: 10.1038/nature17964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Liao M, Cao E, Julius D, Cheng Y. Structure of the TRPV1 ion channel determined by electron cryo-microscopy. Nature. 2013;504:107–112. doi: 10.1038/nature12822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Baconguis I, Gouaux E. Structural plasticity and dynamic selectivity of acid-sensing ion channel-spider toxin complexes. Nature. 2012;489:400–405. doi: 10.1038/nature11375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Dawson RJ, Benz J, Stohler P, Tetaz T, Joseph C, Huber S, Schmid G, Hugin D, Pflimlin P, Trube G, Rudolph MG, Hennig M, Ruf A. Structure of the acid-sensing ion channel 1 in complex with the gating modifier Psalmotoxin 1. Nat Commun. 2012;3:936. doi: 10.1038/ncomms1917. [DOI] [PubMed] [Google Scholar]
- 74.Baconguis I, Bohlen CJ, Goehring A, Julius D, Gouaux E. X-ray structure of acid-sensing ion channel 1-snake toxin complex reveals open state of a Na(+)-selective channel. Cell. 2014;156:717–729. doi: 10.1016/j.cell.2014.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Tilley DC, Eum KS, Fletcher-Taylor S, Austin DC, Dupre C, Patron LA, Garcia RL, Lam K, Yarov-Yarovoy V, Cohen BE, Sack JT. Chemoselective tarantula toxins report voltage activation of wild-type ion channels in live cells. Proc Natl Acad Sci U S A. 2014;111:E4789–4796. doi: 10.1073/pnas.1406876111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Cobb MM, Austin DC, Sack JT, Trimmer JS. Cell Cycle-dependent Changes in Localization and Phosphorylation of the Plasma Membrane Kv2.1 K+ Channel Impact Endoplasmic Reticulum Membrane Contact Sites in COS-1 Cells. J Biol Chem. 2015;290:29189–29201. doi: 10.1074/jbc.M115.690198. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.