

Abstract
Tumor cell invasion, whether penetrating through extracellular matrix (ECM) or crossing a vascular endothelium, is a critical step in the cancer metastatic cascade. Along the way from primary tumor to a distant metastatic site, tumor cells interact actively with the microenvironment either via biomechanical (e. g. ECM stiffness) or biochemical (e.g. secreted cytokines) signals. Increasingly, it is recognized that the tumor microenvironment (TME) is a critical player in tumor cell invasion. A main challenge for the mechanistic understanding of tumor cell–TME interactions comes from the complexity of the TME, which consists of extracellular matrices, fluid flows, cytokine gradients and other cell types. It is difficult to control TME parameters in conventional in vitro experimental designs such as Boyden Chambers, or in vivo such as in mouse models. Microfluidics has emerged as an enabling tool for exploring TME parameter space because of its ease in recreating the complex and physiologically realistic three dimensional TME with well-defined spatial and temporal control. In this Perspective, we will discuss designing principles for modeling the biophysical microenvironment (biological flows and ECM) for tumor cells using microfluidic devices, and the potential microfluidic technology holds in recreating physiologically realistic tumor microenvironment. The focus will be on applications of microfluidic models in tumor cell invasion.
Graphical Abstract
Microfluidic model for the physical tumor microenvironment: intramural and interstitial flows, and the extracellular matrices (ECMs).
Introduction
Cancer metastasis of solid tumors is a physical process where tumor cells generate sufficient forces to break away from the primary tumor, invade through the interstitial extracellular matrix (ECM), squeeze through vascular vessels, and establish a secondary tumor at a distant organ 1–3 (Figure 1). It is now well accepted that the tumor microenvironment (TME) plays an important role, similar to the genetic makeup of the tumor cells, in determining tumor cell invasiveness. While tumor genetics has always been the focus of tumor biology, it is only in recent decades that the critical roles of the TME have been recognized widely in the context of tumor cell invasion 4–9.
Figure 1.
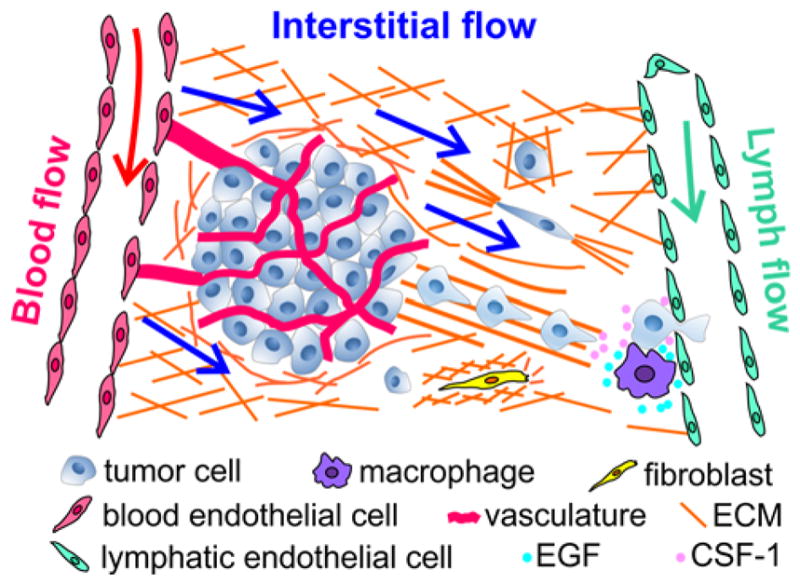
Important biophysical parameters in the tumor microenvironment (TME): intramural (blood and lymph) flows, interstitial flow, and the architectural support of extracellular matrices (ECM). Important biochemical parameters: cytokine gradients, nutrients, and oxygen, and multiple other cell types (stromal, immune, and endothelial cells).
Broadly speaking, the complex TME can be classified into biochemical (e.g. cytokines secreted by cells and nutrients) and biophysical cues (e.g. fluid flows and ECM), as illustrated in Figure 1. Current tools for recreating the TME for tumor cell invasion are primarily Boyden chambers 10–12 and animal models 13–15. Boyden chambers are straightforward to use, but difficult to recreate the complex TME and only provide population level and endpoint results. Animal models, on the other hand, provide a physiologically realistic environment, but are low throughput and it is difficult to isolate individual TME components. Microfluidic models have emerged to fill this gap 16–21. Microfluidic models can allow for well-defined spatial and temporal arrangements of individual components of the TME, and facilitate quantitative analysis and mathematical modeling. In addition, they are compatible with optical imaging, enabling studies of single and collective cell dynamics in real time. We note that dynamic analysis is important for tumor cell invasion studies because heterogeneity and plasticity are hallmarks of cancer 22–24. In this Perspective, we will focus on recent progress as well as future direction in the development of microfluidic devices for studying the roles of biophysical cues, specifically, biological flows and ECM, in driving tumor cell invasion. For microfluidic developments in the analysis of biochemical cues with applications in tumor cell invasion, please refer to recent excellent review articles 16, 25–27.
Biophysical drivers in tumor cell invasion
Intramural flow in tumor cell invasion
Biological flows are ubiquitous in living systems and generally can be classified into intramural flows (blood and lymph flow) and interstitial flows (see Figure 1 and Table 1). Blood flow follows the large scale anatomical pattern, mainly aorta and arteries, with high flow speed in the order of tens of centimeters per second 28, and branches out to smaller scale arterioles and capillaries with lower speed, ranging from a few centimeters to a few hundred micrometers per second 29. In addition to nutrients and oxygen transport, shear stress from blood flow is known to be a critical regulator in vascular physiology 30 and morphogenesis during development 31–33, and is tightly linked to endothelial cell function 34–37. Lymph flow, on the other hand, is several orders of magnitude smaller than blood flow, and has been reported to be on the order of a few millimeters per second in lymphatic vessels 38 and tens of micrometers per second in lymphatic capillaries 39.
Table 1.
Intramural and interstitial flow rates and vessel diameters measured in healthy tissue using animal models, and in tumors using both animal and human models.
| Healthy tissue | Tumor | |
|---|---|---|
| Intramural flow |
Arterioles:
29 diameter 15–60μm: 7–12 mm/s Capillaries: 29 diameter 5μm: 0.2 mm/s Venules: 29 diameter 18–72μm: 0.2–2.4 mm/s Microlymphatic vessels: 38 diameter 100 μm: 1–7 mm/s Lymphatic capillaries: 39, 155 diameter 55 μm: 0–29 μm/s |
Blood vessels in mammary carcinoma:
156 diameter 7–63μm: 0–0.8 mm/s Blood vessels in glioma: diameter 8–55μm: 0–0.5 mm/s 156 diameter 1–100μm: 0.001–10 mm/s 157 Blood vessels in adenocarcinoma: 158 arterioles 9–10μm: 0.59–0.7 mm/s capillaries 6–8μm: 0.09–0.27 mm/s venules 10–26μm: 0.09–0.22 mm/s |
| Interstitial flow |
Rabbit ear tissue: 50 0.59±0.16 μm/s |
VX2 carcinoma: 0.55 ± 0.16 μm/s 50 C6-pTET-VEGF tumor: 0.1–0.5 μm/s 159 Cervical carcinoma: 1–7 μm/s 55 Melanoma: 1–9 μm/s Nonmetastatic squamous carcinoma: 5–25 μm/s Metastatic squamous carcinoma: 10–55 μm/s |
Blood/lymph flow within the TME is aberrant and their roles in tumor cell invasion are largely unknown (see Table 1). Similar to normal tissue, blood flow in the TME is essential for tumor perfusion and growth. Different from the flow in normal tissue, blood flow in the TME is difficult to predict because of its dynamic nature. Blood flow rates within the TME are known to change with the abnormal growth of vascular vessels through angiogenesis, the physical stress from the fast growing tumor mass, and altered ECM mechanical properties. In addition, the blood vessels within the tumor are torturous, and often lack functional pericytes, basement membrane, and tight endothelial cell junctions 40–42. Lymph flow within the TME has received much less attention, but is found to be elevated in contrast to healthy tissue 43.
The roles of blood/lymph flow in tumor cell invasion are often attributed to the shear force regulation at the interface with endothelial cell (EC) layers. In order to disseminate to a distant organ, tumor cells must use the blood or lymphatic vessels as a conduit, and thus need to cross the EC layer 44–46. Tumor cells are unlikely to enter/leave via the aortic or arterial vessels as those vessels have thicker walls and high shear stress. In contrast, blood capillaries with thinner walls and lower shear stress are more likely to physically trap circulating tumor cells for subsequent extravasation 3, 44, 47. In parallel, lymphatic capillaries with significant slower flow at the host tissue are also sites where tumor cells prefer to reside 48. Taken together, flow shear stress critically regulates the physiology of the vascular EC layers, and impacts on tumor cell invasion.
Interstitial flow in tumor cell invasion
Interstitial flow is a slow fluid movement through the interstitial space driven by hydrostatic and osmotic pressure differences between the arterial and venous or arterial and lymphatic vessels 49, 50. First measured by the Jain lab using the fluorescence recovery after photo bleaching (FRAP) technique, the flow speed associated with interstitial flow is typically on the order of a few micrometers per second in normal tissue 50. While the main function of interstitial flow has long been known to drain waste fluids to the lymphatic system, recently it has been shown to regulate vascular morphogenesis 51–53.
Interstitial flow within the TME is elevated due to the heightened interstitial fluid pressure 54, 55 (Table 1). High interstitial fluid pressure in the TME, in part, is a result of the fact that excessive interstitial fluids could not be absorbed by the compressed non-functional lymphatic vessels within the tumor. It has been reported that the pressure is particularly high within a malignant tumor, and drops steeply at the tumor-host tissue interface 55, 56. For nearly three decades, high interstitial fluid pressure has been an important biomarker for tumor progression and a significant obstacle for cancer therapeutic delivery into the tumor 57–60.
Recently, the physical consequence of elevated and excessive interstitial fluid flow outward from the tumor to the host tissue has been investigated in the context of tumor cell invasion using in vitro models 61–63. Using a modified Boyden chamber, the Swartz lab discovered that interstitial flow can guide tumor cell invasion along the flow direction. The unique feature of interstitial flow is that it operates in a region where the Peclet number (convective versus diffusive transport) is close to one. In other words, the transport of secreted cytokines is governed by both convective flow and diffusion. Computation of cell secreted cytokine transport shows that interstitial flow leads to flow-induced spatial gradients of secreted cytokines 52. One example is the spatial gradients of lymphatic chemokine CCL19/21 along the flow direction, and breast tumor cells were discovered to follow the flow/gradient direction using a modified Boyden Chamber model 61. More recently, interstitial flow was found to directly impact tumor cell mechanotransduction via CD44 in brain tumor cells 64 and via integrins in breast tumor cells 65. Interstitial flow also has been shown to promote tumor cell invasion via stromal cell mediated matrix remodeling within the TME 66. In summary, mounting evidence demonstrates that interstitial flow critically modulates tumor cell invasion behavior.
Extracellular matrix in tumor cell invasion
The extracellular matrix (ECM) is a major physical component that surrounds and penetrates solid tumors. Important physical ECM characteristics that are critical for tumor cell invasion include ECM overall stiffness and architecture 67, 68. Stiffness, contributed mainly by dense and cross linked ECM fibrillar structures, is a prognostic and risk factor for breast cancer patients 69, 70. Similarly, increasing evidence demonstrates that stiffer ECM promotes tumor cell invasion in in vitro assays 5, 71. It has been found that malignant tumor cells and tumor associated stromal cells secret collagen, which contributes to dense fibrillar structures 71. They also express enzymatic cross linkers, such as lysyl oxidase, which increase the overall stiffness of the ECM 72, 73. Breast tumor has been reported to be twenty times stiffer than the normal breast tissue 5.
Recent developments in intravital imaging reveal that tumor ECM is highly heterogeneous in space, and evolves with time 74–76. This is not surprising because tumor and stromal cells are known to actively pull onto the ECM, and thereby remodel the ECM architecture 66, 77, 78. Collagen bundles have been reported to align perpendicularly to the tumor periphery, possibly by active tumor and stromal cell contraction, facilitate collective tumor cell invasion and correlate with malignancy 79, 80. Micro-tunnels, hollow micro-sized tunnels with diameter range of 1–30 μm, were recently observed to provide tumor cells fast moving pathways both in vitro and in vivo 81–83. These hollow micro-sized tunnels are created by the degradation of collagen matrices via matrix metalloproteinase (MMP) secreted by the tumor and stromal cells. In the context of ECM architecture, the three dimensional nature of the ECM has been discussed extensively in the cell migration community and can be a critical regulator of cell migration 84–86.
Microfluidic modeling of the biophysical parameters in the tumor microenvironment
Microfluidics has emerged to model the TME because of its tight control of flow rates within a 3D ECM. A good microfluidic model needs to be able to recreate blood/lymphatic vessels of appropriate physiological size and shape within a 3D ECM, with controlled flow rates. In addition, the microfluidics should be robust and high throughput. Here, we discuss current efforts in modeling the biophysical aspects of the TME using microfluidic devices. The goal is to create on chip vascular vessels and ECM with architecture, dimensions and flow rates closely mimicking the in vivo measurement results (see Table 1). We emphasize the mathematical modeling of the flow rates/shear stress within the microfluidic platform using the relevant physical parameters including vascular permeability of endothelium layers and hydraulic conductivity of ECM (see Table 2).
Table 2.
Vascular permeability and hydraulic conductivity of ECM measured in vivo and in vitro. Vascular permeability (or permeability coefficient across an endothelium layer), P, is the ratio of solute flux and solute concentration gradient across the endothelium layer 169. Hydraulic conductivity, K′ is the ratio of fluid flux and pressure gradient as defined in Darcy’s law. Specific hydraulic conductivity (K) is K′η, where η is the fluid viscosity.
| In vivo | In vitro modeling | ||
|---|---|---|---|
| Heathy tissue | Tumor | ||
| Permeability across blood and lymphatic endothelium [cm/s] |
Capillaries and postcapillary venules
105: (for dextran MW=150,000) 7.26±3.29 × 10−8 Postcapillary venules 160: (for sodium fluorescein) 1.4±0.11 × 10−5 (for α-Lactalbumin) 4.4±0.5 × 10−7 (for BSA) 4.9±0.32 × 10−8 Postcapillary venules 161: (for 4kDa dextran) 9.2±4.6 × 10−7 (for 10kDa dextran) 3.1±1.3 × 10−7 (for 20kDa dextran) 2.4±1.0 × 10−7 (for 40kDa dextran) 1.9±1.1 × 10−7 (for 70kDa dextran) 1.5±0.5 × 10−7 Venules 162: (for RSA) 3.5±1.0 × 10−7 Collecting lymphatic vessels 162: (for RSA) 4.0±1.0 × 10−7 |
Capillaries and postcapillary venules in VX2 carcinoma
105: (for dextran MW=150,000) 5.7±3.9 × 10−7 Microvascular in adenocarcinoma 163: (for BSA) 6.06±4.30 × 10−7 Arterioles and venules in mammary adenocarcinoma 156: (for BSA) 1.7±0.6 × 10−7 2.9±1.5 × 10−7 1.9±0.5 × 10−7 Arterioles and venules in glioblastoma 156: (for BSA) 3.8±1.2 × 10−7 1.1±0.5× 10−8 Vasculature in squamous carcinoma 164: (for 3.3kDa dextran) 1.54× 10−5 (for 40kDa dextran) 9.5×10−7 (for 2MDa dextran) 1.7× 10−7 (for BSA) 4.9×10−7 |
In transwell: EC monolayer 165: (for Albumin) 5.6 × 10−6 In microfluidics: Hollow EC lumen with tumor cells 110: (for 10kDa) 4.08±1.11 × 10−5 (for 70kDa) 7.5±0.93 × 10−6 Single blood EC tube 87, 91 (for BSA) 5.5±3.5 × 10−6 7.9±3.5 × 10−6 (for 70kDa dextran) 4.1±0.5 × 10−6 (for 332Da fluorescein) 7.0±1.5 × 10−6 Self-assembled blood vessels 101, 107: (for 70kDa dextran) 8.9±3.1 × 10−7 (for 70kDa dextran) 4.5 × 10−7 (for 150kDa dextran) 1.2 × 10−7 |
| Hydraulic conductivity of ECM |
Hydraulic conductivity (K′) [cm2/mmHg · s] (From references 166 and 167) |
Specific hydraulic conductivity (K) [cm2] | |
|
Abdominal muscle: 1.5×10−7 to 7.8×10−7 Dermis: 5.3×10−8 Tail skin: 7×10−7 to 1.5×10−6 |
HSTS 26T sarcoma: 9×10−8 U87 glioblastoma: 6.5×10−7 LS174T carcinoma: 4.5×10−7 MCaIV carcinoma: 2.5×10−6 |
Type I collagen matrices
143: 3mg/mL: 3 × 10−7 8mg/mL: 3.5 × 10−8 10mg/mL: 7.5 × 10−9 15mg/mL: 9 × 10−10 20mg/mL: 1.5 ×10−10 Type I collagen matrices 168: 10mg/mL: 1 × 10−11 |
|
Microfluidic models for intramural flow in tumor cell invasion
A critical step for modeling intramural flow in the context of tumor cell invasion is to engineer a perfusable and functional vascular tube/network within a 3D ECM. Motivated by the development of vascularized biomaterial, early work began with the engineering of an EC tube in one single microfluidic channel within dense type I collagen matrices. The microfluidic channel was created through a micro-molding method, in which a hypodermic needle or a microfabricated PDMS positive channel feature was used as a template, and hydrogels as molding materials. After removing the template, endothelial cells were introduced into the channel and formed an EC monolayer 87–92. A recent alternative method for creating a single EC tube is through introducing a less dense fluid into a dense fluid (un-polymerized collagen) via a process known as viscous fingering 93–95. This method does not require microfabrication thus is cost effective, the downside being that it lacks precise control over the size of the vessel.
More complex 3D vascular networks embedded within synthetic or naturally derived matrices have been developed recently using a sacrificial method 96–100. Here, one first creates an interconnecting fiber network using either microfabrication or 3D printing with sacrificial materials that are compatible with cell culture (e.g. gelatin and carbohydrate glass). The polymerized gelatin or solidified carbohydrate glass fiber network is then submerged within synthetic or naturally derived gels. After removing the sacrificial layers, ECs are introduced into the network to form lumens. We note that autonomous formation of microfluidic vascular networks by placing ECs in biomatrices along with a set of well-defined reagents and/or fluid flow that promote angiogenesis 101–103 and lymphagiogenesis 104 have been successful and been used in the context of drug delivery. This method can potentially be introduced for the purpose of tumor cell invasion.
An important way that intramural flow regulates tumor cell intravasation/extravasation is through the alteration of EC layer integrity and permeability via flow induced shear stress (See Table 2). A key parameter that is commonly used to characterize the transport of solute across an endothelium layer is the permeability coefficient, P(cm/s), which is defined as Js/∇C, where Js (g/cm2/s) is the solute flux through an unit area, ∇C (g/cm3) is the spatial gradient of solute concentration. In Table 2, we list permeability of both blood and lymphatic vessels measured in healthy and tumor tissues. It is interesting to note that one in vivo study by Gerlowski et al. revealed that the vascular permeability is significantly increased in tumor compared to normal vasculature 105, echoing the notion that tumor vasculature is leakier than healthy vasculature. We also note that error bars for these measurements are typically large. Looking forward, the microfluidic platform allows us to make precise spatial arrangement of solute concentration, obtain temporal information of the concentration field, and thus has the potential to improve the permeability measurements in the future.
Shear stress from the intramural flows are known to alter EC permeability as well as integrity. Flow induced shear stress has been reported to increase the barrier function or lower the permeability coefficient of the EC layer 92, 106, 107 and suppress VEGF-driven EC sprouting 108. Progress has been made to incorporate tumor cell laden ECM into a single engineered microfluidic blood vessel or vascular networks for dynamic tumor trans-endothelial migration 89, 92, 107, 109–111, making it possible to examine quantitatively the impact of intramural flow on tumor cell invasion. Using a microfluidic model, we have learned that blood flow through the capillaries reduces tumor cell extravasation rates 107. This is not surprising because the EC layer junction is strengthened under flow shear stress, and increases its ability to prevent transmigration. It is interesting to note that lymph flow on the other hand, promotes intravasation using a flow chamber modified from the Boyden chamber 112. This discrepancy can likely be explained by the differential responses of blood and lymphatic vessels to flow shear stress. In this context, a tight control over the flow rates, in which microfluidic models have the advantages over other conventional models, will allow us to gain a deeper understanding of the impact of intramural flow on tumor cell transmigration.
Microfluidic models for interstitial flow in tumor cell invasion
Broadly speaking, there are two types of assays for creating interstitial flows. One is the modified Boyden Chamber assay and the other is the microfluidic platform (See Fig. 2). The modified Boyden Chamber assay uses a commercially available platform, and is straightforward to implement, but results are population based and end points. The microfluidic platform, on the other hand, allows for dynamic and single cell imaging, but is typically difficult to make, requiring engineering training for the users. Here, we discuss the evolution of several platforms developed to date for creating interstitial flows. We will highlight biological insights gained using these platforms in the context of tumor cell invasion.
Figure 2.

Modeling interstitial flows in tumor cell invasion studies. A, Modified Boyden chamber platform. Tumor cell embedded biomatrix is introduced into a Boyden Chamber insert, which is placed in a well. The gravitational pressure, provided by the fluid level difference between the fluid within the insert and that in the surrounding well, drives the interstitial flow. The invasion rate is marked by the number of cells transmigrated through the porous membrane at the bottom of the insert. B,C,D: Three different microfluidic platforms for modeling interstitial flows. B. In this device, lines of pillars with circular cross section of diameter 500 μm are used to confine collagen. Interstitial flow is driven by gravity along the horizontal direction. Numerically simulated flow field is shown in the right panel of B. C: In this device, lines of square pillars, each with a cross section of 250 μm x 250 μm, are used to confine collagen. Gravity driven flow is introduced horizontally across collagen matrices. D: In this device, contact lines with cross section of 10 μm x 5 μm are used to confine collagen within the cell channel. Interstitial flow is introduced using a syringe pump in the horizontal flow channel. Image on the right side of panel A is reproduced from reference 61, with permission from Elsevier. Images in B are reproduced from reference 116 with permission from the Royal Society of Chemistry. Images in C are reproduced from references 65, 115 with permission from the Proceedings of the National Academy of Sciences, and images in D are reproduced from reference 118 with permission from the Royal Society of Chemistry.
Pioneering work on roles of interstitial flows in tumor cell invasion was carried out in a modified Boyden chamber 61. A layer of tumor cell embedded biomatrix was introduced into the insert of a Boyden chamber. Interstitial flow is gravity driven, facilitated by the fluid level difference between the fluid within the insert and the cup surrounding it (Fig. 2A). Type I collagen, a main structural component of mammalian tissue, has been used in most of the current studies for its ability to mimic in vivo ECM architecture and compatibility with cell invasion assay 83. In addition, the mechanical properties 78, 113, 114 and specific hydraulic conductivities (see Table 2) of collagen gels have been studied and documented extensively in the literature from the biomaterials community. Here, the readout is the number of cells transmigrated through the porous membrane. Using the modified Boyden chamber, a number of biological insights have been gained 61, 63, most notably the autologous chemotaxis of breast tumor cells mediated by flow induced lymphoid chemokine gradients.
Although the modified Boyden chamber assay is straightforward to use, the results are population based and end points. One of the hallmarks of tumor cells is the heterogeneity of a single cell population. It is thus important to develop assays that are amenable to single cell analysis. Microfluidic platforms have emerged to model interstitial flows. A critical component for modeling interstitial flow through 3D ECM in a microfluidic platform is to confine natively derived or synthetic ECM within an area where fluid flow can be applied through the ECM in a controlled way. To create a microfluidic platform for tumor cell invasion in the presence of interstitial flow, a number of labs have developed unique microfabrication methods to pattern type I collagen in a three parallel channel configuration 65, 115–118. The common feature of all the devices is to develop micro-patterned structures to confine biomatrices in designated places. In the work of Hassler et al. or Polacheck et al., tumor cells embedded in collagen (unpolymerized) are introduced into a channel lined with two parallel lines of micropillars (see red or green channel in Figure 2. B and C). Interstitial flow is introduced in the horizontal direction after gel polymerization. This configuration has advantages over the early engineering method for vascular tube using micro-molding method for its flexibility of the channel layout, and also for its ease of integrating tumor cells within ECM. The use of spaced micropillars utilizes surface tension to confine the un-polymerized collagen solution, while leaving space between the micropillars for interstitial flow to pass through the polymerized collagen matrices. This method offers robust confinement of the ECM, but the micropillars block a significant amount of the flow and complicate the spatial distribution of the flow field (see right panel in Figure 2. B). To overcome this limitation, our lab confined cell-embedded collagen using a contact line pinning method 117, 118. In our work, parallel PDMS microridges (or contact lines) with a cross section of 10 μm by 5 μm were fabricated to confine collagen within a wall-less channel. This method allows interstitial flow to run through the collagen matrices with over 80% spatial uniformity in the area of interest. For details of the device designs, see Figure 2D.
Darcy’s law is often useful for computing interstitial flows during the device design stage. For a given layout of the device, one can compute accurately the flow rates in different parts of the channels by applying the Brinkman equation in a multi-physics software package such as COMSOL. A key parameter required is the hydraulic conductivity, K′, of the biomatrices. Hydraulic conductivity is defined through Darcy’s law, K′ = J/∇P, where J is the fluid flux, ∇P is the spatial pressure gradient. Because of the small dimension of the system, fluid flows are slow, and Darcy’s law is typically valid 117. Values of hydraulic conductivity of healthy and tumor tissue, as well as reconstituted ECM are listed in Table 2. We note that hydraulic conductivity times fluid viscosity, K′η, named specific hydraulic conductivity (K), is typically reported in in vitro ECM measurements.
Using the microfluidic platform developed, we have started to learn how interstitial flows modulate tumor cell heterogeneity and plasticity. This information is difficult to obtain in population level assays such as the Boyden Chamber assay. Haessler et al. showed that interstitial flow at flow speed of 10 μm/s enhanced a subpopulation of breast tumor cell migration, highlighting the heterogeneous nature of tumor cells within a single population 116. Work in our lab demonstrated that interstitial flow at flow speed of 2 μm/s promotes amoeboid over mesenchymal motility of breast cancer cell invasion by carrying away adhesion molecules such as fibronectin. Cells lacking adhesion contacts prefer to migrate via amoeboid motility 118. These studies revealed critical information on how interstitial flow influences tumor cell invasion at the single cell level and the dynamic interaction of tumor cells with their environment. The Kamm lab learned that interstitial flow can impact tumor cell migration directly via chemosensing or mechanosensing molecules. In the case of chemosensing, Polacheck et al. demonstrated that interstitial flow at flow speeds of 0.3 and 3.0 μm/s induced directional migration of breast tumor cells along/against the flow direction in a chemokine receptor CCR7 dependent manner 115. In later work from the Kamm lab, they reported that the directional migration of breast tumor cells along/against interstitial flow was triggered by mechanosensing molecules 65.
Taken together, quantitative understanding how interstitial flow impacts tumor cell invasion within 3D ECM has just begun. Results from these studies are far from converging into a coherent theoretical understanding. Current reported results are very sensitive to experimental conditions, and differ from lab to lab. In particular, it is known that the conditions under which collagen is polymerized is critical in determining collagen fiber architecture, including PH 119, polymerization temperature 120, as well as the actual reagents used. It is thus of paramount importance to carefully record all the detailed cell culture condition for an eventual unified understanding. Looking ahead, many questions remain to be explored. The comparative roles of chemosensing or mechanosensing in the presence of interstitial flow remain to be investigated, as well as whether interstitial flow drives collective cell migration 121. The latter is important because increasing evidence has suggested that collective cell migration may contribute to cancer cell survival and successful metastasis 122–124. Current studies are limited to mainly breast tumor cells, but similar microfluidic platforms can be easily extended to study other cell types such as brain tumor cells 63, 64 and ovarian tumor cells 125.
Microfluidic models for coupling intramural and interstitial flows in tumor cell invasion
In vivo, both interstitial flow and intramural flow coexist. Recent progress has been made in recreating both interstitial and intramural flows within one experimental setup in the context of tumor cell invasion. Tung et al. developed a microfluidic device that allows for the introduction of intramural flow, and at the same time controlling interstitial flow. This device can potentially be used to study tumor cell invasion and transmigration in the present of both flow types 117. The Swartz lab developed an experimental platform that integrates a Boyden chamber and a flow channel, in which interstitial flow was introduced through the Boyden chamber and intramural flow was introduced through a flow channel underneath the Boyden chamber 112. Using this platform, they revealed for the first time that interstitial flow and lymph flow synergistically increase tumor cell transmigration rates through lymphatic vessels. We note that currently, microfluidic platforms that include both interstitial and intramural flows for tumor cell transmigration are at early stages of development.
Microfluidics for modeling ECM architecture in tumor cell invasion
Microfabricated devices provide a unique opportunity for decoupling the contributions of individual ECM properties (e.g. alignment and pore size) from tumor cell invasion, enabling a mechanistic understanding of cell-ECM mechanical interactions.
Two important features within the TME that promote fast and persistent tumor cell invasion are aligned collagen fibers and embedded hollow micro-sized tunnels. To investigate molecular mechanisms that cells use to migrate along 1D collagen fibers, synthetic 1D tracks have been fabricated, including nano/microfabricated topographic lines and fibronectin lines that have line widths ranging from submicrometer to tens of micrometers 84, 126, 127. While these engineered lines are straightforward to produce and easy to manipulate, the disadvantage is that these lines are made on a 2D surface which do not recapitulate the 3D in vivo situation where cells are supported by fiber network around all surfaces. Alternative methods have been developed to circumvent this limitation. One way of producing aligned collagen fibers within a 3D ECM is to use shear stress. One can flow un-polymerized collagen through a micro-sized channel in microfluidics, and the flow shear force has been shown to be able to align collagen fibers along the flow direction 128–131. A second method is to flow a less dense collagen solution (low concentration) into a dense solution (Matrigel solution at high concentration) within a channel to form aligned collagen fibers at the gel interface 132. A third method is to repeatedly stretch and relax a collagen fiber network already polymerized on a 2D substrate 133. Recently, self-assembled micro-sized magnetic bead strings embedded within hydrogel have been used as mimics of 3D collagen fibers for the purpose of tumor cell invasion studies 134. These studies revealed that alignment promote directional cell migration by limiting the cellular protrusion sites within the line/fiber or contact guidance. In addition to 1D topography, confined micro-tunnels have been discussed extensively as a highway for tumor cell migration recently. Microfabricated tunnels/channels surrounded by collagen provide a controlled way to study molecular mechanisms that cells use to migrate through these tunnels. Interestingly, MMPs are required for making these micro-tunnels in the first place, but then existing microfabricated tunnels facilitate MMP independent tumor cell invasion 135, 136.
A limiting factor for tumor cell migration within the 3D ECM network is the nucleus, which is the stiffest part of a cell. It has been reported that tumor cells fail to migrate when their nucleus size is larger than the ECM pore size 137. To understand the nuclear mechanics, microfluidic models have been developed to precisely mimic the constrictions presented to the cells by the ECM pores. Here, an array of micropillars with a few micrometers spacing is fabricated on a substrate 138–140. Nuclear envelope rupture and DNA damage is observed when tumor cells squeeze through gaps with spacing smaller than the nucleus. Interestingly, the majority of tumor cells are seen to self repair and survive 140, 141.
With the rapid progress in biomaterials, we can now fine tune the stiffness of type I collagen as well as its architecture individually or together, enabling a mechanistic understanding of the role of mechanical properties of ECM in tumor cell invasion. Increasing collagen concentration is a simple and straightforward method for increasing gel stiffness, however, this generates significant smaller pore sizes at the same time 137, 142, 143. A number of other techniques have been developed to increase the collagen stiffness without altering the concentration, they include varying polymerization temperature 120, 144 and pH 119, nonenzymatic cross-linking of the fibers 145, 146, and applying mechanical tension 129, 147. Future development requires integrating these biomaterial manipulation techniques with microfabricated device to pattern precisely the matrix stiffness or pore size in space for high throughput studies of tumor cell-ECM interactions.
Opportunities, challenges and future perspective
The biophysical microenvironment critically regulates tumor cell invasion. To date, the majority of the cell and drug screening experiments in vitro are carried out in static conditions, whereas biological flows are everywhere in the human body. Incorporating flows in tumor cell invasion experiments and beyond could potentially revolutionize our understanding in cancer biology. One can foresee the incoming impact will be similar to the impact brought by using 3D ECM in comparison to 2D platform for in vitro tumor cell invasion studies. Looking ahead, several challenges require our immediate attentions.
Recapitulating the complex tumor biophysical microenvironment
Microfluidic models have enabled us to learn the individual roles of ECM mechanical properties, intramural and interstitial flows in tumor cell invasion. However, synergistic roles of multiple biophysical factors remain to be explored. Microfluidic devices provide a unique opportunity here, because one can easily introduce biophysical parameters into the platform in a reconfigurable way. An example of such a device is illustrated in Fig. 3, where biophysical parameters including intramural and interstitial flows can be added or subtracted with ease. Here, two vascular vessels are created mimicking blood and lymphatic vessels respectively, with tumor cells embedded within ECM placed adjacent to the vascular vessels. In this platform, one can fine tune intramural, interstitial flows and ECM stiffness or pore sizes while observing tumor cell dynamic behavior at the same time. This capability allows for a basic understanding of how these three physical parameters synergistically influence tumor cell invasion within a complex and well-defined TME.
Figure 3.

Illustration of a microfluidic model recreating the biophysical microenvironment for tumor cell invasion studies. ECM or tumor cell embedded ECM (pink) is introduced into three wall-less channels confined by contact lines (yellow rectangular, not to scaled). Blood vessel (red) and lymphatic vessel (green) are formed by growing a layer of blood and lymphatic EC cells respectively. Blood and lymph flows are introduced into two vascular vessels (direction marked by x), and interstitial flow is introduced horizontally (direction marked by an arrow).
Biochemical environments such as cytokine gradients are traditional driving forces for tumor cell invasion, and are coupled with the biophysical environment. Towards this end, it will be important to first learn how biochemical gradients influence tumor cell invasion 148, and incorporate this information into the biophysical models proposed. Microfluidic models such as the one shown in Figure 3 can be easily used to generate cytokine gradients to meet this purpose. For example, we can flow cytokine and buffer respectively into the arteriole and venule channels, and a cytokine gradient can be established within the tumor embedded ECM space. The integration of biochemical and biophysical environments will be an important step towards a physiologically realistic microfluidic model.
Creating robust and highly reproducible platforms
Despite the aforementioned many advantages of using microfluidic models, microfluidic technology is still at an early stage, and is not widely accepted by many cancer biology labs. The bottlenecks are availability, ease of use, robustness and reproducibility. Most microfluidic models require engineering training for their use. To make a higher impact in cancer biology, it is important to design devices that are simple in concept, easy to use, and compatible with conventional biology labs. Partnering with industry is one possible route to make microfluidic models available to wider communities 19. Various agencies such as National Institute of Health provide special funds (e. g. SBIR) to encourage industry/academy partnerships. It is commendable that many research groups have now taken the lead in translating their microfluidic tools into the commercial space. Recent development in 3D printing technologies enables production of assembly-free microfluidics and eliminates the need of microfabrication for devices at micrometer or millimeter length scale 149. For ease of use, applying the physical principle of fluids (viscous fingering) to make an EC tube or vascular network within a channel is an alternative method to fabricating complicated features on a device 93. In addition, a design with gravitational driven flow will be more favorable than using pumps to create biological flows through a channel.
Developing quantitative analysis tools for understanding cell-environment interaction
Microfluidic devices allow us to collect large amounts of quantitative information. However, to make sense of the data, or to come up with a theoretical framework of the subject being investigated, it is important to develop quantitative analysis tools. For understanding the roles of physical parameters in tumor cell invasion, one useful tool is to track tumor cells in 3D in an automatic way. Commercial software such as Imaris has made significant progress in this direction, however, it still cannot handle the dynamic variability of tumor cells. A particularly difficult situation is when a cell dynamically changes its shape and merge and then dissociate with another cell in a trajectory - the tracking program does not have the ability to follow the cells. Recent advances in computer vision and machine learning could potentially resolve tracking of the shape and position of the moving cancer cells in space and time. A second tool is cell traction force measurements in 3D. The key regulator for cell-physical environment interaction is cell generated force. A number of 3D single cell traction force microscopy methods have been developed for measuring cell generated forces, but are far from easy to use, and are limited to a few labs around the world at the moment 78, 150, 151.
Outlook
Cancer cells are known to be heterogenous phenotypically and genetically. The altered tumor physical microenvironment, e.g. matrix spatial restriction 152, is a highly relevant contributor to the phenotypic and genetic instability of the invading tumor cells. An ultimate solution to the problem is to reveal molecular and cell level information simultaneously with a controlled microenvironment at single cell level. Recent developments in biosensors (mechanosensors and chemosensors) has made this possible. In addition, microfluidics with on chip polymerase chain reaction capability can be incorporated to further study the gene expression for those invading single tumor cells 153, 154. This platform will be powerful to illustrate the link between gene expression, molecular signaling, and phenotypes of single cancer cells and the altered biophysical microenvironment. Finally, to make an impact in our understanding on how biophysical cues in tumor cell invasion, a close collaboration among microsystems engineers and cancer biologists is critical for moving this field forward. This will require both parties to explore outside their field, attend meetings that they don’t normally go to, and not be afraid to ask questions.
Acknowledgments
This work is supported by grants from the National Center for Research Resources (5R21RR025801-03) and the NIH (8R21 GM103388-03, R21CA138366, R01CA221346, U54CA143876, and P01CA100324). We thank Hall for his drawing of the device in the Table of Contents entry.
Footnotes
Conflict of interest
There are no conflicts to declare.
References
- 1.Chambers AF, Groom AC, MacDonald IC. Nat Rev Cancer. 2002;2:563–572. doi: 10.1038/nrc865. [DOI] [PubMed] [Google Scholar]
- 2.Steeg PS. Nat Med. 2006;12:895–904. doi: 10.1038/nm1469. [DOI] [PubMed] [Google Scholar]
- 3.Wirtz D, Konstantopoulos K, Searson PC. Nat Rev Cancer. 2011;11:512–522. doi: 10.1038/nrc3080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Paszek MJ, Weaver VM. J Mammary Gland Biol Neoplasia. 2004;9:325–342. doi: 10.1007/s10911-004-1404-x. [DOI] [PubMed] [Google Scholar]
- 5.Paszek MJ, Zahir N, Johnson KR, Lakins JN, Rozenberg GI, Gefen A, Reinhart-King CA, Margulies SS, Dembo M, Boettiger D, Hammer DA, Weaver VM. Cancer cell. 2005;8:241–254. doi: 10.1016/j.ccr.2005.08.010. [DOI] [PubMed] [Google Scholar]
- 6.Kumar S, Weaver VM. Cancer and Metastasis Reviews. 2009;28:113–127. doi: 10.1007/s10555-008-9173-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Joyce JA, Pollard JW. Nat Rev Cancer. 2009;9:239–252. doi: 10.1038/nrc2618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bissell MJ, Hines WC. Nat Med. 2011;17:320–329. doi: 10.1038/nm.2328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pickup MW, Mouw JK, Weaver VM. EMBO reports. 2014;15:1243–1253. doi: 10.15252/embr.201439246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Marshall J. Methods in molecular biology (Clifton, NJ) 2011;769:97–110. doi: 10.1007/978-1-61779-207-6_8. [DOI] [PubMed] [Google Scholar]
- 11.Hulkower KI, Herber RL. Pharmaceutics. 2011;3:107–124. doi: 10.3390/pharmaceutics3010107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Albini A, Noonan DM. Curr Opin Cell Biol. 2010;22:677–689. doi: 10.1016/j.ceb.2010.08.017. [DOI] [PubMed] [Google Scholar]
- 13.Roussos ET, Condeelis JS, Patsialou A. Nat Rev Cancer. 2011;11:573–587. doi: 10.1038/nrc3078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhou ZN, Boimel PJ, Segall JE. Drug discovery today Disease models. 2011;8:95–112. doi: 10.1016/j.ddmod.2011.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cekanova M, Rathore K. Drug design, development and therapy. 2014;8:1911–1921. doi: 10.2147/DDDT.S49584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kim BJ, Wu M. Annals of biomedical engineering. 2012;40:1316–1327. doi: 10.1007/s10439-011-0489-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Polacheck WJ, Li R, Uzel SGM, Kamm RD. Lab on a chip. 2013;13:2252–2267. doi: 10.1039/c3lc41393d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sung KE, Beebe DJ. Advanced Drug Delivery Reviews. 2014;79–80:68–78. doi: 10.1016/j.addr.2014.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sackmann EK, Fulton AL, Beebe DJ. Nature. 2014;507:181–189. doi: 10.1038/nature13118. [DOI] [PubMed] [Google Scholar]
- 20.Tien J. Current Opinion in Chemical Engineering. 2014;3:36–41. [Google Scholar]
- 21.Boussommier-Calleja A, Li R, Chen MB, Wong SC, Kamm RD. Trends Cancer. 2016;2:6–19. doi: 10.1016/j.trecan.2015.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hanahan D, Weinberg Robert A. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 23.Friedl P, Alexander S. Cell. 2011;147:992–1009. doi: 10.1016/j.cell.2011.11.016. [DOI] [PubMed] [Google Scholar]
- 24.Meacham CE, Morrison SJ. Nature. 2013;501:328–337. doi: 10.1038/nature12624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wu J, Wu X, Lin F. Lab on a Chip. 2013;13:2484–2499. doi: 10.1039/c3lc50415h. [DOI] [PubMed] [Google Scholar]
- 26.Moraes C, Mehta G, Lesher-Perez SC, Takayama S. Ann Biomed Eng. 2012;40:1211–1227. doi: 10.1007/s10439-011-0455-6. [DOI] [PubMed] [Google Scholar]
- 27.Young EW. Integrative biology : quantitative biosciences from nano to macro. 2013;5:1096–1109. doi: 10.1039/c3ib40076j. [DOI] [PubMed] [Google Scholar]
- 28.GABE IT, GAULT JH, ROSS J, MASON DT, MILLS CJ, SCHILLINGFORD JP, BRAUNWALD E. Circulation. 1969;40:603–614. doi: 10.1161/01.cir.40.5.603. [DOI] [PubMed] [Google Scholar]
- 29.Popel AS, Johnson PC. Annual review of fluid mechanics. 2005;37:43–69. doi: 10.1146/annurev.fluid.37.042604.133933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hahn C, Schwartz MA. Nature reviews Molecular cell biology. 2009;10:53–62. doi: 10.1038/nrm2596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.le Noble F, Moyon D, Pardanaud L, Yuan L, Djonov V, Matthijsen R, Breant C, Fleury V, Eichmann A. Development (Cambridge, England) 2004;131:361–375. doi: 10.1242/dev.00929. [DOI] [PubMed] [Google Scholar]
- 32.Culver JC, Dickinson ME. Microcirculation (New York, NY. 1994;17:164–178. doi: 10.1111/j.1549-8719.2010.00025.x. 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lucitti JL, Jones EAV, Huang C, Chen J, Fraser SE, Dickinson ME. Development (Cambridge, England) 2007;134:3317–3326. doi: 10.1242/dev.02883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Davies PF. Physiol Rev. 1995;75:519–560. doi: 10.1152/physrev.1995.75.3.519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Resnick N, Yahav H, Shay-Salit A, Shushy M, Schubert S, Zilberman LCM, Wofovitz E. Progress in Biophysics and Molecular Biology. 2003;81:177–199. doi: 10.1016/s0079-6107(02)00052-4. [DOI] [PubMed] [Google Scholar]
- 36.Li YSJ, Haga JH, Chien S. Journal of Biomechanics. 2005;38:1949–1971. doi: 10.1016/j.jbiomech.2004.09.030. [DOI] [PubMed] [Google Scholar]
- 37.Chien S. American journal of physiology Heart and circulatory physiology. 2007;292:H1209–1224. doi: 10.1152/ajpheart.01047.2006. [DOI] [PubMed] [Google Scholar]
- 38.Dixon JB, Zawieja DC, Gashev AA, Coté GL. BIOMEDO. 2005;10:064016–064016–064017. doi: 10.1117/1.2135791. [DOI] [PubMed] [Google Scholar]
- 39.Berk DA, Swartz MA, Leu AJ, Jain RK. The American journal of physiology. 1996;270:H330–337. doi: 10.1152/ajpheart.1996.270.1.H330. [DOI] [PubMed] [Google Scholar]
- 40.Jain RK. Cancer research. 1988;48:2641–2658. [PubMed] [Google Scholar]
- 41.Stylianopoulos T, Martin JD, Snuderl M, Mpekris F, Jain SR, Jain RK. Cancer research. 2013;73:3833–3841. doi: 10.1158/0008-5472.CAN-12-4521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jain RK, Martin JD, Stylianopoulos T. Annual review of biomedical engineering. 2014;16:321–346. doi: 10.1146/annurev-bioeng-071813-105259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wiig H, Swartz MA. Physiological Reviews. 2012;92:1005–1060. doi: 10.1152/physrev.00037.2011. [DOI] [PubMed] [Google Scholar]
- 44.Wong SY, Hynes RO. Cell cycle (Georgetown, Tex) 2006;5:812–817. doi: 10.4161/cc.5.8.2646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Paduch R. Cellular Oncology. 2016;39:397–410. doi: 10.1007/s13402-016-0281-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chambers AF, Naumov GN, Varghese HJ, Nadkarni KV, MacDonald IC, Groom AC. Surgical oncology clinics of North America. 2001;10:243–255. vii. [PubMed] [Google Scholar]
- 47.Koumoutsakos P, Pivkin I, Milde F. Annual Review of Fluid Mechanics. 2013;45:325–355. [Google Scholar]
- 48.Swartz MA, Skobe M. Microscopy research and technique. 2001;55:92–99. doi: 10.1002/jemt.1160. [DOI] [PubMed] [Google Scholar]
- 49.Jain RK. Cancer research. 1987;47:3039–3051. [PubMed] [Google Scholar]
- 50.Chary SR, Jain RK. Proceedings of the National Academy of Sciences of the United States of America. 1989;86:5385–5389. doi: 10.1073/pnas.86.14.5385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ng CP, Helm CL, Swartz MA. Microvascular research. 2004;68:258–264. doi: 10.1016/j.mvr.2004.08.002. [DOI] [PubMed] [Google Scholar]
- 52.Helm CLE, Fleury ME, Zisch AH, Boschetti F, Swartz MA. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:15779–15784. doi: 10.1073/pnas.0503681102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Helm CL, Zisch A, Swartz MA. Biotechnology and bioengineering. 2007;96:167–176. doi: 10.1002/bit.21185. [DOI] [PubMed] [Google Scholar]
- 54.Butler TP, Grantham FH, Gullino PM. Cancer research. 1975;35:3084–3088. [PubMed] [Google Scholar]
- 55.Hompland T, Ellingsen C, Øvrebø KM, Rofstad EK. Cancer research. 2012;72:4899–4908. doi: 10.1158/0008-5472.CAN-12-0903. [DOI] [PubMed] [Google Scholar]
- 56.Jain RK, Baxter LT. Cancer research. 1988;48:7022–7032. [PubMed] [Google Scholar]
- 57.Boucher Y, Baxter LT, Jain RK. Cancer research. 1990;50:4478–4484. [PubMed] [Google Scholar]
- 58.Jain RK. Science. 1996;271:1079–1080. doi: 10.1126/science.271.5252.1079. [DOI] [PubMed] [Google Scholar]
- 59.Heldin CH, Rubin K, Pietras K, Ostman A. Nat Rev Cancer. 2004;4:806–813. doi: 10.1038/nrc1456. [DOI] [PubMed] [Google Scholar]
- 60.Lunt SJ, Fyles A, Hill RP, Milosevic M. Future Oncology. 2008;4:793–802. doi: 10.2217/14796694.4.6.793. [DOI] [PubMed] [Google Scholar]
- 61.Shields JD, Fleury ME, Yong C, Tomei AA, Randolph GJ, Swartz MA. Cancer cell. 2007;11:526–538. doi: 10.1016/j.ccr.2007.04.020. [DOI] [PubMed] [Google Scholar]
- 62.Adrian CS, Melody AS. Physical Biology. 2011;8:015012. doi: 10.1088/1478-3975/8/1/015012. [DOI] [PubMed] [Google Scholar]
- 63.Munson JM, Bellamkonda RV, Swartz MA. Cancer research. 2013;73:1536–1546. doi: 10.1158/0008-5472.CAN-12-2838. [DOI] [PubMed] [Google Scholar]
- 64.Kingsmore KM, Logsdon DK, Floyd DH, Peirce SM, Purow BW, Munson JM. Integrative biology : quantitative biosciences from nano to macro. 2016 doi: 10.1039/c6ib00167j. [DOI] [PubMed] [Google Scholar]
- 65.Polacheck WJ, German AE, Mammoto A, Ingber DE, Kamm RD. Proceedings of the National Academy of Sciences. 2014;111:2447–2452. doi: 10.1073/pnas.1316848111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Shieh AC, Rozansky HA, Hinz B, Swartz MA. Cancer research. 2011;71:790–800. doi: 10.1158/0008-5472.CAN-10-1513. [DOI] [PubMed] [Google Scholar]
- 67.Lu P, Weaver VM, Werb Z. The Journal of Cell Biology. 2012;196:395–406. doi: 10.1083/jcb.201102147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Pathak A, Kumar S. Integrative biology : quantitative biosciences from nano to macro. 2011;3:267–278. doi: 10.1039/c0ib00095g. [DOI] [PubMed] [Google Scholar]
- 69.Chang JM, Moon WK, Cho N, Yi A, Koo HR, Han W, Noh DY, Moon HG, Kim SJ. Breast Cancer Research and Treatment. 2011;129:89–97. doi: 10.1007/s10549-011-1627-7. [DOI] [PubMed] [Google Scholar]
- 70.Boyd NF, Lockwood GA, Byng JW, Tritchler DL, Yaffe MJ. Cancer Epidemiol Biomarkers Prev. 1998;7:1133–1144. [PubMed] [Google Scholar]
- 71.Acerbi I, Cassereau L, Dean I, Shi Q, Au A, Park C, Chen YY, Liphardt J, Hwang ES, Weaver VM. Integrative biology : quantitative biosciences from nano to macro. 2015;7:1120–1134. doi: 10.1039/c5ib00040h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kirschmann DA, Seftor EA, Fong SF, Nieva DR, Sullivan CM, Edwards EM, Sommer P, Csiszar K, Hendrix MJ. Cancer research. 2002;62:4478–4483. [PubMed] [Google Scholar]
- 73.Levental KR, Yu H, Kass L, Lakins JN, Egeblad M, Erler JT, Fong SFT, Csiszar K, Giaccia A, Weninger W, Yamauchi M, Gasser DL, Weaver VM. Cell. 2009;139:891–906. doi: 10.1016/j.cell.2009.10.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Condeelis J, Segall JE. Nat Rev Cancer. 2003;3:921–930. doi: 10.1038/nrc1231. [DOI] [PubMed] [Google Scholar]
- 75.Weigelin B, Bakker GJ, Friedl P. Intravital. 2012;1:32–43. doi: 10.4161/intv.21223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Alexander S, Weigelin B, Winkler F, Friedl P. Current Opinion in Cell Biology. 2013;25:659–671. doi: 10.1016/j.ceb.2013.07.001. [DOI] [PubMed] [Google Scholar]
- 77.Gehler S, Baldassarre M, Lad Y, Leight JL, Wozniak MA, Riching KM, Eliceiri KW, Weaver VM, Calderwood DA, Keely PJ. Molecular Biology of the Cell. 2009;20:3224–3238. doi: 10.1091/mbc.E08-12-1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Hall MS, Alisafaei F, Ban E, Feng X, Hui C-Y, Shenoy VB, Wu M. Proceedings of the National Academy of Sciences. 2016 doi: 10.1073/pnas.1613058113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Provenzano PP, Eliceiri KW, Campbell JM, Inman DR, White JG, Keely PJ. BMC Medicine. 2006;4:38. doi: 10.1186/1741-7015-4-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Conklin MW, Eickhoff JC, Riching KM, Pehlke CA, Eliceiri KW, Provenzano PP, Friedl A, Keely PJ. Am J Pathol. 2011;178:1221–1232. doi: 10.1016/j.ajpath.2010.11.076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Gaggioli C, Hooper S, Hidalgo-Carcedo C, Grosse R, Marshall JF, Harrington K, Sahai E. Nat Cell Biol. 2007;9:1392–1400. doi: 10.1038/ncb1658. [DOI] [PubMed] [Google Scholar]
- 82.Gritsenko PG, Ilina O, Friedl P. The Journal of pathology. 2012;226:185–199. doi: 10.1002/path.3031. [DOI] [PubMed] [Google Scholar]
- 83.Wolf K, Alexander S, Schacht V, Coussens LM, von Andrian UH, van Rheenen J, Deryugina E, Friedl P. Seminars in Cell & Developmental Biology. 2009;20:931–941. doi: 10.1016/j.semcdb.2009.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Doyle AD, Wang FW, Matsumoto K, Yamada KM. J Cell Biol. 2009;184:481–490. doi: 10.1083/jcb.200810041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Friedl P, Sahai E, Weiss S, Yamada KM. Nature reviews Molecular cell biology. 2012;13:743–747. doi: 10.1038/nrm3459. [DOI] [PubMed] [Google Scholar]
- 86.Chang Stephanie S, Guo W-h, Kim Y, Wang Y-l. Biophysical Journal. 2013;104:313–321. doi: 10.1016/j.bpj.2012.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Chrobak KM, Potter DR, Tien J. Microvascular research. 2006;71:185–196. doi: 10.1016/j.mvr.2006.02.005. [DOI] [PubMed] [Google Scholar]
- 88.Nguyen DHT, Stapleton SC, Yang MT, Cha SS, Choi CK, Galie PA, Chen CS. Proceedings of the National Academy of Sciences. 2013;110:6712–6717. doi: 10.1073/pnas.1221526110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Wong AD, Searson PC. Cancer research. 2014;74:4937–4945. doi: 10.1158/0008-5472.CAN-14-1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Jimenez-Torres JA, Beebe DJ, Sung KE. Methods in molecular biology (Clifton, NJ) 2016;1458:59–69. doi: 10.1007/978-1-4939-3801-8_5. [DOI] [PubMed] [Google Scholar]
- 91.Zheng Y, Chen J, Craven M, Choi NW, Totorica S, Diaz-Santana A, Kermani P, Hempstead B, Fischbach-Teschl C, López JA, Stroock AD. Proceedings of the National Academy of Sciences. 2012;109:9342–9347. doi: 10.1073/pnas.1201240109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Buchanan CF, Verbridge SS, Vlachos PP, Rylander MN. Cell Adhesion & Migration. 2014;8:517–524. doi: 10.4161/19336918.2014.970001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Bischel LL, Lee SH, Beebe DJ. Journal of laboratory automation. 2012;17:96–103. doi: 10.1177/2211068211426694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Bischel LL, Young EWK, Mader BR, Beebe DJ. Biomaterials. 2013;34:1471–1477. doi: 10.1016/j.biomaterials.2012.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Bischel LL, Sung KE, Jiménez-Torres JA, Mader B, Keely PJ, Beebe DJ. The FASEB Journal. 2014;28:4583–4590. doi: 10.1096/fj.13-243733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Golden AP, Tien J. Lab Chip. 2007;7:720–725. doi: 10.1039/b618409j. [DOI] [PubMed] [Google Scholar]
- 97.Bellan LM, Singh SP, Henderson PW, Porri TJ, Craighead HG, Spector JA. Soft Matter. 2009;5:1354–1357. [Google Scholar]
- 98.Miller JS, Stevens KR, Yang MT, Baker BM, Nguyen D-HT, Cohen DM, Toro E, Chen AA, Galie PA, Yu X, Chaturvedi R, Bhatia SN, Chen CS. Nat Mater. 2012;11:768–774. doi: 10.1038/nmat3357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Kolesky DB, Truby RL, Gladman AS, Busbee TA, Homan KA, Lewis JA. Advanced Materials. 2014;26:3124–3130. doi: 10.1002/adma.201305506. [DOI] [PubMed] [Google Scholar]
- 100.Bertassoni LE, Cecconi M, Manoharan V, Nikkhah M, Hjortnaes J, Cristino AL, Barabaschi G, Demarchi D, Dokmeci MR, Yang Y, Khademhosseini A. Lab on a Chip. 2014;14:2202–2211. doi: 10.1039/c4lc00030g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Sobrino A, Phan DTT, Datta R, Wang X, Hachey SJ, Romero-López M, Gratton E, Lee AP, George SC, Hughes CCW. Scientific Reports. 2016;6:31589. doi: 10.1038/srep31589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Moya ML, Hsu Y-H, Lee AP, Hughes CCW, George SC. Tissue Engineering Part C: Methods. 2013;19:730–737. doi: 10.1089/ten.tec.2012.0430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Phan DTT, Wang X, Craver BM, Sobrino A, Zhao D, Chen JC, Lee LYN, George SC, Lee AP, Hughes CCW. Lab on a Chip. 2017;17:511–520. doi: 10.1039/c6lc01422d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Kim S, Chung M, Jeon NL. Biomaterials. 2016;78:115–128. doi: 10.1016/j.biomaterials.2015.11.019. [DOI] [PubMed] [Google Scholar]
- 105.Gerlowski LE, Jain RK. Microvascular research. 1986;31:288–305. doi: 10.1016/0026-2862(86)90018-x. [DOI] [PubMed] [Google Scholar]
- 106.Price GM, Wong KHK, Truslow JG, Leung AD, Acharya C, Tien J. Biomaterials. 2010;31:6182–6189. doi: 10.1016/j.biomaterials.2010.04.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Jeon JS, Bersini S, Gilardi M, Dubini G, Charest JL, Moretti M, Kamm RD. Proceedings of the National Academy of Sciences. 2015;112:214–219. doi: 10.1073/pnas.1417115112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Song JW, Munn LL. Proceedings of the National Academy of Sciences. 2011;108:15342–15347. doi: 10.1073/pnas.1105316108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Zhang Q, Liu T, Qin J. Lab on a Chip. 2012;12:2837–2842. doi: 10.1039/c2lc00030j. [DOI] [PubMed] [Google Scholar]
- 110.Zervantonakis IK, Hughes-Alford SK, Charest JL, Condeelis JS, Gertler FB, Kamm RD. Proceedings of the National Academy of Sciences. 2012;109:13515–13520. doi: 10.1073/pnas.1210182109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Ehsan SM, Welch-Reardon KM, Waterman ML, Hughes CC, George SC. Integrative biology : quantitative biosciences from nano to macro. 2014;6:603–610. doi: 10.1039/c3ib40170g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Pisano M, Triacca V, Barbee KA, Swartz MA. Integrative Biology. 2015;7:525–533. doi: 10.1039/c5ib00085h. [DOI] [PubMed] [Google Scholar]
- 113.Roeder BA, Kokini K, Sturgis JE, Robinson JP, Voytik-Harbin SL. J Biomech Eng. 2002;124:214–222. doi: 10.1115/1.1449904. [DOI] [PubMed] [Google Scholar]
- 114.Storm C, Pastore JJ, MacKintosh FC, Lubensky TC, Janmey PA. Nature. 2005;435:191–194. doi: 10.1038/nature03521. [DOI] [PubMed] [Google Scholar]
- 115.Polacheck WJ, Charest JL, Kamm RD. Proceedings of the National Academy of Sciences. 2011;108:11115–11120. doi: 10.1073/pnas.1103581108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Haessler U, Teo JC, Foretay D, Renaud P, Swartz MA. Integrative biology : quantitative biosciences from nano to macro. 2012;4:401–409. doi: 10.1039/c1ib00128k. [DOI] [PubMed] [Google Scholar]
- 117.Tung CK, Krupa O, Apaydin E, Liou JJ, Diaz-Santana A, Kim BJ, Wu M. Lab Chip. 2013;13:3876–3885. doi: 10.1039/c3lc50489a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Huang YL, Tung C-k, Zheng A, Kim BJ, Wu M. Integrative Biology. 2015;7:1402–1411. doi: 10.1039/c5ib00115c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Raub CB, Unruh J, Suresh V, Krasieva T, Lindmo T, Gratton E, Tromberg BJ, George SC. Biophysical Journal. 2008;94:2361–2373. doi: 10.1529/biophysj.107.120006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Yang YL, Motte S, Kaufman LJ. Biomaterials. 2010;31:5678–5688. doi: 10.1016/j.biomaterials.2010.03.039. [DOI] [PubMed] [Google Scholar]
- 121.Piotrowski-Daspit AS, Tien J, Nelson CM. Integrative Biology. 2016;8:319–331. doi: 10.1039/c5ib00282f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Friedl P, Wolf K. Nat Rev Cancer. 2003;3:362–374. doi: 10.1038/nrc1075. [DOI] [PubMed] [Google Scholar]
- 123.Friedl P, Locker J, Sahai E, Segall JE. Nat Cell Biol. 2012;14:777–783. doi: 10.1038/ncb2548. [DOI] [PubMed] [Google Scholar]
- 124.Patsialou A, Bravo-Cordero JJ, Wang Y, Entenberg D, Liu H, Clarke M, Condeelis JS. Intravital. 2013;2:e25294. doi: 10.4161/intv.25294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Rizvi I, Gurkan UA, Tasoglu S, Alagic N, Celli JP, Mensah LB, Mai Z, Demirci U, Hasan T. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:E1974–E1983. doi: 10.1073/pnas.1216989110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Nelson MT, Short A, Cole SL, Gross AC, Winter J, Eubank TD, Lannutti JJ. BMC Cancer. 2014;14:825. doi: 10.1186/1471-2407-14-825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Milano Daniel F, Ngai Nicholas A, Muthuswamy Senthil K, Asthagiri Anand R. Biophysical Journal. 2016;110:1886–1895. doi: 10.1016/j.bpj.2016.02.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Lee P, Lin R, Moon J, Lee LP. Biomedical microdevices. 2006;8:35–41. doi: 10.1007/s10544-006-6380-z. [DOI] [PubMed] [Google Scholar]
- 129.Riching KM, Cox BL, Salick MR, Pehlke C, Riching AS, Ponik SM, Bass BR, Crone WC, Jiang Y, Weaver AM, Eliceiri KW, Keely PJ. Biophys J. 2014;107:2546–2558. doi: 10.1016/j.bpj.2014.10.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Sung KE, Su G, Pehlke C, Trier SM, Eliceiri KW, Keely PJ, Friedl A, Beebe DJ. Biomaterials. 2009;30:4833–4841. doi: 10.1016/j.biomaterials.2009.05.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Lanfer B, Freudenberg U, Zimmermann R, Stamov D, Korber V, Werner C. Biomaterials. 2008;29:3888–3895. doi: 10.1016/j.biomaterials.2008.06.016. [DOI] [PubMed] [Google Scholar]
- 132.Han W, Chen S, Yuan W, Fan Q, Tian J, Wang X, Chen L, Zhang X, Wei W, Liu R, Qu J, Jiao Y, Austin RH, Liu L. Proceedings of the National Academy of Sciences. 2016;113:11208–11213. doi: 10.1073/pnas.1610347113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Vader D, Kabla A, Weitz D, Mahadevan L. PloS one. 2009;4:e5902. doi: 10.1371/journal.pone.0005902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Kim J, Staunton JR, Tanner K. Advanced materials (Deerfield Beach, Fla) 2016;28:132–137. doi: 10.1002/adma.201503950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Ilina O, Bakker GJ, Vasaturo A, Hofmann RM, Friedl P. Phys Biol. 2011;8:015010. doi: 10.1088/1478-3975/8/1/015010. [DOI] [PubMed] [Google Scholar]
- 136.Kraning-Rush CM, Carey SP, Lampi MC, Reinhart-King CA. Integrative biology : quantitative biosciences from nano to macro. 2013;5:606–616. doi: 10.1039/c3ib20196a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Wolf K, te Lindert M, Krause M, Alexander S, te Riet J, Willis AL, Hoffman RM, Figdor CG, Weiss SJ, Friedl P. The Journal of Cell Biology. 2013;201:1069–1084. doi: 10.1083/jcb.201210152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Isermann P, Davidson PM, Sliz JD, Lammerding J. Current protocols in cell biology / editorial board, Juan S Bonifacino ... [et al] 2012;CHAPTER(Unit22.16-Unit22.16) doi: 10.1002/0471143030.cb2216s56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Davidson PM, Sliz J, Isermann P, Denais C, Lammerding J. Integrative Biology. 2015;7:1534–1546. doi: 10.1039/c5ib00200a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Denais CM, Gilbert RM, Isermann P, McGregor AL, te Lindert M, Weigelin B, Davidson PM, Friedl P, Wolf K, Lammerding J. Science. 2016 doi: 10.1126/science.aad7297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Skau CT, Fischer RS, Gurel P, Thiam HR, Tubbs A, Baird MA, Davidson MW, Piel M, Alushin GM, Nussenzweig A, Steeg PS, Waterman CM. Cell. 2016;167:1571–1585.e1518. doi: 10.1016/j.cell.2016.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 142.Miron-Mendoza M, Seemann J, Grinnell F. Biomaterials. 2010;31:6425–6435. doi: 10.1016/j.biomaterials.2010.04.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Cross VL, Zheng Y, Won Choi N, Verbridge SS, Sutermaster BA, Bonassar LJ, Fischbach C, Stroock AD. Biomaterials. 2010;31:8596–8607. doi: 10.1016/j.biomaterials.2010.07.072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Raub CB, Suresh V, Krasieva T, Lyubovitsky J, Mih JD, Putnam AJ, Tromberg BJ, George SC. Biophys J. 2007;92:2212–2222. doi: 10.1529/biophysj.106.097998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Roy R, Boskey A, Bonassar LJ. Journal of biomedical materials research Part A. 2010;93:843–851. doi: 10.1002/jbm.a.32231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Mason BN, Starchenko A, Williams RM, Bonassar LJ, Reinhart-King CA. Acta biomaterialia. 2013;9:4635–4644. doi: 10.1016/j.actbio.2012.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Cassereau L, Miroshnikova YA, Ou G, Lakins J, Weaver VM. Journal of Biotechnology. 2015;193:66–69. doi: 10.1016/j.jbiotec.2014.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Kim BJ, Hannanta-anan P, Chau M, Kim YS, Swartz MA, Wu M. PloS one. 2013;8:e68422. doi: 10.1371/journal.pone.0068422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Bhattacharjee N, Urrios A, Kang S, Folch A. Lab on a Chip. 2016;16:1720–1742. doi: 10.1039/c6lc00163g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Gjorevski N, Nelson CM. Biophys J. 2012;103:152–162. doi: 10.1016/j.bpj.2012.05.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Legant WR, Miller JS, Blakely BL, Cohen DM, Genin GM, Chen CS. Nature methods. 2010;7:969–971. doi: 10.1038/nmeth.1531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Irianto J, Xia Y, Pfeifer CR, Athirasala A, Ji J, Alvey C, Tewari M, Bennett RR, Harding SM, Liu AJ, Greenberg RA, Discher DE. Current Biology. 2017;27:210–223. doi: 10.1016/j.cub.2016.11.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Shembekar N, Chaipan C, Utharala R, Merten CA. Lab on a Chip. 2016;16:1314–1331. doi: 10.1039/c6lc00249h. [DOI] [PubMed] [Google Scholar]
- 154.Ahrberg CD, Manz A, Chung BG. Lab on a Chip. 2016;16:3866–3884. doi: 10.1039/c6lc00984k. [DOI] [PubMed] [Google Scholar]
- 155.Fischer M, Franzeck UK, Herrig I, Costanzo U, Wen S, Schiesser M, Hoffmann U, Bollinger A. American Journal of Physiology - Heart and Circulatory Physiology. 1996;270:H358–H363. doi: 10.1152/ajpheart.1996.270.1.H358. [DOI] [PubMed] [Google Scholar]
- 156.Yuan F, Salehi HA, Boucher Y, Vasthare US, Tuma RF, Jain RK. Cancer research. 1994;54:4564–4568. [PubMed] [Google Scholar]
- 157.Kamoun WS, Chae S-S, Lacorre DA, Tyrell JA, Mitre M, Gillissen MA, Fukumura D, Jain RK, Munn LL. Nature methods. 2010;7:655–660. doi: 10.1038/nmeth.1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Leunig M, Yuan F, Menger MD, Boucher Y, Goetz AE, Messmer K, Jain RK. Cancer research. 1992;52:6553–6560. [PubMed] [Google Scholar]
- 159.Dafni H, Israely T, Bhujwalla ZM, Benjamin LE, Neeman M. Cancer research. 2002;62:6731–6739. [PubMed] [Google Scholar]
- 160.Fu BM, Shen S. Microvascular research. 2004;68:51–62. doi: 10.1016/j.mvr.2004.03.004. [DOI] [PubMed] [Google Scholar]
- 161.Yuan W, Lv Y, Zeng M, Fu BM. Microvascular research. 2009;77:166–173. doi: 10.1016/j.mvr.2008.08.004. [DOI] [PubMed] [Google Scholar]
- 162.Scallan JP, Huxley VH. J Physiol. 2010;588:243–254. doi: 10.1113/jphysiol.2009.179622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Yuan F, Leunig M, Berk DA, Jain RK. Microvascular research. 1993;45:269–289. doi: 10.1006/mvre.1993.1024. [DOI] [PubMed] [Google Scholar]
- 164.Dreher MR, Liu W, Michelich CR, Dewhirst MW, Yuan F, Chilkoti A. J Natl Cancer Inst. 2006;98:335–344. doi: 10.1093/jnci/djj070. [DOI] [PubMed] [Google Scholar]
- 165.Albelda SM, Sampson PM, Haselton FR, McNiff JM, Mueller SN, Williams SK, Fishman AP, Levine EM. Journal of Applied Physiology. 1988;64:308–322. doi: 10.1152/jappl.1988.64.1.308. [DOI] [PubMed] [Google Scholar]
- 166.Swartz MA, Fleury ME. Annu Rev Biomed Eng. 2007;9:229–256. doi: 10.1146/annurev.bioeng.9.060906.151850. [DOI] [PubMed] [Google Scholar]
- 167.Levick JR. Quarterly journal of experimental physiology (Cambridge, England) 1987;72:409–437. doi: 10.1113/expphysiol.1987.sp003085. [DOI] [PubMed] [Google Scholar]
- 168.Ramanujan S, Pluen A, McKee TD, Brown EB, Boucher Y, Jain RK. Biophysical Journal. 2002;83:1650–1660. doi: 10.1016/S0006-3495(02)73933-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Michel CC, Curry FE. Physiol Rev. 1999;79:703–761. doi: 10.1152/physrev.1999.79.3.703. [DOI] [PubMed] [Google Scholar]