

Abstract
Magnetic relaxation has been used extensively to study and characterize biological tissues. In particular, spin-lattice relaxation in the rotating frame (T1ρ) of water in protein solutions has been demonstrated to be sensitive to macromolecular weight and composition. However, the nature of the contribution from low frequency processes to water relaxation remains unclear. We have examined this problem by studying the water T1ρ dispersion in peptide solutions (14N- and 15N-labeled), glycosaminoglycan solutions, and samples of bovine articular cartilage before and after proteoglycan degradation. We find in model systems and tissue that hydrogen exchange from NH and OH groups to water dominates the low frequency water T1ρ dispersion, in the context of the model used to interpret the relaxation data. Further, low frequency dispersion changes are correlated with loss of proteoglycan from the extra-cellular matrix of articular cartilage. This finding has significance for the noninvasive detection of matrix degradation.
Keywords: rotating frame‖T1ρ‖extra-cellular matrix‖osteoarthritis
Protein degradation with a loss of proteoglycan (PG) from the extra-cellular matrix is thought to be an initiating event of early osteoarthritis (1). A noninvasive imaging method that can monitor the progression of the disease would be highly desirable for the longitudinal evaluation of disease progression and the utility of therapeutic interventions. Because of the excellent soft tissue contrast and its noninvasive nature, MRI is an attractive modality for imaging cartilage. Unfortunately, currently available conventional MRI methods are unable to detect the earliest stages of the disease when biochemical changes occur without gross tissue damage (2). Recently, several MRI methods have been proposed to detect PG loss from cartilage (3, 4). In particular, spin-lattice relaxation in the rotating frame (T1ρ) has been demonstrated to be elevated in PG-depleted cartilage (5).
T1ρ relaxation is sensitive to molecular motions that have correlation times (τ) such that τωSL∼1, where ωSL = γBSL is the strength of the spin-lock field (6). T1ρ increases with the strength of the spin-lock field, a phenomenon termed dispersion. T1ρ measurements can therefore provide information about the biophysical mechanisms underlying magnetic relaxation. It has been demonstrated that water T1ρ relaxation and dispersion (in the 0.1–10 kHz regime) are sensitive to macromolecule–water interactions in protein solutions and possibly also in biological tissues (7–9). Low frequency (0.1–3 kHz) T1ρ dispersion has been observed in several systems such as protein solutions (7), bovine articular cartilage (5), human patellar cartilage (10), rodent brain (11), and murine tumor tissue (9). However, the exact nature of T1ρ dispersion in biological tissues remains unclear. The range of spin-lock strengths that can be used for in vivo measurements is 0.1–3 kHz (depending on the duration of the spin-lock pulse), without exceeding power deposition limits. Therefore, we have focused our investigations on the low frequency dispersion in biological systems. The goal of this work is to investigate the biophysical mechanisms underlying T1ρ relaxation in biological tissues. Such experiments may help to improve existing MRI methods and provide a basis for the quantitative interpretation of relaxation in normal and diseased cartilage.
Proton exchange between chemically shifted NH and OH groups and the solvent water, along with quadrupolar relaxation effects of the 14N (spin = 1) and 17O (spin = 5/2) modulated by the scalar coupling and proton exchange, contribute to the observed low frequency water T1ρ relaxation in articular cartilage. We have investigated these potential mechanisms underlying bulk water T1ρ dispersion by studying peptide solutions (a model for protein in cartilage), chondroitin sulfate (CS) type C solutions (a major component of cartilage PG), and bovine articular cartilage before and after sequential PG depletion. By using an unstructured polypeptide with many exchangeable NH protons but few hydroxyl groups, the effects of NH groups on water T1ρ dispersion were studied. 15N- to 14N-isotope substitution experiments were performed to determine the effect of 14N quadrupolar relaxation on bulk water relaxation. The effects of hydroxyl group exchange were investigated by measuring the T1ρ dispersion of CS solutions, which have many exchangeable OH groups. These data were used to give some general interpretations for the observed T1ρ dispersion profile of normal and PG-depleted cartilage.
Methods
Measurements on Model Solutions.
All NMR experiments were performed on a 2T magnet interfaced to a custom-built console at room temperature (22°C), unless otherwise stated. A 1-cm diameter solenoidal coil tuned to 86 MHz was used. Spectroscopic relaxation rates of water were measured as described (5). Water proton T1 was measured with an inversion recovery sequence [repetition time (TR) = 12 s, 1,024 data points, 5 kHz bandwidth, with 20 free induction decays (FIDs)], and the inversion time was varied from 400–8,000 ms in steps of 400 ms. T1ρ relaxation times were measured with a spin-locking sequence, with the length of the spin-lock pulse varied between 0.5–10 s in increments of 0.5 s, and the TR time was set to be greater than 5 T1 s. All other parameters were as given previously. T1ρ dispersion was characterized by varying the amplitude of the spin-lock pulse. Phosphate buffer (200 mM, pH 7.4) was used for all peptide NMR experiments. Polyethylene glycol (PEG) [average molecular weight = 1,450; 0.1% wt/vol (pH 7.4); Sigma] has no exchangeable protons, thus it provides a convenient control to determine the effects of proton exchange on T1ρ dispersion.
The peptide based on the calmodulin-binding domain of smooth muscle myosin light-chain kinase peptide from chicken [smMLCKp (GSARRKWQKTGHAVRAIGRLS)] was prepared as described in Escherichia coli BL21(DE3) cells (12). 15N-labeled peptide was prepared from E. coli grown on minimal media with 15NH4Cl as the only nitrogen source. Peptide solution purity was 99.9% (HPLC analysis). For NMR, the peptide was in phosphate buffer at the desired concentration.
CS type C was obtained from Sigma and used without further purification. CS solutions of various concentrations (2, 5, and 10% wt/vol) were in PBS (137 mM NaCl/2.7 mM KCl/10 mM phosphate, pH 7.4; Sigma). Collagen type II, from bovine Achilles tendon (Sigma), was used without further purification to make collagen suspensions (10% wt/vol) in PBS.
Measurements on Bovine Cartilage.
Bovine patellae were obtained from a local butcher. Cartilage discs (9-mm diameter) were cored from the articular surface of the patellae, underlying bone was removed, and the tissue was placed in PBS on ice for 2 h before NMR measurements. Enzymatic digestion was performed with trypsin (0.1 mg/ml; Sigma) at 26°C in PBS at pH 7.4. T1ρ dispersion profiles of bovine articular cartilage were measured before and after enzymatic digestion. The loss of PG from the extra-cellular matrix of the tissue was determined by the spectrophotometric assay of Frandale et al. (13). Any collagen loss induced by the trypsin digestion was determined according to the analysis of Bank et al. (14). T1ρ measurements used repetition time (TR) = 5 s, and the length of the spin-lock pulse varied from 20–600 ms in 30 equal increments. T1 measurements used a standard inversion-recovery sequence (TR = 6 s, 1,024 data points, 5 kHz bandwidth, 20 free induction decays); different interpulse delays were used to compute T1 values.
Relaxation times were determined by fitting the signal amplitude to a monoexponential decay as a function of the spin-locking time. The dispersion analysis was performed by fitting the relaxation rate (R1ρ = 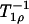 ) as a function of the spin-lock pulse strength (ωSL) according to the following Lorentzian equation:
) as a function of the spin-lock pulse strength (ωSL) according to the following Lorentzian equation:
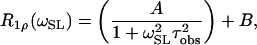 |
1 |
where A and B are constants, and τobs is the effective correlation time for T1ρ dispersion. This function describes the spectral character of any stochastic process and is maximized when ωSLτobs ≈ 1. This empirical function assumes that chemical exchange influences dispersion with an exponential autocorrelation function. A similar expression was shown to be applicable to relaxation caused by proton exchange (15). Dispersion profiles from cartilage were fit to a bi-Lorentzian function.
Results
Water R1ρ dispersion increased with the smMLCKp peptide concentration (Fig. 1). Isotopic substitution of 15N for 14N in the peptide reduced the R1ρ dispersion by only 10%, indicating that the effect of 14N quadrupolar relaxation on water is small at physiologic pH in this peptide system. The effective correlation time (τobs) that characterizes the dispersion was 0.58 ms, yielding a rate of ≈1,700 s−1 (Eq. 1). To test whether this dispersion was caused by a viscosity effect, the R1ρ dispersion of a polyethylene glycol (PEG) solution (0.1% wt/vol) was also measured and found not to increase dispersion (data not shown).
Figure 1.

Dependence of water R1ρ dispersion on peptide concentration. The dispersion of the buffer (▴), attributed to natural abundance H217O effects, increases in 0.9 mM 14N-peptide solution (■) and 1.6 mM 14N-peptide solution (⧫). The dispersion of the 1.6 mM 15N-peptide solution (●) is only 10% less than that of 1.6 mM 14N-peptide solution, indicating that 14N relaxation is not the dominant mechanism modulating the interaction between NH and water protons.
The dependence of R1ρ on solute concentration was also established with CS solutions (Fig. 2). The amplitude of the dispersion curve increased with the CS concentration. The effective correlation rate associated with water dispersion in the CS solutions was ≈1,200 s−1. It is evident that all of the curves do not asymptote to the same value. This shift in relaxation rate at high spin-lock fields is attributed to changes in T1 relaxation associated with increased concentrations of CS. Water R1ρ was found to linearly depend on CS concentration in vitro (Fig. 2B). The magnitude of this dependence varied with the used spin-lock amplitude, indicating that the relaxation effect of CS on water is frequency-dependent.
Figure 2.

Dependence of water R1ρ dispersion on CS concentration. (A) The dispersion of the buffer (▴) is less than that of 2 (⧫), 5 (●), and 10% (■) solutions of CS. The correlation time of these dispersion plots is in agreement with literature values for hydroxyl exchange times, under similar conditions. B shows the dependence of R1ρ with CS concentration at various spin-lock amplitudes: 314 rad/s (●), 930 rad/s (■), 4,650 rad/s (▴), and 1.1 × 104 rad/s (⧫).
In contrast to the model systems described hitherto, the water R1ρ dispersion profile of bovine articular cartilage was best described with a bi-Lorentzian function in the 0.1–2 kHz frequency range (Fig. 3). The parameters that characterize the dispersion profile before and after degradation are given in Table 1. The change in the slow correlation time, obtained by pooling the data from 5 samples each degraded through 3 sequential digestions (giving 15 data points), correlated with PG loss (P < 0.005, r = −0.74, Spearman's product moment correlation). The dispersion profile of the collagen suspension was also better characterized by a bi-Lorentzian than a single Lorentzian function (data not shown).
Figure 3.

Dependence of water R1ρ relaxation and dispersion in articular cartilage on PG loss. This figure shows the water dispersion profile of a representative sample of cartilage before (●), after 28% PG depletion (■), and after 60% PG depletion (▴). The error bar of measurement is about 0.5%. Solid lines represent fits to a bi-Lorentzian function. The low frequency dispersion is attributed to proton exchange from NH and OH groups, whereas the high frequency dispersion is the result of the exchange of entire water molecules (see text).
Table 1.
The characteristic parameters of the water R1ρ dispersion curve of articular cartilage in 5 separate specimens
| Average proteoglycan loss, % | Amplitude of slow component ×103, a.u. | Correlation time of slow component, ×10−6 sec*** | Amplitude of fast component ×104, a.u. | Correlation time of fast component, ×10−6 sec |
|---|---|---|---|---|
| 0 | 6.1 ± 1.3 | 443 ± 16 | 17 ± 6 | 23 ± 7 |
| 39.6 | 7.0 ± 1.2 | 387 ± 81 | 15 ± 6 | 25 ± 8 |
| 57.1 | 5.8 ± 1.6 | 353 ± 64 | 18 ± 2 | 20 ± 2 |
The data are reported as mean ± SD. The asterisks indicate that the decrease in the slow correlation time with proteoglycan loss was statistically significant (P < 0.005, r = −0.74).
Hydroxyproline was extruded into the digestion media after 1 h of trypsin digestion, presumably from collagen degradation. Subsequent digestion did not significantly increase collagen loss (P = 0.13). These data indicate that trypsin treatment caused only a minimal initial loss of collagen from the tissue. It is interesting to note that although the entire dispersion curve seems to be uniformly altered after the initial digestion (in which both PG and collagen were depleted), only the low frequency dispersion component is altered on subsequent digestion (in which PG was lost but collagen content was maintained).
Discussion
Despite ongoing research efforts, there is no consensus view of water relaxation in biological systems (16, 17). There are several theories to explain the magnetic field dependence of T1 relaxation (17), including one which proposes the exchange of a small number of well defined water molecules buried inside the protein with the bulk water on a submicrosecond time scale (16). Although this model explains the dispersion of T1 with field strength, the time scale is too fast to account for low field T1ρ dispersion.
T1ρ measurements are sensitive to slower motions and have been used to investigate the biophysical characteristics of protein solutions and biological tissues (18). However, most of these studies have been performed with spin-locking fields in the 2–30 kHz regime. Knispel et al. (9) have shown that a model which invokes a range of correlation times accounted best for the observed dispersion in the 2 kHz to 30 MHz regime, suggesting that several relaxation mechanisms are present in this frequency range. Virta et al. (7) found that T1ρ dispersion of protein solutions at low frequency (<8.5 kHz) is sensitive to protein content, denaturation, and cross-linking. A cross-relaxation mechanism by which magnetization is transferred from the protein system to the water by means of a “dipolar energy overlap” was proposed to explain these results (7).
We propose that the proton exchange of labile NH and OH protons with bulk water is a significant contributor to the low frequency T1ρ dispersion in biological systems. There are two elements to this relaxation mechanism. (i) The efficient relaxation of the NH and OH protons caused by the fast quadrupolar relaxation of the spin = 1 14N (19, 20) and spin = 5/2 17O is mediated by scalar coupling between 14N-1H or 17O-1H and chemical exchange with the bulk water to affect bulk water T1ρ relaxation (15). (ii) The proton exchange of the chemically shifted NH/OH moieties with water can also lead to water relaxation (21, 22). The phenomena of chemical exchange and fast quadrupolar relaxation contribute to water proton relaxation through the first mechanism. However, only hydrogen exchange can cause water relaxation through the second mechanism by modulating the chemical shift difference between NH/OH protons and water.
The transverse relaxation of water under the conditions of fast exchange has been given (23).
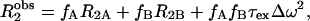 |
2 |
where 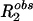 is the observed relaxation rate; fA,B is the mole fraction of species A and B, R2A,B is the transverse relaxation rate of the species A and B, respectively; τex is the chemical exchange time; and Δω is the chemical shift difference between the sites. An analogous expression has been derived to account for T1ρ relaxation in paramagnetic solutions by Chopra et al. (24). In the limit of low spin-lock amplitudes, T1ρ approximates T2. We can use Eq. 2 to interpret the low frequency T1ρ data presented in this article by using typical values for NH chemical shifts, relaxation times, and concentrations.
is the observed relaxation rate; fA,B is the mole fraction of species A and B, R2A,B is the transverse relaxation rate of the species A and B, respectively; τex is the chemical exchange time; and Δω is the chemical shift difference between the sites. An analogous expression has been derived to account for T1ρ relaxation in paramagnetic solutions by Chopra et al. (24). In the limit of low spin-lock amplitudes, T1ρ approximates T2. We can use Eq. 2 to interpret the low frequency T1ρ data presented in this article by using typical values for NH chemical shifts, relaxation times, and concentrations.
We have evaluated the role of proton exchange by studying two relevant systems, smMLCKp, to measure the NH contribution within a peptide system, and CS, to measure the OH contribution from PG. The unblocked smMLCKp provides a meaningful model for NH-mediated water relaxation effects. Each molecule has a total of 54 exchangeable NH protons and only 3 hydroxyl groups (the C-terminal hydroxyl group is deprotonated at experimental conditions) on the peptide, therefore the water R1ρ dispersion of the smMLCKp solution should be dominated by NH-mediated interactions. In contrast, CS has 3 exchangeable hydroxyl protons but only 1 exchangeable NH proton. The OH/NH ratio and the slow exchange of this amide proton [∼25 s−1 at pH 7.4 and 22°C (25, 26)] indicate that hydroxyl groups should dominate the water R1ρ dispersion in CS solutions. Thus, the dispersion profiles of these model systems allow us to evaluate separately the effects of amide and hydroxyl exchange on water relaxation (Figs. 1 and 2).
Let us consider the quadrupolar relaxation of natural abundance 14N and 17O nuclei, in the context of amide and hydroxyl group rotations. We assume for the present discussion that the lower limit of the rotational correlation time of NH and OH moieties is roughly equal to that of methyl group rotations in protein solutions, i.e., ∼50 ps at 30°C in millimolar solutions (27). As the quadrupole-coupling constant of 14N in amino acids varies between 0.8–3.4 MHz (28), we have used an approximate value of 2.5 MHz. In the fast motion regime, the longitudinal relaxation rate can be estimated as (29)
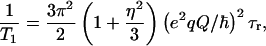 |
3 |
where (e2qQ/ℏ) is the nuclear quadrupole coupling constant, η is the asymmetry parameter, and τr is the rotational correlation time. The quadrupole coupling constant has been measured for many quadrupolar nuclei in various molecules and environments. The asymmetry parameter ranges between 0 and 1, providing a minimal contribution to the overall relaxation rate.
We therefore estimate the T1 of 14N nuclei in NH groups to be ∼200 μs (0.1–2 ms, for the range of coupling constant values given previously). The small difference between the 15N-labeled peptide, which does not exhibit quadrupolar relaxation, and the 14N peptide, which does, indicates that quadrupolar relaxation of nitrogen nuclei plays a small role in the low frequency bulk water T1ρ dispersion for this peptide system. We can attribute this to the fact that the T1 of 14N nuclei is much faster than the proton exchange rate and does not significantly influence the relaxation of the NH protons.
Similarly we can estimate the contribution from quadrupolar relaxation of 17O nuclei in 17O-H groups. The quadrupolar coupling constant of 17O in OH groups in organic compounds is ∼9 MHz (30). By using Eq. 3 and the assumptions as above, we estimate the T1 of 17O in CS to be ∼16 μs. This extremely fast relaxation time and the low natural abundance of 17O (0.037%) eliminates the quadrupolar relaxation of 17O nuclei as a substantial relaxation mechanism for water relaxation in both the peptide and CS systems.
Indeed, the isotopic substitution of 15N in the smMLCKp demonstrates that the quadrupolar relaxation of 14N has a relatively minor effect on the water relaxation. Because 15N nuclei do not exhibit quadrupolar relaxation, the dispersion observed in the 15N-labeled peptide solution is attributed to the chemical exchange of N-H protons with the bulk water. Because we have precluded quadrupolar relaxation of 17O from being a significant T1ρ dispersive mechanism, proton exchange of hydroxyl groups remains as the leading dispersive mechanism in CS solutions. Hills and coworkers (31, 32) have demonstrated that the exchange of hydroxyl protons contributes to transverse relaxation of water protons and that T2 measurements could be used to estimate the proton exchange rate and chemical shift in protein and sugar solutions.
Similarly, we can interpret the T1ρ dispersion data from 15N-peptide solutions by using Eq. 2. Assuming values of 3 s for the T2 of pure water, 1.2 s for the T2 of 15N-H protons in the unstructured peptide (33), a chemical shift difference of 1.8 ppm for the NH protons relative to on-resonance water, an exchange rate of 700 s−1 between NH protons and water, and the concentrations presented earlier, we can estimate the observed water T2 in the peptide solutions. The water T2 in our peptide solutions is predicted to be ∼1 s, whereas that extrapolated from Fig. 1 is 1.5 s. This agreement suggests that proton exchange between chemically shifted species is the dominant mechanism for relaxation in our model systems. In fact, bulk water T2 is relatively independent of the NH proton T2 but is dominated by the chemical shift separation, accounting for the small difference in water relaxation observed in 14N- and 15N-peptide solutions.
If we model the effective correlation rate of water dispersion in the peptide solutions to be the sum of the correlation rates of the different relaxation mechanisms, we can give some physical meaning to the measured correlation times. In our case, the individual relaxation mechanisms are (i) exchange modulation of chemical shift of the amide protons and (ii) exchange modulation of scalar coupling from natural abundance H217O (15). Assuming that these mechanisms are independent of each other, we can write
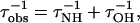 |
4 |
where τobs is the correlation time of the water dispersion curve, and τOH and τNH are the correlation times that maximize the spectral density function associated with the OH- and NH-associated relaxation processes, respectively. Because quadrupolar relaxation effects are small, τNH and τOH are directly related to the exchange times of the NH and hydroxyl protons with water.
We can evaluate the observed correlation rate of the water dispersion in the peptide and CS solutions by using Eq. 4. The overall NH to water proton exchange rate for the smMLCKp was calculated according to published methods and determined to be 700 s−1 at pH 7.4 and 22°C (25, 26). Briefly, the overall exchange rate is calculated as the weighted sum of the exchange rates from individual NH groups. The exchange rate of each group is calculated as the sum of the acid-, base-, and water-catalyzed rates as determined from previous measurements (25, 26). The calculated overall exchange rate is in agreement with the literature (34). The correlation rate of buffer dispersion was determined to be 1,100 s−1, which is in agreement with the results of Meiboom (15). Therefore, the correlation rate in our peptide system is predicted to be 1,800 s−1 (using Eq. 4), which is in good agreement with the observed rate of 1,700 s−1.
Similarly, the observed correlation rate in CS solutions, ∼1200 s−1, is in good agreement with the results of Hills (32), who reported a proton exchange rate of 1,400 s−1 at neutral pH. The observed result that the hydroxyl groups in CS exchange with water with a rate similar to water–water hydrogen exchange at neutral pH [1100 s−1 (15)] suggests that the exchange mechanism might be similar.
The bi-Lorentzian shape of the water R1ρ dispersion profile in cartilage indicates that there are at least two distinct dispersive processes in the 0.1–6 kHz frequency range. The low frequency correlation rate increases as the tissue is degraded with trypsin and loses PG, but the high frequency component does not change significantly. The low frequency dispersion component is particularly important for in vivo experiments, because this is the range of spin-lock amplitudes that can be achieved on MRI systems.
The measured correlation rate for the low frequency dispersion in cartilage is within 25% of the sum of the peptide and CS correlation rates according to Eq. 4, suggesting that the slow dispersion component in cartilage is derived from proton exchange of both NH and OH groups with water. The increase in the low frequency correlation rate with PG loss could be the result of increased proton exchange rates. In fact, it has been shown that because of the fixed charge density in cartilage (caused by negatively charged PG molecules), the sodium content is higher in the tissue than in the surrounding fluid as a result of Donnan equilibrium (35). Similar arguments have been used to suggest that cartilage fluid is more acidic than the surrounding environment (36). The loss of PG from the matrix leads to an increase in the basicity of the cartilage fluid component. For our conditions, the cartilage fluid pH should increase from ∼7.0 in native tissue to ∼7.2 in 60% PG-depleted tissues, using literature values for sodium content in normal and degraded tissues (37, §). This increase in pH would increase the exchange rates from hydroxyl and amide groups 1.6-fold. The slow correlation rate increased by ∼26% (Table 1), in agreement with the predicted increase. In fact, the T1ρ dispersion of bovine cartilage in the 0–3 kHz regime has been shown to be pH-sensitive (18), indicating that chemical exchange is responsible for the low field dispersion in cartilage. Leipinsh and Otting (38) reported that proton exchange from amino acids to water, at physiologic conditions, might occur at a range that could very well explain the T1ρ dispersion properties of cartilage.
According to our model, proton exchange seems to be the dominant low frequency T1ρ dispersion mechanism in peptide and CS solutions and cartilage. The proton exchange model accounts for 70% of the effective correlation time of water dispersion profiles in cartilage. Therefore, we propose that hydrogen exchange, between NH groups that are chemically shifted by ∼1.8 ppm from water protons and water, contributes heavily to the low frequency water T1ρ dispersion in biological systems.
It should be noted that Eq. 4 provides an empirical model to interpret our observations. Because the correlation times referred to in Eq. 4 are derived from the maximization of a model-dependent spectral density function, the relaxation model used will influence the interpretation of these values. For our description, both the proton exchange rate and the chemical shift difference between the labile groups and water determine the effective correlation time. Proton exchange between NH groups and water depends on several factors, including primary structure and hydrogen bonding. By experiment, we can observe only the overall exchange rate. The distribution of exchange rates from different moieties on the molecules (side chain NH vs. backbone amide, for example) along with the distribution of chemical shift values leads us to interpret the effective correlation times not as exchange times per se, but as indicators of proton exchange-induced relaxation.
We have focused the discussion on the low frequency dispersion in cartilage because the range of spin-lock strengths that are useful for diagnostic imaging lie in the 0.1–1.5 kHz regime. We can also offer a plausible interpretation for the higher frequency cartilage dispersion (τ ∼ 20 μs) in the context of the current literature. It has been shown that the exchange of entire water molecules between “bound” (i.e., associated with a macromolecule) and “free” sites can be a relaxation mechanism (39). The residence time of water molecules in the hydration layer has been estimated to be ∼10 μs in tissue, and water molecules closely associated with the tissue matrix have short relaxation times as a result of motional restriction (40). The higher frequency dispersion component in cartilage may therefore arise from the exchange of entire water molecules, closely associated with the tissue matrix, with the solvent water. This explanation is consistent with the observation that PG loss did not significantly affect the higher frequency dispersion in cartilage, because PG does not have well defined water-binding sites based on magnetization transfer measurements (41).
Our measurements demonstrate the existence of at least two processes that contribute to water dispersion in bovine type II collagen (based on the bi-Lorentzian T1ρ dispersion of collagen solutions). The low frequency dispersion component likely reflects the contribution of proton exchange. The high frequency dispersion is attributed to the exchange of water molecules between bound and free states. Therefore, collagen may also contribute to water T1ρ dispersion in cartilage. We postulate that collagen influences water molecules through collagen–water and collagen–PG interactions. Because of the latter, T1ρ measurements may be sensitive to macromolecular interactions between collagen and PG. At this time, we have not isolated the individual contributions from PG and collagen to bulk water T1ρ dispersion. However, the good correlation obtained between PG loss and low frequency T1ρ dispersion shows that T1ρ is sensitive to cartilage PG content.
The observed correlation between low frequency dispersion and PG loss suggests that T1ρ measurements may be particularly useful for the longitudinal evaluation of cartilage disease, and for noninvasively monitoring the efficacy of therapy (42). A relatively small change in the relaxation or correlation time may produce a noticeable effect in T1ρ-weighted images. Because the current measurements are spectroscopic and represent the global effects of trypsin digestion, they should be viewed as the lower limit of the sensitivity of this technique. In fact, trypsin digestion is known to produce a heterogeneous pattern of PG loss, with laminae at the tissue edges having maximal PG loss. Initial imaging experiments show that the effects of PG loss on T1ρ are magnified on T1ρ-weighted images, and degradation-induced signal laminae can be observed.¶ Importantly, the correlation time measurements reported here allow us to study the biophysical mechanisms underlying T1ρ relaxation and dispersion in cartilage. These measurements will be useful for developing methods to noninvasively map PG content in cartilage.
Conclusion
Our data suggest that proton exchange, from NH groups on simple peptide molecules with water, contribute to bulk water T1ρ relaxation in a concentration-dependent fashion. Hydroxyl groups in CS can also contribute. The apparent sensitivity of the low frequency dispersion (in the 0.1–1.5 kHz regime) to PG loss from cartilage suggests that T1ρ-based imaging schemes may be used to probe cartilage integrity.
Acknowledgments
This work was performed at a National Institutes of Health-supported Regional Research Center by National Institutes of Health Grants RR02305 (to J.S.L.), MH11960 (to U.D.), AR45242 (to R.R.), GM81347 (to S.W.E.), and DK39806 (to A.J.W.).
Abbreviations
- T1ρ
spin-lattice relaxation in the rotating frame
- PG
proteoglycan
- CS
chondroitin sulfate
- smMLCKp
smooth muscle myosin light-chain kinase peptide from chicken
Footnotes
Shapiro, E. M., Borthakur, A., Kaufman, J. H., Kudchodkar, S. B., Kneeland, J. B., Leigh, J. S. & Reddy, R. (2000) Osteoarthritis Cartilage 8, S12 (abstr.).
Akella, S. V. S., Regatte, R., Borthakur, A., Shapiro, E., Duwuri, U., Kneeland, J. B., Leigh, J. S. & Reddy, R. (2001) Proc. Int. Soc. Magn. Reson. Med. (Glasgow, Scotland) 3, 2108 (abstr.).
References
- 1.Hall B K, Newman S. Cartilage: Molecular Aspects. Boca Raton, FL: CRC; 1991. [Google Scholar]
- 2.McCauley T R, Disler D G. Radiology (Easton, Pa) 1998;209:629–640. doi: 10.1148/radiology.209.3.9844653. [DOI] [PubMed] [Google Scholar]
- 3.Insko E K, Kaufman J H, Leigh J S, Reddy R. Magn Reson Med. 1999;41:30–34. doi: 10.1002/(sici)1522-2594(199901)41:1<30::aid-mrm6>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- 4.Bashir A, Gray M L, Hartke J, Burstein D. Magn Reson Med. 1999;41:857–865. doi: 10.1002/(sici)1522-2594(199905)41:5<857::aid-mrm1>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- 5.Duvvuri U, Reddy R, Patel S D, Kaufman J H, Kneeland J B, Leigh J S. Magn Reson Med. 1997;38:863–867. doi: 10.1002/mrm.1910380602. [DOI] [PubMed] [Google Scholar]
- 6.Redfield A G. Phys Rev. 1955;98:1787–1809. [Google Scholar]
- 7.Virta A, Komu M, Kormano M. Magn Reson Med. 1997;37:53–57. doi: 10.1002/mrm.1910370109. [DOI] [PubMed] [Google Scholar]
- 8.Sepponen R. In: Magnetic Resonance Imaging. Stark D D, Bradley W G, editors. Vol. 1. St. Louis: Mosby; 1992. pp. 204–218. [Google Scholar]
- 9.Knispel R R, Thompson R T, Pintar M M. J Magn Reson. 1974;14:44–51. [Google Scholar]
- 10.Duvvuri U, Charagundla S R, Kudchodkar S B, Kaufman J H, Kneeland J B, Rizi R, Leigh J S, Reddy R. Radiology (Easton, Pa) 2001;220:822–826. doi: 10.1148/radiol.2203001662. [DOI] [PubMed] [Google Scholar]
- 11.Rizi R R, Charagundla S R, Song H K, Reddy R, Stolpen A H, Schnall M D, Leigh J S. J Magn Reson Imaging. 1998;8:1090–1096. doi: 10.1002/jmri.1880080514. [DOI] [PubMed] [Google Scholar]
- 12.Lee A L, Kinnear S A, Wand A J. Nat Struct Biol. 2000;7:72–77. doi: 10.1038/71280. [DOI] [PubMed] [Google Scholar]
- 13.Farndale R W, Sayers C A, Barrett A J. Connect Tissue Res. 1982;9:247–248. doi: 10.3109/03008208209160269. [DOI] [PubMed] [Google Scholar]
- 14.Bank R A, Krikken M, Beekman B, Stoop R, Maroudas A, Lafeber F P, te Koppele J M. Matrix Biol. 1997;16:233–243. doi: 10.1016/s0945-053x(97)90012-3. [DOI] [PubMed] [Google Scholar]
- 15.Meiboom S. J Chem Phys. 1965;34:375–388. [Google Scholar]
- 16.Venu K, Denisov V P, Halle B. J Am Chem Soc. 1997;119:3122–3134. [Google Scholar]
- 17.Bryant R G. Annu Rev Biophys Biomol Struct. 1996;25:29–53. doi: 10.1146/annurev.bb.25.060196.000333. [DOI] [PubMed] [Google Scholar]
- 18.Chen E-L. Ph.D. thesis. Philadelphia: Univ. of Pennsylvania; 1998. [Google Scholar]
- 19.Solomon I. C R Acad Sci (Paris) 1959;248:92–94. [Google Scholar]
- 20.Solomon I. C R Acad Sci (Paris) 1959;248:1631–1632. [Google Scholar]
- 21.Hills B P. Mol Phys. 1992;76:509–523. [Google Scholar]
- 22.Hills B P. Mol Phys. 1992;76:489–508. [Google Scholar]
- 23.McLaughlin A C, Leigh J S. J Magn Reson. 1973;9:296–304. [Google Scholar]
- 24.Chopra S, McClung R E D, Jordan R B. J Magn Reson. 1984;59:361–372. [Google Scholar]
- 25.Bai Y, Milne J S, Mayne L, Englander S W. Proteins. 1993;17:75–86. doi: 10.1002/prot.340170110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Connelly G P, Bai Y, Jeng M-F, Englander S W. Proteins. 1993;17:87–92. doi: 10.1002/prot.340170111. [DOI] [PubMed] [Google Scholar]
- 27.Lee A L, Flynn P F, Wand A J. J Am Chem Soc. 1999;121:2891–2902. [Google Scholar]
- 28.Hunt M J, Mackay A L. J Magn Reson. 1976;22:295–301. [Google Scholar]
- 29.Abragam A. The Principles of Nuclear Magnetism. Oxford: Clarendon; 1961. [Google Scholar]
- 30.Hsieh Y, Koo J C, Hahn E L. Chem Phys Lett. 1972;13:563–566. [Google Scholar]
- 31.Hills B P, Wright K M, Belton P S. Mol Phys. 1989;67:1309–1326. [Google Scholar]
- 32.Hills B P. Mol Phys. 1991;72:1099–1121. [Google Scholar]
- 33.Cavanagh J, Fairbrother W J, Palmer A G, III, Skelton N J. Protein NMR Spectroscopy: Principles and Practice. San Diego: Academic; 1996. [Google Scholar]
- 34.Ehrhardt M R, Urbauer J L, Wand A J. Biochemistry. 1995;34:2731–2738. doi: 10.1021/bi00009a001. [DOI] [PubMed] [Google Scholar]
- 35.Lesperance L M, Gray M L, Burstein D. J Orthop Res. 1992;10:1–13. doi: 10.1002/jor.1100100102. [DOI] [PubMed] [Google Scholar]
- 36.Kaufman J H. Ph.D. thesis. Philadelphia: Univ. of Pennsylvania; 2000. [Google Scholar]
- 37.Shapiro E M, Borthakur A, Dandora R, Kriss A, Leigh J S, Reddy R. J Magn Reson. 2000;142:24–31. doi: 10.1006/jmre.1999.1932. [DOI] [PubMed] [Google Scholar]
- 38.Leipinsh E, Otting G. Magn Reson Med. 1996;35:30–42. doi: 10.1002/mrm.1910350106. [DOI] [PubMed] [Google Scholar]
- 39.Koening S H, Hallenga K, Shporer M. Proc Natl Acad Sci USA. 1975;72:2667–2671. doi: 10.1073/pnas.72.7.2667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Diegel J G, Pintar M M. Biophys J. 1975;15:855–860. doi: 10.1016/S0006-3495(75)85861-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kim D K, Ceckler T L, Hascall V C, Calabro A, Balaban R S. Magn Reson Med. 1993;29:211–215. doi: 10.1002/mrm.1910290209. [DOI] [PubMed] [Google Scholar]
- 42.Potter K, Buttler J J, Horton W E, Spencer R G S. Arthritis Rheum. 2000;43:1580–1590. doi: 10.1002/1529-0131(200007)43:7<1580::AID-ANR23>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]