

Abstract
Gasdermins mediate inflammatory cell death after cleavage by caspases or other, unknown enzymes. The cleaved N-terminal fragments bind to acidic membrane lipids to form pores, but the mechanism of pore formation remains unresolved. Here we present the cryo-electron microscopy structures of the 27-fold and 28-fold single-ring pores formed by the N-terminal fragment of mouse GSDMA3 (GSDMA3-NT) at 3.8 and 4.2 Å resolutions, and of a double-ring pore at 4.6 Å resolution. In the 27-fold pore, a 108-stranded anti-parallel β-barrel is formed by two β-hairpins from each subunit capped by a globular domain. We identify a positively charged helix that interacts with the acidic lipid cardiolipin. GSDMA3-NT undergoes radical conformational changes upon membrane insertion to form long, membrane-spanning β-strands. We also observe an unexpected additional symmetric ring of GSDMA3-NT subunits that does not insert into the membrane in the double-ring pore, which may represent a pre-pore state of GSDMA3-NT. These structures provide a basis that explains the activities of several mutant gasdermins, including defective mutants that are associated with cancer.
The gasdermin (GSDM) family, expressed in the skin, mucosa and immune antigen-presenting cells, triggers inflammatory programmed cell death (pyroptosis) and inflammatory cytokine secretion1–8. There are six human GSDMs (GSDMA, GSDMB, GSDMC, GSDMD, GSDME (also known as DFNA5) and DFNB59 (also known as pejvakin)), and ten in mice, including three GSDMAs. GSDMs are cleaved by regulated processing that removes an inhibitory C-terminal fragment (GSDM-CT) to allow the N-terminal fragment (GSDM-NT) to bind to acidic lipids in the inner leaflet of mammalian cell membranes or on bacterial membranes to form pores. GSDMD is a substrate of inflammatory caspases1–8, which are activated by inflammasomes that recognize invasive infection or intracellular danger signals9,10, and GSDME is activated by caspase-311. The stimuli and proteases that activate the other GSDMs are largely unknown. To elucidate the mechanism of GSDM pore formation, we determined the cryo-electron microscopy (cryo-EM) structure of the mouse GSDMA3-NT pore.
Cryo-EM structure determination
We formed human GSDMD-NT and mouse GSDMA3-NT pores by cleaving the full-length proteins in the presence of phosphatidylserine- containing and cardiolipin-containing liposomes, respectively (Fig. 1a, Extended Data Fig. 1a, b). A detergent screen identified sodium cholate and C12E8 as suitable solubilizing agents for GSDMA3-NT and GSDMD-NT pores, respectively. Because GSDMA3-NT pores were more homogeneous in size and shape than GSDMD-NT pores (Extended Data Fig. 1c, d), we collected cryo-EM data from native GSDMA3-NT pores using a Talos Arctica microscope (Extended Data Fig. 1e), and from pores treated with HgCl2 using a Titan Krios microscope (Extended Data Fig. 1f). HgCl2 treatment was intended to label free Cys residues for validation of sequence assignment to the cryo-EM map.
Fig. 1. Structure determination of GSDMA3 pores.
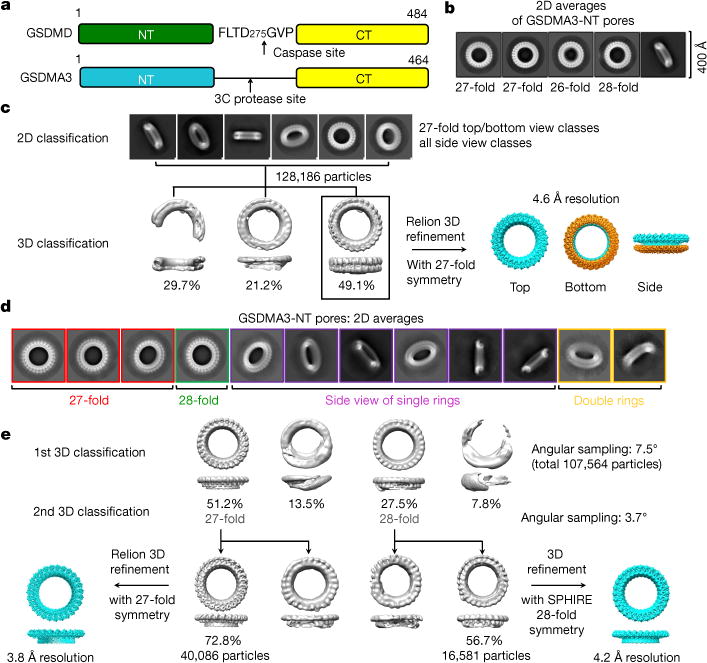
a, Cartoon diagram of human GSDMD and mouse GSDMA3 constructs used for in vitro pore reconstitution. b, 2D classification of GSDMA3 pores showing the presence of 26-, 27- and 28-fold symmetric pores. Each image shows an area of 400 × 400 Å2. c, 3D reconstruction of the 27-fold symmetric double-ring pore to 4.6 Å resolution. d, 2D classes of HgCl2-treated GSDMA3 pores showing mostly single rings and a minor population of double rings. e. 3D reconstruction of 27- and 28-fold symmetric single-ring GSDMA3 pores to 3.8 and 4.2 Å, respectively.
For both datasets, top views of two-dimensional (2D) classified and averaged images showed mostly 27-fold symmetry (around 62% for native pores and around 70% for HgCl2-treated pores), but there were also substantial numbers of pores with 26- (around 16% for native pores) and 28-fold (around 22% for native pores and around 30% for HgCl2-treated pores) symmetry, implying heterogeneity of oligomerization (Fig. 1b–e). For the native dataset, top-view classes of pores with 27-fold symmetry and all side views were used to perform three-dimensional (3D) classification without imposing any symmetry. One major class was further 3D-refined with 27-fold symmetry to a global resolution of 4.6 Å by gold-standard Fourier shell correlation (FSC) in Relion12 (Fig. 1c, Extended Data Fig. 2a, Extended Data Table 1). For the HgCl2-treated dataset, two rounds of 3D classification followed by 3D refinement led to maps at 3.8 Å and 4.2 Å resolution for the 27-fold-and 28-fold-symmetry pores, respectively (Fig. 1d, e, Extended Data Fig. 2b, c, Extended Data Table 1). Unexpectedly, the 4.6 Å cryo-EM map from native pores contains two rings with 27-fold-symmetry, with the top ring representing a membrane pore and the bottom ring with no membrane insertion (Fig. 1c). By contrast, the cryo-EM maps from HgCl2-treated pores contain only the top ring (Fig. 1e). Local resolution distribution calculated in ResMap13 showed better resolution at the putative membrane-inserted region, which contained extended β- hairpins, in comparison to the juxtamembrane globular domain (Extended Data Fig. 2d, e).
Overall structure of the membrane pore
Analysis of the cryo-EM maps of the 27-fold membrane pore revealed a very large structure with an inner diameter of around 180 Å, an outer diameter of around 280 Å and a height of around 70 Å. We focus our discussion on the 3.8 Å-resolution pore, which had clearly defined secondary structures and large side chains (Fig. 2a, Extended Data Fig. 3, Supplementary Video 1). On the basis of the top views of 2D averages (Fig. 1b, d), the 26- and 28-fold pores would have similar dimensions that are only marginally smaller and larger, respectively. However, previously reported GSDMA3 pores assembled on cardiolipincontaining monolayer membranes appear to have 16-fold symmetry and a smaller interior width of 100–140 Å, despite the similar outer diameter of around 30 nm, as determined from 2D averages in negative- staining electron microscopy1. Since native GSDMs assemble on lipid bilayers, which are present in our liposomes, rather than monolayers, and the determination of symmetry and dimensions from low-resolution averages may be inaccurate, the structure we obtained here is more likely to resemble the architecture of the pore in the plasma membrane.
Fig. 2. Structure and conformational changes of the GSDMA3 pore.
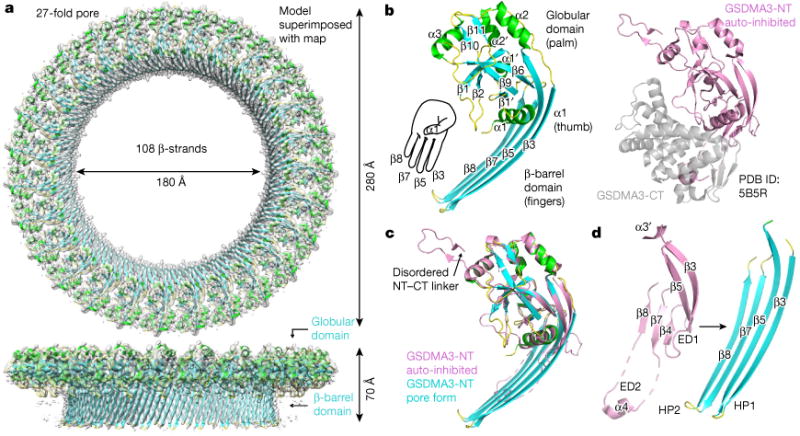
a, Cryo-EM map (grey) superimposed onto the atomic model of the 27-fold symmetric GSDMA3 pore at 3.8 Å resolution. b, Ribbon diagram of GSDMA3-NT in the pore conformation (left) and crystal structure of auto-inhibited GSDMA31 (right). c. Superposition of the auto-inhibited form and the pore form of GSDMA3-NT. d, Structural transitions that accompany the formation of the two β-hairpins HP1 and HP2 in the pore conformation. ED1 and ED2, extension domains 1 and 2, respectively.
The predominant GSDMA3 pore with 27-fold symmetry is a complete anti-parallel β-barrel composed of 108 β-strands (Fig. 2a). Each subunit in the pore contributes four clearly separated long β-strands as two anti-parallel β-hairpins that align in a manner similar to the digits of a left hand to form the transmembrane region (Fig. 2a, b). The β-strands that line the pore are from 15 to 22 residues long, and define a central β-barrel about 50 Å high, sufficient to traverse a lipid bilayer composed of lipid acyl chains with a thickness of about 30–40 Å. A previous analysis of transmembrane β-strands suggested that β-strands of between 6 and 22 residues in length can span a membrane14. The GSDMA3-NT globular region, or the ‘palm’ of the hand, shapes the rim of the pore and contains both α- and β-elements, with the prominent α1 helix acting as the thumb (Fig. 2b).
Conformational transition upon membrane insertion
The conformation of the GSDMA3-NT pore exhibits a radical conformational change in comparison to its structure in the auto-inhibited, uncleaved protein (Protein Data Bank (PDB) ID: 5B5R)1 (Fig. 2b–d). We named the secondary structures in the pore form on the basis of the auto-inhibited structure to facilitate comparison (Extended Data Fig. 4). Superposition of the two structures resulted in a root-mean-square deviation (r.m.s.d.) of 4.95 Å for 161 aligned Cα atoms (Fig. 2c). Whereas the globular domain is largely unaltered, with a superimposed r.m.s.d. of 0.95 Å, marked conformational changes are associated with the formation of the two membrane-inserted β-hairpins. The entire β3–β4–β5 region extends into the first tight transmembrane β-hairpin, and the α3′ and the disordered region preceding it in the auto-inhibited conformation are pulled into the new β3 strand (Fig. 2c, d, Supplementary Video 2). The entire β7–α4–β8 region stretches out into the second transmembrane β-hairpin, including the associated loops and disordered segments in the auto-inhibited structure (Fig. 2c, d, Supplementary Video 2). The α4 helix that binds to the C-terminal fragment (GSDMA3-CT) in the uncleaved structure (Fig. 2d) forms part of the tip of the second β-hairpin. Because of the structural transformation, we named the β4 and α3′ region ‘extension domain 1’ (ED1), the α4 region ‘extension domain 2’ (ED2), and the newly formed long β-strands ‘hairpin 1’ (HP1) and ‘hairpin 2’ (HP2) (Fig. 2d).
Acidic lipid binding by the α1 helix
Analysis of the electrostatic surface of the GSDMA3-NT pore revealed that in the transmembrane region, the side of the pore that faces the membrane is largely hydrophobic, as expected, whereas hydrophilic residues form positively and negatively charged stripes on the inner side of the pore conduit (Fig. 3a). A positively charged surface patch also forms adjacent to the proposed transmembrane region, in the globular domain of the pore (Fig. 3a). When we used a difference density map to locate additional densities, we found a strong density at 6.6σ adjacent to the α1 helix, which fits well to the double phosphate head group of cardiolipin (Fig. 3b–d), the acidic lipid we used to reconstitute the pore on liposomes. We hypothesize that the modelled cardiolipin head group interacts with the basic residues on helix α1 (Fig. 3d).
Fig. 3. Mechanism of lipid recognition.
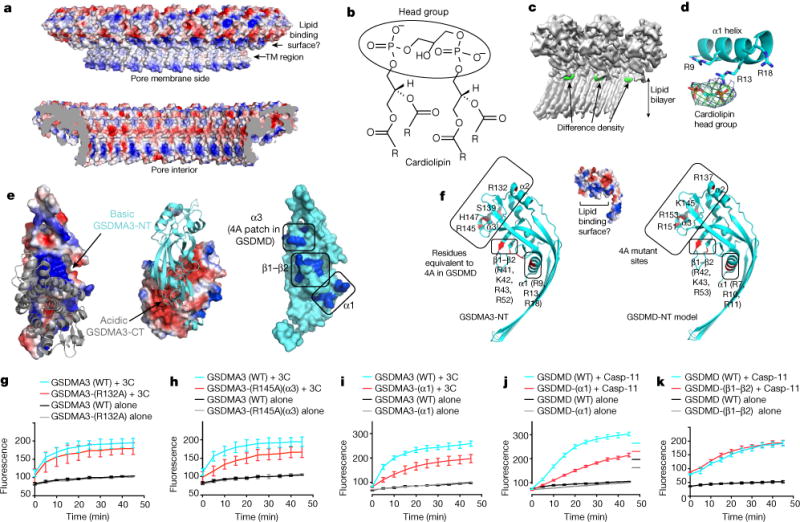
a, Charge distribution in the GSDMA3 pore, viewed from the membrane side (top) and the pore interior (bottom). TM, transmembrane. b, Chemical structure of cardiolipin showing two phosphatidic acid moieties in the head group. The four R groups represent C17H33. c, Cryo-EM map of the GSDMA3 pore (grey) with the difference density map for potential bound lipid (green). d, Cardiolipin head group fitted into the lipid density in proximity to basic residues in the α1 helix. e, Potential lipid-binding patch. Left and middle, the auto-inhibited GSDMA3 structure1, shown in electrostatic surface for the N-terminal domain and grey ribbon for the C-terminal domain (left), and in cyan ribbon for the N-terminal domain and electrostatic surface for the C-terminal domain (middle). Right, the N-terminal domain in its auto-inhibited form shown as a surface diagram with the positively charged patches in blue. f, The basic patch in the pore form contains residues from helices α1 and α3 and the β1–β2 region. Relevant residues are labelled for GSDMA3-NT (left) and for a model of human GSDMD-NT (right). g–k, Effects of GSDMA3 (g–i) and GSDMD (j, k) mutations on liposome-leakage activities, monitored by measuring dipicolinic acid (DPA)-chelating-induced fluorescence of released Tb3+ ion (n = 3 biological replicates). Error bars denote mean ± s.d.
To confirm the role of helix α1 in lipid interaction, we inspected the membrane-proximal, positively charged surface of the GSDMA3 pore, which is made up of basic residues from helix α1, helix α3 and strands β1 and β2 of the pore form of GSDMA3-NT (Fig. 3e, Extended Data Fig. 4). These regions are masked by GSDMA3-CT in the auto-inhibited full-length structure (Fig. 3e), consistent with binding of GSDM-NT, but not full-length GSDMA3 or GSDMA3-CT, to acidic lipids1–4,8. Mutation to Ala of a cluster of four conserved basic residues on helices α2 and α3 in mouse GSDMD (GSDMD(4A)) that correspond to R137, K145, R151 and R153 of human GSDMD (Fig. 3f, right) compromised lipid binding, liposome leakage and pyroptosis2. When these mutations were combined, the loss of both R151 and R153 on α3 was the most defective combination, whereas the combined loss of R137 and K145 only minimally impaired cell death2. Two of the four basic residues in GSDMD(4A) are conserved in GSDMA3 as R132 in α2 and R145 in α3 (Fig. 3f, left). To test the importance of these residues, we generated R132A and R145A mutants of GSDMA3. Liposome leakage assays showed that the R132A mutation on α2 barely compromised pore formation (Fig. 3g), whereas the R145A mutation on α3 or the R132A/R145A double mutation weakened liposome disruption (Fig. 3h, Extended Data Fig. 5a), consistent with the defective phenotype of corresponding mutations on GSDMD2.
To test the effect of the positively charged juxtamembrane patches formed from residues in helix α1 and the β1–β2 region, we generated a triple mutant of GSDMA3 containing R9A, R13A and R18A (GSDMA3(α1)), and a quadruple mutant containing R41A, K42K, R43A and R52A (GSDMA3(β1–β2)) (Fig. 3f, left). The triple mutant was expressed and could be cleaved by the 3C protease with similar kinetics to those of the wild-type protein (Extended Data Fig. 5b), suggesting that the mutation did not radically alter the structure of the full-length protein, whereas the quadruple mutant could not be evaluated because it was not expressed. GSDMA3-α1 cleaved by 3C protease had reduced membranolytic activity in the liposome leakage assay (Fig. 3i). Corresponding Ala mutations were also introduced into human GSDMD at R7, R10 and R11 (GSDMD(α1)), and at R42, K43 and R53 (GSDMD(β1–β2)) (Fig. 3f, right). Both mutants were expressed and could be purified. Liposome leakage assays showed that similar to GSDMA3(α1), the GSDMD(α1) mutant also showed compromised pore formation in comparison to the wild-type protein (Fig. 3j). The importance of the α1 helix is also supported by the lack of cell death when part of α1 is deleted4. By contrast, the GSDMD(β1–β2) mutation did not decrease liposome leakage (Fig. 3k).
Thus, these mutational studies suggest that the α1 and α3 juxtamembrane basic patches, but not basic residues in the β1–β2 region, are potential candidates for lipid binding. Because α3 also participates in oligomerization between subunits (see below), and is further away from the membrane than α1, we argue that the α1 helix is likely to be the major lipid-binding site for GSDM pore formation. The difference density near α1 may represent the acidic head group of cardiolipin, which may interact strongly with GSDMA3-NT and stay associated with the pore after detergent extraction. We do not know whether the acidic lipid interaction is only important for inducing conformational changes in GSDM or whether it is also essential for stabilizing the membrane pore.
Oligomerization interface
Analysis of the oligomerization interface of the GSDMA3-NT pore revealed extensive interactions of both the inserted β-strands and the associated globular domains (Fig. 4a). The β-strands have a tilt angle of about 20° relative to the pore height direction. If one draws the β-strand hydrogen bonding pattern across the β-barrel, there is a two-residue shift from one subunit to the next subunit (Extended Data Fig. 6), giving rise to a total displacement of residues, or a shear number15, of 27 × 2 = 54 for the entire 27-fold symmetric pore. Each subunit interface buries a total of 1,600 Å2 surface area, with key interaction areas dividable into three patches, I, II and III (Fig. 4b, Extended Data Fig. 4). In the first patch, residues between the neighbouring globular domains form both hydrophobic and charged interactions (Fig. 4c). The interaction contains mainly residues from helix α3 of one subunit and the region around α2 and β11 of its neighbouring subunit. In the second patch, the α1 helix from one subunit juxtaposes end-on with the α1 helix from the next subunit through hydrogen-bonding and hydrophobic interactions (Fig. 4d). The third major subunit interface runs along the neighbouring β3 and β8 strands between the subunits (Fig. 4e). Notably, in the inserted β-strands, residues facing the membrane are hydrophobic, while those facing the pore are mostly hydrophilic or charged (Fig. 4e).
Fig. 4. Mechanism of oligomerization and membrane insertion.
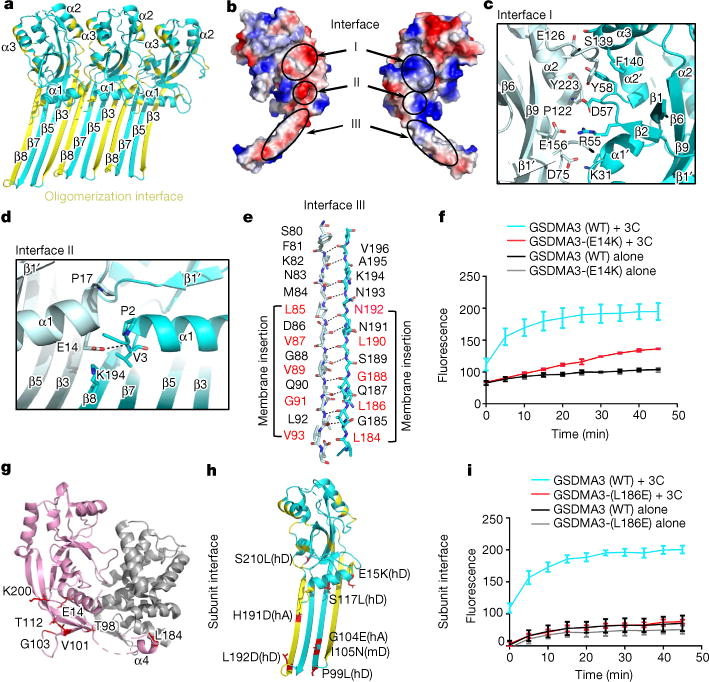
a, Three contiguous subunits in the GSDMA3 pore. Residues that participate in oligomerization are labelled in yellow. b, Electrostatic potential map of a GSDMA3-NT subunit in two side views showing the oligomerization interface with three major patches labelled.
c–e, Main residues on interface I (c), interface II (d) and interface III (e). f, Oligomerization mutant E14K exhibits compromised pore formation compared to wild-type (WT) GSDMA3 (n = 3 biological replicates). Error bars denote mean ± s.d. g, h, The locations of previously reported and cancer-associated mutations on both auto-inhibited (g) and pore (h) conformations on GSDMA3. hA, human GSDMA; hD, human GSDMD; mD, mouse GSDMD. i, Mutation of L186 in the membrane insertion region abolishes pore formation and liposome leakage (n = 3 biological replicates). Error bars denote mean ± s.d.
Previous studies showed that E15K and L192D mutations of GSDMD-NT, and the corresponding mutations E14K and L184D of GSDMA3-NT (Extended Data Fig. 4), strongly reduced pyroptosis1. Consistent with these results, we found that the E14K mutant of GSDMA3 compromised liposome leakage in vitro (Fig. 4f). The inhibitory effects of E14 and L184 mutations on GSDMA3 cannot be explained by its full-length structure because E14 is close to the N-terminal domain–C-terminal domain interface and L184 interacts closely with the C-terminal domain (Fig. 4g), and mutations in the C-terminal domain near E14 or L184 constitutively activate GSDMA3 and GSDMD1,6. However, in the GSDMA3-NT pore structure, E14 and L184 of GSDMA3 are involved in oligomerization at the globular domain and the β-barrel domain, respectively (Fig. 4d, e, g, h); this explains how these mutations compromise pore formation, even though they reduce auto-inhibition. L184 is also involved in membrane insertion and its mutation to an acidic residue could therefore also directly affect this process. To further test the structural model, we mutated another oligomerization or insertion residue L186 to obtain the L186E mutant. As we predicted, the mutation almost completely eliminated the liposome leakage activity of proteolytically cleaved GSDMA3 (Fig. 4i).
A forward genetic screen with randomly mutated mice identified an I105N mutation of mouse GSDMD as defective in the intracellular LPS response in vivo, and in membrane permeabilization in vitro7,8. The I105N mutation in mouse GSDMD corresponds to I104N in human GSDMD and V101N in GSDMA3 (Extended Data Fig. 4). V101 localizes at β5 of the pore form and is involved in membrane insertion (Fig. 4h), explaining the impaired function in the mutant GSDMD. The OASIS cancer genome site (http://www.oasis-genomics.org/) lists a number of point mutations in human GSDMA and GSDMD from patient samples, including mutations in GSDMA that correspond to G103E and N192D in GSDMA3, and GSDMD mutations E15K, E15Q, P99L, S117L and S210L that correspond to mutations on residues E14, T98, T112 and K200 in GSDMA3 (Fig. 4d, g, h). E14 of GSDMA3 is at the oligomerization interface of the pore globular domains, whereas the remaining residues are all involved in membrane insertion and/or oligomerization at the β-barrel domain (Fig. 4h), suggesting a potential mechanism of tumour potentiation through reduced pyroptotic activity of GSDMs.
Comparison with cytolysins
The large β-barrel architecture of the GSDMA3 pore described here shares mechanistic similarities with pores formed by bacterial cytolysins, complement proteins and the pore-forming protein perforin from cytotoxic T lymphocytes, which belong to the ‘membrane attack complex perforin-like’/‘cholesterol dependent cytolysin’ (MACPF/CDC) family16–18. Whereas many structures in the family have been determined in their soluble forms, the only high-resolution structure of the pore form is that of pneumolysin19,20. A DALI search21 using the GSDMA3-NT domain structures in both the full-length context and the pore form identified weak structural homology at the central β-sheets to these proteins, including the soluble form and the pore form of pneumolysin19,20 (Fig. 5a, b), with the highest structural homology to perfringolysin O, with a r.m.s.d. of 7.3 Å for 134 aligned residues.
Fig. 5. Comparison with MACPF/CDC proteins.
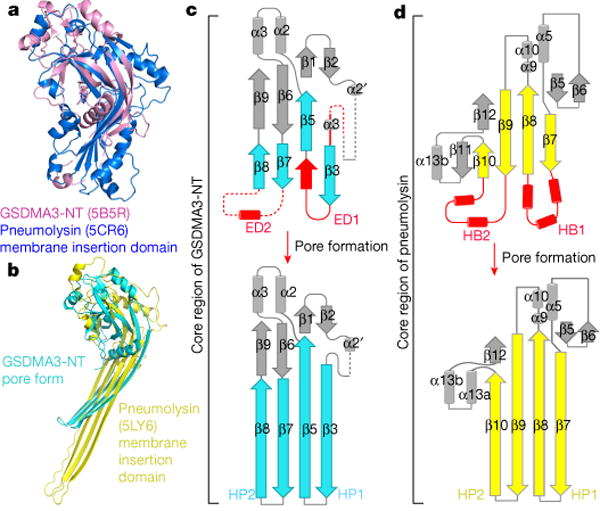
a, b, Superposition of GSDMA3-NT with the membrane insertion domain of pneumolysin in the soluble form (a) and in the pore conformation (b), showing the alignment of the central β-sheets. The C-terminal tail of GSDMA3-NT that links to GSDMA3-CT and part of the pneumolysin structure are omitted for clarity. c, d, Different structural topologies of GSDMA3-NT (c) and pneumolysin (d) in both soluble and pore forms. ED, extension domain; HB, helix bundle; HP, β-hairpin. Regions that are straightened by the insertion are highlighted in red.
Despite the structural similarity at the central β-sheets, we hypothesize that the GSDM and MACPF/CDC families either evolved independently or are so distantly related that divergent evolution at the sequence level and unexpected conserved structural features are not detected. We present three major differences that support this idea. First, previous analysis showed that the MACPF and CDC family proteins contain three conserved glycine residues22, which are absent in GSDMs (Extended Data Fig. 7). Second, the structural homology between MACPF and CDC spreads from the central β-sheets to the clusters of α-helices that switch to the membrane-spanning β-conformation, yielding much lower r.m.s.d. for many more aligned residues than with GSDMA322. Third, the membrane insertion mechanism is different. For GSDMA3, one β-hairpin is formed from straightening the β7 and β8 strands into the connected α4 and disordered loops, while the other hairpin comes from both the elements in between β3 and β5, and elements preceding β3 that include the α3′ helix and a disordered loop in the auto-inhibited conformation (Fig. 5c). By contrast, in pneumolysin, straightening into the connected cluster of helices from the central β-sheet alone is responsible for the formation of the long β-hairpins for membrane insertion and β-barrel formation19 (Fig. 5d, Supplementary Video 3).
The soluble pore may represent a pre-pore
The native cryo-EM map of GSDMA3-NT shows the existence of an additional pore, creating a yo-yo-shaped double-ring pore (Figs. 1c, 6a, b). Unlike the membrane pore, the bottom ring does not insert into the membrane, and we therefore call it the soluble pore. We do not know the physiological significance of the double-ring structure. On the other hand, most HgCl2-treated GSDMA3 pores contain only the membrane-inserted ring (Fig. 1e), suggesting that the double-ring structure is sensitive to the biochemical condition, and could be an artefact of in vitro reconstitution.
Fig. 6. A possible GSDMA3 pre-pore conformation.
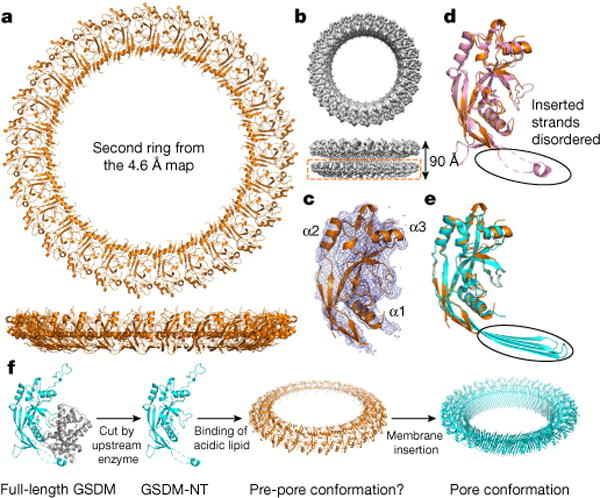
a, b, Model of the additional ring (a) and its cryo-EM density in the double-ring GSDMA3 pore architecture (b). c, Cryo-EM density of an individual subunit superimposed on the model. d, e, Superposition of the subunit structure in the additional ring with that of GSDMA3-NT in the auto-inhibited conformation (pink)1 (d) and the pore conformation (cyan) (e). f, A hypothetical model for GSDM pore formation. Upon cleavage by an activating enzyme, GSDM-NT monomers may first bind to membrane lipids and then oligomerize to form a soluble pre-pore before membrane insertion.
Although the cryo-EM density for the additional ring is much poorer, we could fit the N-terminal part of the full-length auto-inhibited GSDMA3 structure1 rigidly into the density (Fig. 6c). Consistent with lack of membrane insertion, most of the β-strands in the transmembrane region of the pore form, as well as the N-terminal–C-terminal linker that harbours the 3C cleavage site, are disordered in this fitted GSDMA3-NT structure (Fig. 6d, e). Although detailed analysis of the GSDMA3-NT conformation and its subunit association in the soluble pore would require improved resolution of this region, the existence of soluble pre-pore conformations for some MACPF/CDC family members19 suggests that the soluble GSDMA3-NT pore could represent a pre-pore state. Notably, a mutant of aerolysin forms asymmetric pores with a stacked quasi-pore and a soluble pore23,24 that resemble our double-ring native GSMDA3 pore structure.
Summary
The cryo-EM structure of the GSDMA3-NT pore revealed that the N-terminal fragment of GSDM undergoes marked conformational changes to form the pore structure upon binding to acidic inner leaflet or bacterial lipids. Difference density and structure-based mutagenesis suggested that helix α1 is the major site of acidic lipid interaction, which is required for pore formation. We do not know the function of the second ring or whether it is part of the physiological pore in cells or an artefact of in vitro reconstitution; however, we speculate that it may represent a pre-pore structure that precedes membrane insertion (Fig. 6f).
In some circumstances, membrane damage caused by bacterial cytolysins, complement proteins or perforin can be repaired by the ubiquitous plasma membrane repair pathway, which is triggered by a calcium influx from the extracellular milieu into the damaged cell25,26. In the case of perforin, repair enables target cells to die by apoptosis rather than necrosis, which occurs when plasma membrane damage is not repaired. Recent reports have described situations in which inflammasome activation and GSDMD cleavage lead to IL-1β release without cell death27, suggesting that damage to the cell membrane caused by the GSDMD-NT pore can also be repaired under some circumstances. If the double-ring pore exists physiologically, it is possible that under these conditions the second ring could affect whether repair occurs or might even have a signalling function in these activated cells.
Online content
Any Methods, including any statements of data availability and Nature Research reporting summaries, along with any additional references and Source Data files, are available in the online version of the paper at https://doi.org/10.1038/s41586-018-0058-6.
Methods
Cloning, expression and purification of recombinant mouse GSDMA3 and human GSDMD
Full-length mouse GSMDA3 sequence was cloned into a modified pET28a vector with an N-terminal 6× His–SUMO tag, followed by mutagenesis to insert a human rhinovirus 3C protease recognition sequence (LEVLFQGP) immediately after residue E262. The human GSDMD sequence was cloned into the pDB.His.MBP vector to append an N-terminal 6× His–MBP tag followed by a tobacco etch virus (TEV) cleavage site. All mutagenesis was performed using the QuikChange Site-Directed Mutagenesis Kit (Stratagene). All plasmids were verified by DNA sequencing.
To obtain recombinant GSDMA3, Escherichia coli BL21 (DE3) cells were transfected with the vector and grown in LB medium supplemented with 50 μg/ml kanamycin. Protein expression was induced overnight at 18 °C with 0.5 mM isopropyl-β-d-thiogalactopyranoside after optical density at 600 nm reached 0.8. Cells were harvested and resuspended in a lysis buffer containing 25 mM Tris-HCl at pH 8.0 and 150 mM NaCl. The protein was first purified by affinity chromatography using Ni-NTA beads (Qiagen) and the 6× His–SUMO tag was removed by overnight Ulp1 protease digestion at 4 °C. The cleaved GSDMA3 was further purified by HiTrap Q ion-exchange chromatography and Superdex 200 gel filtration chromatography (GE Healthcare Life Sciences). Human GSMSD was expressed and purified with similar procedures, except that the N-terminal 6× His–MBP tag was removed by overnight TEV cleavage.
Reconstitution of GSDMA3 and GSDMD pores
1-Palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) and 1′, 3′-bis[1,2-dioleoyl-sn-glycero-3-phospho]-sn-glycerol (sodium salt) (cardiolipin) (Avanti Polar Lipids) dissolved in chloroform were mixed in a glass tube at a mass ratio of 3:1, and the solvent was evaporated under a stream of N2 gas. Buffer composed of 25 mM Tris-HCl at pH 8.0 and 150 mM NaCl was added to yield a final lipid concentration of 10 mg/ml. Lipid suspension was then vortexed continuously for 5 min. To obtain unilamellar vesicles, liposomes were extruded through a mini-extruder device (Avanti) for 21 passes.
To form GSDMA3 pores on liposomes, purified GSDMA3 was incubated with 3C protease and POPC–cardiolipin liposomes for 4 h on ice. Liposomes were harvested by ultracentrifugation at 60,000g at 4 °C for 30 min and resuspended in 0.5 ml lysis buffer containing 50 mM sodium cholate. After centrifugation at the maximum speed of an Eppendorf centrifuge for 30 min, the supernatant containing the solubilized pores was further purified over a Superose 6 gel-filtration column (GE Healthcare Life Sciences) equilibrated with lysis buffer containing 15 mM sodium cholate. GSDMD pores were formed in the same way but with recombinant caspase-11 in liposomes containing 20% (w/w) phosphatidylserine, and 2% C12E8 was used to solubilize the pores.
Negative staining electron microscopy
For negative staining, 10 μl GSDMA3 or GSDMD pores was placed onto a glow-discharged copper grid (Electron Microscopy Sciences) coated with a layer of thin carbon, washed twice with H2O, stained with 2% uranyl formate for 40 s and air-dried. The grids were imaged on the Tecnai G2 Spirit BioTWIN electron microscope and recorded with an AMT 2k CCD camera (Harvard Medical School Electron Microscopy Facility).
Cryo-EM data collection
A 3-μl drop of native GSDMA3 pores at 2 mg/ml was applied to a glow-discharged Quantifoil grid (R 1.2/1.3 400 mesh, copper, Electron Microscopy Sciences), blotted for 3 s in 100% humidity at 4 °C and plunged into liquid ethane using an FEI Vitrobot Mark IV. The cryo-grids were imaged in an FEI Talos Arctica microscope operating at an acceleration voltage of 200 kV and equipped with a cryo-autoloader (University of Massachusetts Cryo-EM Core Facility). Cryo-EM data were collected automatically on a K2 Summit direct detector camera (Gatan) in super-resolution counting mode, with 8.0 s total exposure time and 200 ms per frame. This resulted in movies each containing 40 frames and an accumulated dose of 41.1 electrons per Å2. The super-resolution pixel size is 0.5843 Å. The defocus level in the data collection was set in the range of −1.0 to −3.0 μm. A total of 3,010 movies was collected.
For mercury treatment to label free Cys residues, GSDMA3 pores at 2 mg/ml (~75 μM) were incubated with MgCl2 at a molar ratio of 1:10 at 4 °C for 2 h. The cryo-grids were plunged in the same way as for the native pores. A total of 1,583 movies was recorded using an FEI Titan Krios electron microscope (National Cryo-electron Microscopy Facility, National Cancer Institute) operating at 300 kV on a K2 Summit direct electron camera operating in super-resolution counting mode, with 12 s total exposure and 300 ms per frame. The defocus range was from −0.75 to −2.0 μm. The resulting movies each contain 40 frames and an accumulated dose of 40.0 electrons per Å2. The super-resolution pixel size is 0.66 Å.
Image processing
For data of native GSDMA3 pores, raw movies were corrected by gain reference and for beam-induced motion, and summed into motioncorrected images using MotionCor228. The CTFFIND4 program was used to determine the actual defocus level of each micrograph29. We initially selected 462,878 particles from the micrographs with a combination of manual and automatic particle picking in RELION12. The picked particles were binned two times (pixel size 1.1686 Å) and then subjected to reference-free 2D classification. Good class averages in different orientations were selected for the reconstruction of an initial model in RELION. The resulting initial model, low-pass filtered to 60.0 Å, was used as the input reference to conduct unsupervised 3D classifications in RELION without assumption of any symmetry. The particle set used contained all the side views and top views with apparent C27 symmetry, but not top views with apparent C26 or C28 symmetry. A best 3D class showing symmetric features was selected for further refinement. Upon imposing C27 symmetry, the reconstructions reached a resolution of 4.6 Å, as measured by FSC of a gold-standard refinement in which two halves of the dataset were refined separately and combined only when building the final map12.
For mercury-soaked GSDMA3 pores, the same procedures were used to process the dataset but two rounds of 3D classification were performed after 2D classification. In brief, 446,553 particles were selected by automatic particle picking in RELION12. Next, 107,564 particles containing good 2D classes of all side views, top views of C27 and C28 classes, and top views of classes with uncertain symmetry were selected for the first round of 3D classification using an angular sampling angle of 7.5° without applying symmetry. This 3D classification yielded four classes, of which two had apparent C27 and C28 symmetry, respectively. Subsequent 3D classification was performed using an angular sampling angle of 3.7° for both the C27 and C28 classes. From a 3D class of C27-fold symmetry, 40,086 particles were selected for auto-refinement by imposing symmetry in RELION12. For the C28-fold symmetry pore, 16,581 particles from the 3D class with the best resolution were extracted and refined using SPHIRE30. Final resolutions for C27 and C28 pores are 3.8 Å and 4.2 Å, respectively. Local resolution distribution of all maps was determined by ResMap13.
Model building and analysis
The 3.8 Å map with 27-fold symmetry from the mercury-soaked data set and the crystal structure of full length GSDMA3 (PDB ID: 5B5R) were used for model building. The model of the N-terminal domain was docked into the map as a rigid body in Chimera31. The fitted model accounted well for most of the density in the globular domain but required extensive remodelling in the β-barrel region using Coot32. PHENIX was used to refine the model against the cryo-EM density in real space and to ensure proper geometry33. All structural and density representations were generated using either Chimera31 or Pymol (https://www.pymol.org). Model versus map FSC curves were calculated using Phenix.mtriage to obtain an estimate of the final resolution for the models.
Liposome leakage assay
The leakage of liposomes encapsulating TbCl3 was determined by an increase in fluorescence intensity when Tb3+ bound to dipicolinic acid (DPA) in the external buffer. Tb3+-entrapped liposomes were prepared using a similar procedure as detailed in ‘Reconstitution of GSDMA3 and GSDMD pores’, with the exception that the buffer contained 20 mM HEPES at pH 7.4, 150 mM NaCl, 50 mM sodium citrate and 15 mM TbCl3. The suspension was loaded onto a Superose 6 gel filtration column to remove unincorporated TbCl3. Purified liposomes were supplemented with 50 μM DPA before addition of a recombinant GSDM protein (0.5 μM) and caspase-11 or 3C protease (0.2 μM). Fluorescence at 545 nm after excitation at 276 nm was recorded continuously for 45 min at 30-s intervals using a Molecular Devices SpectraMax M5 plate reader.
Reporting summary
Further information on experimental design is available in the Nature Research Reporting Summary linked to this paper.
Data availability
The cryo-EM maps have been deposited in the Electron Microscopy Data Bank under accession numbers EMD-7449 (HgCl2-treated, C27 membrane pore), EMD-7450 (HgCl2-treated, C28 membrane pore) and EMD7451 (native, double-ring pore). The atomic structure coordinates have been deposited in the Protein Data Bank under the accession number 6CB8. All other data can be obtained from the corresponding author upon reasonable request.
Extended Data
Extended Data Fig. 1. Reconstitution of GSDMA3 pores in vitro.
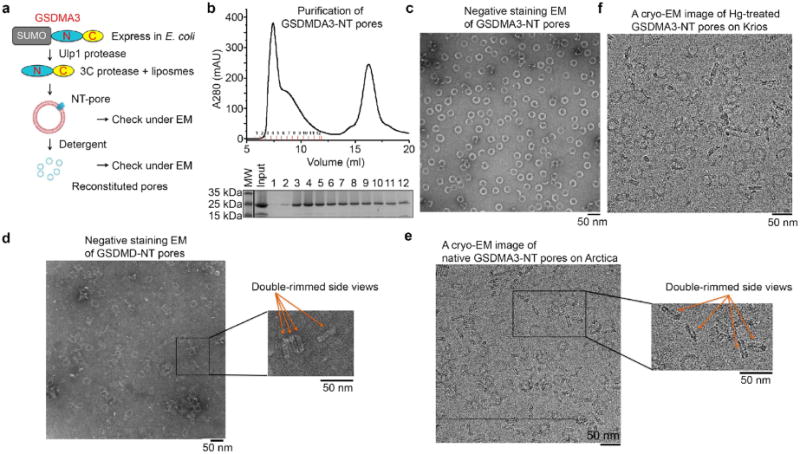
a, A general procedure for GSDMA3 pore reconstitution. b, Size-exclusion chromatography of GSDMA3 pores extracted from liposomes (top) and Coomassie blue-stained SDS–PAGE of the collected fractions. c, Representative negative-stain electron microscopy images of GSDMA3. d, Representative negative-stain electron microscopy images of human GSDMD pores with double-rimmed side views. e, f, Representative cryo-EM images of GSDMA3 pores with double-rimmed side views (e) and HgCl2-treated GSDMA3 pores (f). Scale bar, 50 nm (c–f). All results were confirmed at least three times as technical replicates.
Extended Data Fig. 2. Cryo-EM analysis of double- and single-ring GSDMA3 pores.
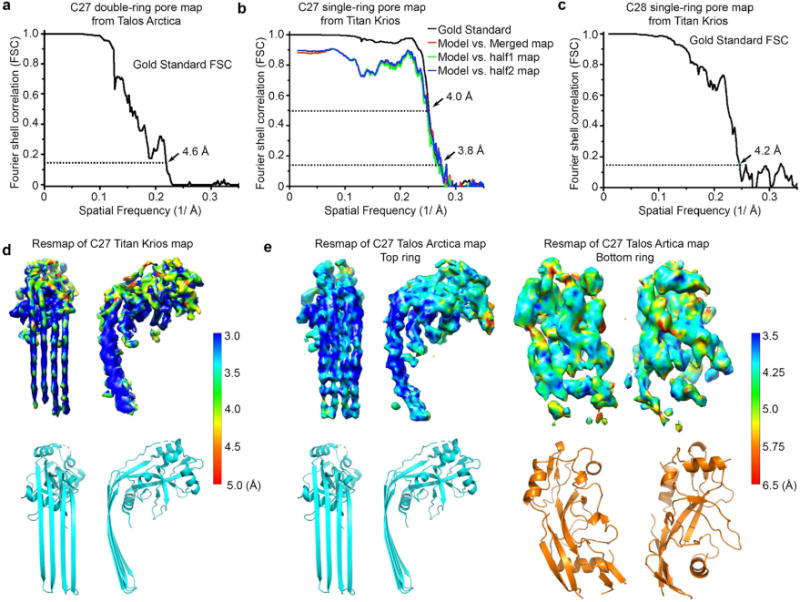
a–c, Gold-standard FSC plots from two half-reconstructions refined separately in RELION for the 27-fold double-ring pore map (a), the 27-fold single-ring pore map (b) and the 28-fold single-ring pore map (c). Model-to-map correlations are shown for the 27-fold single-ring pore (b). d, e, Local resolution estimation generated by ResMap on the two half maps separately refined in a gold-standard procedure in RELION. Local resolutions are colour-coded on the densities. Highest resolution is observed at the β-barrel domain for both the single-ring pore (d) and the membrane-inserted ring of the double-ring pore (e). The globular domains and the additional ring exhibit relatively low resolution.
Extended Data Fig. 3. Cryo-EM density validation.
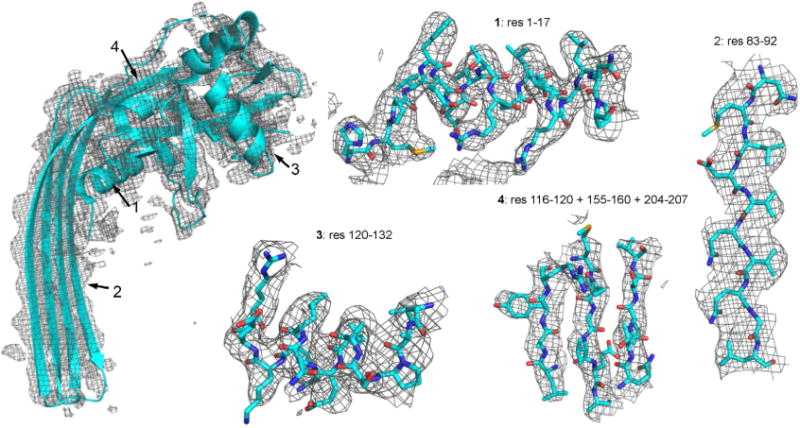
Close-up views of GDSMA3 subunit model fitted into the cryo-EM density map at four locations denoted by residue numbers.
Extended Data Fig. 4. Aligned sequences in the N-terminal region among GSDMA3, human GSDMA, mouse GSDMD and human GSDMD.
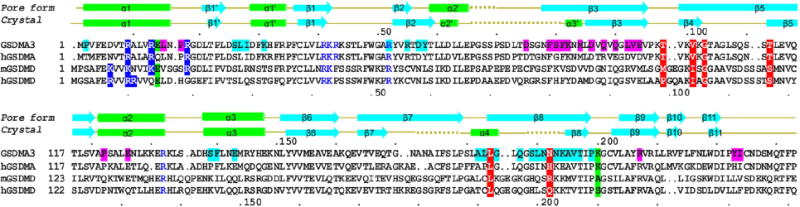
Secondary structures are shown for both the auto-inhibited conformation1 and the pore conformation of GSDMA3. Dotted lines represent disordered regions. Residue numbers are shown as dots every ten residues above the alignment for GSDMA3 and below for human GSDMD. Blue highlights, potential positively charged residues for acidic lipid binding; blue, tested residues that did not affect pore formation; magenta and cyan highlights, residues involved in the oligomerization or membrane insertion of the two adjacent subunits; red and green highlights, previously reported and cancer-associated mutations localized on the oligomerization interface of the globular domains (green) and β-barrel domain (red).
Extended Data Fig. 5. Lipid binding by GSDM.
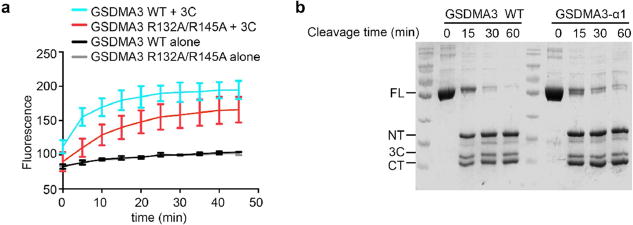
a, Effect of the GSDMA3 R132A/R145A mutation on the liposome-leakage activity, monitored by measuring DPA-chelating-induced fluorescence of released Tb3+ ion (n = 3 biological replicates). Error bars denote mean ± s.d. b, Equal processing of GSDMA3 by the 3C protease for the wild type and the α1 mutant. A representative gel of two independent experiments is shown.
Extended Data Fig. 6. Two-residue shift in the hydrogen-bonding pattern in each GSDMA3 subunit.
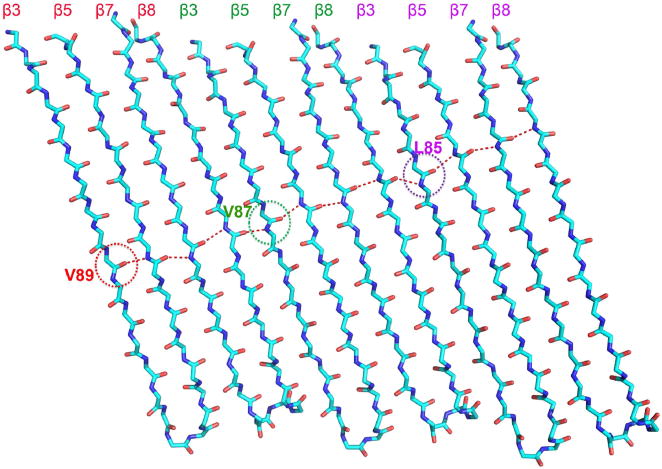
The shift results in a shear number of 27 × 2 = 54 for a 27-fold symmetric pore.
Extended Data Fig. 7. Structure-based sequence alignment.

The N-terminal region of GSDMA3 crystal structure (PDB ID: 5B5R) aligned with structures of perfringolysin O (5DIM), pneumolysin (4QQQ), haemolysin (3HVN), perforin-1 (3NSJ), complement C8 alpha (2QQH) and Plu-MAPCF (2QP2) and using the distance alignment matrix method (DALI). Helices are coloured in green, strands in cyan and loops in yellow. Vertical yellow bars indicate disordered regions in the GSDMA3-NT structure. Vertical red bars denote gaps in alignment of Plu-MACPF. Identical residues conserved among at least three proteins are highlighted in yellow. Glycines conserved among MACPF/CDCs are not conserved in GSDMA3, as highlighted in deep red.
Extended Data Table 1.
Cryo-EM data collection, refinement and validation statistics
| #1 Hg-treated, C27-symmetry (EMDB-7449) (PDB 6CB8) |
#2 Hg-treated, C28-symmetry (EMDB-7450) |
#3 Native, C27-symmetry Stacked double pore (EMDB-7451) |
|
|---|---|---|---|
|
| |||
| Data collection and processing | |||
|
| |||
| Voltage (kV) | 300 | 300 | 200 |
|
| |||
| Electron exposure (e–/Å2) | 40.0 | 40.0 | 41.1 |
|
| |||
| Defocus range (μm) | −0.75 to −2.0 | −0.75 to −2.0 | −0.1 to −3.0 |
|
| |||
| Super-resolution pixel size (Å) | 0.66 | 0.66 | 0.5843 |
|
| |||
| Symmetry imposed | C27 | C28 | C27 |
|
| |||
| Initial particle images (no.) | 446,553 | 446,553 | 462,878 |
|
| |||
| Final particle images (no.) | 40,086 | 16,581 | 64.248 |
|
| |||
| Map resolution (Å) | 3.8 | 4.2 | 4.6 |
|
| |||
| FSC threshold | 0.143 | 0.143 | 0.143 |
|
| |||
| Map resolution range (Å) | 60.0–3.8 | 60.0–4.2 | 60.0–4.6 |
|
| |||
| Refinement | |||
|
| |||
| Initial model used (PDB code) | 5B5R | ||
| Model resolution (Å) | 4.0 | ||
| FSC threshold | 0.5 | ||
|
| |||
| Model resolution range (Å) | 84.5–3.8 | ||
|
| |||
| Map sharpening B factor (Å2) | −131.255 | ||
|
| |||
| Model composition | |||
| Non-hydrogen atoms | 1,844 | ||
| Protein residues | 230 | ||
| Ligands | 14 | ||
|
| |||
| B factors (Å2) | |||
| Protein | 101.86 | ||
| Ligand | 92.69 | ||
|
| |||
| R.m.s. deviations | |||
| Bond lengths (Å) | 0.009 | ||
| Bond angles (°) | 1.122 | ||
|
| |||
| Validation | |||
| MolProbity score | 2.46 (99th percentile)* | ||
| Clashscore | 16.22 (97th percentile)* | ||
| Poor rotamers (%) | 0.00 | ||
|
| |||
| Ramachandran plot | |||
| Favored (%) | 86.28 | ||
| Allowed (%) | 13.72 | ||
| Disallowed (%) | 0.00 | ||
100th is the best among the structures of comparable resolution, 0th is the worst.
Supplementary Material
Acknowledgments
This work was supported by US NIH grant DP1HD087988 (H.W.), R01Al124491 (H.W.), R01AI123265 (J.L.) and Charles A. King Trust Postdoctoral Fellowship Program (J.R., X.L.). We thank the University of Massachusetts Cryo-EM Core Facility and NCI National Cryo-EM Facility at the Frederick National Laboratory for Cancer Research for data collection, and D. Ni and M. Liao for discussions.
Footnotes
Reviewer information
Nature thanks H. Saibil, J. Whisstock and B. Zuber for their contribution to the peer review of this work.
Author contributions
J.R., H.W. and J.L. conceived the study. J.R., S.X. and H.W. designed the experiments and analysed the data. J.R. reconstituted the pores, performed cryo-EM experiments and refined the structure. J.R. and S.X. analysed structure-based mutants. J.R., H.W. and S.X. analysed the structure. H.W., J.R., S.X. and J.L. wrote the manuscript. X.L. provided advice and comments on the manuscript.
Competing interests
The authors declare no competing interests.
Additional information Extended data is available for this paper at https://doi.org/10.1038/s41586-018-0058-6.
Supplementary information is available for this paper at https://doi.org/10.1038/s41586-018-0058-6.
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Ding J, et al. Pore-forming activity and structural autoinhibition of the gasdermin family. Nature. 2016;535:111–116. doi: 10.1038/nature18590. [DOI] [PubMed] [Google Scholar]
- 2.Liu X, et al. Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature. 2016;535:153–158. doi: 10.1038/nature18629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sborgi L, et al. GSDMD membrane pore formation constitutes the mechanism of pyroptotic cell death. EMBO J. 2016;35:1766–1778. doi: 10.15252/embj.201694696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chen X, et al. Pyroptosis is driven by non-selective gasdermin-D pore and its morphology is different from MLKL channel-mediated necroptosis. Cell Res. 2016;26:1007–1020. doi: 10.1038/cr.2016.100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Russo HM, et al. Active caspase-1 induces plasma membrane pores that precede pyroptotic lysis and are blocked by lanthanides. J Immunol. 2016;197:1353–1367. doi: 10.4049/jimmunol.1600699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shi J, et al. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature. 2015;526:660–665. doi: 10.1038/nature15514. [DOI] [PubMed] [Google Scholar]
- 7.Kayagaki N, et al. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature. 2015;526:666–671. doi: 10.1038/nature15541. [DOI] [PubMed] [Google Scholar]
- 8.Aglietti RA, et al. GSDMD p30 elicited by caspase-11 during pyroptosis forms pores in membranes. Proc Natl Acad Sci USA. 2016;113:7858–7863. doi: 10.1073/pnas.1607769113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rathinam VA, Vanaja SK, Fitzgerald KA. Regulation of inflammasome signaling. Nat Immunol. 2012;13:333–342. doi: 10.1038/ni.2237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lamkanfi M, Dixit VM. Inflammasomes and their roles in health and disease. Annu Rev Cell Dev Biol. 2012;28:137–161. doi: 10.1146/annurev-cellbio-101011-155745. [DOI] [PubMed] [Google Scholar]
- 11.Wang Y, et al. Chemotherapy drugs induce pyroptosis through caspase-3 cleavage of a gasdermin. Nature. 2017;547:99–103. doi: 10.1038/nature22393. [DOI] [PubMed] [Google Scholar]
- 12.Scheres SH. RELION: implementation of a Bayesian approach to cryo-EM structure determination. J Struct Biol. 2012;180:519–530. doi: 10.1016/j.jsb.2012.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kucukelbir A, Sigworth FJ, Tagare HD. Quantifying the local resolution of cryo-EM density maps. Nat Methods. 2014;11:63–65. doi: 10.1038/nmeth.2727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jacoboni I, Martelli PL, Fariselli P, De Pinto V, Casadio R. Prediction of the transmembrane regions of β-barrel membrane proteins with a neural network-based predictor. Protein Sci. 2001;10:779–787. doi: 10.1110/ps.37201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Murzin AG, Lesk AM, Chothia C. Principles determining the structure of β-sheet barrels in proteins. I. A theoretical analysis. J Mol Biol. 1994;236:1369–1381. doi: 10.1016/0022-2836(94)90064-7. [DOI] [PubMed] [Google Scholar]
- 16.Law RH, et al. The structural basis for membrane binding and pore formation by lymphocyte perforin. Nature. 2010;468:447–451. doi: 10.1038/nature09518. [DOI] [PubMed] [Google Scholar]
- 17.Rossjohn J, Feil SC, McKinstry WJ, Tweten RK, Parker MW. Structure of a cholesterol-binding, thiol-activated cytolysin and a model of its membrane form. Cell. 1997;89:685–692. doi: 10.1016/s0092-8674(00)80251-2. [DOI] [PubMed] [Google Scholar]
- 18.Köster S, et al. Crystal structure of listeriolysin O reveals molecular details of oligomerization and pore formation. Nat Commun. 2014;5:3690. doi: 10.1038/ncomms4690. [DOI] [PubMed] [Google Scholar]
- 19.van Pee K, et al. CryoEM structures of membrane pore and prepore complex reveal cytolytic mechanism of pneumolysin. eLife. 2017;6:e23644. doi: 10.7554/eLife.23644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Marshall JE, et al. The crystal structure of pneumolysin at 2.0 Å resolution reveals the molecular packing of the pre-pore complex. Sci Rep. 2015;5:13293. doi: 10.1038/srep13293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Holm L, Sander C. Dali: a network tool for protein structure comparison. Trends Biochem Sci. 1995;20:478–480. doi: 10.1016/s0968-0004(00)89105-7. [DOI] [PubMed] [Google Scholar]
- 22.Rosado CJ, et al. A common fold mediates vertebrate defense and bacterial attack. Science. 2007;317:1548–1551. doi: 10.1126/science.1144706. [DOI] [PubMed] [Google Scholar]
- 23.Degiacomi MT, et al. Molecular assembly of the aerolysin pore reveals a swirling membrane-insertion mechanism. Nat Chem Biol. 2013;9:623–629. doi: 10.1038/nchembio.1312. [DOI] [PubMed] [Google Scholar]
- 24.Iacovache I, et al. Cryo-EM structure of aerolysin variants reveals a novel protein fold and the pore-formation process. Nat Commun. 2016;7:12062. doi: 10.1038/ncomms12062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Idone V, et al. Repair of injured plasma membrane by rapid Ca2+-dependent endocytosis. J Cell Biol. 2008;180:905–914. doi: 10.1083/jcb.200708010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Keefe D, et al. Perforin triggers a plasma membrane-repair response that facilitates CTL induction of apoptosis. Immunity. 2005;23:249–262. doi: 10.1016/j.immuni.2005.08.001. [DOI] [PubMed] [Google Scholar]
- 27.Evavold CL, et al. The pore-forming protein gasdermin D regulates interleukin-1 secretion from living macrophages. Immunity. 2018;48:35–44. doi: 10.1016/j.immuni.2017.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zheng SQ, et al. MotionCor2: anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nat Methods. 2017;14:331–332. doi: 10.1038/nmeth.4193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rohou A, Grigorieff N. CTFFIND4: Fast and accurate defocus estimation from electron micrographs. J Struct Biol. 2015;192:216–221. doi: 10.1016/j.jsb.2015.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Moriya T, et al. High-resolution single particle analysis from electron cryo-microscopy images using SPHIRE. J Vis Exp. 2017;123 doi: 10.3791/55448. https://doi.org/10.3791/55448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pettersen EF, et al. UCSF Chimera—a visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 32.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 33.Adams PD, et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The cryo-EM maps have been deposited in the Electron Microscopy Data Bank under accession numbers EMD-7449 (HgCl2-treated, C27 membrane pore), EMD-7450 (HgCl2-treated, C28 membrane pore) and EMD7451 (native, double-ring pore). The atomic structure coordinates have been deposited in the Protein Data Bank under the accession number 6CB8. All other data can be obtained from the corresponding author upon reasonable request.