

1 Introduction
Neurofilaments, which are the intermediate filaments of nerve cells, are long flexible rope-like cytoskeletal polymers composed of internexin, peripherin and neurofilament proteins L, M and H (low, medium and high molecular mass) (Laser-Azogui et al., 2015; Perrot and Eyer, 2013). Like other intermediate filament proteins, these proteins all share a conserved alpha-helical central domain called the rod domain that assembles to form the filament backbone flanked by variable amino- and carboxy-terminal domains that regulate subunit assembly and filament interactions. However, neurofilaments can co-assemble in various combinations and stoichiometries so the precise composition of neurofilaments depends on which of the neurofilament proteins are expressed and on their relative level of expression, both of which vary depending on the neuronal cell type and stage of development.
The principal known function of neurofilaments is to expand axon caliber, which is a major determinant of axonal conduction velocity (Hoffman, 1995). The space-filling properties of these polymers are maximized by the carboxy-terminal domains of the subunit proteins, particularly of neurofilament proteins M and H, which project outward from the filament backbone. The extended and unstructured nature of these highly charged domains forms a dense polyelectrolyte brush border that appears to define a zone of exclusion around the backbone of the polymer much like the bristles on a bottle brush, increasing the spacing between neurofilaments and neighboring structures (Mukhopadhyay et al., 2004).
In addition to their structural role, neurofilaments are cargoes of axonal transport that move along microtubule tracks, propelled by microtubule motor proteins (Brown, 2000; Brown, 2009; Wang et al., 2000). The observed rate of movement depends on the time frame of observation. Radioisotopic pulse labeling experiments in laboratory animals on a time scale of days or weeks have revealed that neurofilaments are conveyed in the slow component of axonal transport at average rates of approximately 0.3–3 mm/day. In contrast, live-cell fluorescence imaging in cultured neurons on a time scale of seconds or minutes has revealed that neurofilaments move at average rates of ~1–3 μm/s, but these rapid bouts of movement are intermittent and highly asynchronous. Thus, the axonal transport of neurofilaments is a “stop-and-go” motility characterized by rapid infrequent movements interrupted by prolonged pauses. The velocity is slow on long time scales because the polymers spend most of their time pausing. It is now generally assumed that other cargoes of slow axonal transport are transported in a similar stop-and-go manner, but this remains to be proven.
Neurofilaments are also of clinical interest because mutations in neurofilament protein L can cause peripheral neuropathy and because neurofilaments accumulate abnormally and excessively in many different neurodegenerative diseases and toxic neuropathies (Lariviere and Julien, 2004). In extreme cases, the neurofilament accumulations lead to large balloon-like swellings of the affected axons. These accumulations are thought to be caused by an impairment of the axonal transport of these cytoskeletal polymers, but how this occurs is poorly understood.
In spite of considerable progress over the past 15 years, there are still many unanswered questions regarding the mechanism of neurofilament transport. For example, how do motors interact with neurofilaments, how do these motors coordinate bidirectional motility, how is the organization of neurofilaments in axons established and maintained, what determines when neurofilaments move and pause, what regulates the accumulation of neurofilaments in developing axons, and what causes neurofilaments to accumulate abnormally in axons in neurodegenerative disease? Cultured neurons offer researchers a special opportunity to address these questions because of their amenability to observation by high-resolution live-cell fluorescence imaging. However, a major challenge is that these polymers measure just 10 nm in diameter and are often packed close together, just tens of nanometers apart. Since the diameter and spacing of these polymers are well below the optical diffraction limit of light microscopes, the movement of single neurofilament polymers can be difficult to detect. A further challenge is that the movement is very infrequent, with single neurofilaments capable of pausing for hours between successive bouts of movement. In this article, we describe fluorescence imaging strategies that we have developed to overcome these and other challenges. Each of these approaches has its strengths and weaknesses, but taken together they form a powerful set of complementary tools which to investigate the kinetics of neurofilament transport in a variety of neuronal cell types.
2 Culturing neurons
2.1 General considerations
We prefer to use primary neuronal cultures, which are cultures of differentiated nerve cells isolated from neuronal tissue. Since these neurons are postmitotic differentiated cells, each batch of cultures yields a fixed number of neurons. Typically we establish cultures on a weekly or biweekly basis depending on our needs, though some cultures may be maintained for weeks. For our live-cell imaging studies of neurofilament transport we have used autonomic motor neurons from superior cervical ganglia (SCG), sensory neurons from dorsal root ganglia (DRG), or neurons from the cerebral cortex (“cortical neurons”). We establish these cultures from either rats or mice, depending on our needs. We purchase timed pregnant rats or mice from Harlan (Indianapolis, IN) or Taconic Biosciences (Cambridge City, IN). If we have a choice we generally prefer to use rats, but in general (except where noted) the procedures that we use are identical for mice. We focus here on our own current methods. Similar or alternative methods for culturing these neuronal cell types, which include detailed descriptions of the tissue dissection and culturing procedures, can be found elsewhere (Goslin et al., 1998; Hawrot and Patterson, 1979; He and Baas, 2003; Higgins et al., 1991; Johnson and Argiro, 1983; Johnson et al., 1981; Kaech and Banker, 2006; Kleitman et al., 1998; Mahanthappa and Patterson, 1998).
To establish and maintain these primary neuronal cultures, we use fine dissecting instruments, a binocular dissecting microscope capable of 8–25x magnification, a horizontal laminar flow hood and a tissue culture incubator with temperature and CO2 control and active or passive humidification. The fine dissection and all subsequent steps are performed under sterile conditions in the hood to avoid bacterial or fungal contamination (Freshney, 2010). Hoods with a vertical flow design are not suitable because they cannot accommodate a dissection microscope.
With the exception of myelinating co-cultures (Section 2.6), we generally prefer to plate the cells at low densities. At higher densities the axons tend to fasciculate, which makes axon tracing more challenging. In addition, axons in dense cultures often do not lie directly against the glass coverslip, making it hard to keep moving neurofilaments in focus. Higher cell densities also result in more non-neuronal cells and more cell debris, leading to higher background fluorescence. However, since neurons fare much better in high-density cultures, culturing them at low density requires that extra care and attention be paid to the culture procedures and conditions. In all cases we maintain the cultures in an incubator at 37°C and 95% relative humidity in an atmosphere of either ambient or 5% CO2, depending on the culture medium.
2.2 Culture media
Media for superior cervical ganglion neuron cultures
For short-term cultures (up to 1 week or so) we use a medium based on the formulation of Bray (1991) which contains Leibovitz’s L-15 culture medium (phenol red-free) supplemented with 10% (v/v) adult rat serum, 0.6% (w/v) glucose, 2 mM L-glutamine, 0.5% (w/v) hydroxypropylmethylcellulose (Methocel™) and 50 ng/ml Nerve Growth Factor (NGF; 2.5S subunit purified from mouse salivary glands) (L-15 Culture Medium; Table 1). L-15 medium is designed to buffer its pH at atmospheric CO2 so it is more convenient for observation and microinjection of cells on the microscope stage than media with bicarbonate buffering systems. An additional benefit is that non-neuronal cells proliferate far less in this medium than in bicarbonate-buffered media, thereby eliminating the need for the addition of anti-mitotic agents. NGF is required to support the survival of the neurons. For longer term cultures, we have used a serum-free DMEM/F12-based medium based on the N2 formulation of Bottenstein and Sato (1979) as modified by Higgins et al. (1991) (DMEM Culture Medium; Table 1). For low-density cultures, this medium can be supplemented with 10% adult rat serum. A defined serum-free L-15 based culture medium can also be used (Hawrot and Patterson, 1979; Mahanthappa and Patterson, 1998). With good sterile technique it is not necessary to use antibiotics, which can have deleterious side-effects on neurons.
Table 1.
Composition of SCG neuron culture media. The L-15 culture medium is maintained at atmospheric CO2 and the DMEM/F12 culture medium is maintained at 5% CO2. DMEM/F12 is a 1:1 (v/v) mixture of Dulbecco’s Modified Eagle and Ham’s F-12 nutrient media. 100 μg/ml bovine transferrin can be substituted with 20 μg/ml rat transferrin.
| L-15 CULTURE MEDIUM | |
|---|---|
| Component | Source |
| Leibovitz’s L-15 medium, phenol red-free | Gibco Life Technologies |
| 10% (v/v) adult rat serum | Harlan Laboratories |
| 0.6% (w/v) D-glucose (33.3 mM) | Sigma |
| 0.29 mg/ml L-glutamine (2 mM) | Sigma |
| 0.5% (w/v) Methocel™ (F4M Premium grade) | Dow Chemical Company |
| 50 ng/ml NGF, 2.5S subunit | BD Biosciences |
| DMEM/F12 CULTURE MEDIUM | |
|---|---|
| Component | Source |
| DMEM/F12 medium, phenol red-free | Gibco Life Technologies |
| 100 μg/ml apo-transferrin, bovine | Sigma |
| 10 μg/ml insulin, bovine | Sigma |
| 5 ng/ml sodium selenite (30 nM) | Sigma |
| 0.2 mg/ml L-glutamine (1.4 mM) | Sigma |
| 0.5% (w/v) bovine serum albumin, Fraction V | EMD Millipore |
| 0.5% (w/v) Methocel™ (F4M Premium grade) | Dow Chemical Company |
| 50 ng/ml NGF, 2.5S subunit | BD Biosciences |
Adult rat serum can be prepared by the method of Hawrot & Patterson (1979) or purchased from a commercial source. We sterilize the serum by syringe filtration with a 0.2 μm filter and store it frozen in aliquots to minimize repeated freezing and thawing. Often the serum forms a fine precipitate after a few days at 37°C. This precipitate can be mistakenly identified as microbial contamination, but it is actually innocuous and does not harm the cells. With serum from some commercial sources, the precipitate can become so dense that it obscures the cells. In this case, switch to a different commercial source or prepare the serum yourself. Fetal bovine serum (FBS) can also be used, but it is our impression that both rat and mouse SCG neurons are healthier and attach better to the substrate in the presence of adult rat serum.
The function of the Methocel™ is to increase the viscosity of the culture medium. It is not critical, but the cells appear to attach better to the substrate when it is present. Since Methocel™ solutions are too viscous to sterilize by filtration, the powder must be autoclaved. We weigh out 200 mg aliquots of Methocel™ powder in 50 ml disposable polypropylene centrifuge tubes and then autoclave with the caps loosely attached. After cooling, the caps can be tightened and the tubes can be stored indefinitely at room temperature. To dissolve the Methocel™, we add 40 ml sterile L-15 and shake the tube overnight at 37°C. The resulting solution contains insoluble particulate material, which can be removed by filtration using a 5 μm syringe filter (Millex-SV, EMD Millipore).
Media for cortical neuron cultures
The cortical neurons are cultured with a glial feeder layer. To expand and maintain the glia we use a medium consisting of Minimum Essential Medium (MEM) supplemented with 10% (v/v) horse serum, 0.7% (w/v) D-glucose and 0.5 μg/ml gentamicin (Glia MEM Culture Medium; Table 2). The neurons are cultured in NbActiv4™ medium (BrainBits LLC, Springfield, IL) with or without additional salt depending on the desired osmolarity (NbActiv4™ Culture Medium; Table 2). NbActiv4™ is identical to Neurobasal/B27™ culture medium except for three additional supplements: creatine, estrogen and cholesterol. NbActiv4™ medium has been reported to yield hippocampal neuron cultures with more synapses, increased electrical activity and less metabolic stress compared to Neurobasal/B27™ medium (Brewer et al., 2009). In our experience we obtain improved cell health and viability using this medium. To enhance neuronal recovery after plating, improve cell attachment, and minimize the risk of bacterial contamination arising from the initial dissection, we supplement the NbActiv4™ medium with 5% (v/v) FBS and 0.5 μg/ml gentamicin for plating (NbActiv4™ Plating Medium; Table 2). On the next day, we replace it with NbActiv4™ Culture Medium (which lacks FBS and gentamicin) and then maintain the cultures in this medium. With good sterile technique it is not necessary to use gentamicin. Around 1 week after plating, we increase the osmolarity of the medium from ~230–245 mOs to ~280–295 mOs by addition of NaCl to a final concentration of 37.5 mM, which we have found to improve long-term cell viability (Section 6.2). While it is possible to culture cortical neurons in this medium for at least 1 month, all of our own work has been performed within two weeks of plating.
Table 2.
Composition of cortical neuron and glia culture media. These media are maintained at 5% CO2.
| Glia MEM CULTURE MEDIUM | |
|---|---|
| Component | Source |
| Minimum Essential Medium (MEM) | Gibco Life Technologies |
| 10% (v/v) horse serum | Gibco Life Technologies |
| 0.7% (w/v) D-glucose (39 mM) | Sigma |
| 5 μg/ml gentamicin | Sigma |
| NbActiv4™ PLATING MEDIUM | |
|---|---|
| Component | Source |
| NbActiv4™ medium | BrainBits |
| 5% (v/v) fetal bovine serum (FBS) | Hyclone, GE Healthcare Life Sciences |
| 5 μg/ml gentamicin | Sigma |
| NbActiv4™ CULTURE MEDIUM | |
|---|---|
| Component | Source |
| NbActiv4™ medium | BrainBits |
| 37.5 mM NaCl (included ~1 week after plating) | Sigma |
Media for dorsal root ganglion neuron cultures
For long-term myelinating co-cultures of DRG neurons and Schwann cells we use NbActiv4™ Myelination Medium (phenol red-free) consisting of NbActiv4™ supplemented with 100 ng/ml NGF or 25 ng/ml Neurotrophin-3 (NT-3; Table 3). As with SCG neurons, a neurotrophic factor is required to support neuron survival. NGF supports the survival of cutaneous sensory neurons, which have relatively small axon diameters, whereas NT-3 supports the survival of the proprioceptive sensory neurons that innervate the skeletal muscles. The NT-3 dependent neurons contain more neurofilaments, have larger axons, and myelinate more readily and more continuously in culture. Five days after plating the cells, the medium is replaced with fresh medium containing a one-time supplement of 1:100 dilution of Matrigel™ (BD Biosciences). Three days later, half the medium is removed and replaced with fresh medium lacking Matrigel™ and containing 50 μg/ml ascorbic acid. Ascorbic acid is an essential cofactor in collagen biosynthesis, which is required for Schwann cells to form a basal lamina, which is in turn required for efficient myelination (Eldridge et al., 1989; Eldridge et al., 1987). The cultures can be maintained for up to three months, with a half medium change every 2–3 days, and a full medium change once each week. For short-term non-myelinating cultures ascorbic acid can be omitted or the cultures can be maintained in the L-15 Culture Medium described above for SCG neurons (Alami et al., 2009) (Table 1).
Table 3.
Composition of culture medium for myelinating DRG cultures. This medium is maintained at 5% CO2. Note that to prevent gelation and clumping, the Matrigel™ should be thawed on a slurry of ice and water to prevent gelation and then diluted into ice-cold NbActiv4™ medium, phenol red-free. After mixing, the medium can then be warmed and the other components can be added.
| NbActiv4™ MYELINATION MEDIUM | |
|---|---|
|
| |
| Component | Source |
|
| |
| NbActiv4™ medium, phenol red-free | BrainBits |
|
| |
| Either | |
| 100 ng/ml NGF, 2.5S subunit | BD Biosciences (NGF) |
| Or | |
| 25 ng/ml NT-3 | PeproTech (NT-3) |
|
| |
| 1% (v/v) Matrigel ™, phenol red-free (one time application ~5 days after plating) | BD Biosciences |
|
| |
| 50 μg/ml ascorbic acid (initiated ~8 days after plating) | Sigma |
2.3 Coverslips and culture dishes
Since the highest quality microscope objectives have large numerical apertures and short working distances, it is preferable to perform live-cell imaging using an inverted microscope, i.e. one in which the objective is positioned beneath the culture dish focusing up on the cells through the bottom of the dish. For high resolution imaging, it is necessary to culture the cells on glass coverslips that match the optical correction of high numerical aperture microscope objectives. Usually these objectives are corrected for imaging through a thickness of glass equal to 170 μm, so the optimal coverslips are ones designated #1.5, which corresponds to a thickness of 0.16–0.19 mm.
We routinely acid-wash all coverslips for cell culture regardless of whether they were acid-washed by the manufacturer. To do this, we soak the coverslips in concentrated nitric acid for at least 18 hours and then rinse extensively with deionized water over a period of several days. To dry the coverslips, we dip them in ethanol and then flame them.
To culture the cells on coverslips, we use one of two approaches. The simplest is to culture the cells in glass-bottomed plastic Petri dishes so that the cells can be imaged in the same dish in which they were cultured. Alternatively, we culture the cells on loose coverslips in plastic Petri dishes and then transfer the coverslips to an imaging chamber prior to live-cell imaging. Each of these strategies has pros and cons, which will be discussed later (Section 6.3).
For glass-bottomed dishes we use 22mm × 22mm #1.5 square coverslips (Fisherfinest™ Premium brand). When culturing cells on loose coverslips the shape and dimensions of the coverslip that is required depends on the shape and dimensions of the imaging chamber that will be used (40 mm #1.5 round coverslips for the Bioptechs FCS2 chamber, 22mm × 22mm #1.5 square coverslips for the Warner Instruments RC-21B or RC-30HV chambers; Section 6.3).
Glass-bottomed dishes
Glass-bottomed dishes are plastic Petri dishes with a glass coverslip base (Bray, 1991) (Fig. 1). The cells are plated and cultured on the coverslip and can be imaged by placing the dish on the stage of an inverted microscope. There are many commercially available options (e.g. MatTek Corporation, Ashland, MA) but they are expensive so we make our own.
Fig. 1.
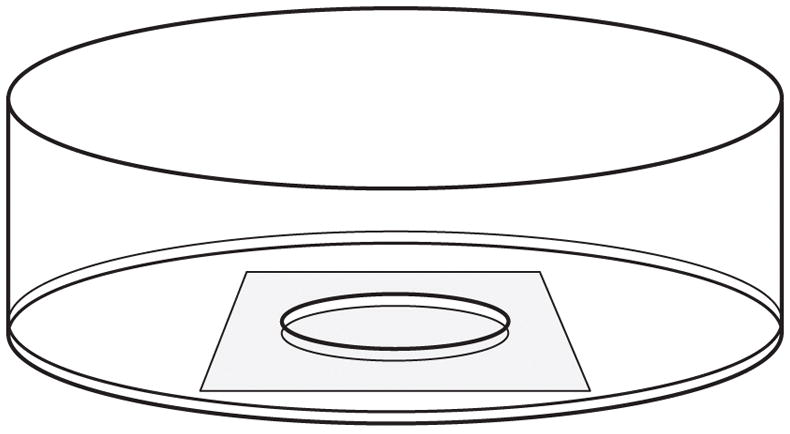
Drawing of a glass-bottomed culture dish. A hole is drilled in the bottom of a 35 mm plastic Petri dish and a glass coverslip is affixed to the base of the dish using wax or silicone adhesive. The cells are plated onto the coverslip in the shallow well created by the hole in the bottom of the dish.
Materials required for drilling the holes:
35mm plastic Petri dishes. We use Nunc™ brand tissue culture dishes with vented lids (Nalge Nunc International, Rochester, NY), but any similar dish should be suitable.
A drill press with a depth stop.
A ⅛″ - ½″ step drill bit with 1/32″ step increments (McMaster-Carr, Elmhurst, IL).
Though not essential, it is helpful to have a jig to hold the dish so that the holes are centered. We use a specially built jig milled out of aluminum to hold the dish in position on the drill press.
A source of clean compressed air to remove the plastic shards from the drill bit, jig and dishes.
Drilling the holes:
Mount drill bit in drill press and the custom jig on the drill press table.
Adjust the depth stop to ensure a 13/32″ hole in the dishes.
Remove the lids from the dishes and store for later use.
Place a dish in the jig and hold it firmly against the base to prevent it from rotating
Slowly lower the spinning drill bit down onto the dish to cut the hole.
Remove the dish and blow away any plastic shards using a source of clean compressed air.
Repeat for each dish. We usually drill a batch of 500 dishes at a time, which takes about 8 hours.
Notes:
Handle the drill bit and dishes throughout with gloves to keep them clean
The drill bit should be cleaned thoroughly prior to use because it contacts the culture dish. We use hot soapy water and then rinse with water and alcohol.
In step 5, it is important to use slow and steady pressure with a sharp drill bit and to support the dish from underneath to avoid cracking the plastic. The jig facilitates this. The dish may crack during the initial penetration of the drill bit but this does not matter as long as the cracks do not extend beyond the final diameter of the hole. When the drill bit becomes dull it should be replaced.
With a different jig, holes as large as ½″ can be drilled
In step 6, the drill often leaves a slight burr, which must be removed by gently scraping around the holes with a scalpel blade in order to allow coverslips to be attached flush against the plastic (see below).
We attach the coverslips to the dishes using either paraffin wax or a platinum-cured medical-grade silicone adhesive (A-103 Medical Grade Elastomer, Factor II Inc., Lakeside, AZ). The advantage of paraffin wax is that it is easy to remove the coverslips later using a razor or scalpel blade. This is helpful after fixing and immunostaining the cells because it allows the coverslip to be mounted on a glass slide using hardening mounting medium. However, a disadvantage of using paraffin is that it softens at 37°C and is not a strong adhesive so sometimes the coverslip can detach.
Materials required for attaching coverslips using paraffin wax:
A micro electric hotplate (we use an Thermolyne Aluminum-Top Micro Hot Plate; Thermo Fisher Scientific, Waltham, MA)
Retort stand and large retort stand hose clamp.
Round-tipped artist’s paint brush for applying the molten wax.
Attaching the coverslips using paraffin wax:
Invert the hot plate and mount it on a retort stand using a large retort stand hose clamp.
Invert the dishes on a clean work surface so that the bases with the holes drilled in them are facing upward
Using the paint brush, apply a ring of hot molten paraffin wax in a circle around the hole on each dish, taking care to keep the wax at least a few millimeters away from the edge of the hole.
After the paraffin has cooled, use forceps to place a clean 22mm × 22mm square #1.5 acid-washed glass coverslip on the base of each inverted dish so as to cover the hole and the wax surrounding it.
Raise the inverted dish with the coverslip laying on it to bring it close to the inverted hot plate and hold it in place until the wax melts and spreads out to fill the space between the coverslip and the base of the inverted plastic dish.
Move the dish away from the hot plate and cool it in a gentle stream of air to solidify the wax before it flows into the well.
Repeat for all the dishes.
Notes:
The dishes can be stored indefinitely before use.
In step 3, be careful not to apply the paraffin too close to the hole or to use too much paraffin or too much heat. Insufficient paraffin may result in gaps or bubbles between the coverslip and the base of the dish. Too much paraffin or heat may result in molten wax flowing into the hole or over the edge of the dish, or may result in the coverslip not laying evenly.
In step 5 it is best to avoid melting the paraffin too quickly. For best results, set the temperature of the hot plate to 60°C.
After step 6, incubating the dishes overnight at 37°C before storing them at room temperature appears to improve the seal and reduce the likelihood of future leaks.
Getting steps 3, 5 and 6 just right takes some trial and error but with practice the procedure becomes routine.
The dishes can be stored indefinitely before use.
To prepare dishes using silicone adhesive, mix the silicone elastomer and cross-linker according to the manufacturer’s instructions and then dispense onto the bases of the dishes using a syringe with a narrow plastic tip. Apply the coverslips using gentle pressure, and then cure at room temperature or 5 hours at 40°C. The adhesive is transparent.
Sterilizing and coating coverslips and glass-bottomed dishes
Prior to plating neurons it is necessary to coat the coverslips, whether loose or in glass-bottomed dishes, to promote cell adhesion and axon outgrowth. For cortical neuron cultures, we coat with poly-D-lysine. For DRG and SCG neurons, we then coat the poly-D-lysine with extracellular matrix proteins. We like to use Matrigel™ (BD Biosciences), which is a heterogeneous basement membrane extract containing laminin, Type IV collagen, entactin and heparan sulfate proteoglycans secreted by Engelbreth-Holm-Swarm mouse tumor cells. It is an excellent substrate for SCG and DRG neurons, but it tends to form sticky clumps that can increase background staining in immunofluorescence applications. If this problem is encountered, laminin is a good substitute. Both Matrigel™ and laminin are stored in aliquots at −80°C. To prevent gelation, they are thawed on a slurry of ice and water on or before the day of the dissection, and diluted into ice-cold L-15 medium. After dilution, the L-15 is allowed to warm to room temperature.
Procedure for sterilizing and coating glass-bottomed dishes:
On the day before the dissection, place the required number of glass-bottomed culture dishes and lids in a laminar flow hood and sterilize them by filling them to the brim with 70% (v/v) ethanol and letting them stand for 45 minutes.
Aspirate the ethanol and allow the dishes and lids to dry.
Treat the coverslips with 1 mg/ml poly-D-lysine hydrobromide (Mw 70–150,000, Sigma) in 0.1M sodium borate buffer, pH 8.5, for 3 hours (150 μl per well) as described by Higgins et al. (1991).
After the poly-D-lysine treatment, rinse the coverslips six times with sterile water (~5 minutes per rinse) to remove poly-D-lysine that is not bound to the glass.
For SCG and DRG neuron cultures and for glial cultures, add a few milliliters of sterile water to each dish, cover with a lid, and store overnight at room temperature. For cortical neuron cultures, use NbActiv4™ Plating Medium (Table 2) and store overnight in an incubator with 5% CO2.
For SCG neuron cultures: on the morning of the dissection, treat the poly-D-lysine coated coverslips with 10 μg/ml phenol red-free Matrigel or 10 μg/mL mouse laminin (BD Biosciences) in L-15. We apply 150 μl of Matrigel™ or laminin solution per coverslip well and incubate for 4 hours in the incubator (37°C, 95% relative humidity, atmospheric CO2).
For myelinating DRG cultures: treat the poly-D-lysine coated coverslips with Matrigel™ diluted to ~50 μg/ml in L-15 medium and leave the lids off the dishes in the hood until the Matrigel™ has dried (4–5 hours). The dried dishes can be stored at 4°C for up to two months, so the dishes can be prepared well in advance of the dissection if required.
Notes:
For step 3, we make up a 10 mg/ml stock solution in borate buffer and freeze it in aliquots. Prior to use, the aliquots are diluted in borate buffer and sterilized by syringe filtration with a 0.2 μm filter.
In step 5, the coverslips can be rinsed three times and then stored in the fourth rinse for up to 1 week, rinsing them an additional three times immediately before use.
In step 6, the dissection is performed during the 4 hour incubation period and then the coverslips are rinsed once with L-15 before plating.
In step 7, drying the Matrigel™ reduces the chance that it peels off the coverslip, which sometimes happens after several weeks in culture.
2.4 Superior cervical ganglion neuron cultures
We refer here to superior cervical ganglion (SCG) cultures from rats, but the procedures also work well for mice. We purchase timed pregnant female Sprague Dawley rats and sacrifice the neonatal (P0–P1) pups using an approved method of euthanasia. We prefer not to use decapitation because the SCG are located in the neck. Older pups can also be used, but we obtain the best cell viability and fewer non-neuronal cells when we use neonates. The two ganglia obtained from a single pup are sufficient for one batch of cultures.
Sacrifice the animals and then transfer them to the laminar flow hood, where all subsequent procedures are performed.
Dissect the ganglia together with the associated carotid artery and vagus nerve as described by Higgins et al. (1991) and He & Baas (2003).
Transfer the dissected ganglia to a 35 mm plastic dish containing Leibovitz’s L-15 medium (Gibco Life Technologies).
Under the dissection microscope, remove the ganglia from the associated tissue using fine forceps (we prefer Dumont #5).
Transfer the cleaned ganglia to a sterile 15 ml disposable polystyrene centrifuge tube containing fresh L-15 and allow them to settle to the bottom of the tube under gravity.
Remove the L-15 solution, rinse the ganglia once with phosphate buffered saline (PBS), then add 2.5 mg/ml collagenase (Type 3, Worthington Biochemicals) in PBS (sterile-filtered), cap the tube, and incubate at 37°C for 1 hour.
Remove the collagenase solution, rinse the ganglia twice with PBS, add 2.5 mg/ml trypsin (crystallized, Worthington Biochemicals) in PBS (sterile-filtered), cap the tube, and incubate at 37°C for 30 minutes.
Remove the trypsin solution and add L-15 containing 20% (v/v) FBS to inactivate the trypsin. Incubate at 37°C for 15 minutes.
Rinse the ganglia twice with L-15 medium containing 0.5% (w/v) bovine serum albumin (BSA; EMD Millipore Cat# 126609, Fraction V, fatty acid, nuclease and protease-free), then triturate in 2 ml L-15 containing 0.5% (w/v) BSA by repeatedly drawing the ganglia in the solution into a Pasteur pipette and then expelling them back into the tube, taking care to minimize frothing. Usually 5 or 6 passages are sufficient to disperse the cells.
Dilute an aliquot of the cell suspension to the desired concentration in L-15 containing 0.5% (w/v) BSA and then plate the suspension onto glass-bottomed dishes coated with poly-D-lysine and Matrigel™ (150 μl cell suspension per well) (Section 2.3).
Gently move the culture dishes to the incubator (37°C, 95% relative humidity, atmospheric CO2) and leave for 45 minutes to allow the cells to settle down onto the coverslips under gravity, then move them back to the hood and add 1.5 ml culture medium (Table 1) to each dish.
Return the dishes to the incubator and replace the medium with fresh medium every 3–4 days.
Notes
For each solution change or rinse, the ganglia are swirled gently in the added solution and then allowed to settle under gravity. Once settled, a Pasteur pipette is used to remove the supernatant, leaving just enough to cover the ganglia at the base of the tube.
When establishing cultures from embryonic ganglia, the collagenase treatment can be omitted.
In step 2, contamination of the cultures with non-neuronal cells can be reduced by removing the thin connective tissue capsule that ensheaths the ganglia. This requires forceps with very fine (Biologie) tips. If grasped successfully, the capsule can be peeled off the ganglia like an onion skin. After this the ganglia are very fragile.
In step 9, the purpose of the BSA is to reduce adhesion of the ganglia to the sides of the tube because the ganglia are very sticky after trypsin treatment.
In steps 6 and 8, any submerged ganglia that do adhere to the sides of the tube can be dislodged by squirting solution on them using a Pasteur pipette.
In step 9, the trituration is critical and requires some care. Variables include the force use to expel the solution from pipette (determined by how fast the pipette bulb is squeezed), the proximity of the pipette tip to the base or side of the tube (placing it against the base will generate the most shear), and the number of passages. Insufficient trituration will result in incomplete dispersal of the cells, whereas excessive trituration will reduce cell viability and increase cell debris. We fire-polish the tip of the pipette in a flame in the hood prior to use in order to ensure that the end is smooth and in order to constrict the orifice slightly. Normally it should not be necessary to exceed 5 or 6 passages. Inspect the solution by eye between passages by holding the tube up to the light in the hood. When dispersed, the cell suspension will have a homogenous and turbid appearance due to light scattering by the cells. The ganglia should disperse easily; avoid the temptation to triturate more if you observe residual particulate material in the suspension after 5 or 6 passages because any particle that persists after that amount of trituration is probably non-neuronal tissue.
In steps 6 and 7, prepare the enzyme solutions fresh in PBS immediately before use and sterilize by syringe filtration with a 0.2 μm filter.
In step 8 the duration of trypsinization and the concentration of the trypsin are important variables. Too little can decrease cell viability due to the greater shear forces required to disperse the cells, and too much can decrease cell viability due to the effects of proteolytic cleavage on plasma membrane integrity and physiology.
In step 10, the cell density can be measured using a hemocytometer but we find it more convenient to estimate it in units of ganglia/ml. In our hands, a concentration of dissociated cells equivalent to 0.03–0.06 rat ganglia/ml or 0.1–0.2 mouse ganglia/ml yields approximately 25–50 neurons per dish. At this density, two ganglia yield enough cells to plate more than 100 dishes, though we typically plate only 8–12.
In step 12, we aspirate the medium from the dish but not from the well (which has a volume of about 85 μl) prior to adding fresh medium, so as to avoid disturbing the cells on the coverslip.
When culturing in L-15 Culture Medium (Table 1), we typically image the cells within one week after plating.
2.5 Cortical neuron cultures
Neurons from the central nervous system tend to fare poorly when cultured at low density, but can be maintained by co-culturing with glia. However, such co-cultures are not suitable for live-cell imaging of axonal transport because the axons grow on top of the glia and thus do not lie flat, and because the glia are a source of elevated background fluorescence. To solve this problem, we use the glial sandwich technique developed by Gary Banker for culturing hippocampal neurons (Goslin et al., 1998; Kaech and Banker, 2006). In the original Banker method, neurons are cultured on coverslips that are inverted and suspended over a monolayer of glial cells (predominantly astrocytes). This way, the neurons are immersed in the glia-conditioned culture medium without actually contacting the glial cells. For our studies, we prefer to culture and image the neurons in glass-bottomed dishes (Section 2.3) so we culture the glia on coverslips that are then inverted and suspended over the neurons (Fig. 2). The latter arrangement works well for imaging experiments because the glia provide trophic support to the neurons, but can be removed for imaging.
Fig. 2.

Diagram of the coverslip sandwich assembly used for cortical neuron cultures. The coverslip bearing a glial monolayer is inverted and suspended above the well in a glass-bottomed dish, supported by paraffin dots at the corners of the coverslip and immersed in culture medium (grey shading).
The methods described below work well for cortical neurons from rats or mice, though we prefer to use rat glial feeder layers for both because this results in more consistently healthy cultures. We purchase timed pregnant female Sprague Dawley rats, timed pregnant C57BL6 or ICR mice, or pregnant mice that we breed in-house. Pups are sacrificed using an approved method. If the pups are not born by the expected time, we sacrifice the mother and remove the pups by C-section. Embryonic day 18–20 (E18–20) rat embryos, E17–18 mouse embryos, neonatal (P0) or postnatal day 1–2 (P1–2) pups can also be used though the cell viability decreases with increasing age. However, we prefer to use neonatal pups because they exhibit the optimal balance of neurofilament number and gap size (Section 5.2).
Preparation of coverslips for glial sandwich cultures
Sterilize 22 × 22 mm square acid-washed coverslips (Section 2.3) by soaking in 70% (v/v) ethanol for 45 min, then flame them one-by-one in the laminar flow hood to dry them.
Place each coverslip in a sterile 35mm diameter plastic Petri dish (we use Nunc™ brand vented cell culture dishes with lids) in the laminar flow hood.
Using the tip of a Pasteur pipette tip dipped in autoclaved hot molten paraffin wax (Fisher Scientific Cat # P31-500), place paraffin dots on the four corners of each coverslip.
Place the lids on the dishes and leave them overnight at 37°C.
Return the dishes to the laminar flow hood, add 500 μl of 1 mg/ml poly-D-lysine to each dish (sufficient to immerse the coverslips), and incubate at room temperature for at least 4 hours.
Rinse the coverslips 6 times with sterile water (5 minutes per rinse).
Notes
In step 4, the overnight incubation improves the adherence of the paraffin dots to the glass when they are subsequently immersed in liquid.
In step 6, the coverslips can be rinsed three times and then stored in the fourth rinse for up to 1 week, rinsing them an additional three times immediately before use.
Preparation of a suspension of cortical neurons and glia
Our protocol allows for the preparation of both glia and neurons on the same day from the same cell suspension. However, the glia must be expanded for 1-2 weeks prior to use, so the glial cultures established one week are used to support the neuronal cultures established 1–2 weeks later. This means that when establishing cortical cultures for the first time, or after a hiatus, only glial cultures are established in the first and second weeks (weeks 1 and 2). Thereafter (weeks 3, 4 etc.) both neuronal and glial cultures are established. The glial cultures from week 1 are used to support the neuronal cultures established in week 3, and the glial cultures established in week 2 are used to support the neuronal cultures established in week 4, etc. (Figure 3).
Fig. 3.

Flowchart showing the protocol for preparing cortical neuron sandwich cultures. A single prep generates enough cells to establish both neuronal and glial cultures, with the glial cultures that are established one week being used to support the neuronal cultures established 1–2 weeks later.
Decapitate 4–6 pups in the laminar flow hood, allowing the heads to drop onto sterile gauze in a large sterile plastic Petri dish.
For each pup, remove the skin over the skull and then make a longitudinal cut, bisecting the cranium. Dissect out the entire brain and place it in a 60 mm diameter dish containing cold Hank’s Balanced Salt Solution (HBSS).
Under a dissecting microscope placed in the laminar flow hood, remove the olfactory bulbs, cerebellum, midbrain, hindbrain and thalamic regions including the hippocampus, leaving only the cerebral hemispheres, and strip away all the meninges.
Transfer the hemispheres to a new culture dish containing 2–3 ml HBSS and chop the tissue into small pieces with a sterile scalpel blade.
Transfer the HBSS containing the minced tissue to a disposable 15 ml polystyrene centrifuge tube and allow the tissue to settle to the bottom of the tube under gravity.
Remove as much of the HBSS as possible, rinse once with 5 ml PBS, then add 5 ml pre-warmed PBS containing 2.5 mg/ml trypsin, 0.01% (w/v) EDTA and 1 mg/ml DNase I (solution prepared fresh and sterile-filtered before use). Cap the tube and incubate for 10 minutes at 37°C.
Remove the excess enzyme solution, rinse the tissue twice with 5 ml NbActiv4™ Plating Medium (Table 2), then remove most of the medium to leave 1–2 ml covering the tissue.
Triturate the tissue in the NbActiv4™ Plating Medium by repeatedly drawing it into a Pasteur pipette and then expelling it back into the tube. 10–20 passages should be sufficient to disperse the cells.
Dilute the cell suspension with 4–6 ml NbActiv4™ Plating Medium.
Spin at 80–100 × g for 2–5 minutes, then remove the supernatant.
Re-suspend the cells in 400–600 μl NbActiv4™ Plating Medium using a 1 ml pipette tip (5–6 gentle passages).
Place the cell suspension on ice. Some of this suspension will be used to establish neuronal cultures and the remainder will be used to establish glial cultures for use 1–2 weeks later (see below).
Notes
In step 3, it is important to strip away the meninges completely to prevent contamination of the cultures with fibroblasts, transferring the brains to dishes containing clean HBSS when the solution gets too cloudy.
In step 4, it is important to be quick. Don’t take too much time and don’t cut the pieces too small because either can result in more cell death.
For the rinses in steps 6 and 7, allow the tissue to settle to the bottom of the tube under gravity then remove the supernatant.
In step 7, we find that the 5% FBS in the NbActiv4 Plating Medium is sufficient to inactivate the trypsin.
In step 8 we fire-polish the Pasteur pipette tip in a flame prior to use to ensure that it is smooth. As with the SCG cultures (Section 2.4), the trituration step is critical. Insufficient trituration results in incomplete dispersal of the cells whereas too much trituration reduces cell viability and results in more cell debris.
In step 9, the actual cell density can be determined using a hemocytometer. The yield varies depending on the age of the animals. For cultures established from rat embryos, we obtain about 1–2 × 107 cells per brain. For cultures established from neonatal rat pups we obtain 4×106 cells per brain.
In step 10, if we want to transfect the cells by electroporation prior to establishing neuronal cultures then we aliquot the suspension into 1.5 ml microcentrifuge tubes and resuspend the pelleted cells in 100 μl Lonza Nucleofector™ solution provided by the manufacturer of our Lonza Nucleofector™ II electroporation device (Section 4.3). One rat brain yields enough cells for one Nucleofector™ electroporation.
Preparation of cortical neuron cultures
Dilute an aliquot of the cell suspension obtained above (step 12) to the desired concentration with NbActiv4™ Plating Medium and then plate on glass-bottomed dishes (1.5 ml per dish) coated with poly-D-lysine (Section 2.3) and leave the dishes in an incubator for 1 hour.
Aspirate the medium from each dish and add 3 ml fresh NbActiv4™ Plating Medium (sufficient to cover the coverslip with the glial feeder layer).
Transfer one coverslip bearing glia (established the previous week according to the procedure below) into each dish, inverting the coverslip and submerging it in the medium so that it is suspended above the neurons by the paraffin dots at the corners with the glia facing down.
Maintain these cultures in an incubator at 37°C in an atmosphere of 5%CO2 and 95% relative humidity.
On the next day, aspirate the medium and replace with fresh NbActiv4™ Culture Medium (Table 2).
Feed the cultures every other day by removing half the medium and replacing with an equal volume of fresh medium.
Three days after plating, we add 5–10 μM cytosine β-D-arabinofuranoside (AraC) to the culture medium to curb glial proliferation. Seven days after plating, we revert to feeding with NbActiv4™ Culture Medium lacking AraC.
Notes
In step 6, replacing only half the medium with each feeding avoids abrupt changes in osmolarity that can arise due to evaporation loss in the incubator, and also retains some glial-conditioned medium. Cortical neurons are very sensitive to abrupt changes in the culture medium.
We typically image the cells 1–2 weeks after plating.
Preparation of glial sandwich cultures
Dilute the remaining cell suspension obtained above (i.e. what remains after plating the neuronal cultures) with 10 ml NbActiv4™ Plating Medium, plate in a T75 plastic tissue culture flask, and place in an incubator.
30 minutes later, tap the bottom of the flask a couple of times to dislodge neurons and other non-adhered cells, then draw off the medium and replace with 10 ml fresh Glia MEM medium (Table 2).
On the next day, aspirate the medium and replace with fresh Glia MEM medium. Repeat every other day.
Maintain the cultures in the incubator.
1–4 days before neuronal cultures will be established (we typically establish them every week), rinse the glia in the T75 flask twice with warm PBS, add 1 ml fresh warm sterile-filtered trypsin solution (0.05% (w/v) trypsin and 0.02% (w/v) EDTA in PBS) to the flask, and incubate at 37°C. Check the flask after ~1 minute. If the cells are not all detached, return to the incubator for another minute or so. The cells should detach within 1–2 minutes.
Stop the trypsinization by adding 5 ml Glia MEM medium to the flask, then draw the medium up into a pipet and expel it gently over the cells, repeating 3–4 times to dislodge the cells.
Gently transfer the cell suspension to a 50 ml disposable plastic centrifuge tube and centrifuge at ~ 120 × g for 5–7 min.
Carefully remove the supernatant and resuspend the cells in 1 ml Glia MEM medium by gently drawing the cells in and out of a 1ml pipette 5–6 times.
Dilute the suspension to 9 ml and plate 1.5 ml into each of six 35 mm plastic tissue culture dishes, each containing one coverslip with paraffin dots (Section 2.2)
Maintain the cells in the incubator, changing the medium every 2 days.
On the day that the glial coverslips will be added to the neuronal cultures, replace the Glia MEM medium with NbActiv4™ Plating Medium (Table 2) and return to the incubator for several hours.
Notes
The above steps for establishing glial cultures should be initiated 1–2 weeks before establishing neuronal cultures. With a weekly culture schedule, glial cultures established one week are used to support the neuronal cultures established the 1–2 weeks later as described above.
In step 1, if we plate four brains worth of cell suspension into the T75 flask, the cells reach 90% confluency after 1 week.
In step 2, the glia adhere to the plastic readily so most of the floating cells are not glia and can be discarded.
In step 3, swirl and shake the flask to unsettle the loosely attached or unattached cells before aspirating off the medium. This step will reduce cell debris and contamination with non-glial cells. A low cell density is normal at this point. The glia proliferate rapidly.
In step 4, the glial cultures can be maintained for up to 2 weeks.
In step 5, ideally the glia should be harvested before exceeding 90% confluency but in practice we have found that this is not critical.
In step 10, the time that the glial cultures can be maintained on the coverslips before the day of neuronal culture depends on the glial cell density. To provide optimal trophic support for the neurons, the glial feeder layer should be at least 70% confluent by the time of use.
2.6 Dorsal root ganglion neuron cultures
Until recently, our studies of neurofilaments and neurofilament transport in DRG neurons were all performed on low-density non-myelinating cultures maintained for up to ~1 week in the L-15 Culture Medium described above for SCG neuron cultures (Table 1) (e.g. Alami et al., 2009; Brown, 1997; Brown, 1998; Koehnle and Brown, 1999). More recently, we have been working predominantly with high-density long-term myelinating co-cultures of DRG neurons and Schwann cells, which are maintained for up to several months in NbActiv4™ Myelination Medium (Table 3) (e.g. Monsma and Brown, 2012; Monsma et al., 2014). Given how well the myelinating cultures fare in the NbActiv4™ Culture Medium, we suspect that this medium may be superior to the L-15 Culture Medium for short-term low-density cultures also, but we have not yet tested this. We describe here the protocol for preparing myelinating co-cultures and then explain briefly how this protocol can be varied to establish short-term non-myelinating cultures.
Long-term myelinating DRG co-cultures
A method for the establishment and study of myelinating co-cultures from DRGs was pioneered by Richard and Mary Bunge (Kleitman et al., 1998). This method involves the separate production of neuronal cultures from dissociated DRGs and of Schwann cell cultures from DRG explants, and relies heavily on the use of antimitotic agents to eliminate Schwann cells and fibroblasts from the neuronal cultures and to eliminate fibroblasts from the Schwann cell cultures. Once pure cultures are obtained, the Schwann cells are harvested and added back to the neuron cultures. These steps are necessary to avoid contamination of the co-cultures with fibroblasts, which proliferate rapidly in the serum-containing culture medium that the authors used. The method works well and is used widely but we prefer a much simpler approach based on the method of Fex Svenningsen et al. (2003) which involves simply dissociating DRGs to create a mixed co-culture. The key difference compared to the Bunge method is that the cultures are maintained in serum-free medium throughout. Fibroblasts do not proliferate in the absence of serum, allowing the production of myelinating co-cultures with no additional measures. We originally used Neurobasal™ medium, as reported by Fex Svenningsen et al. (2003), but have discovered recently that myelination is much more robust when this is substituted with NbActiv4™ medium. The procedures described below refer to cultures established from rats, with which we have most experience, but they also work well for mouse cultures when NbActiv4™ medium is used.
Sacrifice a pregnant E16.5 rat using an approved method and shave the fur off the abdomen.
Dissect out 2–3 embryos from the uterine lining, place them in Petri dishes containing Leibovitz’s L-15 medium (Gibco Life Technologies), and transfer the dishes to the laminar flow hood where all subsequent procedures will be performed.
Under the dissecting microscope, remove the spinal cord using illumination from below and transfer to a dish containing fresh L-15 medium.
Switch to dark field illumination from below and pluck the DRGs from the spinal cord using fine forceps. Pool the ganglia in an area of the dish off to the side.
Transfer the ganglia into a 15 ml disposable polystyrene centrifuge tube containing L-15 medium containing 0.5% (w/v) BSA using a pipette with a siliconized 200 μl pipette tip (cut the tip off with a sterile blade to widen it). Expel the ganglia directly into the medium in order to prevent them from adhering to the meniscus.
Remove the L-15, rinse the ganglia three times with PBS, then add 2.5 mg/ml trypsin (crystallized, Worthington Biochemicals) in PBS (sterile-filtered), cap the tube, and incubate at 37°C for 20 minutes.
Remove the trypsin solution and add L-15 containing 25% (v/v) FBS to inactivate the trypsin. Incubate at 37°C for 15 minutes.
Rinse the ganglia twice with L-15 medium containing 0.5% (w/v) BSA.
Dissociate the cells by trituration in L-15 medium containing 0.5% (w/v) BSA using a fire-polished Pasteur pipet as described in Section 2.4.
Measure the density of viable cells by Trypan Blue exclusion using a hemocytometer, then dilute the cells to the desired concentration in NbActiv4™ Myelination Medium (Table 3).
Plate the cell suspension onto dry Matrigel™-coated glass-bottomed dishes (Section 2.3) and maintain the cultures at 37°C in an atmosphere of 5% CO2 with 95% relative humidity.
About 5 days after plating, replace all of the media in the dish with NbActiv4™ Myelination Medium including 1% (v/v) Matrigel™ (Table 3).
About 8 days after plating, replace half of the medium in each dish with NbActiv4™ Myelination Medium lacking Matrigel™ but including 50 μg/ml ascorbic acid (Table 3).
For the lifetime of the culture, replace half of the medium with fresh NbActiv4™ Myelination Medium containing ascorbic acid every 2–3 days, with a full medium change once each week. Myelination is first apparent at approximately 3 weeks after plating.
Notes
When rinsing the ganglia in steps 6 and 8, swirl them gently in the added solution and then allow them to settle under gravity. Once settled, remove the supernatant using a Pasteur pipette, leaving just enough to cover the ganglia at the base of the tube.
In step 2, two embryos are enough to yield ~100 ganglia.
In step 4, ganglia that are not attached to the spinal cord when it is removed can be plucked directly from the inner surface of the vertebral column.
In step 5, the siliconization prevents adhesion of the ganglia to the inside of the pipette tip and the BSA reduces adhesion of the ganglia to the sides of the tube.
In step 6, collagenase treatment is recommended before trypsinization for older embryos or postnatal pups. Treat with 2.5 mg/ml collagenase (Type 3, Worthington Biochemicals) in L-15 (sterile filtered) at 37°C for 1 hour. Postnatal pups also require a longer incubation time (45 minutes) in trypsin.
In step 6, refer to the cautionary notes on trypsinization in Section 2.4.
In step 9, refer to the cautionary notes on trituration in Section 2.4.
In step 11, we plate the cells at a density of 5,000–15,000 cells/cm2.
Depending on the application, we supplement the NbActiv4™ Myelination Medium with either NGF or NT-3 (Table 3; Section 2.2).
Short-term non-myelinating DRG cultures
To establish and maintain short-term non-myelinating DRG neuron cultures, the above procedure can be modified as follows: (1) the coverslips can be coated with a lower concentration of Matrigel™ (10μg/ml), (2) there is no need to dry the Matrigel™ down, (3) there is no need to add Matrigel™ or ascorbic acid to the culture medium, and (4) the cells can be plated at a much lower density.
3 Neurofilament fusion proteins
All of our live-cell imaging of neurofilaments over the past 15 years has relied exclusively on the use of fluorescent fusion proteins consisting of a fluorescent protein fused in-frame to the N- or C- terminus of a neurofilament protein. A very real concern is the potential of the fluorescent protein domain to interfere with the assembly or interactions of the neurofilament protein to which it is linked. Since the precise molecular structure of intermediate filaments is not known, a rational design strategy based on structural considerations is not possible so we are forced to approach the problem empirically.
3.1 Choice of neurofilament subunit
The subunit composition of neurofilaments varies with neuronal cell type and developmental stage. Cultured cortical neurons from embryonic or neonatal rats or mice express predominantly NFL, NFM and internexin, whereas neurofilaments in cultured SCG and DRG neurons from embryonic or neonatal rats or mice express predominantly NFL, NFM, internexin and peripherin. NFH expression is very low in embryonic and neonatal cortical and SCG cultures, which reflects the fact that this neurofilament protein is expressed primarily postnatally in the central and peripheral nervous system (Carden et al., 1987; Nixon and Shea, 1992). However, NFH is detectable in cultured DRG neurons. Immunostaining of single neurofilaments in all three neuronal cell types indicates that each neurofilament contains all of the expressed proteins and that each of these proteins is present along the entire length of every neurofilament. Though internexin and peripherin are capable of forming homopolymers in vitro, they form heteropolymers when other neurofilament proteins are present. Thus, neurofilaments can be tagged using fluorescent fusions of any of the neurofilament proteins. Our lab has imaged neurofilament movement using fluorescent fusions of NFL, M and H, though for historical reasons we have the most experience with NFM. The NFH fusion protein incorporates throughout the neurofilaments just like other neurofilament fusion proteins, even in cells that express very little endogenous NFH.
The structure of neurofilaments is not well understood. However, by analogy with intermediate filament polymers in other cell types, neurofilaments are expected to have a linear mass density of about 32 polypeptides per 65 nm of polymer (which corresponds to the length of the Unit Length Filaments that are intermediates in the filament assembly) (Koster et al., 2015). For live-cell imaging with a camera that has 16 × 16 μm pixels (corresponding to 160 × 160 nm pixels in the image plane at 100x objective magnification), we would thus expect ~80 polypeptides per pixel along the length of each polymer. It is likely that only a small proportion of these polypeptides need to be fluorescent fusions for us to detect neurofilaments in neurons, so the fusion protein load on the filaments need not be high, but we have not yet quantified this.
3.2 Choice of fluorescent protein
Recent progress in the engineering of new and improved monomeric or tandem dimeric fluorescent proteins has greatly increased the options available for making fluorescent fusion constructs (Chudakov et al., 2010; Day and Davidson, 2009). To choose the best fluorescent protein, it is important to consider the spectral properties (excitation and emission), oligomerization state (monomeric, dimeric, tetrameric), brightness (determined by the extinction coefficient and quantum yield), folding efficiency and photostability. To ensure that the fusion protein co-assembles into neurofilaments freely, and to minimize the possibility that the fusion protein disrupts the structure or interactions of the assembled neurofilaments, it is important to select fluorescent proteins that are monomeric, i.e. with low tendency for dimerization or oligomerization.
For green fluorescence we use the popular human codon-optimized red-shifted F64L/S65T variant of green fluorescent protein (GFP) (Tsien, 1998). This variant is sometimes referred to as enhanced GFP (EGFP). For red fluorescence, we have used DsRed2 (Bevis and Glick, 2002) but now prefer to use mCherry (Shaner et al., 2004) or TagRFP-T (Shaner et al., 2008) because DsRed2 is excited weakly by blue light, resulting in cross-talk in dual wavelength imaging applications. The brightest red fluorescent protein that we have used is tdTomato, which is a tandem dimer of the dimeric fluorescent protein Tomato (Shaner et al., 2004). The tandem dimer adds bulk but minimizes the formation of intermolecular dimers which would most likely disrupt neurofilament assembly. For cyan and yellow fluorescence, we have used the Cerulean variant of CFP (Rizzo et al., 2004) and the Venus variant of YFP (Nagai et al., 2002). In spite of the many new alternatives, GFP remains our overall favorite because of its brightness and photostability which permit long-term time-lapse imaging at low light levels with minimal fluorescence photobleaching. mCherry has good brightness and photostability though in our hands mCherry-neurofilament fusions sometimes have a slight tendency to form small non-filamentous aggregates. If this problem is encountered, then TagRFP-T is a suitable alternative that does not exhibit this tendency.
For photoactivation or photoconversion applications, we have used photoactivatable green fluorescent protein (PAGFP) extensively (Patterson and Lippincott-Schwartz, 2002). In its naïve state, PAGFP is a green fluorescent protein that excites in the violet (but not in the blue) and emits in the green. After illumination with violet light it undergoes a photoconversion to a form that excites in the blue (in addition to the violet), effectively “activating” the fluorescence excitation at this wavelength. PAGFP can be photoconverted efficiently using a mercury arc lamp with a violet filter cube (385–425 nm excitation bandwidth), or using a violet (405nm) laser (Section 6.13). For green-to-red photoactivatable or photoswitchable proteins, we have used Dendra2 (Chudakov et al., 2007) but now prefer mEos2 (McKinney et al., 2009), which is a green fluorescent protein (excites in the blue, emits in the green) that is converted to a red fluorescent protein (excites in the green, emits in the red) by illumination with violet light. Though reported to form dimeric or oligomeric aggregates at high concentration, we have not observed this for neurofilament fusions. Newer variants of this protein such as mEos3.1 and mEos3.2 may be superior alternatives (Zhang et al., 2012).
3.3 Design of fusion and expression construct
Neurofilament fusion constructs are generated by cloning the cDNA for a neurofilament protein in-frame into the multiple cloning site of a suitable mammalian expression vector. By convention, when we fuse the fluorescent protein to the N-terminus of the neurofilament protein we use the abbreviation for the fluorescent protein as a prefix, e.g. GFP-NFL, and when we fuse the fluorescent protein to the C-terminus of the neurofilament protein we use it as a suffix, e.g. NFL-GFP. Though the N-terminus of intermediate filaments is known to be critical for filament assembly, both N- and C-terminal fusions of neurofilaments have been found to assemble readily (Colakoglu and Brown, 2009).
In principle, the length and structure of the peptide that links the fluorescent protein to the neurofilament protein in each fusion protein could be a critical determinant of the functionality and utility of the fluorescence fusion protein as a neurofilament tag. In practice, this linker is formed by the remnants of the multiple cloning site into which the neurofilament protein is cloned and its sequence is therefore arbitrary. For fusions of fluorescent proteins to the N-terminus of neurofilament proteins we have used linkers ranging from 7–25 amino acids in length (e.g. Fig. 4). For fusions to the carboxy terminus, we have used linkers from 5–6 residues in length. While we have not examined this systematically, anecdotally it appears that the linker length is not critical. Perhaps this reflects the relatively flexible and unstructured conformation of the neurofilament N- and C-terminal domains.
Fig. 4.

A fluorescent neurofilament fusion protein. A. Diagram of a GFP-NFM fusion protein, which consists of the human codon-optimized red-shifted F64L/S65T variant of green fluorescent protein (GFP) joined to the amino terminus of rat neurofilament protein M (NFM) by a 25-residue peptide linker (Wang et al., 2000). B. Drawing of a single GFP-tagged NFM polypeptide incorporated into a neurofilament polymer. The filament backbone is formed by lateral interactions between the alpha-helical rod domains of the neurofilament proteins, which form coiled coil dimers (represented here as cylinders). The N- and C-terminal domains are shown projecting outward from the filament backbone, though this is only known to be so for the C-terminal domains. The GFP is drawn to approximate scale, but the precise organization of the polypeptides in the filament is speculation because the molecular structure of the intermediate filament is not known.
By default, all our fluorescent fusion protein expression constructs use a minimal human cytomegalovirus (CMV) immediate early gene enhancer-promoter and the SV40 early mRNA polyadenylation signal because they were constructed by insertion into the standard Clontech fluorescent fusion protein plasmid backbone (Clontech Laboratories, Mountain View, CA). In our hands such plasmid expression constructs express at suitable levels in SCG, cortical or DRG neurons transfected by a variety of methods. We have little experience with the use of other promoters or polyadenylation signals. Some laboratories prefer to use the chicken β-actin promoter (Niwa et al., 1991).
3.4 Testing the constructs for assembly competence
We routinely test each new fusion construct for its capacity to assemble into neurofilaments both in SW13vim- cells and in neurons in primary culture. SW13vim- cells are a human adrenal carcinoma cell line in which the expression of vimentin (the endogenous cytoplasmic intermediate filament of these cells) has been transcriptionally silenced, resulting in no cytoplasmic intermediate filaments (Sarria et al., 1990; Yamamichi-Nishina et al., 2003). Transfection of these cells with tagged or untagged neurofilament proteins results in the formation of cytoplasmic neurofilaments that can be imaged at high resolution (Colakoglu and Brown, 2009; Yan et al., 2007) (Fig. 5). Since neurofilament triplet proteins (NFL, NFM and NFH) are obligate heteropolymers (Ching and Liem, 1993; Lee et al., 1993) and these cells express no other neurofilament proteins, it is necessary to co-transfect fusions of these proteins with another untagged neurofilament protein (e.g. NFM or NFH with GFP-NFL, or NFL with GFP-NFM or GFP-NFH). If the construct co-assembles into filaments in SW13 vim- cells with no signs of filamentous or amorphous aggregates, then we confirm that it also assembles to form neurofilaments in neurons by expressing it in primary neuronal cultures. Using such approaches, we have found neurofilament protein assembly to be remarkably forgiving of fluorescent protein fusions to both the N- and C-termini. This includes a 2x fusion of NFM to the Cerulean variant of cyan fluorescent protein (CFP-CFP-NFM) as well as a tandem dimer fusion of the red fluorescent protein dTomato to NFM (tdTomato-NFM).
Fig. 5.

Examples of SW13vim- cells expressing neurofilaments co-assembled from fluorescent neurofilament fusion proteins. A. Filaments assembled from GFP-NFM and NFL. Live imaging of the GFP fluorescence. B. Filaments assembled from mCherry-NFM and NFL-PAGFP. Live imaging of the mCherry fluorescence. Both images were acquired with spinning disk confocal microscopy using an Andor Ultra EMCCD camera (Section 6). Scale bar= 10 μm.
We have noted some tendency of C-terminal fusions of NFM (fluorescent protein fused to the neurofilament protein C-terminus) to form filaments that aggregate in SW13vim- cells. However, both N- and C-terminal fusions of NFL and NFM appear to assemble efficiently into filaments in cultured neurons, as do N-terminal fusions of NFH (we have not tested C-terminal fusions of NFH). The explanation for this difference may be that the fusion protein represents a smaller proportion of the total neurofilament subunit protein in neurons, where it is effectively “diluted out” by the multiple endogenous neurofilament proteins with which it co-assembles, whereas in SW13vim- cells the fusion protein may represent as much as 50% of the protein in the filament. Thus, the effective “fusion load” on the filaments is expected to be much higher in SW13vim- cells than when these proteins are expressed at low levels in neurons. For this reason, SW13vim- cells are a particularly stringent test of assembly competence.
4 Transfecting neurons
4.1 General considerations
A variety of methods are now available for expressing fluorescent fusion proteins in cultured neurons including electroporation, microinjection, viral or retroviral delivery, lipofection, magnetofection, and calcium phosphate (Craig, 1998). Transfection efficiencies tend to be much lower than for mitotic cells. However, this is not a problem for live-cell imaging studies on neurofilament transport because it is usually necessary to image single axons in isolation, and a lot of data can be obtained from a small number of transfected cells. Here we describe four methods that have worked well for us, depending on the culture system and experimental application: electroporation, lipofection, magnetofection and nuclear injection. Each method has its advantages and disadvantages (Table 4).
Table 4.
Four methods for transfecting neurons in primary neuronal cultures. Viability is defined as the proportion of the cells that survive the transfection procedure. Transfection efficiency is defined as the proportion of surviving cells that express the transfected plasmid.
| Transfection method | Viability | Transfection efficiency | Advantages | Disadvantages |
|---|---|---|---|---|
| Electroporation | ~ 60–80% (depending on the cell density) | ~5–10% for cortical neurons. <1% for DRG neurons in long-term myelinating co-cultures. | Simple and effective. Consistent and moderate expression levels. | Requires an electroporation device. The commercially optimized reagents can be very expensive. Not selective for neurons. Low yield of viable cells. |
| Lipofection | ~40–60% (depending on the cell density and DNA concentration) | <1% for DRG neurons in long-term myelinating co-cultures (typically ~5–10 neurons per culture dish). | Simple and inexpensive. No special equipment required. | Hard to control expression. Low transfection efficiencies. Not selective for neurons. |
| Magnetofection | ~50–70% (depending on the cell density) | ~5–10% for cortical neurons. | Simple and inexpensive. Better transfection efficiency than lipofection. | Hard to control expression. Requires a magnetic plate. Not selective for neurons. |
| Nuclear injection | ~80–90% (with a skilled operator) | ~70–90% for SCG neurons depending on the culture age. 50% for DRG neurons in long-term myelinating co-cultures. | Works for hard-to-transfect cells. Permits selection of the cells to be transfected. | Labor-intensive. Requires specialized equipment, training and practice. |
4.2 Plasmid purification
It is important to use DNA that is pure and free of endotoxins. We purify the plasmids using QIAGEN EndoFree™ Plasmid kits and store the purified DNA at 2–2.5 mg/ml in Tris/EDTA buffer (10 mM Tris-HCl, 1 mM EDTA, pH 8.0) in aliquots at −20°C. DNA purity should be measured by the ratio of the absorbance at 260 to 280 nm (A260/A280). The A260/A280 ratio should be at or above 1.8. During the plasmid preparation we take care to use disposable plastic whenever possible. If it is necessary to re-use plastic ware such as centrifuge tubes, care should be taken to remove all traces of detergent after washing. We wash our centrifuge tubes with ES-7X detergent (MP Biomedicals) and/or Triton-X 114 (Sigma) and rinse thoroughly.
4.3 Electroporation
Electroporation uses an electrical pulse to create transient pores in the plasma and nuclear membranes of cells, allowing DNA in the culture medium to gain entry to the nucleus. The method is fast, simple and effective but does require an electroporation device. We use the Lonza Nucleofector™II (Lonza, Allendale, NJ), which has optimized programs and solutions for all commonly used primary neuronal cell types. The device and solutions are expensive and the solution composition is proprietary, but they work well in our experience. For example, electroporation of cortical neurons using the Nucleofector™ device and solutions yields cells with suitably moderate expression levels (high expression levels are not desirable) and transfection efficiencies in the range of 5–10% (defined as the proportion of the surviving cells that express fluorescent protein). We do observe cell death associated with the electroporation, but this is not a problem for us because a single cortical, SCG or DRG neuron prep can yield more than enough cells for a single batch of low-density cultures (Sections 2.4, 2.5 & 2.6).
Lonza supplies two kind of transfection kits depending on the cell number: regular Nucleofector™ kits requiring 100 μl of cell suspension containing 1–5 × 106 cells per transfection, and Small Cell Number (SCN) Nucleofector™ kits requiring 20 μl cell suspension containing 2 × 104 cells per transfection. For optimal viability and transfection efficiencies it is desirable to use the recommended cell densities. For neonatal rat cortical neuron cultures we obtain ~4–8 × 106 cells from one brain, which is enough for one transfection using the regular kit. For rat DRG neuron cultures, we obtain ~1 × 106 cells from 2 embryos (~100 ganglia) and also use a regular kit. For rat SCG neuron cultures we obtain ~2–3 × 104 cells from 8–10 pups and use a Small Cell Number kit.
Electroporation protocol:
Aliquot 10 μl of the cortical, SCG or DRG neuron cell suspension onto a hemocytometer slide and dilute 100–200 μl of Trypan Blue (0.4% (w/v) solution, Gibco Life Technologies).
Measure the density of viable cells (cells that exclude the Trypan Blue dye) and estimate their total number.
For each separate transfection, transfer 1 ml of the cell suspension into a 1.5 ml microcentrifuge tube and centrifuge at 80–100 × g for 2–5 minutes.
Remove the supernatant, measure the density of viable cells, and estimate their total number.
Estimate the number of viable cells in the pellet by subtracting the total number obtained in step 4 from the total number obtained in step 2.
Resuspend the pelleted cells in 100 μl of the Nucleofector™ solution that is supplied with the regular Nucleofector™ kit (or 20 μl when using the Small Cell Number kit), add the appropriate amount of plasmid(s), and mix well by tapping the tube.
Transfer the mixture to a Nucleofector™ cuvette and electroporate using the appropriate program. The electroporation takes seconds.
Transfer the sample to a 1.5 ml microcentrifuge tube, add 500 μl warm culture or plating medium, and incubate for 10 min at 37°C.
Count the number of viable cells again, then dilute the suspension to the required density in warm medium and plate onto glass-bottomed culture dishes coated with the desired substrate.
Notes
In steps 1 and 2, the number of cells obtained from a given number of brains or ganglia is fairly reproducible so with experience it is possible to skip the cell counting step and estimate the cell number.
In step 3, 1 ml of suspension should contain sufficient cells for one electroporation reaction.
In step 6, use the Small Cell Number Nucleofector™ kit if the cell concentration is lower than 1 × 105 cell/ml
In step 6, we recommend using 1–5 μg plasmid DNA. The plasmid amount can be increased up to 10 μg if higher transfection efficiencies are required, but this also decreases cell viability. As a guide, we usually use 2–3 μg DNA for cortical neuron cultures and 5 μg DNA for DRG neuron cultures.
In step 7,we use program O-005 for mouse cortical or DRG neurons, and program O-003 or G-013 for rat cortical or DRG neurons.
4.4 Lipofection
Lipofection relies on the use of synthetic cationic lipid micelles or liposomes as vehicles for the delivery of plasmid DNA into neurons. The method is simple, the reagents are relatively inexpensive, and no special equipment is required. We use Lipofectamine 2000™ (Invitrogen) and follow the manufacturer’s instructions except that we have found that it is not essential to use Opti-MEM medium; media such as Neurobasal or L-15 without serum and antibiotics also work well. In addition, we find that cell viability is improved with no loss of transfection efficiency if we use less than the recommended amount of DNA, while keeping the recommended DNA:lipofection reagent ratio the same. The lipofection can be performed in cell suspension before plating but the cell viability is much better if it is performed on adherent cells after plating. The transfection efficiency for neurons is very low, but is enough for our high-density myelinating DRG co-cultures because even a few transfected cells per dish can be enough for live-cell imaging.
4.5 Magnetofection
Magnetofection relies on the use of magnetic nanoparticles and a magnetic field. We use the NeuroMag™ Transfection Reagent (OZ Biosciences, San Diego, CA) which consists of magnetic nanoparticles (140–200 nm in diameter) coated with specific proprietary cationic macromolecules. The particles are complexed with the DNA and then applied to adherent cells. Subsequently the culture dish is placed on a magnetic plate which draws the particles down onto the cells, facilitating the uptake of the DNA. In our experience this method results in higher transfection efficiencies than lipofection, and also higher levels of expression than obtained with electroporation. In neurons with low levels of endogenous neurofilament expression, such as cortical neurons, this can result in diffuse fluorescence (presumably because there are insufficient endogenous neurofilament protein subunits to co-assemble with the fluorescent fusion protein). To overcome this problem it is possible, depending on the experiment, to co-transfect the fluorescent fusion protein with an untagged neurofilament subunit, e.g. GFP-NFM and NFL. For reasons that are not yet clear to us, magnetofection has not worked well for our myelinating co-cultures.
Magnetofection protocol
Thaw the NeuroMag™ solution and vortex well at room temperature.
Add 1 to 10 μg DNA to 100 μl culture medium lacking serum, supplements or antibiotics (e.g. DMEM, L-15 or Neurobasal™ medium, depending on the neuronal cell type).
Add 3.5–35 μl NeuroMag™ solution to the diluted DNA, mix well, and incubate at room temperature for 15–20 minutes to allow the nanoparticles to complex with the DNA.
Add the NeuroMag™/DNA mixture to the culture dish by pipetting into the culture medium.
Place the dish on an OZ Biosciences Super Magnetic Plate for 15 minutes in the culture incubator at 37°C.
Remove the magnetic plate and leave the dish in the incubator for another 45 minutes.
Replace the medium with fresh medium and maintain in the incubator, following the normal feeding schedule, until the day of observation.
Notes
In step 2, we usually use Neurobasal™ medium without B27 supplements.
In step 3, use 3.5 μl NeuroMag™ solution per 1 μg of DNA.
In step 3, the solution can be mixed by vigorous pipetting or brief vortexing.
In step 4, move the pipette around while dispensing and then tilt the dish gently to ensure mixing.
In step 7, the medium change is optional, but we have the impression that it improves cell viability.
4.6 Nuclear injection
In the early days of our studies on neurofilament transport we relied exclusively on nuclear injection of our plasmid expression constructs to transfect cultured neurons (Garcia et al., 1992; Wang and Brown, 2001; Wang et al., 2000). This method requires specialized equipment and training, but in experienced hands it is fast and efficient. By minimizing the number and relative proximity of the injected cells in each dish, we can ensure that we are able to trace single axons of expressing neurons for long distances without ambiguity. Nuclear injection is not suitable for transfection of large numbers of cells, but this is not a limitation for our live-cell imaging studies because a considerable amount of data can be obtained from a small number of cells. Prior to injection, the DNA is diluted to 0.06–0.2 mg/ml in 50 mM potassium glutamate, pH 7.2. The cells appear to tolerate injection using this buffer with no apparent adverse effects.
Microinjection requires an inverted microscope equipped with a pressure injection apparatus and a micromanipulator. We currently inject using an Eppendorf Injectman NI2 semi-automated injection system which is attached to a Nikon TE300 inverted microscope mounted on a vibration isolation table (Technical Manufacturing Corporation) (Fig. 6). However, for many years we used a simpler and less expensive manual system comprised of a Narashige 3-axis hydraulic manipulator and Medical Systems Corporation PLI-100 pressure injector (Warner Instruments, Hamden, CT) with equal success. The Eppendorf system uses internally generated compressed air as the pressure source and the Medical Systems Corporation system uses an external source of compressed nitrogen gas. The Eppendorf system is more complex to operate but it has the convenience of semi-automation and, once mastered, it can result in more reliable penetration of the plasma and nuclear membranes because it can be programmed to advance into the cell at rates of up to 7.5 mm/sec.
Fig. 6.

A microinjection workstation. Eppendorf Injectman NI2 semi-automated microinjection system, consisting of a FemtoJet™ air compressor and regulator and an InjectMan™ 3-axis micromanipulator and controller, mounted on a Nikon Diaphot 300 inverted fluorescence microscope.
We pull our own micropipettes from thick-wall borosilicate glass (1.0 mm OD, 0.58 mm ID, 4″ long with internal filament; World Precision Instruments, Sarasota, FL) using a Sutter P-97 Flaming-Brown Micropipette Puller (Sutter Instrument Co., Novato, CA). The internal glass filament facilitates back-loading of the micropipettes by capillary action. The puller heats the glass using a metal heating filament. The puller settings must be determined empirically for a given heating filament geometry, and they may vary with ambient temperature and humidity. The goal is to obtain a tip that is fine enough to penetrate nuclei but not so fine that it frequently becomes blocked. We pull micropipettes with a two-stage pull cycle in order to obtain a short taper. The glass can be sterilized with dry heat (170°C for 3 hours) prior to pulling, but we do not find this to be necessary. A container to hold glass capillaries during dry heat sterilization is commercially available (World Precision Instruments). To load the micropipette, we place approximately 1 μl of injection solution at the opening of the capillary (the end opposite to the micropipette tip) and then suspend the micropipette vertically on ice in a humid environment (to prevent drying out) for a few minutes to allow the solution to flow down to the tip.
The micromanipulator is orientated so that the micropipette is at an angle of about 30–45 degrees to the horizontal. The manipulator can be programmed penetrate the cell axially (advancing the pipette into the cell in the direction that it is pointing) or vertically (lowering the angled pipette into the cell from above). The former generally works better but the latter method is preferable for neurons in dense cultures, such as myelinating DRG co-cultures. For short-term SCG cultures, we typically inject cells 18–48 hours after plating and observe them 1–3 days later. For longer term myelinating DRG co-cultures, we typically inject after 2–3 weeks in culture and observe them 7–14 days later. We inject under phase contrast optics using a 40x dry objective and a 0.52 NA long working distance condenser, which provides sufficient clearance for the micropipette. It is necessary to remove the lid from the culture dish for injection, but with care this rarely results in microbial contamination. If the cells are to be kept out of the incubator for more than 20 minutes, we warm the dish and microscope stage to 37°C using an air stream incubator (Section 6.3). To prevent evaporation loss, we pipette a layer of low viscosity silicone fluid (dimethylpolysiloxane, 5 centistokes, Sigma) over the culture medium (0.75 ml is sufficient for a 35 mm culture dish). The silicone fluid is pre-warmed to 37°C and sterilized by syringe filtration with a 0.2 μm filter. After injection, we generally remove the silicone fluid and the culture medium by aspiration and replace with fresh medium. This may reduce the chance of microbial contamination and improve gaseous exchange, but the condition of the cells does not appear to be compromised in the short term if the silicone fluid is not removed and the medium is not changed. If cells are cultured in bicarbonate-buffered media, then the medium will become alkaline in atmospheric CO2. To solve this problem it is necessary to use media containing a buffering agent such as HEPES (McKenna and Wang, 1989), or to replace the medium with a medium that is designed for use at ambient CO2 concentrations, such as L-15 or Hibernate (Section 6.2).
Microinjection of adherent cells does require some practice, but once the technique is mastered it is possible to manually locate and inject 15 neurons in a single dish within 20 minutes. The principal challenges are to penetrate the nucleus with minimal damage to the cell, and to minimize blockage of the injector. To minimize blockages, we routinely clarify the injection solution by centrifugation or by filtration using an Amicon Ultrafree-MC 0.1 μm centrifugal concentrator (EMD Millipore) prior to loading it into the micropipette. It is essential to include a fluorescent marker in the injection solution so that the flow rate can be adjusted empirically for each micropipette. The fluorescent marker also provides immediate visual confirmation of a successful injection (Fig. 7). We generally use rhodamine-labeled dextran (Mw 10,000 or Mw 70,000, Molecular Probes). The smaller dextran diffuses out of the nucleus within minutes after injection. To maximize cell survival, it is critical to use very low injection pressures. We typically use injection pressures of 7–14 hPa (0.1–0.2 psi) above the balance pressure, though for nuclei it is sometimes preferable to inject using only the balance pressure (see below). Higher pressures can result in damage to the nucleus. We have not measured the volume that is injected under these conditions, but it is small enough that we do not observe any noticeable increase in nuclear volume as judged by phase contrast or differential interference contrast microscopy. Too much pressure or too much volume (e.g. sufficient to cause a visible increase in nuclear volume) will result in cell death.
Fig. 7.
A nucleus injected with fluorescent dextran. Cell body of a cultured neuron imaged by epifluorescence microscopy immediately after injection of the nucleus with Mw 70,000 rhodamine-labeled dextran (Section 4.6). The dark spot within the nucleus represents the nucleolus, which excludes the fluorescent dextran. Scale bar= 4 μm.
It is important to use a pressure injection apparatus that can supply a constant balance pressure to the injector at all times in order to prevent medium being drawn into the micropipette tip by capillary action between injections. The magnitude of the balance pressure should be adjusted empirically for each pipette so that injection solution is trickling out into the medium at all times (the fluorescent dextran in the injection solution allows the flow rate to be observed directly). This constant positive pressure also reduces the frequency with which the pipette tip gets clogged. Cells can be injected by initiating the injection pressure before penetration and terminating the injection pressure after the tip is removed (“continuous flow technique”), or alternatively by initiating the injection pressure after the tip has penetrated the cell and terminating the injection pressure before it is removed. We generally use the former method, which results in less clogging of the pipettes during penetration. However, this method can also result in more leakage of injection solution into the medium, which can temporarily obscure the fluorescence of the injected cell, making it difficult to determine whether the injection was successful. In experienced hands, about 60–80% of the injected cells typically survive and express the injected construct.
5 Strategies for observing movement
5.1 General considerations
We describe below three distinct strategies for observing neurofilament transport in axons of cultured neurons (Fig. 8). The first approach takes advantage of the fact that certain types of cultured neuron at certain stages of development exhibit naturally occurring discontinuities in the axonal neurofilament array which we call gaps. These gaps appear to arise due to low levels of neurofilament protein expression, which results in low numbers of axonal neurofilaments. Neurofilaments that move into the gaps can be tracked because they can be observed in isolation. The second approach is a related technique that employs fluorescence photobleaching to create a gap in the axonal neurofilament array, allowing the observation of unbleached neurofilaments that move through the bleached region. The third approach is a fluorescence photoactivation technique that enables analysis of the long-term pausing behavior of neurofilaments in axons that contain abundant neurofilaments. The photobleaching and photoactivation methods do not rely on the occurrence of gaps in the neurofilament array, so they can be applied to axons that have a higher neurofilament content. Each approach has its advantages and disadvantages, but the natural gap and photobleaching methods yield different kinetic information from the photoactivation method, so they should be considered to be complementary, not alternatives (Table 5).
Fig. 8.

Three strategies for studying neurofilament movement in axons. A. Natural gap technique. Fluorescent neurofilaments can be tracked as they move through gaps in the axonal neurofilament array. B. Photobleaching technique. Fluorescent neurofilaments can be tracked as they move through photobleached gaps in the axonal neurofilament array. C. Photoactivation technique. Fluorescent neurofilaments can be tracked as they depart from a photoactivated segment of axon, or the transport kinetics can be analyzed by measuring the loss of fluorescence in the activated region over time.
Table 5.
Comparison of three strategies for studying neurofilament movement in axons.
| Brief description | Advantages | Disadvantages | |
|---|---|---|---|
| Natural gap method | Track single neurofilaments through gaps in the axonal neurofilament array. | Enables direct observation of neurofilament movement on short time scales with high temporal and spatial resolution. | Limited to neurons that have a relatively low neurofilament content. Not well suited to analysis of the long-term pausing behavior. |
| Photobleaching method | Track single neurofilaments through bleached regions of axons. | Enables direct observation of neurofilament movement on short time scales with high temporal and spatial resolution in axons that have no naturally occurring gaps. | For axons that contain many neurofilaments, it may be hard to bleach the fluorescence enough to detect the movement of single neurofilaments. Photobleaching has the potential to induce photo-oxidative damage. Not well suited to analysis of the long-term pausing behavior. |
| Fluorescence photoactivation method | Track the departure of neurofilaments from a photoactivated region. | Enables analysis of the moving and pausing behavior of neurofilaments on short and long time scales in axons that contain abundant neurofilaments. | Requires computational modeling to extract kinetic data. Not well suited to axons that contain relatively few neurofilaments. |
5.2 Naturally occurring gaps
Cultured SCG or cortical neurons from neonatal rats or mice express relatively low levels of neurofilament proteins, resulting in relatively low numbers of neurofilaments in the axons. In these cells, the asynchronous and infrequent movement of the polymers results in segments of axon that lack neurofilament polymers entirely. We call these discontinuities naturally occurring gaps. They appear to have no functional significance for immature axons growing in culture but they offer a special opportunity for studies on neurofilament transport. Neurofilaments that move into these regions can be observed and tracked in isolation, which is not possible in axonal regions that contain many overlapping neurofilaments because the adjacent polymers tend to be spaced too close together to be resolved by conventional light microscopy. Long-term observations have revealed that the gaps are highly dynamic, appearing and disappearing over time as a result of the stochastic movements of the neurofilament polymers. When a neurofilament that is the sole neurofilament in a segment of axon moves out of that segment, a gap arises. When another neurofilament moves into that segment and pauses, the gap shortens or disappears. Often we have the experience of scanning along a section of axon that has no gaps, only to find a gap when we return to that region later in the same imaging session.
In neonatal SCG neurons it is not hard to find gaps that measure tens of micrometers in length, whereas in neonatal cortical neurons, which can contain even fewer neurofilaments, it is not unusual to encounter gaps that stretch for hundreds of micrometers. However, the number and length of the gaps depends on the age of the animals from which the cultures are established as well as the age of the neurons in culture. For example, we find that gaps are more prevalent in cortical neurons from neonatal (P0–P1) animals than from late stage (E18–20) embryos, although the cell viability is higher for embryonic cultures, and that they are more prevalent in young cultures (<2 weeks) compared to older cultures (>2 weeks). Importantly, gaps are never observed in the axons of cultured DRG neurons because the high neurofilament content of their axons makes the probability of gaps arising exceedingly low. Thus, studies on neurofilament transport in cultured DRG neurons require the use of photobleaching or photoactivation strategies (Sections 6.12 and 6.13).
To detect movement using the natural gap method, a GFP-tagged neurofilament protein is expressed in cultured SCG or cortical neurons and allowed to incorporate throughout the endogenous neurofilament array (we wait at least several days), and then gaps in the axonal neurofilament array are located and observed by time-lapse fluorescence imaging. Single neurofilaments can be detected as they move through these gaps because the gaps are devoid of other neurofilaments (Fig. 9). Essentially all of the neurofilament proteins are polymerized in these neurons (Black et al., 1986), so there should be negligible diffusible neurofilament subunits. The presence of diffuse fluorescence in the gaps is indicative of elevated expression levels; generally we avoid using such cells for our studies.
Fig. 9.
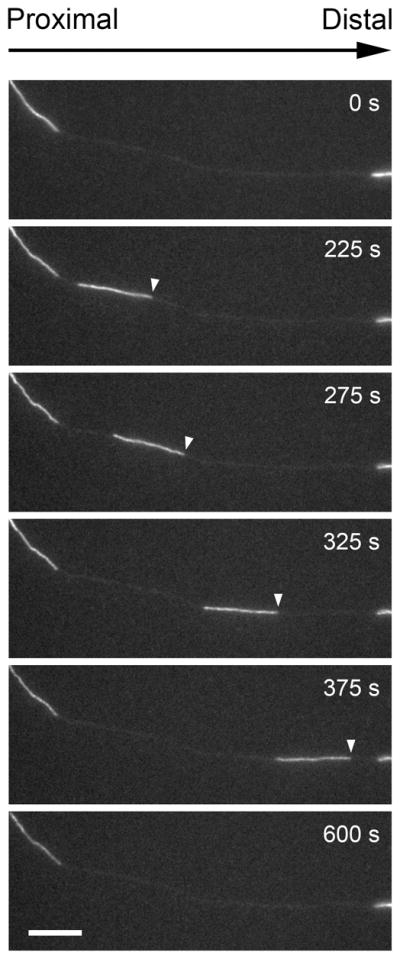
Example of a neurofilament moving through a naturally occurring gap in the axonal neurofilament array. Images excerpted from a time-lapse movie of a cultured neonatal mouse cortical neuron expressing GFP-NFM. Proximal is left, distal is right. The white arrowheads mark the leading end of the moving filament. Scale bar = 10 μm. Data from Wang & Brown (2010).
5.3 Fluorescence photobleaching
In axons or axonal regions that lack naturally occurring gaps, the movement of single neurofilaments can be observed by creating “artificial gaps” using fluorescence photobleaching. A fluorescent neurofilament fusion protein is expressed in cultured neurons and allowed to incorporate throughout the endogenous neurofilament array, and then the fluorescence is bleached in a segment of axon and the bleached region is observed by time-lapse fluorescence imaging. Neurofilaments that move into the bleached region are fluorescent (and can thus be detected) because they originate from the flanking non-bleached regions of axon (Fig. 10). The key requirements of this approach are: (1) the use of thin axons, which can be bleached sufficiently to allow the detection of the moving polymers without causing photodamage, and (2) the use of long bleached regions (20–60 μm in length), which enables the entire length of the neurofilaments to be observed while they are moving. For example, we have used photobleaching to detect the movement of GFP-tagged neurofilament proteins in axons of cultured SCG neurons (Wang and Brown, 2001). Comparison of the kinetics of neurofilament movement in photobleached and naturally occurring gaps indicates that bleaching can be performed without impairing the movement, at least in thin axons (Wang and Brown, 2001).
Fig. 10.
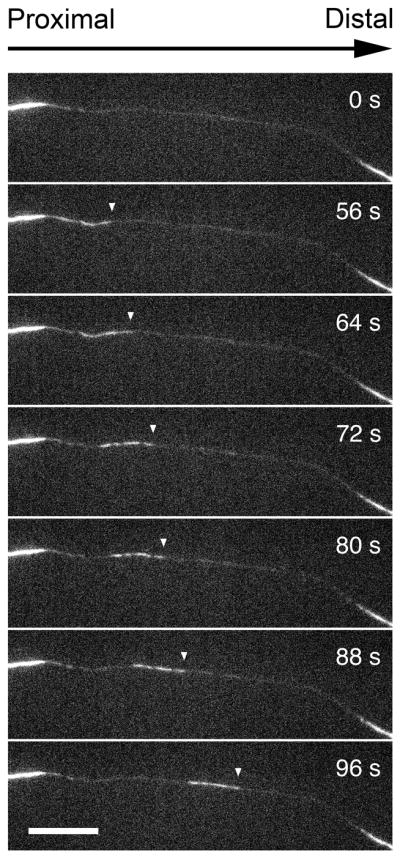
Movement of a neurofilament through a photobleached region of an axon. Images excerpted from a time-lapse movie of a cultured rat SCG neuron expressing GFP-NFM. Proximal is left, distal is right. The white arrowheads mark the leading end of the moving filament. Scale bar = 10 μm. Data from Wang & Brown (2001)
5.4 Fluorescence photoactivation
When fluorescently tagged neurofilaments enter natural occurring or photobleached gaps they can be resolved because they are the only fluorescent structures. However, it is important to note that this method tends to underestimate the long-term pausing behavior of the filaments because only those filaments that move into the gaps can be tracked. Moreover, photobleaching may not be effective for axons that have very large numbers of neurofilaments because it may be hard to bleach the fluorescence enough to detect the movements of single neurofilaments without inducing photodamage. To address these problems, we developed a complementary fluorescence photoactivation pulse-escape technique that permits a population-level analysis of neurofilament transport kinetics in single axons that contain many neurofilaments. This method relies on the use of neurofilament polymers tagged with a photoactivatable or photoconvertible fluorescent protein. We have the most experience using photoactivatable GFP (PAGFP) (Patterson and Lippincott-Schwartz, 2002) (Section 3.2).
To analyze neurofilament transport using the fluorescence photoactivation technique, we co-transfect cultured neurons with a PAGFP-tagged neurofilament protein and a red fluorescent protein (as a marker of transfection). After allowing several days for the fusion protein to incorporate throughout the axonal neurofilament array, we photoactivate a short segment of axon (e.g. 5–10 μm), creating a discrete population of fluorescent filaments. Over time, the filaments depart the activated region, but this happens infrequently because they spend most of their time pausing. If we acquire time-lapse images at time intervals on the order of seconds, it is possible to capture the motile behavior of the single fluorescent neurofilaments that occasionally depart the activated region and move into the flanking non-fluorescent regions (Fig. 11). This single-filament tracking strategy yields information similar to that obtained by tracking neurofilaments moving through naturally occurring and photobleached gaps (Trivedi et al., 2007). However, the real power of this technique is that it allows us to analyze not what leaves but what remains behind. To do this, we image the activated regions at time intervals on the order of minutes or tens of minutes instead of seconds. This allows us to monitor the regions for several hours with minimal fluorescence photobleaching (Fig. 12). Given that there is little soluble neurofilament protein in axons, there is no measurable loss of fluorescence due to diffusion (provided that the level of fusion protein expression is low) and therefore, the decay kinetics are due entirely to neurofilament transport. Since the average rate of neurofilament movement is on the order of 0.5 μm/s, it takes only seconds for a neurofilament to move out of the activated region. Thus, the kinetics of loss of fluorescence from the activated region on long time scales are dictated largely by the time spent pausing.
Fig. 11.

Example of a neurofilament escaping from a photoactivated region. Images excerpted from a time-lapse movie of a cultured SCG neuron co-expressing DsRed2 (to permit observation of the axons prior to photoactivation) and PAGFP-NFM. Proximal is left, distal is right. The white arrowheads mark the leading end of the moving filament. The diffuse axonal fluorescence that can be seen outside of the activated region was present before photoactivation, and is caused by weak excitation of the DsRed2 by the blue light used to excite GFP. These days we avoid this problem by using mCherry, which exhibits minimal excitation at this wavelength (see Section 6.13). Scale bar =10 μm. Data from Trivedi et al. (2007).
Fig. 12.
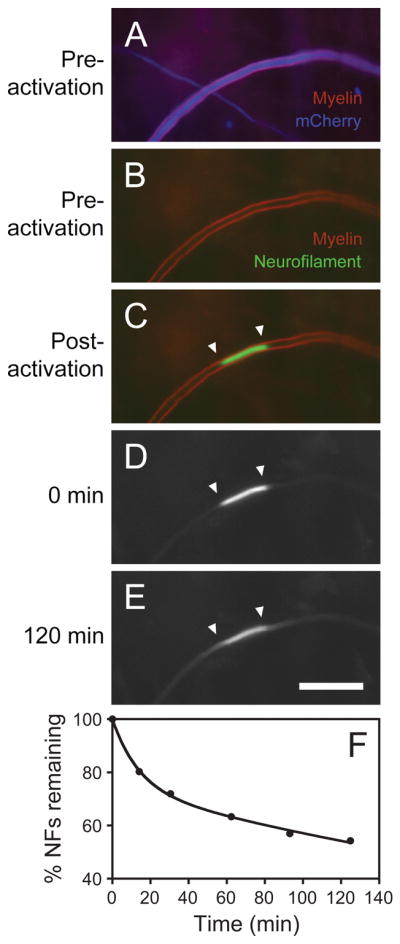
A fluorescence photoactivation pulse-escape experiment. Images excerpted from a time-lapse movie of a cultured DRG neuron in a long-term myelinating co-culture. The neuron is co-expressing mCherry (to permit observation of the axons prior to photoactivation) and PAGFP-NFM. The culture was stained with FluoroMyelin™ Red, which is a vital fluorescent marker for compact myelin (Monsma and Brown, 2012). A, B. The mCherry fluorescence is shown in blue to distinguish it from the FluoroMyelin™ Red fluorescence, which is shown in red. The myelin sheath appears purple in A because of FluoroMyelin™ Red cross-talk on the mCherry channel. We avoid mCherry cross talk on the FluoroMyelin™ Red channel by using a custom filter that excites the FluoroMyelin™ Red with blue light, taking advantage of this dye’s unusually large Stokes shift. C. The PAGFP-NFM fluorescence (green) and FluoroMyelin™ Red fluorescence (red) before photoactivation. D, E. Images of the PAGFP-NFM fluorescence at t = 0 and 120 min after photoactivation. F. Graph of the average pulse-escape kinetics for 15 axons. Note that a significant fraction of the fluorescence remains in the activated region after two hours, reflecting the prolonged pausing behavior of these polymers. Scale bar = 15 μm. Data from Monsma et al. (2014).
6 Live-cell imaging
6.1 General considerations
The dynamics of intermediate filament assembly in cells is not well understood, but it is likely that the mechanism of fusion protein incorporation into neurofilaments involves de novo filament assembly, subunit exchange along the length of pre-existing filaments (which we have termed intercalary subunit exchange (Colakoglu and Brown, 2009)), and also cyclical annealing and severing mechanisms (Uchida et al., 2013). Regardless of the imaging strategy to be used, it is preferable to allow time for expression and incorporation of the fluorescent fusion protein throughout the axonal neurofilament array before imaging. Using immunostaining to compare the distribution of the fusion protein to endogenous neurofilament proteins, we have found that this takes a minimum of several days. For SCG neurons transfected by nuclear injection 2 days after plating, we usually image 3–5 days later (Wang and Brown, 2001; Wang et al., 2000), whereas for cortical neurons transfected by electroporation we usually image 7–14 days later (Wang et al., 2010; Uchida et al., 2013). With enough time, the fusion protein should incorporate along the entire length of all neurofilaments throughout the axonal neurofilament array. For example, in one study in which we transfected mouse cortical neurons with GFP-tagged neurofilament protein H (GFP-NFH) by electroporation, on average the fusion protein was incorporated along 92% of the filament length throughout the axons by 13 days after plating (Wang & Brown, unpublished data). However, at earlier times, some neurofilaments may lack fluorescence along all or part of their length. For example, in one study in which we transfected rat SCG neurons with GFP-NFM by nuclear injection 2 days after plating, there was incomplete incorporation along the filaments 3 days later, though all neurofilaments contained GFP-NFM along at least part of their length (Yan and Brown, 2005).
6.2 Imaging media
Care must be taken in the choice of medium for live-cell fluorescence imaging because most commonly used culture media contain compounds that are autofluorescent. This can result in elevated background fluorescence and a substantial degradation of image quality. One obvious culprit is phenol red dye, which is included in most media as a pH indicator. It is necessary to use phenol red-free media formulations for live-cell fluorescence imaging. Other culprits include riboflavin and folic acid. We currently use a low fluorescence formulation of Hibernate™ medium (BrainBits). This medium lacks phenol red, riboflavin and folic acid and is supplied in two different osmolarities: Hibernate E™ (~230–240 mOs) for embryonic cells and Hibernate A™ (~250–60 mOs) for postnatal and adult cells. Hibernate™ media are designed for the maintenance of neural tissue or cells at ambient CO2 levels, so an added advantage is that they permit observation of the cells on the microscope stage without the need to supply CO2. A cheaper alternative is phenol red-free Leibovitz’s L-15 medium (Gibco Life Technologies), but we find Hibernate™ to be superior. Since cortical neurons fare better in higher osmolarity media after 7 days in culture (Section 2.2), we add 37.5 mM NaCl to the Hibernate™ media when imaging cortical neuron cultures of this age. This increases the osmolarity to ~290–300 mOs for Hibernate E™ and ~310–320 mOs for Hibernate A™. For long-term observation (> 3 hours) we recommend supplementing the Hibernate EB™ or AB™ media, which include 2% (v/v) B27 supplement mixture (Invitrogen) and 0.5 mM Glutamax (Sigma). With these supplements, it is possible to keep the cells healthy on the microscope stage for at least 48 hours.
6.3 Controlling temperature, CO2 and humidity
To observe living cells on the microscope stage it is necessary to maintain the temperature at 37°C. Tight control of this temperature is also important to ensure stable focus. Since warming the media leads to evaporation, which in turn increases osmolarity, it is also necessary to minimize evaporation loss. Some media also require an atmosphere of 5% CO2. Thus, environmental control is an important requirement for live-cell imaging. We use several different methods for controlling the environment on the microscope stage, each of which has advantages and disadvantages (Table 6).
Table 6.
Comparison of three options for environmental control on the microscope stage.
| Chamber/dish | Advantages | Disadvantages |
|---|---|---|
| Stage-top incubator | Permits stable control of temperature, humidity and optionally CO2. Glass-bottomed dishes can be transferred directly from the incubator to the stage top and imaged immediately. Adapters can allow the chamber to accommodate a variety of dish sizes as well as glass-bottomed multi-well plates. | The most expensive option. An objective heater is preferable for use with oil-immersion objectives. |
| Imaging and perfusion chamber | Permits stable control of temperature with no evaporation loss. Perfusion is possible. | Less expensive than the stage-top incubator option. Depending on the chamber design, there is a possibility of leaks if assembled incorrectly. Cells must be cultured on free coverslips of a specified dimension and then assembled into the chamber prior to observation. An objective heater is preferable for use with oil-immersion objectives. |
| Air-stream incubator | Simplest and least expensive option. Can heat both the dish and objective. Can be used with any dish size. | Requires an enclosure to provide good temperature or focus stability. Heats the room (or microscope, if an enclosure is used). |
Stage-top incubators
For imaging the cells in 35mm glass-bottomed dishes on an inverted microscope, the most convenient and effective solution is a stage-top incubator. This is essentially a low profile heated metal box with a heated glass lid and a hole in the bottom to allow access for the microscope objective. We use a Model H301 stage-top incubator made by Okolab (Ottaviano, Italy). This model contains a reservoir inside the chamber that can be filled with water to humidify the space around the dish. Alternatively, evaporation loss can be prevented by floating a layer of silicone fluid over the medium. We use low viscosity polydimethylsiloxane fluid (5 centistokes, Sigma) warmed to 37°C (0.75 ml is sufficient to cover the medium in a 35 mm glass-bottomed culture dish). For imaging in media such as Hibernate™ that buffer the pH at ambient CO2, there is no need to supply CO2. For imaging in media that require CO2 control, we use an Okolab gas controller to supply 5% CO2. The gas is humidified before entering the chamber by bubbling it through heated water using an Okolab humidity module.
Imaging chambers
An alternative to stage-top incubators is to use a Focht chamber such as those supplied by Bioptechs (Butler, PA). We use the FCS2 closed-bath imaging chamber. Advantages include very uniform and stable heating, no evaporation loss due to closed design, inlet and outlet ports for perfusion applications, and a laminar flow that permits rapid solution changes. However, the cells must be grown on 40mm diameter round glass coverslips sold by Bioptechs and the coverslip must be assembled into the chamber, which takes a few minutes. Warner Instruments has imaging and perfusion chambers (Series 20 and Series 30) that accommodate standard square or round coverslips. We have used the model RC-21B and RC-30HV chambers. In general, these chambers are a little more fiddly to assemble without leaks and they are not as thermally stable as the Bioptechs chambers. The Series 30 chambers have a lower profile, which allows them to be used with a high NA oil-immersion condenser for applications requiring high resolution imaging with bright field or differential interference contrast (DIC) microscopy. When we use the Warner Instruments chambers we get the best thermal stability if we perfuse medium that is pre-heated with a Warner Instruments in-line solution heater.
Air stream incubators
The least expensive approach is to use an air stream incubator (e.g. the ASI 400, Nevtek, Williamsville, VA), which maintains the temperature of the cells by blowing warm air across the microscope stage. Feedback regulation and proportional electronics prevent temperature overshoots. Silicone fluid is layered over the culture medium to prevent evaporation (see above). Because the fan generates vibration, it must be isolated mechanically from the microscope. We place it on an equipment shelf supported above the vibration isolation table top. Depending on how the air is directed, the air stream is wide enough to warm the stage, dish and objective. However, the temperature varies with distance from the blower, can fluctuate and is difficult to control precisely, resulting in focus drift and sometimes stage drift during imaging.
Another downside of air stream incubators is that they heat the entire room. A solution to this problem, which also gives better temperature stability, is to enclose the stage or entire microscope in a plexiglass cabinet and use the incubator to heat the air inside this enclosure. Such cabinets are available for all major microscope brands. However, we feel that stage-top incubators and chambers are a superior solution.
Objective heaters
To observe neurofilament transport, we use high NA oil-immersion objectives (Section 6.5). Since the use of immersion media can allow the microscope objective to become a heat sink, locally cooling the cells on the coverslip, we recommend the use of an objective heater when using a stage-top incubator or imaging chamber. We use objective heaters supplied by Okolab or Bioptechs. With appropriate calibration, the use of these heaters in combination with a stage-top incubator or imaging chamber can maintain exceptionally stable temperature and focus on the microscope stage for prolonged periods.
6.4 Choice of microscope
An inverted microscope is preferable for live-cell imaging of cultured cells because it permits cells in glass-bottomed dishes to be imaged from beneath with high quality oil-immersion objectives. Fig. 13 shows the configuration for a glass-bottomed dish. For optimal image stability, the microscope should be placed on a vibration isolation table. For imaging neurofilament movement in axons in low-density cultures, confocal microscopy offers no benefit because the axons are so thin that they lie within a single focal plane. Thus, for these cultures, an inverted microscope with wide-field epifluorescence illumination is ideal (we use Nikon TiE or TE2000 microscopes). However, for imaging in the cell bodies of these neurons or in myelinating co-cultures, which are very dense and can be tens of micrometers in thickness, a multi-point laser scanning confocal microscope is preferable (we use an Andor Revolution WD spinning disk confocal, which incorporates a Yokogawa CSU-W1 confocal scanning unit attached to a Nikon TiE inverted microscope). The choice of appropriate filters and dichroics is important; modern so-called “hard-coated” filters and dichroics have much higher transmission and reflection efficiencies than the older “soft-coated” filters. We use ET (“sputter coated”) filters from Chroma Technology (Bellows Falls, VT) such as ET-EGFP filter set number 49002 for GFP, and ET-mCherry filter set number 49008 for mCherry. For a standard microscope filter set, which consists of excitation and emission filters and a dichroic mirror, substituting ET filters and dichroics can increase the light throughput by about two-fold.
Fig. 13.

Imaging of cells in glass-bottomed dishes on an inverted microscope. The cells on the coverslip can be imaged from below using high-NA oil-immersion objectives.
6.5 Choice of objective
For imaging single neurofilaments, which are just 10 nm in width, it is necessary to use a 100x oil-immersion objective with high numerical aperture (ideally 1.4 NA or higher). We use a Nikon 100x/1.4 NA Plan Apo VC DIC objective, which is corrected for spherical and chromatic aberration across the visible spectrum. Phase contrast objectives are not optimal because the phase ring reduces light throughput. For fluorescence photoactivation pulse-escape studies using wide-field fluorescence illumination that do not require the detection of single neurofilaments, we use a Nikon 40x/1.0 NA Plan Apo oil-immersion objective; the lower numerical aperture of this objective results in greater depth of focus, which is helpful for the myelinated cultures. For fluorescence photoactivation pulse-escape studies on myelinating co-cultures using spinning disk confocal microscopy we use a 40x/1.15 NA Apo Lambda S LWD or 60x/1.2 NA Plan Apo VC water immersion objective. For thinner cultures, we can use higher NA objectives that have shorter working distances, such as a 40x/1.3 NA Plan Fluor or 60x/1.4 NA Plan Apo VC oil-immersion objective.
6.6 Choice of camera
In selecting a camera for live-cell fluorescence imaging, three features are of paramount importance: sensitivity, resolution and speed. Generally there is a trade-off between speed and resolution on the one hand and sensitivity on the other. Current options are conventional cooled charge coupled device (CCD) cameras, electron multiplying cooled CCD (EMCCD) cameras, and scientific complementary metal oxide semi-conductor (sCMOS) cameras. Sensitivity is determined by the quantum efficiency and read noise, resolution is determined by the pixel size on the detector, and speed is determined by the readout rate. However, for imaging at low light levels the sensitivity of the camera can be a more critical determinant of the rate of image acquisition than the actual readout time. In other words, a camera with a faster readout rate may not necessarily result in faster image acquisition if longer exposures and/or frame averaging are required to detect the fluorescent signal. Modern sCMOS cameras are the fastest available and have the highest resolution and largest fields of view, but they do not yet have the sensitivity of EMCCD cameras at low light levels. While speed and resolution are important, high sensitivity is paramount for live-cell imaging because it allows for the use of lower intensities of exciting light, which results in less photobleaching and photo-oxidative damage (Section 6.10).
For imaging neurofilament transport, we currently use an Andor iXon Ultra 897 EMCCD camera (Andor Technology, Belfast, UK), which has a 512 × 512 array of 16 × 16 μm pixels. With a 100x objective, this yields a field of view of ~82 × 82 μm and a pixel size in the image plane of 160 × 160nm. With sufficient exciting light, this camera is capable of streaming images of neurofilaments at up to 56 frames per second (i.e. with exposures of less than ~18 milliseconds). According to the Nyquist sampling theorem, the best possible image resolution is obtained when the pixel size of the detector is less than half the optical resolution of the microscope objective (Young, 1989). The maximum theoretical resolution of an objective lens is given by 0.61 λ/NA (where λ is the wavelength of the light and NA is the numerical aperture of the objective). For a 100x/1.4 NA objective in green light (510 nm), this corresponds to 222 nm. Thus, an optimal detector for our application would have a pixel size no greater than 111 × 111 nm. While the larger pixels size of our EMCCD camera does not satisfy this criterion, the actual resolution in our images is undoubtedly less than the theoretical limit, and in practice the pixelation of the image is an acceptable trade-off for the improved sensitivity of this camera compared to sCMOS and conventional CCD cameras.
For applications in which a larger field of view is required, we use an Andor Neo sCMOS camera, which has a 2560 × 2160 array of 6.5 × 6.5 μm pixels. With a 100x objective, this yields a field of view of ~166 × 140 μm and a pixel size in the image plane of 65 × 65nm, which satisfies the Nyquist sampling criterion. In our hands, both sCMOS and EMCCD cameras outperform conventional front-illuminated interline CCD cameras for imaging neurofilaments at moderate light levels (Fig. 14). However EMCCD cameras come into their own at lower light levels.
Fig. 14.
Images of neurofilaments acquired using EMCCD and sCMOS cameras compared to images acquired using a conventional CCD camera. Wide-field epifluorescence microscopy of cultured rat cortical neurons expressing GFP-NFM. Camera exposure time=100 milliseconds. A. An axonal neurofilament imaged with an Andor Neo sCMOS camera (circa 2013). A′. The same neurofilament imaged with a Photometrics CoolSNAP HQ camera (circa 2002). B. An axonal neurofilament imaged with an Andor Ultra 897 EMCCD camera (circa 2013). B′. The same neurofilament imaged with a Photometrics CoolSNAP HQ camera (circa 2002). Note that both the EMCCD and sCMOS cameras result in a significant improvement in image contrast due to the higher signal-to-noise ratio. The cameras have different pixel sizes so the images are different sizes; the EMCCD image (160 nm pixels) was enlarged two-fold to make it similar to the sCMOS (65 nm pixels) and CCD (64.5 nm pixels) images. To avoid resampling, the images were not resized further. Scale bars = 4 μm.
6.7 Selection of cells and axonal regions for imaging
To observe movement, fluorescent cells are imaged at high magnification using a 40x, 60x or 100x objective. We select cells that have a normal healthy overall morphology and (if single filaments can be resolved) neurofilaments with continuous fluorescence along their length. We avoid cells that are overly bright because of the potential for artifacts associated with high concentrations of exogenous protein. Signs of overexpression also include fluorescent aggregates or diffuse fluorescence within gaps in the neurofilament array (Section 5.2). To find gaps in the neurofilament array or to locate axons that are suitable for photobleaching or photoactivation, we scan quickly along the axons using epifluorescence and looking through the microscope eyepieces, always taking care to observe the fluorescence as little as possible so as to minimize photobleaching prior to imaging. Gaps tend to be less frequent in proximal axonal regions, where axons tend to be thicker and the neurofilament content can be greater. For wide-field microscopy, we stop down the iris field diaphragm in the epifluorescence illumination light path prior to imaging to prevent exposing areas outside the field of view of the camera. For imaging the movement of single neurofilaments in cortical or SCG cultures, the neurons are cultured at low density (Sections 2.4 & 2.5) so that axons can be traced from the cell body to the growth cone. This is necessary in order to establish the proximal and distal orientation of the axons in the time-lapse movies. The low density also helps to minimize axonal fasciculation.
In some cases, such as in cortical neuron cultures, it may be necessary to confirm the orientation of the axons by co-transfection with a fluorescently tagged +TIP protein that tracks the growing plus ends of microtubules (e.g. mCherry-EB3). Since axonal microtubules are orientated with their plus ends predominantly distal, the +TIP comets should all track in an anterograde direction. To confirm the directionality of the comets, we acquire a 2-minute time-lapse movie at 2 second time intervals and then construct a kymograph along the axon as described in Section 7.4. The comets appear as diagonal streaks in the kymograph (Fig. 15).
Fig. 15.

Example of a kymograph used to determine axon orientation in a cultured cortical neuron. The cell was transfected with GFP-NFM (to visualize neurofilaments) and mCherry-EB3 (to visualize the plus ends of growing microtubules). The kymograph was created from a short time-lapse movie of the mCherry fluorescence. The horizontal dimension represents distance (proximal is left, distal is right) and the vertical dimension represents time (from top to bottom). Since axonal microtubules are oriented uniformly with their plus-ends pointing distally, the mCherry-EB3 comets all track in a proximal-to-distal direction (white arrow). Thus the trajectories of the comets in the kymograph reveal the orientation of the axon. Scale bar = 5 μm.
6.8 Time-lapse or streaming image acquisition
To observe neurofilament movement in naturally occurring or photobleached gaps, we acquire movies using time-lapse or by real-time streaming image acquisition. The principal factors limiting the duration of imaging are the sensitivity of the camera and the brightness and photostability of the fluorescent protein. For wide-field epifluorescence using an EMCCD camera, we typically attenuate the mercury light source by 4–30 fold (~75–97%) with neutral density filters to minimize photobleaching (Section 6.10) and use exposures in the range of ~30–200 milliseconds. Under these conditions, we are able to acquire time-lapse or streaming movies that are 400–15,000 images in length with minimal photobleaching. However, there is a trade-off between the intensity of the exciting light and the exposure time. Higher exciting light intensities allow for shorter exposures but result in more photobleaching (Section 6.10), whereas longer exposures allow for the use of lower light intensities but can result in significant motion blurring during bouts of rapid movement due (caused by movement of the filament during the exposure period). For time-lapse image acquisition, the choice of time interval depends on the purpose of the experiment. Shorter intervals provide greater temporal resolution of the kinetics on fast time scales, whereas longer intervals allow for longer tracking times, which can provide more information about the kinetics (movements and pauses) on slow time scales. As a general guide, 1–5 second intervals represents a good range for many studies. For streaming image acquisition, there is a trade-off between speed and image quality (Section 6.6). To achieve the fastest streaming rates, it is necessary to stream to RAM (saving to disk later) and to set the software to update the image window infrequently (e.g. every 100th frame). Since the movements are very infrequent, it is not unusual to acquire movies in which little or no movement is observed, especially if the duration of the movie is short. In cultured SCG and cortical neurons, we typically observe about 0.1–0.3 moving neurofilaments per minute (Uchida et al., 2009; Wang and Brown, 2001; Wang and Brown, 2010; Wang et al., 2000).
6.9 Focus drift
A problem that is often encountered during time-lapse or streaming movie acquisition is focus drift, which requires constant manual focus adjustment to maintain correct focus. The causes of focus drift are either mechanical (e.g. instability in the focusing mechanism of the microscope) or thermal (e.g. expansion and contraction of the microscope stage and/or optics). In our hands, thermal instability appears to be the major culprit because we observe substantial improvement by using a thermally stable stage-top incubator or imaging chamber in combination with an objective heater (Section 6.3). However, active focus drift compensation systems such as the Nikon Perfect Focus™ (PFS) system that correct for focus drift automatically in real time have essentially eliminated focus drift as a problem for all practical purposes. On our Nikon microscopes, we now routinely engage the Perfect Focus™ for all our time-lapse and streaming image acquisition whenever possible.
6.10 Minimizing photobleaching and photodamage
One of the challenges associated with live-cell fluorescence imaging is to minimize photobleaching and photodamage. One obvious way to accomplish this is to limit the illumination of the cells. With conventional lamp light sources, it is helpful to use electronic shutters controlled by the imaging software. With LED light sources, the bulbs can be turned on and off fast enough that a shutter may not be necessary. To limit illumination of the specimen during direct observation through the microscope eyepieces (e.g. when surveying the dish and selecting cells prior to imaging) we configure our shutters and LED light sources with foot switches. However, the most important way to minimize photobleaching and photodamage is to reduce the intensity of the exciting light. The total illumination is not as important as the intensity. For example, imaging for 1 second with no attenuation of the exciting light will result in significantly more photobleaching than imaging for 10 seconds with a 10-fold attenuation of the exciting light. Generally we try to use the lowest possible illumination intensity, which is limited primarily by the brightness (quantum efficiency) of the fluorescent protein and the sensitivity of the camera. For wide-field imaging using an EMCCD camera and mercury arc lamp illumination, we are able to detect single GFP-tagged neurofilaments in axons with exposures as low as 30 milliseconds (~33 frames per second) using 5-fold attenuation of the exciting light, permitting streaming acquisition of 10–15,000 frames over a period of ~5–8 minutes with modest levels of photobleaching (Section 6.8).
Since photobleaching and photodamage are enhanced in the presence of oxygen, we used to image the cells in sealed chambers containing oxygen-depleted imaging medium. To deplete the oxygen we used an oxygen scavenging system called Oxyrase™ (Oxyrase Inc.), which is a bacterial membrane fraction that contains the enzymes and substrates of oxidative phosphorylation (Mikhailov and Gundersen, 1995). However, while this treatment appears to reduce photobleaching of proteins covalently conjugated with fluorochromes such as fluorescein or rhodamine, we no longer use it because it appears to offer no benefit for imaging fluorescent fusion proteins.
6.11 Methods for spatially selective illumination
To perform photobleaching or photoactivation experiments it is necessary to be able to selectively illuminate a sub-region of the field of view with a particular wavelength of light. Here we describe three methods: iris diaphragm, digital multimirror device, and laser scanning (Table 7).
Table 7.
Comparison of spatially selective illumination methods for photobleaching, photoactivation and photoconversion applications.
| Method | Advantages | Disadvantages |
|---|---|---|
| Iris diaphragm | Simple and inexpensive. | Limited control of the size of the illuminated area. No control of the shape or location of the illuminated regions. Not possible to illuminate multiple regions at once. |
| Digital Multimirror Device (DMD) | Allows for the illumination of multiple regions of user-specified shape, size and number. Can be configured with laser or arc lamp light sources. Fast. | More expensive. Illuminated area is limited by the size of the multimirror array. |
| Laser scanner | Allows for the illumination of multiple regions of user-specified shape, size and number. | More expensive. Can be slow for large regions. High laser power is required for rapid scanning, increasing the potential for photodamage. |
Iris diaphragm method
The simplest method to achieve spatially selective illumination is to insert an iris diaphragm into the epifluorescence illumination light path at a position conjugate to the specimen plane so that the edges of the diaphragm are in focus when the specimen is in focus. Although crude, we have used this method successfully in several studies (Colakoglu and Brown, 2009; Trivedi et al., 2007; Wang and Brown, 2001). On our Nikon microscopes, we use the epifluorescence illumination field-limiting iris diaphragm, which can be centered in the field of view using two thumbscrews. However, this diaphragm is a fixed shape (circular) and cannot be closed all the way, so the smallest field of view that can be illuminated using a 100x objective is a circle with a diameter of ~23 μm. The region of axon to be bleached is positioned in the center of the field of view by moving the microscope stage and then the diaphragm is closed down to illuminate only the desired area. To obtain the maximum possible light intensity, we remove all neutral density filters from the epifluorescence illumination light path for the bleaching exposure and then replace them prior to subsequent imaging.
Digital Multimirror Device method
A superior, albeit more expensive, method to achieve spatially selective wide-field illumination is to use a Digital Multimirror Device (DMD), which comprises a two-dimensional array of individually addressable micro-mirrors that can be switched “on and off” (tilted) with microelectromechanical “hinge” elements. By reflecting light off this array, it is possible to selectively illuminate user-defined subregions in the specimen plane. We use the Andor Mosaic Digital Diaphragm with a 100W mercury arc lamp light source attached to a Nikon TiE wide-field epifluorescence microscope, but a laser light source or higher wattage lamp can be used if greater light intensities are required. The device is controlled using the Targeted Illumination drop-in in the MetaMorph™ software package (Molecular Devices, Sunnyvale, CA). After calibration of the camera to the DMD, which is required to map the mirrors in the DMD mirror array to the pixels in the CCD array, it is possible to draw one or more regions on the image in MetaMorph™ and then selectively illuminate only those areas.
Laser scanning method
A third method for achieving spatially selective illumination is to use a laser scanner. On our spinning disk confocal microscope, we use an Andor FRAPPA laser galvanometer (“laser galvo”) mirror scanner which scans a diffraction-limited spot of light across the field in a raster pattern using mirrors controlled by a dual galvanometer scan head, and an AOTF to attenuate the beam in areas that fall outside the user-defined illumination regions. This method has similar versatility to the DMD method, but one cautionary note is that high beam intensities are required for rapid scanning, which increases the possibility of photodamage.
6.12 Fluorescence photobleaching experiments
Fluorescence photobleaching is used to analyze the movement of neurofilaments in axons or sections of axon that have no naturally occurring gaps in the neurofilament array. The wavelength of light required to bleach the fluorescence is the excitation wavelength of the fluorescent protein (e.g. blue light for GFP, green light for mCherry). We have most experience using GFP-tagged neurofilament proteins. The extent of bleaching should be sufficient to reduce the fluorescence below the fluorescent intensity of a single neurofilament. Though the bleaching does not have to be complete, any residual unbleached fluorescence will reduce the contrast of the moving filaments. For photobleaching applications using a mercury arc lamp light source (either using the iris diaphragm or a DMD), we need to use continuous illumination for 30–120 seconds. While the long duration of this bleaching exposure makes this approach unsuitable for many photobleaching applications, it can work for studies on neurofilament transport because of the asynchronous and infrequent nature of the movement (Section 1). Since bleaching at this intensity is relatively slow, it is desirable to select thin axons, which can be bleached more readily than thicker axons. Moreover, larger more neurofilament-rich axons may require longer bleach times, which could increase the chances of inducing photodamage. We can bleach GFP-tagged or mCherry-tagged neurofilament protein in axons of cultured SCG neurons for up to 2 minutes without noticeable impairment of neurofilament movement, so we believe that there is minimal photodamage under these conditions (Wang and Brown, 2001). Signs of severe photodamage include blebbing and vesiculation of the axons, but more subtle damage may go unnoticed.
If overexpression is avoided, the fluorescent neurofilament fusion protein should be fully assembled into polymer and therefore there should be no diffusive recovery of the bleached fluorescence; recovery of the bleached fluorescence will depend on neurofilaments moving into the bleached region and pausing there rather than diffusion of neurofilament subunits. This means that the bleached region can normally be observed for tens of minutes or more.
To record neurofilament movement, we use time-lapse imaging or real-time streaming image acquisition (Section 6.8). Imaging can begin immediately after bleaching. For cultured SCG neurons, we observed an average of ~1 neurofilament per 7 minutes of observation (Wang and Brown, 2001). Since moving neurofilaments average about 10 μm in length in cultured neurons, the bleached region should measure at least 30 μm in length. However, longer bleached regions are required to track neurofilaments for longer periods of time, or to track the movement of the longest neurofilaments, which can exceed 25 μm in length.
6.13 Fluorescence photoactivation experiments
For fluorescence photoactivation pulse-escape analyses of neurofilament transport, we co-transfect neurons with photoactivatable GFP (PAGFP) neurofilament fusion protein (e.g. PAGFP-NFM or PAGFP-NFL) and a diffusible red fluorescent protein such as mCherry. The red fluorescent protein allows us to identify transfected cells by their red fluorescence and it also enables us to trace the axons and select regions for analysis prior to photoactivation. To ensure that all cells that express red fluorescence also photoactivate, a dual promoter expression construct can be useful (e.g. Clontech pBI-CMV1).
Cells expressing red fluorescence are located using a high magnification oil-immersion objective (40x, 60x or 100x depending on the application) and axonal regions suitable for activation are located. Since photoactivation of PAGFP requires illumination with violet light, it is preferable to use objectives that are corrected for chromatic aberration in the violet (we use Nikon Plan Apo VC objectives). Prior to activation, an image of the mCherry fluorescence is acquired to record the location and morphology of the axon, and an image of the PAGFP fluorescence is acquired to record the baseline fluorescence with blue light excitation. Then we illuminate a segment of the axon with violet light using one of the spatially selective illumination methods described above (Section 6.11). When using our spinning disk confocal microscope, we use the FRAPPA laser scanner with a 405 nm laser line. When using the iris diaphragm method on our wide-field microscope, we use a 100W mercury arc lamp with violet filter cube (385–425 nm band-pass exciter). When using the Mosaic Digital Diaphragm DMD device with a 100W mercury arc lamp light source on our wide-field microscope, we introduce the device into the epi-illumination light path using a dichroic mirror that transmits in the violet spectrum (385–450 nm), and we direct the light to the specimen using a 495 nm long-pass dichroic mirror (Chroma Technology) in the microscope filter turret. Then we switch to a GFP filter cube in the microscope filter turret to observe the activated fluorescence, acquiring images at intervals of seconds, minutes or tens of minutes over a period of minutes to hours, depending on the experimental application. Typically for the latter two methods, which both use a mercury arc lamp source, we obtain optimal activation with exposures of 1–2 seconds using a 100x/1.4 NA Plan Apo VC oil-immersion objective. For the laser scanning method, the scan time required varies depending on the size and shape of the scanned area, the laser intensity, and the pixel dwell time.
A cautionary note about the use of PAGFP is that activation with violet light can convert some of the proteins to a non-fluorescent “dark state” from which the molecules return slowly (Bancaud et al., 2010). This can result in a delayed increase in the activated fluorescence after the initial activation. In our experience this problem is more pronounced with more intense illumination and can take as long as 45 seconds to recover completely. When activating PAGFP using the FRAPPA laser scanner on our spinning disk confocal, we have observed an increase in the fluorescence of up to 20% within the first 45 seconds after activation. To minimize this problem one can reduce the intensity of the violet illumination, but with a laser scanner this can lead to excessively long activation times so it may be preferable to use a DMD device. Alternatively, one can simply wait one minute after activation before taking the first image and then consider that to be the “zero” time point. This can work for long-term pulse-escape experiments on neurofilament transport because the neurofilaments leave the activated region very infrequently, but it would not be a satisfactory workaround for more rapidly changing events.
7 Software and analysis
7.1 Software
For many years we have used MetaMorph™ image analysis software for most image acquisition, image processing and analysis. This software integrates control of the microscopes and associated hardware (e.g. illumination, shutters, cameras, filter wheels, and motorized stages) into a single imaging workstation on both our wide-field and spinning disk confocal systems, allowing semi-automated control during still-frame, time-lapse and streaming image acquisition.
A major disadvantage of the MetaMorph™ software is that it is expensive to purchase and maintain. For many labs the cost may be prohibitive. An excellent alternative that is free and very powerful is the ImageJ open-source software package that was developed at the NIH (www.imagej.NIH.gov). We use Fiji (www.fiji.sc), which is an implementation of ImageJ that is designed specifically for the life sciences. We have been impressed with the power of this software and its many plugins, which rivals or exceeds the capabilities of commercial imaging software in many respects. When combined with μManager (www.micro-manager.com), which is a freely available and open-source microscope control software package, ImageJ can provide a total solution that will meet the needs of most laboratories. Developed in the laboratory of Ron Vale at UCSF with funding from the NIH, μManager runs as an ImageJ plug-in and can control most microscopes, cameras and related hardware on the market.
For preparation of figures, we perform intensity scaling and then save the images as 8 bit tiff files for subsequent processing in Adobe Photoshop. To show the movement, we save the movies in QuickTime™ format. Typically a play-back rate of 5–10 frames per second works well for time-lapse data, but higher rates can be used for streaming acquisitions. If necessary, the movies can be compressed with a lossy video compression algorithm (“video codec”) such as H.264/MPEG-4. The choice of codec should be empirical based on the image detail that you wish to preserve and the desired size of the compressed file.
7.2 Fixed-field tracking
To analyze the movement of single neurofilaments it is necessary to track their position in successive frames of time-lapse or streaming movies. There are many methods available for automated tracking of intracellular cargoes in time-lapse or real time movies. We have experimented with some ourselves (e.g. Yuan et al., 2012), but significant obstacles remain to the development of a robust method for neurofilaments. Particular challenges include the variable length of these unique cargoes, which can range from <1 μm to >50 μm in length, and their flexibility, which is evident from the complex folding behaviors that are often observed between bouts of movement (Taylor et al., 2012). Other challenges include non-uniform incorporation of fluorescent fusion protein along the polymers and overlap with other moving or pausing polymers, both of which are commonly encountered. Thus, it is still necessary to track the moving filaments manually, which is very labor intensive.
Our preferred manual tracking method is the TrackPoints function in the Motion drop-in of MetaMorph™ software (Fig. 16), though comparable options are available as plugins for ImageJ. This feature allows the user to click on the leading end of the filament in each plane of the movie. The software can be configured to mark the clicked points in various ways and to advance to the next frame automatically after each click to speed up the process. The coordinates and other parameters such as elapsed time, direction and velocity can be logged via Dynamic Data Exchange to a Microsoft Excel spreadsheet for further analysis. Of course, a limitation of this approach is that the decision about the precise location of end of the filament, which is diffraction-limited, is subjective. In addition, the method is vulnerable to user error. Nevertheless, with experience the method can work well.
Fig. 16.
Manual tracking of a neurofilament that moved through a gap in the axonal neurofilament array. Cultured mouse cortical neuron expressing GFP-NFM. Time-lapse imaging at 5 second intervals. The leading end of the neurofilament was tracked through 53 image planes using the TrackPoints function in the Motion drop-in the MetaMorph™ software. A. Image of the neurofilament in the last plane of the movie, just before the filament exited the gap. The white arrowheads mark the leading and trailing ends of the filament and the white arrow shows the direction of movement. B. The tracked positions of the leading end of the filament in each image plane superimposed on the same image (tracked points marked with a green “x”). Scale bar= 10 μm. C. Graph of the distance moved along the axon versus time for all 53 image planes.
Given the inherent subjectivity of manual tracking, it is important to establish objective criteria for which filaments will be tracked and precisely how their position will be recorded. For some studies it may be acceptable to exclude structures if they move less than a certain total distance (e.g. 10 μm) during the course of the movie, that cannot be tracked for a certain minimum number of time intervals, or whose location is ambiguous. Ambiguity can arise if the filament moves out of focus (e.g. if the axon is thick or courses out of the plane of focus) or if it overlaps other moving or pausing filaments. Though small reversals are common, each filament generally exhibits a single preferred direction of movement. We generally track the leading end of the filament in the predominant direction of movement, though of course this end becomes the trailing end whenever the filament reverses. For some applications, we track both the leading and trailing ends, though this makes the task even more laborious. Fiduciary points in the field of view can be used to ensure that there is no stage drift during the movie, though this is only a problem when using a manual (non-motorized) stage, and even then only rarely. If stage drift does occur, the planes in the stack can be registered using the “Align Stack” function in MetaMorph™.
To facilitate detection of the moving structures in the time-lapse movies, it can sometimes help to perform difference imaging. To obtain a rolling-frame difference image, each image in the time-lapse series is subtracted from the image that follows it. To obtain a fixed-frame difference image, a single reference image at the start of the time-lapse series is subtracted from all subsequent images in the series. Both methods selectively enhance changes in fluorescence intensity within the field of view, such as those associated with the movement of fluorescent structures. However, they work best when the frame rate is high.
7.3 Multi-field tracking
A limitation of standard fixed-field tracking strategies is that filaments can only be tracked during the brief time that they move through the camera field of view. For our Andor Ultra 897 EMCCD camera the field of view is about ~82 × 82 μm whereas for our Andor Neo sCMOS camera it is ~166 × 140 μm (Section 6.6). Since neurofilaments move at average velocities of about 0.5 μm/s, this means that most filaments move through the field in a matter of minutes. However, axons in culture can grow to be millimeters in length. To analyze the long-range motile behavior of single filaments, we have developed a multi-field tracking method which keeps the filament within the field of view at all times. This method takes advantage of a motorized stage and the “Move stage to image position” function in the MetaMorph™ software (similar functionality is available in μManager). We enable the “Offset stage position with mouse click” and “Move stage immediately on mouse click” options. The user selects a filament to track during time-lapse image acquisition by observing the acquired images live on the computer monitor and then clicking on that filament whenever it approaches the edge of the camera field of view. When the mouse is clicked, the stage moves to center that position (and thus the filament being tracked) within the field of view. The time-lapse interval must be long enough to allow for the stage to move and for the software to register and update the new location. We have found this to work well with time-lapse intervals as short as 2 seconds. Using this approach, we have been able to follow single filaments for >30 minutes during which they may travel total distances approaching 1 mm (Fig. 17). After the movie has been acquired, the image planes can be stitched together using the x-y coordinates of the stage (saved in the image header) to create what we call a “movie montage”. Briefly, a blank image stack is created with x and y dimensions large enough to span the total distance travelled by the tracked filament, and then each image frame is copied into the corresponding frame of the new stack, shifting the image position to account for the stage movement. When the resulting movie is played back, the camera field appears to skip across the field tracking the moving filament. The montage becomes very large so at least 32 GB RAM is required. With the use of linear encoders, the sub-pixel precision of modern motorized stages is sufficient that no image processing or manual adjustment is required to further align the images. The movement of the filaments in these montaged movies can be analyzed using manual tracking as described above (Section 7.2).
Fig. 17.

Long-term multi-field tracking of neurofilaments. A. Maximum projection of a movie montage showing part of the path of a fluorescent neurofilament tracked across multiple fields of view (yellow squares) along an axon of a cultured rat cortical neuron expressing GFP-NFM. The movie was acquired with time-lapse on a spinning disk confocal using an EMCCD camera, 3 second time intervals and 100 millisecond exposures. The inset (red box) is a single plane from the movie showing the tracked neurofilament, which measured 4.3 μm in length. Scale bar = 20 μm in the montage and 5 μm in the inset. B. Plot of distance moved versus time for this neurofilament. The filament was tracked for 30 minutes, during which it moved anterogradely, pausing occasionally, for a distance of almost 600 μm.
7.4 Kymograph analysis
The increased speed and sensitivity of modern cameras now permits kymograph analysis of neurofilament movement, which is a powerful method for analyzing intracellular cargo transport with high temporal resolution. A kymograph is a two-dimensional graphical representation of one-dimensional movement over time in which one spatial axis represents distance and the other represents time. To construct a kymograph of neurofilament movement, a video is acquired of a neurofilament moving along an axon and then a maximum intensity projection of that video is created to show the path of the filament along the axon. The maximum intensity projection is traced manually or using automated methods to obtain a curvilinear line, which is then used in turn to generate a linear intensity profile along the path of the filament (i.e. the axis of the axon) for each image plane in the movie. Since the neurofilaments are diffraction-limited in width, the precise placement of the line can influence the intensity profile so we generally set a “line width” in the software of 3 or 5 pixels and assign each pixel along the line a value equal to the average or maximum pixel intensity of that pixel plus one or two flanking pixels extending perpendicular to the line on either side. These linear intensity profiles are then represented as rows of pixels and stacked vertically in temporal sequence from top to bottom, resulting in a two-dimensional image in which the horizontal axis represents distance along the axon and the vertical axis represents elapsed time (Fig. 18). When stationary, the filaments in these kymographs generate vertical stripes whose width equals the filament length. When moving, the filaments generate diagonal stripes whose slope yields the velocity. At low expression levels, random incorporation of the fluorescent fusion protein results in a kind of speckling of the filaments that generates dark and light streaks in the kymographs. We refer to this as ”barcoding” because the longer filaments take on the appearance of a commercial barcode. This barcoding can aid in the interpretation of filament folding and unfolding events, and it can also aid in the unambiguous assignment of filament orientation after a reversal or folding event.
Fig. 18.
Kymograph analysis of neurofilament transport. Rat cortical neuron culture transfected with GFP-NFM and imaged by streaming acquisition with 30 millisecond exposures (~33 frames per second) using an Andor Ultra 897 EMCCD camera. The acquisition period was 450 seconds (15,000 frames). A. Maximum projection of the movie planes, revealing the path of the moving filaments along the axon. B. A single plane from the movie, corresponding to the time point marked by the black arrowheads in C in the kymograph below. Note the three filaments whose trajectories can be traced in the kymograph. C. Two portions of the corresponding kymograph. The horizontal dimension represents distance along the axon and the vertical dimension represents time. Each row of pixels in the kymograph represents a linear intensity profile along the axon path (defined by the maximum projection in A) in one plane of the movie. The kymograph is wider than the images in A and B because the axon traces a curved path. The temporal resolution is sufficient to resolve the single processive bouts of the moving filaments (diagonal streaks) separated by pauses (vertical streaks). The slope of the diagonal streaks yields the velocity. The three filaments shown here all move retrogradely. Note the non-uniform incorporation of GFP-NFM along the filaments, which gives their trajectories a “barcode” appearance. Scale bar = 20 μm.
Compared to raw movies, the advantage of kymographs is that they represent the neurofilament movement in a single image in which the problem of motion analysis of a two-dimensional object in time becomes essentially a problem of edge detection in space. This is possible because the movement of neurofilaments in thin axons is essentially one-dimensional (either forwards or backwards). One limitation, however, is that axons in culture are generally curved so the calculation of a linear intensity profile in the software requires some interpolation of the pixel intensities and this can lead to image artifacts when the axon bends tightly. In addition, kymograph analysis only detects axial movement so it may not work well for thicker axons in which there may be potential for movement tangential or perpendicular to the medial axis of the axon. To ensure continuous traces it is preferable to use a fast frame rate so that the displacement of the filament in each time frame is at most a few pixels. With an EMCCD camera that has 160 × 160 nm pixels, we perform streaming (“real time”) image acquisition with 30 millisecond exposures (~33 frames per second) (Section 6.8). Frame rates slower than this have the potential to generate discontinuous trajectories during periods of fast movement.
7.5 Pulse-escape fluorescence photoactivation analysis
The pulse-escape fluorescence photoactivation strategy for studying neurofilament transport relies on quantifying the fluorescence remaining in an activated segment of axon over time. The data are acquired by time-lapse imaging using regularly or irregularly spaced time intervals over a period of tens of minutes or hours. The intensity of the activated PAGFP fluorescence is measured using image processing and analysis software such as MetaMorph™ or Fiji (Section 7.1). A rectangular region of interest (ROI-1) is drawn manually around a segment of axon, encompassing its entire width. Additional flanking regions (ROI-2 and ROI-3) are drawn in the background on either side of the axon, taking care to avoid other fluorescent material or cells (Fig. 19). The contribution of the background fluorescence to ROI-1 is calculated by multiplying the average pixel intensity in ROI-2 and ROI-3 by the area of ROI-1. This value is then subtracted from the total fluorescent intensity in ROI-1 to yield the background-corrected intensity. The resulting background-corrected intensities are then corrected for photobleaching. To calibrate the rate of photobleaching for a given set of illumination conditions, we acquire images of the photoactivated PAGFP fluorescence in rapid succession and then quantify the fluorescence decay.
Fig. 19.

Quantification of photoactivated fluorescence in a segment of axon. Shown here is an axon of a rat DRG neuron in a long-term myelinating culture (see Section 2.6) co-transfected with mCherry (red fluorescence) and PAGFP-NFM. The PAGFP fluorescence was photoactivated in a 5 μm-long segment of the axon (green fluorescence) by illumination with violet light (section 6.13). The central yellow box shows the region of interest used to measure the fluorescence in the activated region (ROI-1) and the flanking boxes show the regions of interest used to measure the average background fluorescence (ROI-2 and ROI-3). Scale bar = 5 μm.
After photoactivation, the rapid intermittent movement of the photoactivated neurofilament polymers causes fluorescent neurofilaments to exit the activated region and non-fluorescent neurofilaments to enter. When the fluorescence intensity in the activated region is plotted over time, we consistently observe a biphasic decay profile that can be fitted with a double exponential decay function (Alami et al., 2009; Monsma et al., 2014; Trivedi et al., 2007). These kinetics suggest the existence of two distinct pausing states for neurofilaments, which we have termed on and off track. We have proposed that neurofilaments cycle between these two states during their transport along the axon, alternating between bouts of rapid intermittent “on track” movement, and prolonged “off track” pauses. According to this interpretation, the initial steep phase of the decay profile is due to the rapid departure of neurofilaments that are mobile (on-track) at the time of activation, whereas the later more shallow phase is due to the slow departure of neurofilaments that are immobile (off-track) at the time of activation and which can only escape the window after they have cycled into the mobile state. Hence, the time course of fluorescence decay at late times reflects the fraction of neurofilaments that are paused off-track at the time of activation and their rate of transition into the mobile states. Since there is little soluble neurofilament protein in axons, there is no measurable contribution of diffusion to the loss of fluorescence and therefore the decay kinetics are due entirely to neurofilament transport.
Based on computational modeling, we have posited that the overall time course of fluorescence decay from the activation window holds complete information about the neurofilament transport kinetics and that tracking the fluorescence decay on a time scale of several hours should therefore reveal the pausing behavior of neurofilaments residing within the activation window on that time scale. In Li et al. (2014) we describe an analytical solution for the decay kinetics in terms of the kinetic parameters of neurofilament transport, and computational methods that can allow extraction of kinetic data from such experiments. See that article for a more detailed discussion.
8 Summary
Neurofilaments are unique among the cargoes of axonal transport because they are flexible protein polymers, just 10 nm in diameter but extending up to 100 μm or more in length. The filaments move in a rapid and asynchronous manner, but the overall rate is slow because the movements are also very infrequent. In other words, these structures spend only a very small proportion of their time moving during their journey down the axon, sometimes pausing for hours or more between their bouts of rapid intermittent movement. These physical and kinetic properties present unique challenges for live-cell imaging. In this chapter we present a suite of complementary approaches that address these challenges using conventional and photoactivatable fluorescent fusion proteins to tag the neurofilaments and epifluorescence microscopy to observe the movement. Collectively, these approaches permit a comprehensive analysis of neurofilament transport on fast and slow time scales in primary cell cultures of a range of different neuronal cell types.
Acknowledgments
Work in the Brown lab is supported by grants from the NINDS and NSF to A.B. We thank the many Brown lab members past and present who have helped develop and refine our imaging strategies over the years, particularly Lei Wang, Yanping Yan, Niraj Trivedi, Nael Alami, Gulsen Colakoglu and Lina Wang.
Abbreviations
- SCG
superior cervical ganglion
- DRG
dorsal root ganglion
- GFP
green fluorescent protein
- PAGFP
photoactivatable GFP
- DIC
differential interference contrast
- NFL
neurofilament protein L
- NFM
neurofilament protein M
- NFH
neurofilament protein H
- FBS
fetal bovine serum
- PBS
phosphate buffered saline
- HBSS
Hank’s Balanced Salt Solution
- NGF
nerve growth factor
- NT3
neurotrophin-3
- DMD
digital multimirror device
References
- Alami NH, Jung P, Brown A. Myosin Va increases the efficiency of neurofilament transport by decreasing the duration of long-term pauses. J Neurosci. 2009;29:6625–6634. doi: 10.1523/JNEUROSCI.3829-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bancaud A, Huet S, Rabut G, Ellenberg J. Fluorescence perturbation techniques to study mobility and molecular dynamics of proteins in live cells: FRAP, photoactivation, photoconversion, and FLIP. Cold Spring Harb Protoc. 2010;2010 doi: 10.1101/pdb.top90. pdb top90. [DOI] [PubMed] [Google Scholar]
- Bevis BJ, Glick BS. Rapidly maturing variants of the Discosoma red fluorescent protein (DsRed) Nat Biotechnol. 2002;20:83–87. doi: 10.1038/nbt0102-83. [DOI] [PubMed] [Google Scholar]
- Black MM, Keyser P, Sobel E. Interval between the synthesis and assembly of cytoskeletal proteins in cultured neurons. J Neurosci. 1986;6:1004–1012. doi: 10.1523/JNEUROSCI.06-04-01004.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bottenstein JE, Sato GH. Growth of a rat neuroblastoma cell line in serum-free supplemented media. Proc Natl Acad Sci U S A. 1979;76:514–517. doi: 10.1073/pnas.76.1.514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bray D. Isolated chick neurons for the study of axonal growth. In: Banker G, Goslin K, editors. Culturing Nerve Cells. MIT Press; Cambridge, MA: 1991. pp. 119–135. [Google Scholar]
- Brewer GJ, Boehler MD, Pearson RA, DeMaris AA, Ide AN, Wheeler BC. Neuron network activity scales exponentially with synapse density. Journal of neural engineering. 2009;6:014001. doi: 10.1088/1741-2560/6/1/014001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown A. Visualization of single neurofilaments by immunofluorescence microscopy of splayed axonal cytoskeletons. Cell Motil Cytoskel. 1997;38:133–145. doi: 10.1002/(SICI)1097-0169(1997)38:2<133::AID-CM3>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- Brown A. Contiguous phosphorylated and non-phosphorylated domains along axonal neurofilaments. J Cell Sci. 1998;111:455–467. doi: 10.1242/jcs.111.4.455. [DOI] [PubMed] [Google Scholar]
- Brown A. Slow axonal transport: stop and go traffic in the axon. Nat Rev Mol Cell Biol. 2000;1:153–156. doi: 10.1038/35040102. [DOI] [PubMed] [Google Scholar]
- Brown A. Slow axonal transport. In: Squire LR, editor. Encyclopedia of Neuroscience. Vol. 9. Academic Press; Oxford: 2009. pp. 1–9. [Google Scholar]
- Carden MJ, Trojanowski JQ, Schlaepfer WW, Lee VM. Two-stage expression of neurofilament polypeptides during rat neurogenesis with early establishment of adult phosphorylation patterns. J Neurosci. 1987;7:3489–3504. doi: 10.1523/JNEUROSCI.07-11-03489.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ching GY, Liem RK. Assembly of type IV neuronal intermediate filaments in nonneuronal cells in the absence of preexisting cytoplasmic intermediate filaments. J Cell Biol. 1993;122:1323–1335. doi: 10.1083/jcb.122.6.1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chudakov DM, Lukyanov S, Lukyanov KA. Tracking intracellular protein movements using photoswitchable fluorescent proteins PS-CFP2 and Dendra2. Nat Protoc. 2007;2:2024–2032. doi: 10.1038/nprot.2007.291. [DOI] [PubMed] [Google Scholar]
- Chudakov DM, Matz MV, Lukyanov S, Lukyanov KA. Fluorescent proteins and their applications in imaging living cells and tissues. Physiol Rev. 2010;90:1103–1163. doi: 10.1152/physrev.00038.2009. [DOI] [PubMed] [Google Scholar]
- Colakoglu G, Brown A. Intermediate filaments exchange subunits along their length and elongate by end-to-end annealing. J Cell Biol. 2009;185:769–777. doi: 10.1083/jcb.200809166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craig AM. Transfecting cultured neurons. In: Banker G, Goslin K, editors. Culturing nerve cells. MIT Press; Cambridge: 1998. pp. 79–111. [Google Scholar]
- Day RN, Davidson MW. The fluorescent protein palette: tools for cellular imaging. Chem Soc Rev. 2009;38:2887–2921. doi: 10.1039/b901966a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eldridge CF, Bunge MB, Bunge RP. Differentiation of axon-related Schwann cells in vitro: II. Control of myelin formation by basal lamina. J Neurosci. 1989;9:625–638. doi: 10.1523/JNEUROSCI.09-02-00625.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eldridge CF, Bunge MB, Bunge RP, Wood PM. Differentiation of axon-related Schwann cells in vitro. I. Ascorbic acid regulates basal lamina assembly and myelin formation. J Cell Biol. 1987;105:1023–1034. doi: 10.1083/jcb.105.2.1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fex Svenningsen A, Shan WS, Colma DRn, Pedraza L. Rapid method for culturing embryonic neuron-glial cell cocultures. J Neurosci Res. 2003;72:565–573. doi: 10.1002/jnr.10610. [DOI] [PubMed] [Google Scholar]
- Freshney RI. Culture of Animal Cells: A Manual of Basic Technique and Specialized Applications. Wiley-Blackwell; 2010. p. 796. [Google Scholar]
- Garcia I, Martinou I, Tsujimoto Y, Martinou JC. Prevention of programmed cell death of sympathetic neurons by the bcl-2 proto-oncogene. Science. 1992;258:302–304. doi: 10.1126/science.1411528. [DOI] [PubMed] [Google Scholar]
- Goslin K, Hannelore A, Banker G. Rat hippocampal neurons in low-density culture. In: Banker G, Goslin K, editors. Culturing nerve cells. MIT Press; Cambridge: 1998. pp. 339–370. [Google Scholar]
- Hawrot E, Patterson PH. Long-term culture of dissociated sympathetic neurons. Methods Enzymol. 1979;58:574–584. doi: 10.1016/s0076-6879(79)58174-9. [DOI] [PubMed] [Google Scholar]
- He Y, Baas PW. Growing and working with peripheral neurons. Methods Cell Biol. 2003;71:17–35. doi: 10.1016/s0091-679x(03)01002-1. [DOI] [PubMed] [Google Scholar]
- Higgins D, Lein PJ, Osterhout DJ, Johnson MI. Tissue culture of mammalian autonomic neurons. In: Banker G, Goslin K, editors. Culturing nerve cells. The MIT Press; Cambridge, Mass: 1991. pp. 177–205. [Google Scholar]
- Hoffman PN. The synthesis, axonal transport, and phosphorylation of neurofilaments determine axonal caliber in myelinated nerve fibers. The Neuroscientist. 1995;1:76–83. [Google Scholar]
- Johnson MI, Argiro V. Techniques in the tissue culture of rat sympathetic neurons. Methods Enzymol. 1983;103:334–347. doi: 10.1016/s0076-6879(83)03022-0. [DOI] [PubMed] [Google Scholar]
- Johnson MI, Iacovitti L, Higgins D, Bunge RP, Burton H. Growth and development of sympathetic neurons in tissue culture. Ciba Found Symp. 1981;83:108–122. doi: 10.1002/9780470720653.ch6. [DOI] [PubMed] [Google Scholar]
- Kaech S, Banker G. Culturing hippocampal neurons. Nat Protoc. 2006;1:2406–2415. doi: 10.1038/nprot.2006.356. [DOI] [PubMed] [Google Scholar]
- Kleitman N, Wood PM, Bunge RP. Tissue culture methods for the study of myelination. In: Banker G, Goslin K, editors. Culturing Nerve Cells. MIT Press; Cambridge, Mass: 1998. pp. 545–594. [Google Scholar]
- Koehnle TJ, Brown A. Slow axonal transport of neurofilament protein in cultured neurons. J Cell Biol. 1999;144:447–458. doi: 10.1083/jcb.144.3.447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koster S, Weitz DA, Goldman RD, Aebi U, Herrmann H. Intermediate filament mechanics in vitro and in the cell: from coiled coils to filaments, fibers and networks. Curr Opin Cell Biol. 2015;32C:82–91. doi: 10.1016/j.ceb.2015.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lariviere RC, Julien JP. Functions of intermediate filaments in neuronal development and disease. J Neurobiol. 2004;58:131–148. doi: 10.1002/neu.10270. [DOI] [PubMed] [Google Scholar]
- Laser-Azogui A, Kornreich M, Malka-Gibor E, Beck R. Neurofilament assembly and function during neuronal development. Curr Opin Cell Biol. 2015;32C:92–101. doi: 10.1016/j.ceb.2015.01.003. [DOI] [PubMed] [Google Scholar]
- Lee MK, Xu Z, Wong PC, Cleveland DW. Neurofilaments are obligate heteropolymers in vivo. J Cell Biol. 1993;122:1337–1350. doi: 10.1083/jcb.122.6.1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Brown A, Jung P. Deciphering the axonal transport kinetics of neurofilaments using the fluorescence photoactivation pulse-escape method. Phys Biol. 2014;11:026001. doi: 10.1088/1478-3975/11/2/026001. [DOI] [PubMed] [Google Scholar]
- Mahanthappa NK, Patterson PH. Culturing mammalian sympathoadrenal derivatives. In: Banker G, Goslin K, editors. Culturing Nerve Cells. MIT Press; Cambridge, MA: 1998. pp. 289–307. [Google Scholar]
- McKenna NM, Wang YL. Culturing cells on the microscope stage. Methods Cell Biol. 1989;29:195–205. doi: 10.1016/s0091-679x(08)60195-8. [DOI] [PubMed] [Google Scholar]
- McKinney SA, Murphy CS, Hazelwood KL, Davidson MW, Looger LL. A bright and photostable photoconvertible fluorescent protein. Nat Methods. 2009;6:131–133. doi: 10.1038/nmeth.1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikhailov AV, Gundersen GG. Centripetal transport of microtubules in motile cells. Cell Motil Cytoskel. 1995;32:173–186. doi: 10.1002/cm.970320303. [DOI] [PubMed] [Google Scholar]
- Monsma PC, Brown A. FluoroMyelin Red is a bright, photostable and non-toxic fluorescent stain for live imaging of myelin. J Neurosci Methods. 2012;209:344–350. doi: 10.1016/j.jneumeth.2012.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monsma PC, Li Y, Fenn JD, Jung P, Brown A. Local regulation of neurofilament transport by myelinating cells. J Neurosci. 2014;34:2979–88. doi: 10.1523/JNEUROSCI.4502-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukhopadhyay R, Kumar S, Hoh JH. Molecular mechanisms for organizing the neuronal cytoskeleton. Bioessays. 2004;26:1017–1025. doi: 10.1002/bies.20088. [DOI] [PubMed] [Google Scholar]
- Nagai T, Ibata K, Park ES, Kubota M, Mikoshiba K, Miyawaki A. A variant of yellow fluorescent protein with fast and efficient maturation for cell-biological applications. Nat Biotechnol. 2002;20:87–90. doi: 10.1038/nbt0102-87. [DOI] [PubMed] [Google Scholar]
- Niwa H, Yamamura K, Miyazaki J. Efficient selection for high-expression transfectants with a novel eukaryotic vector. Gene. 1991;108:193–199. doi: 10.1016/0378-1119(91)90434-d. [DOI] [PubMed] [Google Scholar]
- Nixon RA, Shea TB. Dynamics of neuronal intermediate filaments: a developmental perspective. Cell Motil Cytoskel. 1992;22:81–91. doi: 10.1002/cm.970220202. [DOI] [PubMed] [Google Scholar]
- Patterson GH, Lippincott-Schwartz J. A photoactivatable GFP for selective photolabeling of proteins and cells. Science. 2002;297:1873–1877. doi: 10.1126/science.1074952. [DOI] [PubMed] [Google Scholar]
- Perrot R, Eyer J. Neurofilaments: properties, functions, and regulation. In: Dermietzel R, editor. The Cytoskeleton: Imaging, Isolation, and Interaction. Springer Science+Business Media; 2013. [Google Scholar]
- Rizzo MA, Springer GH, Granada B, Piston DW. An improved cyan fluorescent protein variant useful for FRET. Nat Biotechnol. 2004;22:445–449. doi: 10.1038/nbt945. [DOI] [PubMed] [Google Scholar]
- Sarria AJ, Nordeen SK, Evans RM. Regulated expression of vimentin cDNA in cells in the presence and absence of a preexisting vimentin filament network. J Cell Biol. 1990;111:553–565. doi: 10.1083/jcb.111.2.553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaner NC, Campbell RE, Steinbach PA, Giepmans BN, Palmer AE, Tsien RY. Improved monomeric red, orange and yellow fluorescent proteins derived from Discosoma sp. red fluorescent protein. Nat Biotechnol. 2004;22:1567–1572. doi: 10.1038/nbt1037. [DOI] [PubMed] [Google Scholar]
- Shaner NC, Lin MZ, McKeown MR, Steinbach PA, Hazelwood KL, Davidson MW, Tsien RY. Improving the photostability of bright monomeric orange and red fluorescent proteins. Nat Methods. 2008;5:545–551. doi: 10.1038/nmeth.1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor NJ, Wang L, Brown A. Neurofilaments are flexible polymers that often fold and unfold but they move in a fully extended configuration. Cytoskeleton (Hoboken) 2012;69:535–544. doi: 10.1002/cm.21039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trivedi N, Jung P, Brown A. Neurofilaments switch between distinct mobile and stationary states during their transport along axons. J Neurosci. 2007;27:507–516. doi: 10.1523/JNEUROSCI.4227-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsien RY. The green fluorescent protein. Annu Rev Biochem. 1998;67:509–544. doi: 10.1146/annurev.biochem.67.1.509. [DOI] [PubMed] [Google Scholar]
- Uchida A, Alami NH, Brown A. Tight functional coupling of kinesin-1A and dynein motors in the bidirectional transport of neurofilaments. Mol Biol Cell. 2009;20:4997–5006. doi: 10.1091/mbc.E09-04-0304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uchida A, Colakoglu G, Wang L, Monsma PC, Brown A. Severing and end-to-end annealing of neurofilaments in neurons. Proc Natl Acad Sci U S A. 2013;110:E2696–E2705. doi: 10.1073/pnas.1221835110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Brown A. Rapid intermittent movement of axonal neurofilaments observed by fluorescence photobleaching. Mol Biol Cell. 2001;12:3257–3267. doi: 10.1091/mbc.12.10.3257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Brown A. A hereditary spastic paraplegia mutation in kinesin-1A/KIF5A disrupts neurofilament transport. Molecular Neurodegeneration. 2010;5:52. doi: 10.1186/1750-1326-5-52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Ho CL, Sun D, Liem RKH, Brown A. Rapid movement of axonal neurofilaments interrupted by prolonged pauses. Nat Cell Biol. 2000;2:137–141. doi: 10.1038/35004008. [DOI] [PubMed] [Google Scholar]
- Yamamichi-Nishina M, Ito T, Mizutani T, Yamamichi N, Watanabe H, Iba H. SW13 cells can transition between two distinct subtypes by switching expression of BRG1 and Brm genes at the post-transcriptional level. J Biol Chem. 2003;278:7422–7430. doi: 10.1074/jbc.M208458200. [DOI] [PubMed] [Google Scholar]
- Yan Y, Brown A. Neurofilament polymer transport in axons. J Neurosci. 2005;25:7014–7021. doi: 10.1523/JNEUROSCI.2001-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan Y, Jensen K, Brown A. The polypeptide composition of moving and stationary neurofilaments in cultured sympathetic neurons. Cell Motil Cytoskel. 2007;64:299–309. doi: 10.1002/cm.20184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young IT. Image fidelity: characterizing the imaging transfer function. Methods Cell Biol. 1989;30:1–45. doi: 10.1016/s0091-679x(08)60974-7. [DOI] [PubMed] [Google Scholar]
- Yuan L, Zheng YF, Zhu J, Wang L, Brown A. Object tracking with particle filtering in fluorescence microscopy images: application to the motion of neurofilaments in axons. IEEE Trans Med Imaging. 2012;31:117–130. doi: 10.1109/TMI.2011.2165554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang M, Chang H, Zhang Y, Yu J, Wu L, Ji W, Chen J, Liu B, Lu J, Liu Y, Zhang J, Xu P, Xu T. Rational design of true monomeric and bright photoactivatable fluorescent proteins. Nat Methods. 2012;9:727–729. doi: 10.1038/nmeth.2021. [DOI] [PubMed] [Google Scholar]