

Abstract
Vascular basal cyclooxygenase-2 (COX-2) expression and activity can be induced by endotoxin, hypoxia, or ischemia. During vascular pathologies such as atherosclerosis, increases in COX-2 activity result in prostanoid production, a contributor to the development and progression of vascular inflammation leading to unstable atherosclerotic plaques and increased risk for thrombotic events. Recent studies demonstrate that select free fatty acids, such as palmitate, can act as proinflammatory mediators. However, the effect of palmitate on COX-2 expression and activity, and its impact on the development and progression of vascular inflammation, are not well elucidated. We investigated the effect of palmitate on COX-2 expression and function in human vascular smooth muscle cells. Cells were treated with palmitate, COX-2 protein levels were assessed using Western analysis, and activity was assessed via ELISA. We observed that palmitate dose-dependently increased COX-2 levels and specifically enhanced band intensity of the COX-2 74 kDa band (slowest migrating band). This response was attenuated by N-linked glycosylation inhibition, suggesting that palmitate impacts expression of the fully activated glycoform of COX-2. Palmitate-induced increases in COX-2 levels correlated with an increase in prostaglandin E2 production that was also attenuated by a glycosylation inhibitor. Additionally, palmitate altered cell morphology and increased cell density which were reversed by selective COX-2 inhibition. Thus, we conclude that palmitate acts on COX-2 by two separate mechanisms of action in human vascular smooth muscle. It elicits dose-dependent increases in COX-2 protein expression and modulates regulation of COX-2 activity via modification of posttranslational glycosylation.
Keywords: cyclooxygenase-2; glycosylation; inflammation; palmitate, vascular smooth muscle
INTRODUCTION
The inducible isozyme, cyclooxygenase-2 (COX-2), is a well-studied vascular inflammatory mediator (7, 26). Known officially as prostaglandin-endoperoxide synthase, COX-2 is nominally expressed in vascular tissue under normal physiological conditions but serves as an inflammatory regulator upon induction during pathophysiological conditions (17). COX-2 catalyzes the rate limiting step in the biosynthesis pathway converting arachidonic acid to prostaglandin H2 (PGH2) resulting in downstream production of prostanoids including prostaglandins (e.g., PGE2 and prostacyclin) and thromboxane (29). Prostanoids are responsible for many normal vascular physiological effects, including vasoconstriction, vasodilation, platelet aggregation, and apoptosis (2). However, inflammation-induced COX-2 activity in the blood vessel wall can contribute to unstable atherosclerotic plaques (5–7, 31, 35), leading to higher chances of thromboembolic events like heart attack or ischemic stroke.
The COX-2 enzyme contains five potential N-glycosylation sites, three of which are reported to be continually glycosylated (23). The carbohydrates at each site are believed to be high mannose-type sugar chains, and the variability of glycosylation leads to the production of distinct glycoforms (19). The fifth site is reported to be infrequently glycosylated, and variability at the fourth site produces two distinct glycoforms of 72 and 74 kDa (33). The consequence of glycosylation of the fourth site is unclear, although it has been reported that removal of the site results in an increase in total COX-2 activity and accumulation of the 70- and 72 kDa glycoforms (33) while others have demonstrated that inhibition of glycosylation decreases production of downstream prostaglandins (11).
COX-2 expression is regulated by nuclear factor-κB (NF-κB) (15), an inducible transcription factor present in vascular smooth muscle cells (18) that controls expression of several other genes related to cell growth, apoptosis, and cytokine production (14, 37). COX-2 expression can be induced by cytokines such as IL-1β or by the endotoxin lipopolysaccharide (LPS), promoting activation of the “classical inflammatory pathway” in human vascular smooth muscle (22). Others have demonstrated that mechanisms associated with “sterile inflammatory pathway” activation involve dietary free fatty acids (FFAs) acting as ligands to promote activation of the Toll-like receptor 4 (TLR4)/NF-κB signaling cascade (12, 32). FFAs have been shown to induce proinflammatory mediator expression including tumor necrosis factor-α, inducible nitric oxide synthase, and interleukin-6 in rat vascular smooth muscle cells (36). However, the effect of FFAs on COX-2 expression and/or activity as well as the regulation of COX-2 via transcription or posttranslational modification has not been addressed in the human vasculature.
Investigating the impact of palmitic acid on vascular health is significant since it is the most abundant saturated fatty acid in FFAs (13) and the most widespread natural saturated fat found in plant and animal food sources (16). Its anion, palmitate, is the observed form at physiological pH (16). Since previous studies have shown that palmitate can induce expression of other inflammatory markers in vascular smooth muscle (36) and other cell types like adipose microvascular endothelial cells (24) and mouse myoblast cell lines (3), here we investigated the impact of palmitate on COX-2 expression and potential regulatory mechanisms in cultured primary human vascular smooth muscle cells.
MATERIALS AND METHODS
Cell culture models.
Primary human aortic vascular smooth muscle cells (HAoVSM) (lot no. 0000316660, Lonza) of adult female origin were grown in 5% CO2 and room air at 37°C in phenol red-free basal medium (Lonza) supplemented with a smooth muscle cell growth supplement (insulin, human epidermal growth factor, human fibroblastic growth factor), gentamycin-amphotericin, and 5% FBS. Primary adult male human brain vascular smooth muscle cells (HBVSM) (lot no. ACBRI 405, Cell Systems) and female human coronary vascular smooth muscle cells (HCVSM) (lot no. 0000289727, Lonza) were also grown under the same conditions as the HAoVSM and were included in these studies 1) to assess palmitate’s effect on COX-2 expression in cells from another vascular bed origin and 2) to investigate primary vascular cells from additional donors. Representative light microscopic images of each type of cultured vascular smooth muscle cells studied are shown in Fig. 1A. Vascular phenotype was verified by assessing protein expression of the contractile markers anti-α-smooth muscle actin (Sigma-Aldrich, St. Louis, MO) and smoothelin (Thermo Fisher Scientific) (Fig. 1, B and C). All vendor-purchased cryopreserved subcultures were received at either passage 2 or -3 and seeded in house at a density of 4 to 5 × 105/cm2. Under these conditions, cells reached 70% to 80% confluence within 5 to 7 days. At that time, designated plates were cryopreserved (froze back) for future use or continued in culture and studied at passage 6–7. Characterization for sex was carried out via performing RNA isolation (28) and PCR for SRY-1 gene expression verifying male origin or absence of expression verifying female origin (data not shown). Although vascular cells were studied using two different female donors and one male donor, direct comparisons for sex differences were not assessed in current studies.
Fig. 1.

Primary cultured human vascular cells express contractile proteins. A: representative light micrographs of primary human vascular smooth muscle cell cultures of coronary (HCVSM), brain (HBVSM), and aortic (HAoVSM) origin. B and C: vascular smooth muscle cell phenotype was verified using anti-α-smooth muscle actin (B) or anti-smoothelin (C).
Palmitate preparation and treatment protocol.
Palmitate and drug solutions were freshly prepared on the same day of treatments/experiments under sterile conditions. To enhance the solubility of palmitate, the FFA was complexed with bovine serum albumin (BSA) (20). Briefly, a stock solution of palmitate (100 μM) was solubilized in 50% ethanol and heated to 90°C. This solution was conjugated with 5% BSA at 90°C to create working dilutions needed to achieve final treatment concentrations. The corresponding BSA and ethanol solution was used as a vehicle in the control groups. The BSA/palmitate complex or BSA/vehicle solutions were maintained at 37°C during preparation and during the treatment protocol. In some experiments, to assess the influence of palmitate on N-linked glycosylation of COX-2, vascular smooth muscle cell cultures were treated with palmitate (100 µM), palmitate plus tunicamycin (N-linked glycosylation inhibitor; 1 µg/ml), tunicamycin, or vehicle (5% BSA for palmitate; nanopure water for tunicamycin) for 18 h. Tunicamycin or its vehicle (nanopure water) was administered 30 min before palmitate or BSA treatment. Glucosamine hydrochloride (GS-HCl) was also used to investigate N-linked glycosylation of COX-2. GS-HCl was solubilized in nanopure water to create a stock solution (100 mM) and subsequent working dilutions at room temperature. In experiments using GS-HCl, cell cultures were treated with palmitate (100 µM), palmitate plus GS-HCl (1 mM, 2.5 mM, 5 mM), GS-HCl, or vehicle for 18 h. Similar to tunicamycin, GS-HCl or its vehicle (nanopure water) was administered 30 min before palmitate or BSA treatment.
Immunoblotting.
Levels of COX-2 protein were examined using standard immunoblotting methods, as previously described (22). Briefly, cells were rinsed twice with filtered phosphate buffered solution (PBS), collected using trypsin and trypsin-neutralizer, and centrifuged (Thermo Sorvall Legend RT+) at 1,900 g for 10 min at 4°C. The cell pellet was rinsed with ice-cold PBS, centrifuged again, and resuspended in ice-cold lysis buffer solution (50 mM Tris·HCl, 150 mM NaCl, 0.1% sodium dodecyl sulfate, 1% Nonidet P-40) containing protease inhibitors (20 µM pepstatin, 20 µM leupeptin, 0.1 U/ml aprotinin, 0.1 mM PMSF, 1 mM DTT), phosphatase inhibitors (25 mM β-glycerophosphate, 0.1 mM sodium orthovanadate), 1 mM EDTA, and 1 mM EGTA for 15 min. The solution was vortexed and centrifuged (Fisher Accuspin Micro 17R) at 17,000 g for 10 min at 4°C before collection of the supernatant containing whole cell lysate.
Total protein content was assessed with a bicinchoninic acid protein assay kit (Thermo Fisher Scientific) and measured on a Safire II (Tecan) plate reader. Samples were then diluted in Tris-glycine SDS sample buffer with the reducing agent β-mercaptoethanol (10%) and boiled for 5 min. The diluted samples and fluorescent standards (LI-COR Biosciences) were loaded onto freshly made 8% or 10% polyacrylamide gels. Protein separation occurred by SDS-PAGE in homemade running buffer (25 mM Trizma base, 190 mM glycine, 0.1% SDS) at 125 V using a mini PROTEAN Tetra electrophoresis system (Bio-Rad). Proteins were transferred to nitrocellulose membranes using the same apparatus at 0.25 A for 1 h. Membranes were blocked in 3% nonfat dry milk in PBS containing 1% Tween (TPBS) at room temperature for 1 h to prevent nonspecific binding, then incubated overnight at 4°C in with COX-2 (catalog no. 160126, Cayman Chemical Company), β-actin (catalog no. A5441, Sigma Aldrich), smoothelin (catalog no. OMA1-06020, Thermo Fisher Scientific) or α-smooth muscle actin (catalog no. A5228, Sigma Aldrich) primary antibodies diluted 1:500 in TPBS. After washing with TPBS, membranes were incubated in goat anti-rabbit IRDye 800CW dye (LI-COR) secondary antibody at room temperature for 1 h. Membranes were washed again with TPBS and proteins were visualized using an Odyssey Classic infrared imager (LI-COR). After imaging COX-2 bands, membranes were exposed to a β-actin primary antibody (Sigma Chemical) and goat anti-mouse IRDye 680LT (LI-COR) and visualized for verification of equal loading of protein. Data were analyzed using ImageStudio 3.0 software (LI-COR).
Prostaglandin E2 assay.
Prostaglandin E2 (PGE2) is a metabolite of COX-2 and an indicator of functional activity of COX-2. PGE2 levels were indirectly determined by measuring the inactive metabolite, 13,14-dihydro-15-keto PGE2, in culture media collected at the end of drug treatments using a PGE2 Monoclonal enzyme immunoassay (EIA) Kit (Cayman Chemical). The half-life of PGE2 in the circulatory system is very short lived, after which it is converted to an inactive metabolite (4). The EIA kit converts all major PGE2 metabolites into one single stable derivative for measurement to indirectly assay PGE2 biosynthesis. Assay protocol and analysis were performed per manufacturer’s instructions and absorbance levels were measured using a Safire II plate reader and software (Tecan).
Microscopy.
Cells were observed visually using an Olympus DP71 camera mounted to an Olympus IX71 inverted light microscope. Electronic micrographs were captured using a ×40 objective and total cell counts were obtained by a blinded analyzer using Adobe Photoshop count tool to determine cell density from digital images.
Reagents.
All reagents were purchased from Sigma-Aldrich unless otherwise noted.
Data and statistical analysis.
The PGE2 assay analysis was conducted using a four-parameter logistic nonlinear regression analysis tool (Cayman Chemical Company). Lysates from each treatment group, including vehicle, were run on the same Western blot or 96-well plate (PGE2 assay) for direct comparison. Band densities during Western analysis were normalized to loading control band density (i.e., β-actin) and data are expressed as a fold change relative to vehicle. Experiments were repeated for statistical analysis, performed using Sigma Stat 3.5 (Systat Software). Comparisons were made using a paired t-test, and a Shapiro-Wilk test confirmed that data sets achieved normal distribution. P < 0.05 was considered significant. Values are expressed as means ± SE.
RESULTS
Palmitate increased COX-2 protein levels in human primary vascular smooth muscle cells.
To assess whether FFAs alter vascular proinflammatory mediator COX-2 protein expression, HAoVSM were treated with either vehicle (5% BSA) or palmitate (50 or 100 µM) for 18 h (Fig. 2). Faint COX-2 bands were detected in human vascular smooth muscle cell lysate following vehicle-treated conditions, suggesting basal expression. In comparison to vehicle treatment, palmitate treatment increased COX-2 protein levels and this response was dose-dependent (representative blot; Fig. 2A). Data analysis of total 74/72 kDa COX-2 band density normalized to β-actin (loading control) and plotted as ratio versus vehicle (5% BSA) are graphically illustrated in Fig. 2B. The effect of palmitate on COX-2 levels was also investigated in HBVSM (Fig. 3). Cells were treated with increasing doses of palmitate (10, 50, or 100 µM) or vehicle (5% ethanol in 5% BSA) for 12 h. A representative blot demonstrating a dose-dependent change in COX-2 band intensity at 74/72 kDa (total COX-2) is illustrated in Fig. 3A. Analysis of band density normalized to β-actin (loading control) and expressed as a ratio to vehicle (5% BSA) is plotted in Fig. 3B. Similar to that observed in vascular smooth muscle of aortic origin, palmitate dose dependently increased COX-2 levels compared with vehicle in brain vascular smooth muscle. Similar to the HAoVSM, basal levels of COX-2 were detected during unstimulated control conditions. The level of COX-2 band intensity ratio as a result of low-dose LPS (10 ng/ml) administration to indicate a low grade, subseptic vascular inflammatory state is illustrated in Figs. 2B and 3B. In previous studies we have shown that COX-2 is expressed as heavier basal amounts in rodent testes and vasculatures such as the cerebral circulation (8).
Fig. 2.

Palmitate dose dependently increased cyclooxygenase-2 total protein levels in human aortic vascular smooth muscle cells (HAoVSM). A: representative Western blot of cyclooxygenase-2 (COX-2) levels in HAoVSM treated with vehicle (5% BSA), 50 µM palmitate, or 100 µM palmitate for 18 h. B: summary of the Western blot analysis for COX-2 band intensity normalized to β-actin band intensity (loading control) and expressed as a ratio vs. vehicle. A subseptic dose of lipopolysaccharide (LPS; 10 ng/ml) was used to observe the caliber of low-grade endotoxin-induced COX-2 levels in HAoVSM. Values are expressed as means ± SE. *P < 0.05 vs. vehicle; n = 4/group.
Fig. 3.

Cyclooxygenase-2 total protein levels were dose dependently increased in human brain vascular smooth muscle cells. A: representative Western blot of cyclooxygenase (COX-2) expression in human brain vascular smooth muscle cells (HBVSM) treated with vehicle (5% BSA) or palmitate (10, 50, or 100 µM) for 12 h. B: analysis of COX-2 band intensity is summarized. COX-2 band intensity was normalized to β-actin (loading control) and is expressed as a ratio vs. vehicle. A subseptic dose of lipopolysaccharide (LPS; 10 ng/ml) was used to observe the caliber of low-grade endotoxin-induced COX-2 levels in human brain vascular smooth muscle. Values are expressed as means ± SE. *P < 0.05 vs. vehicle; n = 4/group.
Palmitate increased levels of the 74 kDa glycoform of COX-2.
We observed a COX-2 doublet in palmitate-treated HAoVSM and HBVSM, and band density for both the 74- and 72 kDa glycoforms is summarized in Table 1. Densitometry analysis revealed that the 74 kDa glycoform of COX-2 increased significantly following palmitate treatment in HAoVSM and HBVSM. There were no significant changes in the 72 kDa glycoform in HBVSM, but there was a significant increase in the 72 kDa glycoform, with the highest dose of palmitate treatment in HAoVSM. Since an increase in total COX-2 protein levels was observed in response to palmitate treatment (Figs. 2 and 3) and this increase in intensity was more robust with the 74 kDa band, this suggests that a saturated free fatty acid such as palmitate may regulate active transcription of COX-2 in addition to modulating changes in glycosylation.
Table 1.
Cyclooxygenase-2 protein levels in human vascular smooth muscle cells
| Treatment | 72-kDa Glycoform | 74-kDa Glycoform |
|---|---|---|
| HAoVSM COX-2 band intensity (normalized to β-actin vs. vehicle) | ||
| Vehicle | 1 | 1 |
| 50 µM Pal | 1.80 | 5.99* |
| 100 µM Pal | 3.09* | 17.01* |
| HBVSM COX-2 band intensity (normalized to β-actin vs. vehicle) | ||
| Vehicle | 1 | 1 |
| 10 µM Pal | 1.38 | 1.08 |
| 50 µM Pal | 2.22 | 2.77* |
| 100 µM Pal | 2.39 | 7.53* |
HAoVSM, human aortic vascular smooth muscle, HBVSM, human brain vascular smooth muscle; Pal, palmitate. n = 4;
P < 0.05 vs. vehicle.
Glycosylation inhibitors attenuated palmitate-induced shifts in COX-2 molecular mass.
To test whether enhanced levels of the 74 kDa glycoform of COX-2 are due to palmitate-induced N-linked glycosylation of the isoenzyme COX-2, HAoVSM were treated with palmitate (100 µM) or palmitate in the presence of the glycosylation inhibitor tunicamycin (1 µg/ml) or vehicle for 18 h. A representative blot is illustrated in Fig. 4A. Glycosylation inhibition with tunicamycin shifted COX-2 band migration from 72/74 kDa to ~66 kDa, the aglycosylated form. Similarly, tunicamycin alone shifted the 74 kDa band observed in the vehicle to 66 kDa. In terms of COX-2 protein levels, as predicted, palmitate increased basal total protein levels of the COX-2 compared with vehicle (Fig. 4B). Total COX-2 band intensity was significantly decreased in cells cotreated with palmitate plus tunicamycin compared with those treated with palmitate alone. A separate set of vascular smooth muscle cell treatments was performed using GS-HCl, a less potent inhibitor of N-linked glycosylation (11), to further verify palmitate-induced glycosylation of COX-2. HCVSM cells were treated with vehicle (Veh), palmitate (100 µM), or palmitate plus GS-HCl (1, 2.5, and 5 mM), or GS-HCl (1, 2.5, and 5 mM) for 18 h. Palmitate increased COX-2 levels in HCVSM and GS-HCl cotreatment resulted in a dose-dependent shift in COX-2 band migration from 72 to 74 kDa to 66–70 kDa in the presence of palmitate (Fig. 5A). GS-HCl cotreatment also resulted in a decrease in total COX-2 levels compared with cells treated only with palmitate (Fig. 5B). Together, these data suggest that palmitate alters both COX-2 protein levels and posttranslational glycosylation of COX-2 in human vascular smooth muscle.
Fig. 4.

Cyclooxygenase-2 band migration was altered in the presence of the glycosylation inhibitor tunicamycin. A: representative Western blot of cyclooxygenase (COX-2) isoform levels and their band migration in human aortic vascular smooth muscle cells (HAoVSM) treated with vehicle (Veh; water-5% BSA), palmitate (Pal; 100 µM), palmitate (100 µM) plus tunicamycin (TN, 1 µg/ml), or tunicamycin for 18 h. B: analysis of COX-2 band intensity normalized to β-actin is summarized. Values are expressed as means ± SE. *P < 0.05 vs. Veh; #P < 0.05 vs. palmitate; n = 3/group.
Fig. 5.

Cyclooxygenase-2 band migration was altered in the presence of the glycosylation inhibitor, glucosamine-HCl. A: representative Western blot of cyclooxygenase (COX-2) levels and band migration in human coronary vascular smooth muscle cells (HCVSM). Cells were treated with vehicle (Veh; water-5% BSA), palmitate (100 µM), palmitate plus GS-HCl (1, 2.5, and 5 mM), or GS-HCl (1, 2.5, 5 mM) for 18 h. B: analysis of band intensity is summarized. COX-2 band intensity was normalized to β-actin (loading control). Values are expressed as means ± SE. *P < 0.05 vs. Veh; n = 3/group.
Prostaglandin E2 production increased following palmitate treatment.
In addition to increases in protein expression and modulation of glycosylation, we next investigated whether palmitate alters COX-2 activity. Prostaglandin E2 (PGE2) production was assessed in media collected at the end of timed treatment protocols. PGE2 was used as an indirect indicator of the proinflammatory enzyme activity since PGE2 is one of the functional downstream products of COX-2 and is implicated as a key mediator in the pathophysiology of vascular diseases (7). Following treatment with vehicle or palmitate for 18 h, culture medium from HAoVSM was assayed using a PGE2 Monoclonal EIA Kit. Data in Fig. 6 graphically illustrate that PGE2 production was significantly increased following palmitate treatment compared with vehicle. These findings correlate with the observed increases in protein expression of COX-2 following palmitate exposure. Cotreatment with the glycosylation inhibitor tunicamycin attenuated palmitate-induced PGE2 production, and treatment with tunicamycin alone did not impact PGE2 production in vascular smooth muscle cells in vitro. Thus, these results indicate that attenuating palmitate-induced glycosylation of COX-2, and in turn downstream production of prostaglandins, may be a potential mechanism for the involvement of COX-2 in the progression or development of vascular disease pathology.
Fig. 6.
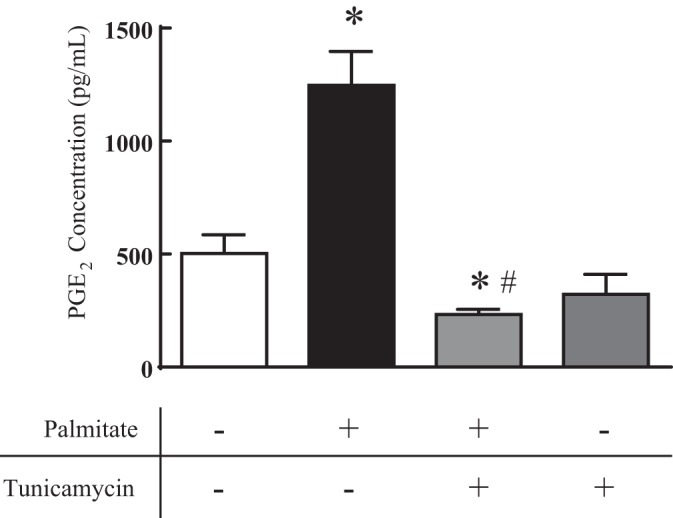
Tunicamycin attenuated palmitate-induced prostaglandin E2 (PGE2) production in human vascular smooth muscle. PGE2 production was measured in response to vehicle (Veh: water-BSA-ethanol), palmitate (100 µM), tunicamycin (1 µg/ml), or palmitate plus tunicamycin (18 h) in human aortic vascular smooth muscle cell (HAoVSM) culture media. n = 6–9; *P < 0.05 vs. vehicle; #P < 0.05 vs. palmitate.
Selective COX-2 inhibition attenuates palmitate-induced changes in vascular smooth muscle cell morphology and density.
Cell culture dishes were routinely screened visually at each passage using light microscopy to assess health, morphology, and growth. Representative micrographs demonstrate that HAoVSM grew well and reached ideal confluence between 5 to 7 days (Fig. 7A, pretreatment: a–d). In addition to pretreatment observations, culture plates were also visualized for cell morphology and culture density following posttreatment conditions (Fig. 7A, e–h). Following an 18 h exposure, vehicle treatment consisting of 5% BSA had no visual effect on normal cell growth and morphology (Fig. 7A, a and e). However, in contrast to vehicle, palmitate exposure (Fig. 7Af) resulted in atypical smooth muscle cell morphology compared with pretreatment conditions (Fig. 7Ab) or to those cells treated with vehicle (Fig. 7Ae). Based on total cell density calculations, palmitate also increased total cell culture density compared with vehicle-treated culture plates (Fig. 7B). To determine if these responses were COX-2 dependent, cells were treated with the selective COX-2 inhibitor NS-398 (25 µM). The addition of selective COX-2 inhibition with palmitate resulted in attenuated FFA-induced morphological changes and increases in FFA-induced cell density. NS-398 alone did not alter cell morphology or growth. These data suggest that saturated FFA exposure may play an important role in altering the morphology and density of human vascular smooth muscle cells and this response may involve enhanced COX-2 activity.
Fig. 7.

Selective cyclooxygenase-2 inhibition attenuates palmitate-induced changes in vascular smooth muscle cell morphology and density. A: representative light micrographs of human aortic vascular smooth muscle (HAoVSM) morphology pre- and posttreatment with palmitate (100 µM), vehicle (5% BSA), and/or the selective cyclooxygenase-2 (COX-2) inhibitor NS-398 (25 µM) for 18 h. Scale bar, 500 μm. B: analysis of cell density in response to palmitate, vehicle, or NS-398. Data are expressed as average cell counts per mm2. *P < 0.05 vs. vehicle; n = 6–9/group.
DISCUSSION
This study provides evidence that the saturated fatty acid anion, palmitate, alters the expression and function of the proinflammatory isozyme, COX-2, in human vascular smooth muscle. In summary, our experimental findings demonstrate that palmitate treatment 1) increased total basal COX-2 protein levels, 2) altered cell morphology accompanied by an increase in cell density and these responses were reversed by selective COX-2 inhibition, 3) enhanced the glycosylation state of COX-2 from the 72 kDa to the 74 kDa glycoform which was attenuated by an N-linked glycosylation inhibition, and 4) increased the production of the COX-2 metabolite PGE2 (indicator of COX-2 activity), and its synthesis was attenuated by glycosylation inhibition.
Multiple studies have elucidated part of the mechanism by which palmitate and other saturated fatty acids affect classic nonsterile activation in vascular smooth muscle. Some have demonstrated that palmitate can act as a proinflammatory ligand in human umbilical vein endothelial cells by activating the TLR4/NF-κB cascade leading to expression of proinflammatory cytokine interleukin-8 (IL-8) (10). Others have shown that palmitate induces IL-8 mRNA expression and NF-κB activity (25). To the best of our knowledge this is the first study to investigate the effect of palmitate on COX-2 in human vascular smooth muscle cells by acting as a proinflammatory modulator altering COX-2 levels and activity in the vasculature.
Previous research has also shown that COX-2 has multiple immunoblot band migrations correlating to different glycosylation states (33). COX-2 undergoes N-linked glycosylation in the endoplasmic reticulum (33, 34), with the completely aglycosylated form of COX-2 weighing ~66 kDa and the posttranslationally modified form, glycosylated at three sites, weighing ~72 kDa. Glycosylation of COX-2 at the fourth site results in the most active form (11) with a molecular mass of 74 kDa. In the vasculature, detection of COX-2 via Western blotting often appears as a distinct or smeared doublet with observed band migration at or between 74 and 72 kDa during both basal and stimulated conditions (9). In our studies, in addition to an increase in total COX-2 protein levels with high fat exposure, we also noted an unequal change in the band intensity of the COX-2 doublet on immunoblotting. In addition, we observed that the slower-migrating band with an estimated molecular mass of 74 kDa was increased with palmitate treatment, while the faster-migrating band with an estimated molecular mass of 72 kDa remained constant. Based on previous published studies, since it has been suggested that the 72 kDa glycoform thrice glycosylated is a functional enzyme and the 74 kDa glycoform is glycosylated at a fourth site is correlated with a possible increase in enzymatic activity (11), we predicted that palmitate treatment was causing additional glycosylation of COX-2 and perhaps altering its enzyme activity. Our hypothesis that palmitate was eliciting additional glycosylation was verified by demonstrating inhibition of N-linked glycosylation with tunicamycin. Specifically, our results demonstrated that the palmitate-induced increase in the 74 kDa glycoform of COX-2 was attenuated with pharmacological N-linked glycosylation inhibition. The 74 kDa glycoform was absent in tunicamycin-treated cells and while an aglycosylated form of COX-2 was present. Therefore, palmitate may contribute to regulation of COX-2 activity through the mechanism of glycosylation. Other methods of assessing glycosylation state involve use of an enzymatic protein deglycosylation kit to remove all N-linked and simple O-linked carbohydrates from glycoproteins in a solution before immunoblotting. As demonstrated in the current study, treatment with tunicamycin inhibits N-linked glycosylation, allowing for similar analysis of glycosylation state. Since tunicamycin can contribute to endoplasmic reticulum stress, we used low doses of tunicamycin to minimize the associated effects on endoplasmic reticulum stress. And we noted similar effects with GS-HCl, also demonstrated to be an N-linked glycosylation inhibitor (11).
In addition to alterations in COX-2 isoform band migration, we also observed that total COX-2 protein levels were decreased in cells cotreated with glycosylation inhibitors. This finding is consistent with a previous study by Jang et al. (11) demonstrating an increase in time-dependent COX-2 turnover in cells treated with N-linked glycosylation inhibitors. Future time-course treatment studies are needed to further investigate the effect of glycosylation inhibition on COX-2 protein turnover. However, based on the totality of the aforementioned findings, we conclude that palmitate can affect COX-2 by at least two mechanisms: increasing COX-2 protein levels and regulating posttranslational modification. To the best of our knowledge, this is the first report of palmitate-induced glycosylation of COX-2 in primary human vascular cells.
COX-2 is the rate-limiting enzyme in the conversion of arachidonic acid to PGH2, which is further metabolized to prostaglandins, prostacyclin, and thromboxane (29). COX-2 serves as a useful inflammatory marker because COX-2 and its downstream metabolite, PGE2, have been shown to be involved in vascular inflammation (1, 7, 8, 21, 22). Treatment with palmitic acid increased the 74 kDa glycoform and total COX-2 protein levels, so we hypothesized that PGE2 levels would be similarly altered. Palmitate-treated cells had significantly elevated PGE2 levels in HAoVSM compared with vehicle-treated cells, an effect attenuated by treatment with tunicamycin. This is consistent with our postulation that palmitate-induced glycosylation increases the proinflammatory activity of COX-2.
In addition to Western blotting and EIA, cells were imaged during culture and treatment to assess for any morphological changes. Images of cells treated with vehicle, palmitate, or a selective COX-2 inhibitor, NS-398, were taken throughout the allocated treatment time. Cells treated with vehicle showed the characteristic phenotypes of healthy vascular smooth muscle cells (27), while those treated with palmitate deviated morphologically and also appeared to be more proliferative, indicated by an apparent increase in total cell density. Treatment with palmitate and NS-398, a selective COX-2 inhibitor, attenuated the changes in cell density and morphology. This suggests that the inflammatory cascade triggered by palmitate treatment may affect the morphology and function of the smooth muscle cell, which can lead to excessive proliferation of vascular smooth muscle cells and contribute to the pathogenesis of atherosclerosis (30). Future studies are needed to determine how palmitate and NS-398 treatment may affect the morphology and cell proliferation of human vascular smooth muscle cells. An additional novel finding is that palmitate had a profound effect on glycosylation of COX-2 in human vascular smooth muscle cells. Although beyond the scope of this study, future studies are also needed to investigate the impact of glycosylation inhibition on vascular cell morphology and density in the presence of high fat exposure.
The proinflammatory effect of palmitate observed in cells isolated from two different vasculatures (brain and aorta) suggests that an inflammatory response to palmitic acid occurs peripherally and centrally. It also reduces the likeliness of the results occurring due to inter-donor variability, as each cell type originated from a separate donor. Primary vascular smooth muscle cell culture is often limited because smooth muscle cells change phenotypically over time in culture; thus experiments were performed at passage 6–7 to minimize any unintended phenotypic changes. Two markers of contractile smooth muscle cell phenotype were present at treatment passage and verified through Western blotting (Fig. 1, B and C): α-smooth muscle actin and smoothelin (27). α-Smooth muscle actin is involved in filament contraction and smoothelin in regulation of contraction.
In summary, we have demonstrated that palmitate increased total COX-2 protein levels in human vascular smooth muscle. An in vitro model of saturated fatty acid exposure suggests that high-fat diet may impact the glycosylation state of COX-2. Inhibition of glycosylation, as with tunicamycin, attenuated increases in palmitate-induced PGE2 production and suggests that a high-fat exposure also alters vascular COX-2 activity. Mechanistically, this indicates increased proinflammatory COX-2 enzymatic activity, likely due to glycosylation. Because PGE2 production was attenuated with tunicamycin treatment, it also demonstrates regulation of COX-2 enzymatic activity via posttranslational modification.
In conclusion, our results suggest that palmitate, and possibly via select high-fat exposure, can trigger inflammation cascades via COX-2 in the smooth muscle cells of the vessel wall, setting the stage for mechanisms associated with atherosclerosis development and progression. Based on our findings in the current study, we conclude that palmitate acts on COX-2 by two separate mechanisms of action in vascular smooth muscle. It increases protein levels of COX-2 and elicits regulation of COX-2 activity via posttranslational modification processes involving glycosylation. As there are many possible mechanisms of palmitate-induced vascular inflammation, future studies are needed to verify the specific mechanism(s) of action in the human vasculature including COX-2 and upstream mediators. An understanding of the regulation of COX-2 stimulated by saturated fatty acids like palmitic acid may guide us to better understand potential therapeutic targets for cardiovascular disease and associated risk factors such as obesity and diets high in saturated fats.
GRANTS
Grant support was derived from a Sarver Heart Center Mentored Medical Student award (P. Raman) and a Valley Research Partnership Grant VRP04 P1 (R. J. Gonzales).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
P.R. and R.J.G. conceived and designed research; P.R. and R.J.G. performed experiments; P.R., L.M., and R.J.G. analyzed data; P.R., L.M., and R.J.G. interpreted results of experiments; P.R. and R.J.G. prepared figures; P.R. and R.J.G. drafted manuscript; P.R., L.M., and R.J.G. edited and revised manuscript; P.R., L.M., and R.J.G. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Trinh Tat for technical assistance with cell culture and Dr. Aparna R. Sertil for thoughtful discussion regarding the tunicamycin experiments.
REFERENCES
- 1.Avendaño MS, Martínez-Revelles S, Aguado A, Simões MR, González-Amor M, Palacios R, Guillem-Llobat P, Vassallo DV, Vila L, García-Puig J, Beltrán LM, Alonso MJ, Cachofeiro MV, Salaices M, Briones AM. Role of COX-2-derived PGE2 on vascular stiffness and function in hypertension. Br J Pharmacol 173: 1541–1555, 2016. doi: 10.1111/bph.13457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Capra V, Bäck M, Angiolillo DJ, Cattaneo M, Sakariassen KS. Impact of vascular thromboxane prostanoid receptor activation on hemostasis, thrombosis, oxidative stress, and inflammation. J Thromb Haemost 12: 126–137, 2014. doi: 10.1111/jth.12472. [DOI] [PubMed] [Google Scholar]
- 3.Chen PY, Wang J, Lin YC, Li CC, Tsai CW, Liu TC, Chen HW, Huang CS, Lii CK, Liu KL. 18-carbon polyunsaturated fatty acids ameliorate palmitate-induced inflammation and insulin resistance in mouse C2C12 myotubes. J Nutr Biochem 26: 521–531, 2015. doi: 10.1016/j.jnutbio.2014.12.007. [DOI] [PubMed] [Google Scholar]
- 4.Fitzpatrick FA, Aguirre R, Pike JE, Lincoln FH. The stability of 13,14-dihydro-15 keto-PGE2. Prostaglandins 19: 917–931, 1980. doi: 10.1016/0090-6980(80)90126-4. [DOI] [PubMed] [Google Scholar]
- 5.George SJ. Tissue inhibitors of metalloproteinases and metalloproteinases in atherosclerosis. Curr Opin Lipidol 9: 413–423, 1998. doi: 10.1097/00041433-199810000-00005. [DOI] [PubMed] [Google Scholar]
- 6.Glass CK, Witztum JL. Atherosclerosis. The road ahead. Cell 104: 503–516, 2001. doi: 10.1016/S0092-8674(01)00238-0. [DOI] [PubMed] [Google Scholar]
- 7.Gomez I, Foudi N, Longrois D, Norel X. The role of prostaglandin E2 in human vascular inflammation. Prostaglandins Leukot Essent Fatty Acids 89: 55–63, 2013. doi: 10.1016/j.plefa.2013.04.004. [DOI] [PubMed] [Google Scholar]
- 8.Gonzales RJ, Duckles SP, Krause DN. Dihydrotestosterone stimulates cerebrovascular inflammation through NFκB, modulating contractile function. J Cereb Blood Flow Metab 29: 244–253, 2009. doi: 10.1038/jcbfm.2008.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Habib A, Créminon C, Frobert Y, Grassi J, Pradelles P, Maclouf J. Demonstration of an inducible cyclooxygenase in human endothelial cells using antibodies raised against the carboxyl-terminal region of the cyclooxygenase-2. J Biol Chem 268: 23448–23454, 1993. [PubMed] [Google Scholar]
- 10.Ishikado A, Nishio Y, Yamane K, Mukose A, Morino K, Murakami Y, Sekine O, Makino T, Maegawa H, Kashiwagi A. Soy phosphatidylcholine inhibited TLR4-mediated MCP-1 expression in vascular cells. Atherosclerosis 205: 404–412, 2009. doi: 10.1016/j.atherosclerosis.2009.01.010. [DOI] [PubMed] [Google Scholar]
- 11.Jang BC, Sung SH, Park JG, Park JW, Bae JH, Shin DH, Park GY, Han SB, Suh SI. Glucosamine hydrochloride specifically inhibits COX-2 by preventing COX-2 N-glycosylation and by increasing COX-2 protein turnover in a proteasome-dependent manner. J Biol Chem 282: 27622–27632, 2007. doi: 10.1074/jbc.M610778200. [DOI] [PubMed] [Google Scholar]
- 12.Kim F, Tysseling KA, Rice J, Pham M, Haji L, Gallis BM, Baas AS, Paramsothy P, Giachelli CM, Corson MA, Raines EW. Free fatty acid impairment of nitric oxide production in endothelial cells is mediated by IKKβ. Arterioscler Thromb Vasc Biol 25: 989–994, 2005. doi: 10.1161/01.ATV.0000160549.60980.a8. [DOI] [PubMed] [Google Scholar]
- 13.Lambertucci RH, Hirabara SM, Silveira LR, Levada-Pires AC, Curi R, Pithon-Curi TC. Palmitate increases superoxide production through mitochondrial electron transport chain and NADPH oxidase activity in skeletal muscle cells. J Cell Physiol 216: 796–804, 2008. doi: 10.1002/jcp.21463. [DOI] [PubMed] [Google Scholar]
- 14.Lim JW, Kim H, Kim KH. Nuclear factor-κB regulates cyclooxygenase-2 expression and cell proliferation in human gastric cancer cells. Lab Invest 81: 349–360, 2001. doi: 10.1038/labinvest.3780243. [DOI] [PubMed] [Google Scholar]
- 15.Lindstrom T, Bennett P. Transcriptional regulation of genes for enzymes of the prostaglandin biosynthetic pathway. Prostaglandins Leukot Essent Fatty Acids 70: 115–135, 2004. doi: 10.1016/j.plefa.2003.04.003. [DOI] [PubMed] [Google Scholar]
- 16.Mangold HK. In: The Lipid Handbook, Second Edition, edited by Gunstone FD, Harwood JL, Padley FB. London: Chapman and Hall, 1995, p. 315–316. [Google Scholar]
- 17.Masferrer JL, Zweifel BS, Colburn SM, Ornberg RL, Salvemini D, Isakson P, Seibert K. The role of cyclooxygenase-2 in inflammation. Am J Ther 2: 607–610, 1995. doi: 10.1097/00045391-199509000-00005. [DOI] [PubMed] [Google Scholar]
- 18.Morinelli TA, Lee MH, Kendall RT, Luttrell LM, Walker LP, Ullian ME. Angiotensin II activates NF-κB through AT1A receptor recruitment of β-arrestin in cultured rat vascular smooth muscle cells. Am J Physiol Cell Physiol 304: C1176–C1186, 2013. doi: 10.1152/ajpcell.00235.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nemeth JF, Hochgesang GP Jr, Marnett LJ, Caprioli RM. Characterization of the glycosylation sites in cyclooxygenase-2 using mass spectrometry. Biochemistry 40: 3109–3116, 2001. doi: 10.1021/bi002313c. [DOI] [PubMed] [Google Scholar]
- 20.Oliveira AF, Cunha DA, Ladriere L, Igoillo-Esteve M, Bugliani M, Marchetti P, Cnop M. In vitro use of free fatty acids bound to albumin: a comparison of protocols. Biotechniques 58: 228–233, 2015. doi: 10.2144/000114285. [DOI] [PubMed] [Google Scholar]
- 21.Omori K, Kida T, Hori M, Ozaki H, Murata T. Multiple roles of the PGE2-EP receptor signal in vascular permeability. Br J Pharmacol 171: 4879–4889, 2014. doi: 10.1111/bph.12815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Osterlund KL, Handa RJ, Gonzales RJ. Dihydrotestosterone alters cyclooxygenase-2 levels in human coronary artery smooth muscle cells. Am J Physiol Endocrinol Metab 298: E838–E845, 2010. doi: 10.1152/ajpendo.00693.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Otto JC, DeWitt DL, Smith WL. N-glycosylation of prostaglandin endoperoxide synthases-1 and -2 and their orientations in the endoplasmic reticulum. J Biol Chem 268: 18234–18242, 1993. [PubMed] [Google Scholar]
- 24.Pillon NJ, Azizi PM, Li YE, Liu J, Wang C, Chan KL, Hopperton KE, Bazinet RP, Heit B, Bilan PJ, Lee WL, Klip A. Palmitate-induced inflammatory pathways in human adipose microvascular endothelial cells promote monocyte adhesion and impair insulin transcytosis. Am J Physiol Endocrinol Metab 309: E35–E44, 2015. doi: 10.1152/ajpendo.00611.2014. [DOI] [PubMed] [Google Scholar]
- 25.Quan J, Liu J, Gao X, Liu J, Yang H, Chen W, Li W, Li Y, Yang W, Wang B. Palmitate induces interleukin-8 expression in human aortic vascular smooth muscle cells via Toll-like receptor 4/nuclear factor-κB pathway (TLR4/NF-κB-8). J Diabetes 6: 33–41, 2014. doi: 10.1111/1753-0407.12073. [DOI] [PubMed] [Google Scholar]
- 26.Renna NF, Diez ER, Lembo C, Miatello RM. Role of Cox-2 in vascular inflammation: an experimental model of metabolic syndrome. Mediators Inflamm 2013: 513251, 2013. doi: 10.1155/2013/513251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rensen SS, Doevendans PA, van Eys GJ. Regulation and characteristics of vascular smooth muscle cell phenotypic diversity. Neth Heart J 15: 100–108, 2007. doi: 10.1007/BF03085963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ribaudo R, Gilman M, Kingston RE, Choczynski P, Sacchi N. . Preparation of RNA from tissues and cells. Curr Protoc Immunol ch. 10: Unit 10.11, 2001. doi: 10.1002/0471142735.im1011s04. [DOI] [PubMed] [Google Scholar]
- 29.Ricciotti E, FitzGerald GA. Prostaglandins and inflammation. Arterioscler Thromb Vasc Biol 31: 986–1000, 2011. doi: 10.1161/ATVBAHA.110.207449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rudijanto A. The role of vascular smooth muscle cells on the pathogenesis of atherosclerosis. Acta Med Indones 39: 86–93, 2007. [PubMed] [Google Scholar]
- 31.Saren P, Welgus HG, Kovanen PT. TNF-α and IL-1β selectively induce expression of 92-kDa gelatinase by human macrophages. J Immunol 157: 4159–4165, 1996. [PubMed] [Google Scholar]
- 32.Schaeffler A, Gross P, Buettner R, Bollheimer C, Buechler C, Neumeier M, Kopp A, Schoelmerich J, Falk W. Fatty acid-induced induction of Toll-like receptor-4/nuclear factor-κB pathway in adipocytes links nutritional signalling with innate immunity. Immunology 126: 233–245, 2009. doi: 10.1111/j.1365-2567.2008.02892.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sevigny MB, Li CF, Alas M, Hughes-Fulford M. Glycosylation regulates turnover of cyclooxygenase-2. FEBS Lett 580: 6533–6536, 2006. doi: 10.1016/j.febslet.2006.10.073. [DOI] [PubMed] [Google Scholar]
- 34.Simmons DL, Botting RM, Hla T. Cyclooxygenase isozymes: the biology of prostaglandin synthesis and inhibition. Pharmacol Rev 56: 387–437, 2004. doi: 10.1124/pr.56.3.3. [DOI] [PubMed] [Google Scholar]
- 35.Warner SJ, Friedman GB, Libby P. Immune interferon inhibits proliferation and induces 2′-5′-oligoadenylate synthetase gene expression in human vascular smooth muscle cells. J Clin Invest 83: 1174–1182, 1989. doi: 10.1172/JCI113998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wu D, Liu J, Pang X, Wang S, Zhao J, Zhang X, Feng L. Palmitic acid exerts pro-inflammatory effects on vascular smooth muscle cells by inducing the expression of C-reactive protein, inducible nitric oxide synthase and tumor necrosis factor-α. Int J Mol Med 34: 1706–1712, 2014. doi: 10.3892/ijmm.2014.1942. [DOI] [PubMed] [Google Scholar]
- 37.Yu LL, Yu HG, Yu JP, Luo HS. Nuclear factor-κB regulates cyclooxygenase-2 expression and cell proliferation in human colorectal carcinoma tissue. Eksp Onkol 26: 40–47, 2004. [PubMed] [Google Scholar]