

Abstract
G protein-gated inwardly rectifying K+ (GIRK) channels are the major inwardly rectifying K+ currents in cardiac atrial myocytes and an important determinant of atrial electrophysiology. Inhibitory G protein α-subunits can both mediate activation via acetylcholine but can also suppress basal currents in the absence of agonist. We studied this phenomenon using whole cell patch clamping in murine atria from mice with global genetic deletion of Gαi2, combined deletion of Gαi1/Gαi3, and littermate controls. We found that mice with deletion of Gαi2 had increased basal and agonist-activated currents, particularly in the right atria while in contrast those with Gαi1/Gαi3 deletion had reduced currents. Mice with global genetic deletion of Gαi2 had decreased action potential duration. Tissue preparations of the left atria studied with a multielectrode array from Gαi2 knockout mice showed a shorter effective refractory period, with no change in conduction velocity, than littermate controls. Transcriptional studies revealed increased expression of GIRK channel subunit genes in Gαi2 knockout mice. Thus different G protein isoforms have differential effects on GIRK channel behavior and paradoxically Gαi2 act to increase basal and agonist-activated GIRK currents. Deletion of Gαi2 is potentially proarrhythmic in the atria.
Keywords: atria, electrophysiology, inhibitory heterotrimeric G protein, G protein-gated potassium channel
INTRODUCTION
Inwardly rectifying K+ (Kir) channels are widely expressed in all chambers of the heart and are important in setting the resting membrane potential. However, there is a dichotomy, with the Kir2.0 family being predominant in the ventricles and His-Purkinje system and the Kir3.0 family in atrial and nodal tissues (7, 18, 24, 45). The Kir3.0 channel family encodes G protein-gated inwardly rectifying K+ (GIRK) channel present in neurons and neuroendocrine tissues in addition to the heart (11, 27). In the heart the channel is thought to consist largely of a heteromultimer of Kir3.1 and Kir3.4 (19). A characteristic of these channels is that they are activated by G protein-coupled receptors linked to inhibitory G proteins and specifically by the free Gβγ subunits (25, 42, 44). For example, in the sinoatrial (SA) node (SAN) activation of the channel by acetylcholine released from the vagus nerve is responsible for heart rate slowing (39, 43). Recently a study has shown a critical role for Kir3.4 in the kinetics of heart rate recovery to resting level after sympathetic activation (30).
Despite GIRK channel activation being mediated by Gβγ directly binding to domains on the channel, activation seems to occur largely via members of the inhibitory G protein family (14, 21, 22, 35). In a series of studies from different laboratories using varied approaches, a more complex model has emerged. It appears that the inhibitory G protein heterotrimer is able to directly interact with the channel, and, on activation, heterotrimer dissociation occurs in a microdomain in or around the channel subunit. The dissociated Gβγ subunit leads to activation (9, 21, 34, 38). However the Gα, as either a monomer or as part of the heterotrimer, may also play an important role leading to inhibition of channel activity, and this process may be isoform dependent (5, 6, 15). Multiple isoforms of inhibitory Gα subunits (Gαi1, Gαi2, Gαi3 and, Gοα) are present in atrial tissue, and their roles in modulating parasympathetic signal transduction remain unclear. The subunits Gαi2 and Gαi3 have been shown to mediate signaling to GIRK in embryonic stem cell-derived cardiomyocytes (42a). Our own work shows that Gαi2 is important for heart rate regulation in vivo (41, 46), but this occurs via modulation in the SA node (not the atria) and might be via a mechanism independent of GIRK.
There is further complexity in that the right (RA) and left (LA) atria may be different and have gradients of channel expression (17). GIRK channels are expressed at higher levels in the RA in mice and humans. It has been proposed that the gradient of GIRK current, combined with the heterogeneous distribution of parasympathetic innervation and adenosine receptor expression in the atria, may contribute to the ability of vagal nerve stimulation to augment dispersion of atrial refractoriness (12, 13, 23, 26, 40). The purpose of this study is to define the type of G proteins involved in the signaling to GIRK in the atria, the possible role of these G proteins in atrial asymmetry, and how they might potentially modulate arrhythmogenesis.
MATERIALS AND METHODS
Gene-targeted mice.
Mice with global deletion of Gαi2 and Gαi1/Gαi3 (deletion of both Gαi1 and Gαi3) maintained on a Sv129 background were compared with wild-type littermate controls. The gene-targeting strategy, genotyping, and confirmation of relevant Gi/oα deletions have previously been described (16, 46). Mice were maintained in an animal core facility under the United Kingdom Home Office guidelines relating to animal welfare. All procedures were approved by the local animal care and use committee and performed in accord with the United Kingdom Home Office regulations (PPL 70/7665). All mice were kept in a temperature-controlled environment (21–24°C) with a 12:12-h light-dark cycle. Animals were allowed ad libitum access to standard rodent chow and drinking water. Mice on a 129/Sv background aged 3–4 mo (20–30 g) were used for this study. Both males and females were used in the study, and there was no gender discrimination. Littermate controls were obtained from the Gαi2 crosses.
Quantitative real-time reverse transcription PCR.
RNA was isolated from the left atria and right atria from 14-wk-old mice with global deletion of Gαi2 (n = 3) maintained on a Sv129 background and wild-type littermate controls (n = 3) using the RNeasy kit (Qiagen). Briefly, hearts were removed from each group of mice [Gαi2+/+ and, Gαi2−/−] and washed with cold PBS, and left atria and right atria were isolated and immediately placed in RNALater. RNA was extracted using the RNeasy kit (catalog no. 74104; Qiagen). cDNA was synthesized using the high-capacity cDNA reverse transcription kit (4368814; Life Technologies) and quantified, and 50 ng of cDNA/20 μl were used for the subsequent real-time expression assay. Real-time PCR was performed using Taqman gene expression assays (Life Technologies). All genes (Mm00434618_m1: Kcnj3, Mm01175829_m1: Kcnj5, Mm00492379_m1: Gnai3, Mm01165301_m1: Gnai1, Mm00494677_m1: Gnb1, Mm00501973_m1: Gnb4, Mm01165191_m1: Gng11, Mm00515876_m1: Gng7) were assayed in triplicate, and GAPDH was used as the housekeeping gene.
Single-cell isolation and electrophysiology.
Atrial and sinoatrial cells were isolated using an adapted method for isolation of sinoatrial cardiomyocytes (29). Briefly, mice were injected with heparin, and beating hearts were removed under pentobarbital sodium (3 ml/kg) and ketamine (1 ml/kg) anesthesia. The left and right atria were excised in normal Tyrode solution containing (in mM): 140 NaCl, 5.4 KCl, 1.8 CaCl2, 1 MgCl2, 5 HEPES-NaOH, and 5.5 d-glucose (pH 7.4). Strips of tissues were enzymatically digested in a low-Ca2+ and low-Mg2+ solution containing (in mM): 140 NaCl, 5.4 KCl, 0.5 MgCl2, 0.2 CaCl2, 1.2 KH2PO4, 50 taurine, 5.5 d-glucose, and 5 HEPES-NaOH, pH 6.9. Collagenase type II (224 U/ml; Worthington), elastase (1.9 U/ml; Worthington), protease (0.9 U/ml; Sigma Aldrich), and bovine serum albumin (BSA, 1 mg/ml) were added. The digestion step was carried out for 20 min or 45 min for atrial and SAN tissue, respectively, under gentle mechanical agitation at 37°C. Tissue strips were then washed out and transferred to a modified “Kraftbrühe” (KB) medium containing (in mM): 70 l-glutamic acid, 20 KCl, 80 KOH, 10 d-β-hydroxybutyric acid, 10 KH2PO4, 10 taurine, 1 mg/ml BSA, and 10 HEPES-KOH, pH 7.4. Single myocytes were manually dissociated in KB solution by employing a fire-polished glass pipette. Finally, extracellular Ca2+ concentration was recovered up to 1.3 mM. A drop of cell suspension was seeded on sterilized laminin-coated coverslips. After 30–45 min, Tyrode solution containing 10% BSA was added, and cells were stored at 37°C until used in humidified 5% CO2-95% air at 37°C. All experiments were performed at room temperature.
Patch-clamp current recordings were performed with an Axopatch 200B amplifier (Axon Instruments) using fire-polished pipettes with a resistance of 3–4 MΩ pulled from filamented borosilicated glass capillaries (1.5 mm OD × 1.17 mm ID; Harvard Apparatus). Data were acquired and analyzed by using a Digidata 1322A interface (Axon Instruments) and pCLAMP software (version 10; Axon Instruments). Action potentials were recorded in the current-clamp mode. Cardiomyocytes were stimulated using a 5-ms current pulse. The resting membrane potential, the magnitude of the initial depolarization, and the action potential duration at which 50 and 90% repolarization (APD50 and APD90, respectively) occurred were measured. The cells were clamped at −60 mV in an extracellular solution containing (in mM): 135 NaCl, 5.4 KCl, 2 CaCl2, 1 MgCl2, 0.33 NaH2PO4, 5 H-HEPES, and 10 glucose (buffered to pH 7.4 with NaOH). The intracellular solution was (in mM): 110 potassium gluconate, 20 KCl, 10 NaCl, 1 MgCl2, 2 MgATP, 2 EGTA, and 0.3 Na2GTP (buffered to pH 7.2 with KOH). The liquid junction potential was +13 mV.
Measurement of atrial electrophysiology using multielectrode arrays.
Using a multielectrode array (MEA; Multichannel Systems), we investigated the effect of ablation of Gαi2 on electrophysiological parameters in ex vivo atrial tissue (33). Left atria were dissected from isolated mouse hearts after mounting in a Langendorff setup and perfused with Krebs solution supplemented with 30 mM 2,3-butanedione monoxime. The tissue was then transferred to the array perfused with Krebs solution (37°C; 95% O2-5% CO2). Experiments were conducted in the absence and the presence of 10 nM–10 µM carbachol. Left atrial electrophysiology was assessed during electrical stimulation using a multielectrode array (MEA) system, which allows noninvasive synchronous multifocal recording of extracellular field potentials. The MEA (MEA2100; Multichannel Systems, Reutlingen, Germany) consists of 60 microelectrodes arranged in an 8 × 8 matrix, with a 20-μm electrode diameter and an interelectrode distance of 200 μm. Myocardial samples were positioned in the center of the MEA dish, held in contact with electrodes by a holder, and continuously superfused with oxygenated Krebs solution at 37°C. Baseline electrical stimulation (bipolar pulses, 2× threshold, 2 s duration, 4 Hz frequency) was applied via one of the MEA microelectrodes. Field potential data were acquired simultaneously from all 60 microelectrodes. S1–S2 train stimulation with a S1–S1 cycle length of 250 ms was used to assess atrial effective refractory period (AERP). To assess conduction properties, isolated atria were sequentially stimulated (4 Hz) from one electrode of each four edges of the array. Field potential recordings obtained in these conditions were processed using LabChart7 (ADInstruments) to define local activation time based on minimum of the derivative of field potential. Average conduction velocity (CV) was calculated by linear regression relating interelectrode distance to activation times, as previously described (33). The slope of the regression line was the average CV. Minimal wave-front cycle length was calculated for each isolated atria as AERP × CV.
Statistical analysis.
The mean and SE are presented. Student’s t-test or one-way ANOVA was used, with a P value <0.05 being statistically significant.
RESULTS
Inward currents in right and left atria.
With the use of a step voltage protocol, current-voltage (I–V) relationships of atrial cardiomyocytes isolated from the right atria (RA) and left atria (LA) were compared in control mice (normal genotype littermates from the Gαi2 crosses) (Fig. 1). Currents were larger and showed greater inward rectification in RA than in the LA in the presence of carbachol (Figs. 1 and 2). Experiments were also performed in the presence of GTPγS in the patch pipette to activate GIRK currents in the absence of receptor stimulation. In these experimental conditions, we still obtained a larger current in RA (−59.8 ± 6.5 pA/pF, n = 5) compared with LA (−27.8 ± 2 pA/pF, n = 5) of Gαi2+/+ atrial myocytes (n = 5 mice).
Fig. 1.

GIRK currents in atrial tissue. A: representative traces of currents measured in atrial myocytes isolated from the left and right atria of Gαi2+/+ mice. B: mean current-voltage relationships. Atrial myocytes were challenged with 10 μM carbachol. GIRK currents were larger in RA compared with LA (n = 10, 6 mice).
Fig. 2.

Deletion of Gαi1/3 affects the gradient of GIRK across the atria. Representative traces and mean current-voltage relationships of currents measured in atrial myocytes isolated from the left and right atria of Gαi1/3−/− mice. There was a loss of GIRK current gradient between the LA and RA (n = 5–6, 4 mice).
In Gαi2−/− mice, the I–V relationship of the RA was altered with larger basal and carbachol-activated inward currents in the RA (Figs. 3 and 4). In mice with the combined deletion of Gαi1/Gαi3 there were reduced carbachol-activated currents in the RA (Figs. 2 and 4). The effects of Gαi2 deletion were more pronounced in the RA, leading to loss of regional difference across the atria. Kinetics of GIRK current activation by carbachol were assessed using a 20-s application of agonist, as we have previously described (2, 3, 31). There were no major changes in activation and rapid desensitization between RA and LA and in the mice with either Gαi2 or Gαi1/Gαi3 deletion (Table 1). In contrast, deactivation was slower in the RA than the LA, but this pattern was not changed in the Gαi2−/− mice or mice with combined Gαi1/Gαi3 deletion (Table 1).
Fig. 3.

Deletion of Gαi2 affects the gradient of GIRK across the atria. Representative traces and mean current-voltage relationships of currents measured in atrial myocytes isolated from the left and right atria of Gαi2−/− mice. Basal GIRK currents were larger in RA compared with Gαi2+/+ (n = 7–8, 4 mice).
Fig. 4.
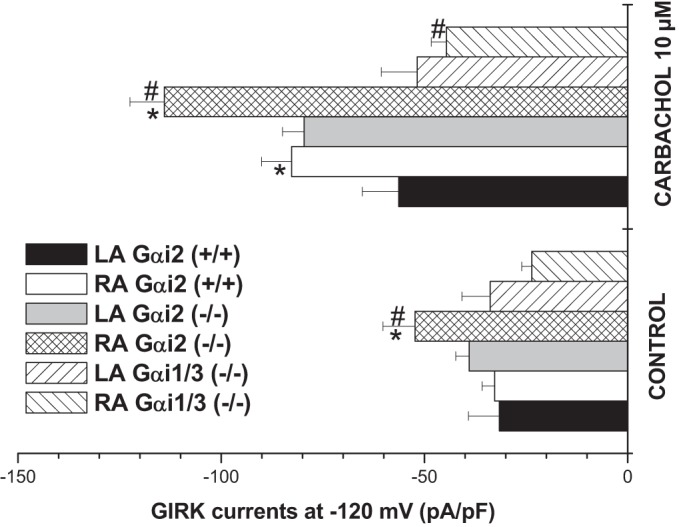
Comparison of GIRK currents in the LA and RA. Bar graph showing maximum GIRK currents measured at −120 mV. In Gαi2+/+, carbachol (10 μM) led to a larger activation of GIRK currents in the RA (n = 10, 6 mice). Deletion of Gαi2 led to larger basal and carbachol-activated currents in the RA (n = 6, 4 mice). Deletion of Gαi1/3 led to smaller carbachol-activated currents in both LA and RA with a marked effect in the RA (n = 8, 4 mice), the consequence being a loss of gradient across the atria. *Comparison between LA and RA, #comparison between KO and WT Gαi2 (+/+) and either Gαi2 (−/−) or Gαi1/3 (−/−).
Table 1.
Deletion of Gαi2 or Gαi1/3 and GIRK current kinetics across the atria
| Gαi2+/+ |
Gαi2−/− |
Gαi1/3−/− |
|||||||
|---|---|---|---|---|---|---|---|---|---|
| LA (n = 9) | RA (n = 8) | SAN (n = 10) | LA (n = 12) | RA (n = 10) | SAN (n = 11) | LA (n = 7) | RA (n = 8) | SAN (n = 5) | |
| Im, pA/pF | −85 ± 8 | −86.5 ± 7.8 | −55.9 ± 8 | −77.6 ± 6.2 | −75.1 ± 7 | −49.7 ± 6.2 | −67 ± 4.2 | −70 ± 9.5 | −58.2 ± 18 |
| GIRK, pA/pF | −106 ± 15 | −162 ± 34.5 | −108.3 ± 20 | −87.8 ± 8.2 | −128 ± 12 | −118.3 ± 14.4 | −92.51 ± 10 | −134.8 ± 24 | −175.2 ± 18 |
| Lag + TTP, s | 1.06 ± 0.06 | 0.84 ± 0.05 | 0.99 ± 0.01 | 0.9 ± 0.05 | 0.82 ± 0.04 | 0.96 ± 0.05 | 1 ± 0.05 | 0.68 ± 0.04 | 0.79 ± 0.07 |
| τac, ms | 188 ± 11.1 | 142 ± 11.3 | 162 ± 24.4 | 156 ± 10.8 | 153 ± 27 | 144 ± 7.2 | 256 ± 45 | 114 ± 13 | 143 ± 13.7 |
| τdeac, ms | 2,996 ± 458 | 811 ± 66.6* | 607 ± 92* | 2,654 ± 438 | 779 ± 137* | 466 ± 40.3* | 3,567 ± 739 | 824 ± 141* | 493 ± 71.3* |
| Lag inac, s | 0.68 ± 0.14 | 0.32 ± 0.06* | 0.21 ± 0.01* | 0.59 ± 0.08 | 0.33 ± 0.03* | 0.23 ± 0.02* | 0.53 ± 0.01 | 0.30 ± 0.07* | 0.26 ± 0.01* |
| % des 20 s | 25.8 ± 2.35 | 25.2 ± 3.34 | 20 ± 1.5 | 29.5 ± 3.4 | 27.1 ± 2.9 | 31 ± 3.2 | 22.5 ± 1.6 | 24.1 ± 2.7 | 14.1 ± 3.5 |
| G | 22.7 ± 1.3 | 18.7 ± 1 | 26.4 ± 2.7 | 25 ± 2.2 | 19.6 ± 1.3 | 22.8 ± 2.4 | 34.4 ± 3.3 | 22.6 ± 2.6 | 17.8 ± 2.2 |
Values are means ± SE. Cells were clamped at −60 mV and carbachol was applied for 20 ms with a fast perfusion system. Characteristics of the GIRK current kinetics are presented for the LA, RA, and SAN region for Gαi2+/+ (n = 10–12 from 4 mice), Gαi2−/− (n = 8–10, 5 mice), and Gαi1/3−/− (n = 5–8, 3 mice). The current inactivation characteristics [τdeac (tau of deactivation) and Lag inac (lag of inactivation)] were faster in the RA and SAN compared with the LA (*P < 0.05). Im, basal current; Lag, time lag; TTP, time to peak; τac, tau of activation; % des 20 s, percentage of desensitization after 20 s; G, cell conductance.
We examined for expression changes of relevant components in the signaling cascade in the RA and LA of Gαi2−/− mice and littermate controls. Using quantitative real-time reverse transcription PCR, we measured the expression of Gαi1 (Gnai1), Gαi3 (Gnai3), some representative Gsβγ (Gnb1, Gnb4, Gng7, and Gng11), and the GIRK channel subunits (Kcnj3 and Kcnj5), and the results are shown in Table 2. Gnb4 and Gng11 were chosen as they have potentially been associated with cardiovascular traits, in particular heart rate, in genomewide association studies (10). In general, the changes between Gαi2−/− mice and littermate controls are modest even when significant. However, in Gαi2−/− mice, GIRK channel subunit kcnj5 expression was increased in both atrial chambers, suggesting that some of the differences in regulation of GIRK channels in Gαi2−/− mice may be related to transcriptional changes in channel expression.
Table 2.
Quantitative real-time reverse transcription PCR to measure gene expression in the RA and LA
| Gene | LA ΔCT |
RA ΔCT |
||
|---|---|---|---|---|
| WT | KO | WT | KO | |
| Gnai1 | 6.75 ± 0.06 | 6.98 ± 0.05 | 8.46 ± 0.35 | 8.15 ± 0.09 |
| Gnai3 | 6.90 ± 0.18 | 6.65 ± 0.08 | 6.98 ± 0.12 | 7.42 ± 0.09 |
| Gnb1 | 4.76 ± 0.14 | 5.72 ± 0.25* | 5.21 ± 0.50 | 5.50 ± 0.07 |
| Gnb4 | 7.98 ± 0.18 | 7.72 ± 0.07 | 8.75 ± 0.18 | 8.60 ± 0.08 |
| Gng7 | 10.8 ± 0.14 | 11.5 ± 0.14 | 11.5 ± 0.51 | 11.7 ± 0.16 |
| Gng11 | 7.04 ± 0.11 | 7.49 ± 0.03* | 7.56 ± 0.21 | 7.43 ± 0.04 |
| Kcnj3 | 2.86 ± 0.14 | 2.30 ± 0.07 | 3.62 ± 0.18 | 3.00 ± 0.05 |
| Kcnj5 | 5.38 ± 0.12 | 4.86 ± 0.06* | 5.51 ± 0.24 | 4.94 ± 0.07* |
Values are means ± SE. Quantitative real-time reverse transcription PCR was performed as described in materials and methods for the genes indicated. Measurements were performed in triplicate from Gαi2−/− mice (n = 3 mice) and littermate controls (n = 3 mice). WT, wild type; KO, knockout.
P < 0.05 using one-way ANOVA.
Comparison with the SAN.
We also isolated and patch clamped SA nodal cells. GIRK currents were of a similar magnitude as that in the RA but rectified more strongly and deactivated more rapidly after carbachol application (Fig. 5 and Table 1). The properties of the currents in the SAN were unaffected in mice with global genetic deletion of Gαi2 or Gαi1/Gαi3.
Fig. 5.

GIRK current in the SAN. Left: representative traces of GIRK currents in control and after activation with 10 μM carbachol. Comparison is made between Gαi2+/+ (n = 18, 8 mice), Gαi2−/− (n = 15, 5 mice), and Gαi1/3−/− (n = 16, 7 mice). Right: mean current-voltage relationships. Atrial myocytes were challenged with 10 μM carbachol. GIRK currents were not affected by Giα deletion.
Single-cell action potentials.
It might be predicted that an increased GIRK current in Gαi2−/− mice might lead to a shortened action potential duration and atrial effective refractory period. We compared atrial action potentials in the RA and LA myocytes in control and Gαi2−/− mice and found that RA atrial myocytes from RA Gαi2−/− mice had a shorter APD90 than control RA myocytes. A similar trend was observed in the LA myocytes, although this was not statistically significant (Fig. 6).
Fig. 6.

Consequence of the deletion of Gαi2 on the action potential of single cardiomyocytes. Single cardiomyocyte action potential (AP) was measured after stimulation of cells by a 5-ms pulse after pacing at 1 Hz for 60 s. In Gαi2+/+, the APD90 were longer in the RA (n = 8–9, 3 mice), although the mean values did not reach significance during analyses with t-test due to the variability of the data values. In Gαi2−/− (n = 6–7, 3 mice), both APD50 and APD90 were significantly reduced in the right atria.
Carbachol (10 µM) led to shortening of APD with a more pronounced effect in the RA. The decrease of the APD90 reached 61 ± 3% in Gαi2+/+ LA (n = 9, n = 3 mice) and 76 ± 3% in Gαi2+/+ RA (n = 8, n = 3 mice, P = 0.006). In Gαi2−/− murine atrial myocytes, carbachol decreased APD90 by 57 ± 7% in the LA (n = 7, n = 3 mice) and by 50 ± 8% in the RA (n = 6, n = 3 mice, not significant).
Tissue electrophysiology.
We performed an analysis of the tissue electrophysiology in isolated LA using a multielectrode array. The analysis of intact RA was complicated by the intrinsic pacemaking activity. In the Gαi2−/− LA, there was a shortened effective refractory period (ERP) and no alteration in conduction velocity in comparison to Gαi2+/+ LA, resulting in a significant decrease in potential path length for reentry (Fig. 7). A similar relative decrease of left atrial ERP was observed in the presence of carbachol (10 nM to 10 µM) between Gαi2−/− and control, with no significant difference in log EC50, i.e., −6.9 ± 0.4 vs. −7.1 ± 0.4, respectively. Carbachol did not alter left atrial CV (n = 7, Fig. 7).
Fig. 7.

Electrophysiology of isolated right and left atria. A–C: atrial effective refractory period (ERP; A), conduction velocity (CV; B), and minimum wave front cycle length (mWFCL; C) in left atrial tissue deficient in Gαi2−/− mice compared with Gαi2+/+. *P < 0.05. D–F: dose response for carbachol on left atrial AERP (D), relative AERP to baseline (E), and CV (F) in Gαi2−/− mice compared with Gαi2+/+. Experiments were performed with 7 mice in each group.
DISCUSSION
Our main findings are that in murine atria specific isoforms of inhibitory G proteins have defined roles in controlling GIRK channel function. Specifically, Gαi2 suppresses the basal and agonist-induced activity of GIRK, whereas Gαi1 and/or Gαi3 mediate muscarinic activation of the current. Our experiments are in agreement with previous work showing larger GIRK currents in the RA and the SAN regions compared with the LA region (12, 13, 23, 26, 40). Deletion of Gαi2 accentuated this chamber asymmetry, but it was attenuated in mice with global genetic deletion of Gαi1 and Gαi3. In keeping with the changes in GIRK currents, the global genetic deletion of Gαi2 resulted in a shortened action potential duration, reduced tissue atrial effective refractory period, and reduced minimum wave front cycle length.
These findings complement our previous work in which we have investigated heart rate regulation in various G protein α-subunit knockout mice (41, 46). Specifically, mice with global and SAN-specific deletion of Gαi2 were tachycardic with impaired high-frequency responses in heart rate variability studies. It is worth stating that the majority of studies reported here were conducted in the atria and reveal differences between the SAN and across the atria in GIRK channel signaling and G protein dependency. Our observations reported here in the SAN show preserved signaling via muscarinic receptors to GIRK channels with deletion of both Gαi2 and combined Gαi1\Gαi3. This suggests that GIRK channel-independent mechanisms may be important in determining the in vivo phenotype in Gαi2 knockout mice. Specifically, the loss of negative coupling to adenylate cyclase, fall in cAMP, and effects on the hyperpolarization-activated cyclic nucleotide-gated channel and/or modulation of protein kinase A regulating the “calcium clock” may be important (20).
Inhibitory G protein α-subunits and GIRK channel function.
GIRK channels are traditionally viewed as an example of a canonical effector activated by Gβγ subunits. However, it is clear that Gα subunits play a role. For example, we demonstrated in heterologous systems that channel activation seemed to be preferentially activated via inhibitory rather than stimulatory G proteins (21). Furthermore, studies have shown that the inhibitory G protein heterotrimer can bind to the channel complex (6, 9, 37). This interaction may have important functional consequences, namely that it suppresses basal current activity (6, 34). In other studies the Dascal laboratory demonstrated that there may be isoform differences in the nature of this behavior between Gαi1 and Gαi3 (15). One issue with a portion of this work is that the conclusions often depend on overexpression of engineered components in heterologous expression systems. It is unclear whether these kinds of effects occur in native settings with physiological levels of G protein and channel expression. In this study in native atrial myocytes, we show that deletion of Gαi2 leads to an unexpected increase in basal and agonist-activated currents. One interpretation of this finding is that inhibitory G protein α-subunits do indeed have an ability in vivo to negatively regulate GIRK currents. However, our data reveal another potential mechanism, namely transcriptional changes in GIRK channel subunit expression engendered by Gαi2 deletion. Specifically, in the Gαi2−/− mice, expression levels for kcnj3 and kcnj5 mRNA, in a statistically significant fashion for the latter, were increased compared with littermate controls, and the magnitude of these effects was comparable to the changes in GIRK current density observed. In contrast, combined deletion of Gαi1 and Gαi3 impairs the magnitude of muscarinic activation, suggesting that one of Gαi1 and Gαi3 or both are important for mediating the agonist-induced response. Although we studied single isolated cardiac cells ex vivo, it is still possible that extracardiac effects could lead to long-lasting changes in myocyte biology.
We also examined if deletion of Gαi2 had effects on the expression of other components in the signaling cascade, namely Gnai1, Gnai3, Gnb1, Gnb4, Gng7, and Gng11. These experiments have substantial practical complications, since there are 5 G protein β-genes and 14 G protein γ-genes. We selected four to examine for compensatory changes in part determined from genomewide association studies in heart rate and our own unpublished studies (10). Although there were some changes, these were modest in magnitude (possible decreases in expression of gnb1 and gng11 in the LA but no significant change in gnb4, gng7, gnai1, or gnai3). Furthermore, in the functional studies in Gαi2−/− mice, carbachol led to increased agonist-induced current activation, suggesting Gβγ expression was not limiting for signal transduction.
Regional differences.
There were regional differences in the nature and coupling profile of GIRK currents in supraventricular tissues. GIRK currents were larger in the right atrium and SAN, and these differences were accentuated in the right atrium by global genetic deletion of Gαi2. Kinetic analysis also showed that GIRK current inactivation is faster in the RA and the SAN regions compared with the left atrium. This fast inactivation of the GIRK currents in the RA and pacemaker regions could reflect differential expression of regulators of G protein signaling that can increase the hydrolysis rate of GTP-bound and active G protein α-subunits (4, 33, 36). Furthermore, GIRK currents were more outwardly rectifying in the SA node and this could contribute to their importance in recovery of heart rate after exercise as explored recently in GIRK4 knockout mice as they may play a more significant role at depolarized potentials (30).
Another interesting finding is that the molecular details of the signaling system differ between closely related regions of the heart. Hence, although Gαi2 and Gαi1/Gαi3 seem to have roles in inhibition and activation, respectively, in the atria, this pattern does not exist in the SA node. Indeed, a significant amount of redundancy is suggested, since GIRK channel activation was little affected in either Gαi2 or Gαi1/Gαi3 knockout mice. This suggests that the specifics of the signaling can be tissue and region dependent. G protein deletion had no effect on the kinetics of signaling, suggesting that this was predominantly determined by other factors such as the expression of regulators of G protein signaling.
G protein deletion and predisposition to arrhythmia.
The increase in GIRK currents in mice with global genetic deletion of Gαi2 might lead to more general effects on single-cell and tissue-level electrophysiology. Indeed, the increase in GIRK currents was sufficient to decrease the action potential duration. Furthermore, in whole left atrial preparations, Gαi2 shortened the atrial effective refractory period without an effect on conduction velocity, leading to a decreased minimum wave front cycle length. This change would be potentially proarrhythmic. We have also previously observed that Gαi2 deletion in the ventricle and silencing of the vagal input increase the predisposition to ventricular arrhythmia (28, 47). The mechanism is different with an effect on calcium channel regulation and expression (47). It is also known that GIRK4 knockout mice are resistant to the induction of atrial fibrillation, whereas RGS6 knockout mice with increased GIRK channel activity are predisposed (18, 36). Other investigators have observed in the dog that Gαi2 and/or Gαi3 knockdown using cell-permeable peptides may suppress vagally mediated atrial fibrillation when delivered into the posterior left atrium (1). The authors did not examine the specifics of the mechanism and whether it was related to GIRK channel activation.
Action potential duration has been shown to decrease with increasing distance from the SAN region (26, 32). In contrast, GIRK currents are larger in the right atrium than left atrium, and this suggests there are other important electrophysiological determinants of the variation in action potential duration across the atria. The shortened action potential duration in the left atrium is potentially important as it may allow the support of higher-frequency rotors in and around the pulmonary veins (40) although in this study the authors found higher GIRK currents in the the left versus right atrium of sheep. Despite this lack of consensus, suppression of GIRK channel activity in the left atrium abrogates reentry and atrial fibrillation (8).
Conclusions.
Although not directly addressing the issue, our studies are compatible with the long-standing view that Gβγ subunits are important for GIRK channel activation. However, they do reveal layers of complexity in how the Gα heterotrimeric G protein subunit might shape this response. A body of work, which we discuss above, has suggested various ways by which this might occur, including direct protein-protein interaction between G protein heterotrimer components and channel domains. However, much of this work is accomplished by expressing components, often at nonphysiological levels, in model cell systems. Here we examine native signaling in various chambers and regions of the heart using mice with global genetic deletion of Gα subunits. Our overall conclusion is that there is much plasticity in the system, with the exact importance of a specific Gα subunit being dependent on tissue region expression. Gα subunits may directly suppress GIRK currents in native systems, but this could be accounted for by additional effects on channel transcription. These observed phenomena may result from variations in Gα subunit expression or compartmentation with the channel in different cardiac regions, and these are topics for future investigation.
GRANTS
This work was supported by the British Heart Foundation (RG/15/15/31742) and the Intramural Research Program of the National Institutes of Health (project Z01-ES-101643). D. Montaigne was supported by a grant from la Fédération Française de Cardiologie.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
M.N., D.M., and S.S. performed experiments; M.N. and D.M. analyzed data; M.N., D.M., S.S., and A.T. interpreted results of experiments; M.N. and D.M. prepared figures; M.N., D.M., and A.T. drafted manuscript; M.N., D.M., and A.T. edited and revised manuscript; M.N., D.M., S.S., L.B., and A.T. approved final version of manuscript.
REFERENCES
- 1.Aistrup GL, Villuendas R, Ng J, Gilchrist A, Lynch TW, Gordon D, Cokic I, Mottl S, Zhou R, Dean DA, Wasserstrom JA, Goldberger JJ, Kadish AH, Arora R. Targeted G-protein inhibition as a novel approach to decrease vagal atrial fibrillation by selective parasympathetic attenuation. Cardiovasc Res 83: 481–492, 2009. doi: 10.1093/cvr/cvp148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Benians A, Leaney JL, Milligan G, Tinker A. The dynamics of formation and action of the ternary complex revealed in living cells using a G-protein-gated K+ channel as a biosensor. J Biol Chem 278: 10851–10858, 2003. doi: 10.1074/jbc.M212299200. [DOI] [PubMed] [Google Scholar]
- 3.Benians A, Leaney JL, Tinker A. Agonist unbinding from receptor dictates the nature of deactivation kinetics of G protein-gated K+ channels. Proc Natl Acad Sci USA 100: 6239–6244, 2003. doi: 10.1073/pnas.1037595100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Benians A, Nobles M, Hosny S, Tinker A. Regulators of G-protein signaling form a quaternary complex with the agonist, receptor, and G-protein. A novel explanation for the acceleration of signaling activation kinetics. J Biol Chem 280: 13383–13394, 2005. doi: 10.1074/jbc.M410163200. [DOI] [PubMed] [Google Scholar]
- 5.Berlin S, Keren-Raifman T, Castel R, Rubinstein M, Dessauer CW, Ivanina T, Dascal N. Gαi and Gβγ jointly regulate the conformations of a Gβγ effector, the neuronal G protein-activated K+ channel (GIRK). J Biol Chem 285: 6179–6185, 2010. doi: 10.1074/jbc.M109.085944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Berlin S, Tsemakhovich VA, Castel R, Ivanina T, Dessauer CW, Keren-Raifman T, Dascal N. Two distinct aspects of coupling between Gαi protein and G protein-activated K+ channel (GIRK) revealed by fluorescently labeled Gαi3 protein subunits. J Biol Chem 286: 33223–33235, 2011. doi: 10.1074/jbc.M111.271056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bettahi I, Marker CL, Roman MI, Wickman K. Contribution of the Kir3.1 subunit to the muscarinic-gated atrial potassium channel IKACh. J Biol Chem 277: 48282–48288, 2002. doi: 10.1074/jbc.M209599200. [DOI] [PubMed] [Google Scholar]
- 8.Bingen BO, Neshati Z, Askar SF, Kazbanov IV, Ypey DL, Panfilov AV, Schalij MJ, de Vries AA, Pijnappels DA. Atrium-specific Kir3.x determines inducibility, dynamics, and termination of fibrillation by regulating restitution-driven alternans. Circulation 128: 2732–2744, 2013. doi: 10.1161/CIRCULATIONAHA.113.005019. [DOI] [PubMed] [Google Scholar]
- 9.Clancy SM, Fowler CE, Finley M, Suen KF, Arrabit C, Berton F, Kosaza T, Casey PJ, Slesinger PA. Pertussis-toxin-sensitive Gα subunits selectively bind to C-terminal domain of neuronal GIRK channels: evidence for a heterotrimeric G-protein-channel complex. Mol Cell Neurosci 28: 375–389, 2005. doi: 10.1016/j.mcn.2004.10.009. [DOI] [PubMed] [Google Scholar]
- 10.den Hoed M, Eijgelsheim M, Esko T, Brundel BJ, Peal DS, Evans DM, Nolte IM, Segrè AV, Holm H, Handsaker RE, Westra HJ, Johnson T, Isaacs A, Yang J, Lundby A, Zhao JH, Kim YJ, Go MJ, Almgren P, Bochud M, Boucher G, Cornelis MC, Gudbjartsson D, Hadley D, van der Harst P, Hayward C, den Heijer M, Igl W, Jackson AU, Kutalik Z, Luan J, Kemp JP, Kristiansson K, Ladenvall C, Lorentzon M, Montasser ME, Njajou OT, O’Reilly PF, Padmanabhan S, St Pourcain B, Rankinen T, Salo P, Tanaka T, Timpson NJ, Vitart V, Waite L, Wheeler W, Zhang W, Draisma HH, Feitosa MF, Kerr KF, Lind PA, Mihailov E, Onland-Moret NC, Song C, Weedon MN, Xie W, Yengo L, Absher D, Albert CM, Alonso A, Arking DE, de Bakker PI, Balkau B, Barlassina C, Benaglio P, Bis JC, Bouatia-Naji N, Brage S, Chanock SJ, Chines PS, Chung M, Darbar D, Dina C, Dörr M, Elliott P, Felix SB, Fischer K, Fuchsberger C, de Geus EJ, Goyette P, Gudnason V, Harris TB, Hartikainen AL, Havulinna AS, Heckbert SR, Hicks AA, Hofman A, Holewijn S, Hoogstra-Berends F, Hottenga JJ, Jensen MK, Johansson A, Junttila J, Kääb S, Kanon B, Ketkar S, Khaw KT, Knowles JW, Kooner AS, Kors JA, Kumari M, Milani L, Laiho P, Lakatta EG, Langenberg C, Leusink M, Liu Y, Luben RN, Lunetta KL, Lynch SN, Markus MR, Marques-Vidal P, Mateo Leach I, McArdle WL, McCarroll SA, Medland SE, Miller KA, Montgomery GW, Morrison AC, Müller-Nurasyid M, Navarro P, Nelis M, O’Connell JR, O’Donnell CJ, Ong KK, Newman AB, Peters A, Polasek O, Pouta A, Pramstaller PP, Psaty BM, Rao DC, Ring SM, Rossin EJ, Rudan D, Sanna S, Scott RA, Sehmi JS, Sharp S, Shin JT, Singleton AB, Smith AV, Soranzo N, Spector TD, Stewart C, Stringham HM, Tarasov KV, Uitterlinden AG, Vandenput L, Hwang SJ, Whitfield JB, Wijmenga C, Wild SH, Willemsen G, Wilson JF, Witteman JC, Wong A, Wong Q, Jamshidi Y, Zitting P, Boer JM, Boomsma DI, Borecki IB, van Duijn CM, Ekelund U, Forouhi NG, Froguel P, Hingorani A, Ingelsson E, Kivimaki M, Kronmal RA, Kuh D, Lind L, Martin NG, Oostra BA, Pedersen NL, Quertermous T, Rotter JI, van der Schouw YT, Verschuren WM, Walker M, Albanes D, Arnar DO, Assimes TL, Bandinelli S, Boehnke M, de Boer RA, Bouchard C, Caulfield WL, Chambers JC, Curhan G, Cusi D, Eriksson J, Ferrucci L, van Gilst WH, Glorioso N, de Graaf J, Groop L, Gyllensten U, Hsueh WC, Hu FB, Huikuri HV, Hunter DJ, Iribarren C, Isomaa B, Jarvelin MR, Jula A, Kähönen M, Kiemeney LA, van der Klauw MM, Kooner JS, Kraft P, Iacoviello L, Lehtimäki T, Lokki ML, Mitchell BD, Navis G, Nieminen MS, Ohlsson C, Poulter NR, Qi L, Raitakari OT, Rimm EB, Rioux JD, Rizzi F, Rudan I, Salomaa V, Sever PS, Shields DC, Shuldiner AR, Sinisalo J, Stanton AV, Stolk RP, Strachan DP, Tardif JC, Thorsteinsdottir U, Tuomilehto J, van Veldhuisen DJ, Virtamo J, Viikari J, Vollenweider P, Waeber G, Widen E, Cho YS, Olsen JV, Visscher PM, Willer C, Franke L, Erdmann J, Thompson JR, Pfeufer A, Sotoodehnia N, Newton-Cheh C, Ellinor PT, Stricker BH, Metspalu A, Perola M, Beckmann JS, Smith GD, Stefansson K, Wareham NJ, Munroe PB, Sibon OC, Milan DJ, Snieder H, Samani NJ, Loos. Identification of heart rate-associated loci and their effects on cardiac conduction and rhythm disorders. Nat Genet 45: 621–631, 2013. doi: 10.1038/ng.2610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hibino H, Inanobe A, Furutani K, Murakami S, Findlay I, Kurachi Y. Inwardly rectifying potassium channels: their structure, function, and physiological roles. Physiol Rev 90: 291–366, 2010. doi: 10.1152/physrev.00021.2009. [DOI] [PubMed] [Google Scholar]
- 12.Hirose M, Carlson MD, Laurita KR. Cellular mechanisms of vagally mediated atrial tachyarrhythmia in isolated arterially perfused canine right atria. J Cardiovasc Electrophysiol 13: 918–926, 2002. doi: 10.1046/j.1540-8167.2002.00918.x. [DOI] [PubMed] [Google Scholar]
- 13.Hirose M, Leatmanoratn Z, Laurita KR, Carlson MD. Partial vagal denervation increases vulnerability to vagally induced atrial fibrillation. J Cardiovasc Electrophysiol 13: 1272–1279, 2002. doi: 10.1046/j.1540-8167.2002.01272.x. [DOI] [PubMed] [Google Scholar]
- 14.Huang CL, Slesinger PA, Casey PJ, Jan YN, Jan LY. Evidence that direct binding of Gβγ to the GIRK1 G protein-gated inwardly rectifying K+ channel is important for channel activation. Neuron 15: 1133–1143, 1995. doi: 10.1016/0896-6273(95)90101-9. [DOI] [PubMed] [Google Scholar]
- 15.Ivanina T, Varon D, Peleg S, Rishal I, Porozov Y, Dessauer CW, Keren-Raifman T, Dascal N. Galphai1 and Galphai3 differentially interact with, and regulate, the G protein-activated K+ channel. J Biol Chem 279: 17260–17268, 2004. doi: 10.1074/jbc.M313425200. [DOI] [PubMed] [Google Scholar]
- 16.Jiang M, Spicher K, Boulay G, Martín-Requero A, Dye CA, Rudolph U, Birnbaumer L. Mouse gene knockout and knockin strategies in application to α subunits of Gi/Go family of G proteins. Methods Enzymol 344: 277–298, 2002. doi: 10.1016/S0076-6879(02)44721-0. [DOI] [PubMed] [Google Scholar]
- 17.Kahr PC, Piccini I, Fabritz L, Greber B, Schöler H, Scheld HH, Hoffmeier A, Brown NA, Kirchhof P. Systematic analysis of gene expression differences between left and right atria in different mouse strains and in human atrial tissue. PLoS One 6: e26389, 2011. doi: 10.1371/journal.pone.0026389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kovoor P, Wickman K, Maguire CT, Pu W, Gehrmann J, Berul CI, Clapham DE. Evaluation of the role of IKACh in atrial fibrillation using a mouse knockout model. J Am Coll Cardiol 37: 2136–2143, 2001. doi: 10.1016/S0735-1097(01)01304-3. [DOI] [PubMed] [Google Scholar]
- 19.Krapivinsky G, Gordon EA, Wickman K, Velimirović B, Krapivinsky L, Clapham DE. The G-protein-gated atrial K+ channel IKACh is a heteromultimer of two inwardly rectifying K+-channel proteins. Nature 374: 135–141, 1995. doi: 10.1038/374135a0. [DOI] [PubMed] [Google Scholar]
- 20.Lakatta EG, Maltsev VA, Vinogradova TM. A coupled SYSTEM of intracellular Ca2+ clocks and surface membrane voltage clocks controls the timekeeping mechanism of the heart’s pacemaker. Circ Res 106: 659–673, 2010. doi: 10.1161/CIRCRESAHA.109.206078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Leaney JL, Milligan G, Tinker A. The G protein α subunit has a key role in determining the specificity of coupling to, but not the activation of, G protein-gated inwardly rectifying K+ channels. J Biol Chem 275: 921–929, 2000. doi: 10.1074/jbc.275.2.921. [DOI] [PubMed] [Google Scholar]
- 22.Leaney JL, Tinker A. The role of members of the pertussis toxin-sensitive family of G proteins in coupling receptors to the activation of the G protein-gated inwardly rectifying potassium channel. Proc Natl Acad Sci USA 97: 5651–5656, 2000. doi: 10.1073/pnas.080572297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li N, Csepe TA, Hansen BJ, Sul LV, Kalyanasundaram A, Zakharkin SO, Zhao J, Guha A, Van Wagoner DR, Kilic A, Mohler PJ, Janssen PM, Biesiadecki BJ, Hummel JD, Weiss R, Fedorov VV. Adenosine-induced atrial fibrillation: localized reentrant drivers in lateral right atria due to heterogeneous expression of adenosine A1 receptors and GIRK4 subunits in the human heart. Circulation 134: 486–498, 2016. doi: 10.1161/CIRCULATIONAHA.115.021165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liu GX, Derst C, Schlichthörl G, Heinen S, Seebohm G, Brüggemann A, Kummer W, Veh RW, Daut J, Preisig-Müller R. Comparison of cloned Kir2 channels with native inward rectifier K+ channels from guinea-pig cardiomyocytes. J Physiol 532: 115–126, 2001. doi: 10.1111/j.1469-7793.2001.0115g.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Logothetis DE, Kurachi Y, Galper J, Neer EJ, Clapham DE. The βγ subunits of GTP-binding proteins activate the muscarinic K+ channel in heart. Nature 325: 321–326, 1987. doi: 10.1038/325321a0. [DOI] [PubMed] [Google Scholar]
- 26.Lomax AE, Rose RA, Giles WR. Electrophysiological evidence for a gradient of G protein-gated K+ current in adult mouse atria. Br J Pharmacol 140: 576–584, 2003. doi: 10.1038/sj.bjp.0705474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lüscher C, Slesinger PA. Emerging roles for G protein-gated inwardly rectifying potassium (GIRK) channels in health and disease. Nat Rev Neurosci 11: 301–315, 2010. doi: 10.1038/nrn2834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Machhada A, Ang R, Ackland GL, Ninkina N, Buchman VL, Lythgoe MF, Trapp S, Tinker A, Marina N, Gourine AV. Control of ventricular excitability by neurons of the dorsal motor nucleus of the vagus nerve. Heart Rhythm 12: 2285–2293, 2015. doi: 10.1016/j.hrthm.2015.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mangoni ME, Traboulsie A, Leoni AL, Couette B, Marger L, Le Quang K, Kupfer E, Cohen-Solal A, Vilar J, Shin HS, Escande D, Charpentier F, Nargeot J, Lory P. Bradycardia and slowing of the atrioventricular conduction in mice lacking CaV3.1/α1G T-type calcium channels. Circ Res 98: 1422–1430, 2006. doi: 10.1161/01.RES.0000225862.14314.49. [DOI] [PubMed] [Google Scholar]
- 30.Mesirca P, Marger L, Toyoda F, Rizzetto R, Audoubert M, Dubel S, Torrente AG, Difrancesco ML, Muller JC, Leoni AL, Couette B, Nargeot J, Clapham DE, Wickman K, Mangoni ME. The G-protein-gated K+ channel, IKACh, is required for regulation of pacemaker activity and recovery of resting heart rate after sympathetic stimulation. J Gen Physiol 142: 113–126, 2013. doi: 10.1085/jgp.201310996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nobles M, Sebastian S, Tinker A. HL-1 cells express an inwardly rectifying K+ current activated via muscarinic receptors comparable to that in mouse atrial myocytes. Pflugers Arch 460: 99–108, 2010. doi: 10.1007/s00424-010-0799-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nygren A, Lomax AE, Giles WR. Heterogeneity of action potential durations in isolated mouse left and right atria recorded using voltage-sensitive dye mapping. Am J Physiol Heart Circ Physiol 287: H2634–H2643, 2004. doi: 10.1152/ajpheart.00380.2004. [DOI] [PubMed] [Google Scholar]
- 33.Opel A, Nobles M, Montaigne D, Finlay M, Anderson N, Breckenridge R, Tinker A. Absence of the regulator of G-protein signaling, RGS4, predisposes to atrial fibrillation and is associated with abnormal calcium handling. J Biol Chem 290: 19233–19244, 2015. doi: 10.1074/jbc.M115.666719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Peleg S, Varon D, Ivanina T, Dessauer CW, Dascal N. G(alpha)(i) controls the gating of the G protein-activated K+ channel, GIRK. Neuron 33: 87–99, 2002. doi: 10.1016/S0896-6273(01)00567-0. [DOI] [PubMed] [Google Scholar]
- 35.Pfaffinger PJ, Martin JM, Hunter DD, Nathanson NM, Hille B. GTP-binding proteins couple cardiac muscarinic receptors to a K channel. Nature 317: 536–538, 1985. doi: 10.1038/317536a0. [DOI] [PubMed] [Google Scholar]
- 36.Posokhova E, Ng D, Opel A, Masuho I, Tinker A, Biesecker LG, Wickman K, Martemyanov KA. Essential role of the m2R-RGS6-IKACh pathway in controlling intrinsic heart rate variability. PLoS One 8: e76973, 2013. doi: 10.1371/journal.pone.0076973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Riven I, Iwanir S, Reuveny E. GIRK channel activation involves a local rearrangement of a preformed G protein channel complex. Neuron 51: 561–573, 2006. doi: 10.1016/j.neuron.2006.08.017. [DOI] [PubMed] [Google Scholar]
- 38.Riven I, Kalmanzon E, Segev L, Reuveny E. Conformational rearrangements associated with the gating of the G protein-coupled potassium channel revealed by FRET microscopy. Neuron 38: 225–235, 2003. doi: 10.1016/S0896-6273(03)00193-4. [DOI] [PubMed] [Google Scholar]
- 39.Sakmann B, Noma A, Trautwein W. Acetylcholine activation of single muscarinic K+ channels in isolated pacemaker cells of the mammalian heart. Nature 303: 250–253, 1983. doi: 10.1038/303250a0. [DOI] [PubMed] [Google Scholar]
- 40.Sarmast F, Kolli A, Zaitsev A, Parisian K, Dhamoon AS, Guha PK, Warren M, Anumonwo JM, Taffet SM, Berenfeld O, Jalife J. Cholinergic atrial fibrillation: IK,ACh gradients determine unequal left/right atrial frequencies and rotor dynamics. Cardiovasc Res 59: 863–873, 2003. doi: 10.1016/S0008-6363(03)00540-6. [DOI] [PubMed] [Google Scholar]
- 41.Sebastian S, Ang R, Abramowitz J, Weinstein LS, Chen M, Ludwig A, Birnbaumer L, Tinker A. The in vivo regulation of heart rate in the murine sinoatrial node by stimulatory and inhibitory heterotrimeric G proteins. Am J Physiol Regul Integr Comp Physiol 305: R435–R442, 2013. doi: 10.1152/ajpregu.00037.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Slesinger PA, Reuveny E, Jan YN, Jan LY. Identification of structural elements involved in G protein gating of the GIRK1 potassium channel. Neuron 15: 1145–1156, 1995. doi: 10.1016/0896-6273(95)90102-7. [DOI] [PubMed] [Google Scholar]
- 42a.Sowell MO, Ye C, Ricupero DA, Hansen S, Quinn SJ, Vassilev PM, Mortensen RM. Targeted inactivation of αi2 or αi3 disrupts activation of the cardiac muscarinic K+ channel, IK+Ach, in intact cells. Proc Natl Acad Sci USA 94:7921-7926, 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wickman K, Nemec J, Gendler SJ, Clapham DE. Abnormal heart rate regulation in GIRK4 knockout mice. Neuron 20: 103–114, 1998. doi: 10.1016/S0896-6273(00)80438-9. [DOI] [PubMed] [Google Scholar]
- 44.Wickman KD, Iñiguez-Lluhl JA, Davenport PA, Taussig R, Krapivinsky GB, Linder ME, Gilman AG, Clapham DE. Recombinant G-protein βγ-subunits activate the muscarinic-gated atrial potassium channel. Nature 368: 255–257, 1994. doi: 10.1038/368255a0. [DOI] [PubMed] [Google Scholar]
- 45.Zaritsky JJ, Redell JB, Tempel BL, Schwarz TL. The consequences of disrupting cardiac inwardly rectifying K+ current (IK1) as revealed by the targeted deletion of the murine Kir2.1 and Kir2.2 genes. J Physiol 533: 697–710, 2001. doi: 10.1111/j.1469-7793.2001.t01-1-00697.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zuberi Z, Birnbaumer L, Tinker A. The role of inhibitory heterotrimeric G proteins in the control of in vivo heart rate dynamics. Am J Physiol Regul Integr Comp Physiol 295: R1822–R1830, 2008. doi: 10.1152/ajpregu.90625.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zuberi Z, Nobles M, Sebastian S, Dyson A, Lim SY, Breckenridge R, Birnbaumer L, Tinker A. Absence of the inhibitory G-protein Gαi2 predisposes to ventricular cardiac arrhythmia. Circ Arrhythm Electrophysiol 3: 391–400, 2010. doi: 10.1161/CIRCEP.109.894329. [DOI] [PMC free article] [PubMed] [Google Scholar]