

Abstract
Blackburn MB, Andrade MA, Toney GM. Hypothalamic PVN contributes to acute intermittent hypoxia-induced sympathetic but not phrenic long-term facilitation. J Appl Physiol 124: 1233–1243, 2018. First published December 19, 2017; doi:10.1152/japplphysiol.00743.2017.— Acute intermittent hypoxia (AIH) repetitively activates the arterial chemoreflex and triggers a progressive increase of sympathetic nerve activity (SNA) and phrenic nerve activity (PNA) referred to as sympathetic and phrenic long-term facilitation (S-LTF and P-LTF), respectively. Neurons of the hypothalamic paraventricular nucleus (PVN) participate in the arterial chemoreflex, but their contribution to AIH-induced LTF is unknown. To determine this, anesthetized rats were vagotomized and exposed to 10 cycles of AIH, each consisting of ventilation for 3 min with 100% O2 followed by 3 min with 15% O2. Before AIH, rats received bilateral PVN injections of artificial cerebrospinal fluid (aCSF; vehicle) or the GABA-A receptor agonist muscimol (100 pmol in 50 nl) to inhibit neuronal activity. Thirty minutes after completing the AIH protocol, during which rats were continuously ventilated with 100% O2, S-LTF and P-LTF were quantified from recordings of integrated splanchnic SNA and PNA, respectively. PVN muscimol attenuated increases of SNA during hypoxic episodes occurring in later cycles (6–10) of AIH (P < 0.03) and attenuated post-AIH S-LTF (P < 0.001). Muscimol, however, did not consistently affect peak PNA responses during hypoxic episodes and did not alter AIH-induced P-LTF. These findings indicate that PVN neuronal activity contributes to sympathetic responses during AIH and to subsequent generation of S-LTF.
NEW & NOTEWORTHY Neural circuits mediating acute intermittent hypoxia (AIH)-induced sympathetic and phrenic long-term facilitation (LTF) have not been fully elucidated. We found that paraventricular nucleus (PVN) inhibition attenuated sympathetic activation during episodes of AIH and reduced post-AIH sympathetic LTF. Neither phrenic burst patterning nor the magnitude of AIH-induced phrenic LTF was affected. Findings indicate that PVN neurons contribute to AIH-induced sympathetic LTF. Defining mechanisms of sympathetic LTF could improve strategies to reduce sympathetic activity in cardiovascular and metabolic diseases.
Keywords: arterial blood pressure, phrenic nerve activity, respiratory-sympathetic coupling, sympathetic nerve activity, synaptic plasticity
INTRODUCTION
Exposure to acute intermittent hypoxia (AIH) is an established means of inducing a form of respiratory neuroplasticity referred to long-term facilitation (LTF) (11, 31). Perhaps the most studied form of AIH-induced LTF is phrenic LTF (P-LTF) in the anesthetized animal (7, 9, 11, 31), which is characterized by a progressive increase of neural inspiratory drive indexed as exaggerated phrenic nerve activity (PNA) lasting for at least several hours after resuming normoxic ventilation. Because early AIH studies demonstrated that P-LTF requires repeated activation of arterial chemoreceptor inputs to the respiratory network and activation of Gq-coupled 5-HT1A/2A receptors in the spinal cord (31, 34), it is generally considered that the progressive post-AIH increase of PNA depends on synaptic and possibly intrinsic plasticity among inspiratory phrenic motor neurons (11).
The respiratory rhythm-generating network strongly entrains the sympathetic rhythm-generating network (32, 45), and hence, early evidence showed that intermittent hypoxia consistently induces P-LTF, as well as sympathetic LTF (S-LTF) (7, 9). The coincidence of P-LTF and S-LTF together with evidence that both are prevented by 5-HT2 receptor blockade (9) led to the conclusion that S-LTF likely occurs secondary to P-LTF and reflects amplification of respiratory network-driven bursts of sympathetic nerve activity (SNA) (9). This interpretation was consistent with then available data, but it was later appreciated that AIH can induce S-LTF without concurrent P-LTF, indicating that nonidentical neural substrates and/or mechanisms underlie S-LTF and P-LTF (49). Still more recently, it was reported that intermittent optogenetic activation of the nucleus tractus solitarius (NTS) without exposure to AIH induces both S- and P-LTF (51), indicating that intermittent depolarization/spiking of putative chemoreceptor sensitive circuitry within and/or downstream of the NTS is sufficient to induce LTF plasticity.
NTS neurons project widely in the central nervous system (CNS) and synapse within multiple brain regions involved in respiratory and sympathetic control (47). There are consequently multiple synaptic targets of NTS that could participate in generating S-LTF and P-LTF signals and transmitting these signals to sympathetic and inspiratory motor neurons. The hypothalamic paraventricular nucleus (PVN) is a major synaptic target of NTS projection neurons (39), and parvocellular PVN neurons dynamically modulate sympathetic and respiratory motor outflows through their brain stem and spinal projections (1, 16, 19, 52). Of special note is that PVN neurons not only participate in the inspiratory and sympathetic limbs of the arterial chemoreflex (37, 38), but also in neurogenic (sympathetic) hypertension (2, 26), including the hypertension induced by chronic intermittent hypoxia (18, 30, 33, 43, 44, 53)—a form of hypertension that critically depends on exaggerated arterial chemoreflex sympathoexcitation (35, 54), but not baroreflex dysfunction (53).
In light of the preceding evidence, the present study was performed to test the hypothesis that PVN neuronal activation during AIH contributes to subsequent generation of S-LTF and P-LTF. Findings indicate that chemical inhibition of the PVN before AIH consistently attenuated sympathetic and variably reduced peak neural inspiratory activation during hypoxic episodes of AIH, suggesting that PVN neurons more robustly contribute to the sympathetic than the inspiratory limb of the arterial chemoreflex during repeated exposures to hypoxia. PVN inhibition also attenuated the development of post-AIH S-LTF, but not the LTF of integrated PNA, indicating that PVN neurons rather selectively participate in the neuroplasticity of AIH-induced S-LTF.
MATERIALS AND METHODS
Ethical approval.
All experimental procedures conformed to the National Institutes of Health “Guide for the Care and Use of Laboratory Animals” and were approved by the University of Texas Health Science Center at San Antonio Animal Care and Use Committee.
Animals.
Male Sprague-Dawley rats (350–450 g; Charles River Laboratories) were housed in a temperature-controlled room (22 ± 1°C) with a 14:10-h light-dark cycle with ad libitum access to standard chow and deionized water.
General procedures.
Rats were anesthetized by intraperitoneal injection of urethane (750 mg/kg) and α-chloralose (75 mg/kg). Polyethylene catheters (PE-50 tubing) were implanted in a femoral artery and vein to record arterial blood pressure (ABP) and administer drugs, respectively. A lead 1 ECG was recorded. To prevent respiratory network entrainment to the pattern of artificial ventilation (see below), vagus nerves were transected at the midcervical level, leaving aortic depressor nerves intact. After tracheal cannulation, rats were artificially ventilated with 100% O2 and paralyzed with gallamine triethiodide (20 mg/ml, 0.25 ml/h iv). End-tidal CO2 was continuously recorded and maintained between 4.5 and 5%. Adequacy of anesthesia was also continuously monitored by stability of expiratory CO2 and systolic arterial pressure, and by periodic (~15-min intervals) noxious pinching of a forepaw to check for the lack of a limb withdrawal reflex before paralysis and for the lack of pressor or sympathoexcitatory responses thereafter. Supplemental anesthesia (10% of initial dose) was given as needed. Body temperature was maintained at 37 ± 0.5°C.
Recording phrenic and sympathetic nerve activity.
Rats were prepared for recording of PNA and splanchnic sympathetic nerve activity (SNA), as previously described (3, 16). Briefly, PNA was recorded by incising the skin and muscle overlying the left scapula, which was retracted to expose the phrenic nerve near the brachial plexus. The phrenic nerve was isolated and transected, and its proximal end was placed on a bipolar silver-wire electrode. To record splanchnic SNA, the left greater splanchnic nerve was exposed through a retroperitoneal incision and placed on a bipolar silver-wire electrode. Signals were full-wave rectified and smoothed using a moving average algorithm offline. The smoothing time constant was ±30 s for quantifying LTF responses of SNA and PNA. A smoothing time constant of ±30 ms was used to resolve individual PNA bursts to quantify changes of burst frequency and amplitude (Figs. 2, 3A, and 4A).
Fig. 2.

Effects of PVN inhibition on PNA responses during hypoxic and normoxic episodes of AIH. Summary data for peak changes in PNA burst frequency (A) and amplitude (B) during each of 10 AIH cycles of hypoxic (left) and normoxic (right) ventilation in aCSF- (open bars; n = 5) and muscimol-injected (solid bars; n = 6) rats. Data are expressed as means ± SE. Note: ANOVA compared absolute values (not changes) of PNA burst frequency (bursts/min) and amplitude (voltage) during each AIH cycle with corresponding values at the Pre-AIH time point. *P < 0.0001. †P < 0.0009.
Fig. 3.

Effects of PVN inhibition on the frequency, amplitude and temporal fidelity of PNA bursts before and after exposure to AIH. A: effects of PVN injections of aCSF vehicle (open bars; n = 5) and muscimol (solid bars; n = 6) on PNA burst frequency (left) and amplitude (right) at quantified time points (see methods) before and after AIH. B: Poincare plots showing effects of PVN inhibition with muscimol on average PNA interburst interval at quantified time points (see methods) before and after AIH exposure. *P < 0.05 vs. within group Pre-AIH time point. ‡P < 0.05, †P = 0.0008 vs. corresponding time point in aCSF group.
Fig. 4.

Effects of PVN inhibition on splanchnic SNA and MAP responses during hypoxic and normoxic episodes of AIH. A: peak changes in splanchnic SNA (top) and MAP (bottom) during hypoxic and normoxic episodes of 10 cycles of AIH in rats receiving PVN injections of aCSF (open bars; n = 7) or muscimol (solid bars; n = 6) B: relationship between splanchnic SNA and MAP changes during hypoxic and normoxic episodes of 10 cycles of AIH. Note: ANOVA compared absolute values (not changes) of splanchnic SNA (voltage) and MAP (mmHg) during each AIH cycle to corresponding values at the Pre-AIH time point. *P < 0.03 vs. within-group Pre-AIH average. †P < 0.03 vs. corresponding cycle number in aCSF group. ‡P < 0.0001 vs. within group Pre-AIH average. **P < 0.05 vs. within group cycle 1 value.
PVN microinjections.
Rats were placed in a stereotaxic head frame with the skull leveled between bregma and lambda. A midline craniotomy was performed and a single-barrelled glass pipette (tip ID = 30–50 µm) was lowered into the PVN at the following coordinates referenced to bregma: 1.8 mm caudal, 7.4–7.6 mm ventral, and 0.2–0.3 mm lateral. Muscimol was dissolved in artificial cerebrospinal fluid (aCSF) comprising the following (in mM): 124 NaCl, 3 KCl, 2.4 CaCl2, 26 NaHCO3, 1.4 MgSO4, 1.4 NaH2PO4, and 11 glucose. Separate groups of rats received aCSF vehicle or muscimol (100 pmol) injections bilaterally into the PVN 20 min before initiating the AIH protocol (2). Each injection was made over a period of ~30 s using a pneumatic pump. The volume of each injection was ~50 nl, as determined by calibrated meniscus movement. Injection sites were marked with rhodamine beads (Lumafluor, Naples, FL), which were added to the injected solution in a final concentration of 0.2%.
Exposure to acute intermittent hypoxia.
To determine the role of PVN neuronal activity in mediating SNA and PNA responses during AIH and in mediating post-AIH induction of S-LTF and P-LTF, rats were ventilated with 100% O2, and baseline values of mean ABP (MAP), splanchnic SNA, and PNA were recorded for 10 min. Muscimol or aCSF was then bilaterally injected into the PVN. Twenty minutes later (3, 43), rats were exposed to our AIH protocol, which consisted of 10 cycles of ventilation alternating every 3 min between 15% O2 (balance N2) and 100% O2. Following AIH, rats were again continuously ventilated with 100% O2, and variables were recorded for an additional 30 min to monitor development of post-AIH LTF. It should be stressed that no standard AIH protocol has been established for investigation of S-LTF (9, 34, 49). Here, hypoxic ventilation for 3 min was selected to mimic patterning used previously in our studies of chronic intermittent hypoxia (42, 43). Ventilation with 15% O2 was selected to ensure robust arterial chemoreflex activation while allowing for MAP to be stably maintained throughout the post-AIH period of LTF development.
Histology.
Injection sites were determined as previously described (42, 43). Briefly, brains were removed at the conclusion of experiments and postfixed in 4% paraformaldehyde for 3–6 days. After being cryoprotected in 30% sucrose-PBS, brains were cut using a sliding microtome into 50-µm-thick sections through the PVN. Rhodamine beads were viewed through a fluorescence microscope equipped with a TRITC filter set (590–650 nm), and their location was mapped bilaterally in relation to the PVN.
Data analysis.
Responses of splanchnic SNA were quantified as 2-min averages with noise subtracted. Noise was determined as a 5-min average of integrated voltage recorded after bolus injection of the ganglionic blocker hexamethonium (30 mg/kg iv). Data were statistically compared at the following time points 1): baseline (BL, before PVN injections), 2) pre-AIH (after PVN injections and immediately before AIH), 3) post-AIH (within 3 min of completing the AIH protocol), and 4) LTF (30 min after completing the AIH protocol). To assess MAP, splanchnic SNA, PNA burst frequency, and amplitude responses during individual cycles of AIH, the average value across a 30-s data segment centered on the peak change was analyzed. Each cycle average was compared with the 2-min baseline average determined after PVN injection of muscimol/vehicle at the Pre-AIH time point.
PNA burst amplitudes were quantified from PNA burst-triggered averages of PNA using 2-min data segments at the four time points analyzed before and after AIH and using 30-s data segments within each hypoxic and normoxic episode of AIH. Peak amplitudes were taken as the voltage difference between the expiratory phase average (pretrigger baseline) and peak value of the posttrigger PNA average, as previously reported (16). PNA burst frequency was determined as the mean frequency of phrenic onset events during the same four analyzed time points before and after AIH and during each of the 10 hypoxia and normoxia cycles of AIH. PNA burst amplitudes are expressed in microvolts, and burst frequency is expressed as bursts per minute, as previously described (22–24).
To determine effects of AIH and PVN inhibition on the fidelity of PNA burst timing, Poincarè plots were constructed using PNA burst interval data from PVN vehicle and muscimol-injected rats at the four indicated time points. Poincarè plots are return maps constructed as x-y plots in which the x coordinate of each point is the duration of the current PNA interval (n), and the corresponding y coordinate is the duration of the subsequent PNA interval (n + 1). The dispersion of points above and below the line of identity (m = 1) is quantified as the standard deviation 1 (SD1) and indexes the average timing variability between consecutive PNA bursts (short-term interval variability) at each analyzed time point among rats comprising each treatment group (aCSF vs. muscimol). The same number of intervals was analyzed at each time point in each group (n = 57). SD1 is indicated by the width of an oval extending above and below the line of identity. Points distributed along the line of identity index PNA-PNA interval variability among all intervals recorded among subjects within a group without regard to their sequence (longer-term interval variability). This variability is quantified as SD2 and is indexed by the length of the oval placed along the line of identity. Variance ovals are centered on the line of identity and placed at the average value of all PNA intervals within each group. Average interval values are represented by dashed lines transecting each oval and running perpendicular to the line of identity (12).
To assess how the change of SNA grew or shrank across the cycles of AIH relative to the prevailing level of MAP, a ratio was calculated with the numerator holding the percent change of SNA and the denominator holding the MAP response, with changes in both variables being relative to their Pre-AIH averages. Changes of SNA and MAP during each AIH cycle were separately normalized to their cycle 1 averages before calculation of the SNA:MAP ratio (see Fig. 4B).
Statistics.
Absolute values of MAP, integrated splanchnic SNA, as well as PNA burst frequency and amplitude at each of the four quantified time points (see Data analysis above), were compared across groups receiving PVN injections of aCSF and muscimol using two-factor repeated-measures ANOVA. Within- and across-group comparisons of AIH cycle-specific responses during episodes of hypoxic and hyperoxic ventilation were compared using one- or two-factor repeated-measures ANOVA. Effects on MAP within each of the 10 cycles of hypoxic/hyperoxic cycles were determined by subtracting cycle-specific MAP values from the Pre-AIH averages of MAP. When significant F values in one-factor ANOVA and significant time-PVN treatment interactions in two-factor ANOVA were achieved, post hoc pairwise comparisons were performed using independent t-tests with layered Bonferroni corrections. All tests were performed using Prism v7.02 (GraphPad Software). An a priori critical value of P < 0.05 was deemed statistically significant. Summary data in figures are expressed as means ± SE.
RESULTS
Effects of PVN inhibition on hemodynamic, SNA, and PNA responses to AIH.
Figure 1A shows representative responses to AIH following bilateral PVN injections of aCSF (left) or the GABA-A receptor agonist muscimol (right). In the aCSF-injected rat, each episode of hypoxic ventilation with 15% O2 increased integrated splanchnic SNA (Fig. 1A, middle trace) and PNA (Fig. 1A, bottom trace), consistent with arterial chemoreflex activation. These were accompanied by reductions of ABP (Fig. 1A, top trace), which is consistent with systemic hypoxia-induced vasodilation. Expanded traces show ~6 s of raw splanchnic SNA and integrated PNA at the Pre-AIH time point, during the 10th hypoxic episode of AIH, and at the LTF time point 30 min after AIH.
Fig. 1.

Effects of paraventricular nucleus (PVN) inhibition on arterial blood pressure, splanchnic sympathetic nerve activity (SNA), and phrenic nerve activity (PNA) responses during and after exposure to acute intermittent hypoxia (AIH). A: representative responses of arterial blood pressure (top), integrated splanchnic SNA (middle), and integrated PNA (bottom) before, during, and after exposure to 10 cycles of AIH in separate rats that received either PVN injections of artificial cerebrospinal fluid (aCSF; left) or the GABA-A receptor agonist muscimol (right). B: summary data of mean arterial pressure (MAP), integrated splanchnic SNA and PNA responses at quantified time points (see methods) before and after AIH in control (open bars; n = 5–7) and muscimol (solid bars; n = 6) injected rats. Data are expressed as means ± SE and compared by one- or two-way ANOVA with Bonferroni post hoc comparison tests. *P < 0.05 vs. baseline. †P < 0.001 vs. aCSF at corresponding time point.
During episodes of normoxic ventilation with 100% O2, splanchnic SNA and PNA each returned toward baseline from elevated levels during hypoxia, consistent with diminished arterial chemoreceptor input. Note, however, that as the AIH protocol progressed, splanchnic SNA progressively failed to return fully to baseline during normoxic episodes (stippled area below trace). Meanwhile, ABP rebounded during normoxic episodes to values that exceeded baseline, suggesting that compensatory pressor mechanisms were activated during hypoxic episodes and were sufficient to support “overshoots” of ABP during subsequent normoxic episodes.
After AIH, the aCSF-injected rat showed robust S-LTF and P-LTF, as indicated by progressive ramp-like increases (gray wedges) of splanchnic SNA (Fig. 1A, middle trace) and PNA (Fig. 1A, bottom trace) during the 30-min period following AIH. By contrast, ABP remained stable after AIH, albeit slightly below the pre-AIH baseline, indicating that S-LTF and P-LTF responses were not associated with progressive unloading of arterial baroreceptors.
Representative traces recorded after PVN injections of muscimol (Fig. 1A, right) show that episodes of hypoxia were accompanied by increases of integrated PNA (Fig. 1A, bottom traces, right vs. left) as a measure of neural minute ventilation. PNA responses during AIH cycles appeared more variable in the muscimol injected rat (right), and this was consistently observed in the PVN inhibited group. PVN muscimol more robustly attenuated increases of splanchnic SNA that occurred during bouts of hypoxia (Fig. 1A, middle traces, right vs. left). These blunted increases of SNA were associated with failure of ABP to “overshoot” baseline during subsequent episodes of normoxia (Fig. 1A, top traces, right vs. left).
Summary data in Fig. 1B were analyzed by repeated-measures two-factor ANOVA. For MAP responses (Fig. 1B, right), significant effects of time (P < 0.0001) and PVN treatment (aCSF vs. muscimol, P = 0.026) were achieved, and there was a significant time-treatment interaction (P = 0.0006). For aCSF-injected rats (open bars), MAP was unchanged at measured time points before and after AIH, but fell progressively in muscimol-injected rats (solid bars), reaching significance relative to baseline both at the post-AIH and LTF time points (P < 0.05). Across-group comparisons revealed that PVN muscimol also significantly reduced MAP at the Post-AIH and LTF time points relative to corresponding time points in the aCSF-injected group (P < 0.001). Statistical analysis of splanchnic SNA data (Fig. 1B, left) likewise revealed significant effects of time (P < 0.0001) and PVN treatment (P = 0.024) and a significant time-treatment interaction (P = 0.018), with pairwise analysis indicating that splanchnic SNA in the aCSF-injected group significantly increased relative to baseline at the LTF time point (P < 0.05) and that PVN muscimol significantly reduced the LTF level of splanchnic SNA relative to aCSF-injected controls (P < 0.001). Analysis of integrated PNA by two-factor ANOVA indicated significant variation in time only (P = 0.0001). Within-group, one-factor ANOVA indicated that integrated PNA at the LTF time point was significantly increased relative to baseline in rats that received PVN injections of aCSF (P < 0.05) or muscimol (P < 0.05).
Effects of PVN inhibition on phrenic burst discharge during cycles of AIH.
The above responses of integrated PNA indicate that AIH strongly modulated neural inspiration, as expected. Contrary to our expectation, data further indicate that PVN neuronal activity was not required for peak PNA responses post-AIH, which prompted further analysis to assess the contribution of PVN neuronal activity to frequency and amplitude responses of PNA within specific episodes of hypoxia and normoxia during exposure to AIH (Fig. 2). The latter was accomplished by integrating the raw PNA signal with a faster time constant (±30 ms). Analysis of PNA burst frequency data by repeated-measures, two-factor ANOVA revealed a significant effect of time (i.e., cycle number, P < 0.0001), but not of PVN treatment (P = 0.5636), and a significant time-treatment interaction (P = 0.0041). Pairwise comparisons revealed that peak PNA burst frequency significantly increased relative to the Pre-AIH level during all hypoxic episodes of AIH (P < 0.0001) in aCSF- (open bars) and muscimol-injected (solid bars) rats (Fig. 2A, left). Analysis of PNA burst frequency during normoxic episodes (Fig. 2A, right) again indicated a significant effect of time/cycle number (P < 0.0001), but not of PVN treatment (P = 0.1594), and no time-treatment interaction (P = 0.4765). Within-group, one-factor ANOVA applied to PNA burst frequency data during normoxic episodes failed to identify specific changes relative to the Pre-AIH level in either aCSF- (P = 0.0678) or muscimol-injected (P = 0.0576) rats. Figure 2B shows peak changes of PNA burst amplitude during hypoxic (left) and normoxic (right) episodes of AIH. Repeated-measures, two-factor ANOVA comparing PNA burst amplitudes during hypoxic episodes relative to the Pre-AIH level revealed significant effects of time/AIH cycle number (P = 0.0363), but not of PVN treatment (P = 0.9927), and a significant time-PVN treatment interaction (P = 0.0037). Pairwise comparisons revealed that burst amplitude was significantly increased only in the aCSF group and only during the hypoxic episode of AIH cycle 1 (P < 0.009). Analysis of PNA amplitudes during normoxic episodes revealed an effect of time/AIH cycle number (P = 0.008), but not of PVN treatment (P < 0.9214), and no time-PVN treatment interaction (P = 0.1791). Within-group, one-factor ANOVA indicated that normoxic levels of PNA burst amplitude were unchanged as a function of time/AIH cycle number both in aCSF- (open bars, P = 0.1396) and muscimol-injected (solid bars; P = 0.2869) rats. Collectively, data in Fig. 2 indicate that PVN neuronal activity does not contribute significantly to increased frequency or peak strength of neural inspiration during acute intermittent bouts of hypoxia.
Effects of PVN inhibition on phrenic burst responses before and after AIH.
Because increased integrated PNA at the LTF time point 30 min after AIH (see Fig. 1B, center) could reflect increased PNA burst frequency, amplitude, or both, further analysis was performed. Comparison of PNA burst frequency data (Fig. 3A, left) by two-factor, repeated-measures ANOVA revealed significant effects of time (P < 0.0001), PVN treatment (P = 0.0038), and a significant time-PVN treatment interaction (P = 0.0294). Pairwise comparisons revealed that PNA burst frequency increased significantly at the LTF time point relative to the Pre-AIH level only in the aCSF-injected group (open bars; P < 0.05). In the muscimol-injected group, PNA burst frequency was significantly reduced at the Post-AIH time point relative to the Pre-AIH level (solid bars; P < 0.05). Compared with corresponding time points in aCSF-injected controls, muscimol significantly reduced PNA frequency at the Pre-AIH (P < 0.05), Post-AIH (P < 0.05), and LTF (P < 0.0008) time points. Comparison of PNA burst amplitude data by repeated-measures, two-factor ANOVA (Fig. 3A, right) revealed an effect of time (P < 0.0001), but not of PVN treatment (P = 0.2883), and no time-treatment interaction (P = 0.6898). Relative to the Pre-AIH value, one-way repeated-measures ANOVA revealed that PNA burst amplitude was unchanged in the aCSF-injected group at any time point. Within the muscimol-injected group, PNA burst amplitude increased significantly relative to the Pre-AIH level both at the Post-AIH and LTF time point (P < 0.05). Collectively, data indicate that under conditions of these experiments, post-AIH P-LTF reflected increased neural minute ventilation brought about mainly by increased PNA frequency with a less robust, and statistically insignificant, increase of burst amplitude.
Effects of PVN inhibition on the fidelity of phrenic burst timing before and after AIH.
PVN neurons target numerous cell groups of the respiratory network and have the potential to influence network rhythmicity. Therefore, we evaluated effects of AIH with and without PVN inhibition on the temporal consistency of PNA burst intervals using Poincarè plots. For clarity, plots in Fig. 3B contain representative data from 3 aCSF- (open circles) and 3 muscimol-injected (solid squares) rats. Statistical comparison of group SD1 and SD2 values revealed no significant effect of time (SD1: P = 0.4322; SD2: P = 0.2124) or PVN treatment (SD1: P = 0.7265; SD2: P = 0.3225) and no time-treatment interaction (SD1: P = 286; SD2: P = 3463). As expected, baseline data (Fig. 3B, top, left) revealed that variability of intervals between consecutive PNA bursts within each group (i.e., SD1) was small (aCSF: 53 ± 10 ms; muscimol: 43 ± 2 ms) relative to the spread of burst intervals among animals of each group (i.e., SD2) (aCSF: 115 ± 18 ms; muscimol: 109 ± 18 ms). Neither PVN muscimol before AIH (Fig. 3B, top, right; Pre-AIH) nor exposure to AIH (Fig. 3B, bottom, left; Post-AIH) significantly changed SD1 or SD2 values relative to baseline (i.e., before PVN injections of aCSF or muscimol). At the LTF time point measured 30 min after AIH when significant P-LTF had developed in both groups (Fig. 3B, bottom, right), timing variability of PNA bursts was effectively unchanged within and across groups, although the average interval was significantly increased in the PVN muscimol group (P < 0.05), consistent with reduced PNA burst frequency at the LTF time point, as shown in Fig. 3A, left). These findings indicate that PVN neuronal activity retards AIH-induced neural tachypnea without altering the temporal fidelity (consistency) of inspiratory rhythmicity.
Effects of PVN inhibition on hemodynamic and sympathetic responses during AIH.
Figure 4A shows peak splanchnic SNA (top) and MAP (bottom) responses during each hypoxic (left) and normoxic (right) episode of AIH in aCSF- (open bars) and muscimol-injected (solid bars) rats. Responses during each cycle were quantified relative to levels recorded at the Pre-AIH time point (i.e., ~20 min after PVN injections). Two-factor ANOVA of splanchnic SNA responses during hypoxic episodes revealed a significant effect of time/AIH cycle number (P < 0.0001), PVN treatment (P < 0.0427), and a significant time-PVN treatment interaction (P < 0.0001). Splanchnic SNA reliably increased during hypoxic episodes in aCSF-injected rats (P < 0.03), except for during cycle 1, but failed to do so in muscimol-injected rats. Across-group comparison revealed that SNA responses in muscimol-injected rats were significantly reduced compared with aCSF controls toward later cycles (6–10) of AIH (P < 0.03), suggesting that repeated bouts of hypoxia normally result in progressive recruitment of the PVN, such that presympathetic neurons play a greater sympathoexcitatory role. Analysis of splanchnic SNA data recorded during normoxic episodes of AIH (Fig. 4A, top, right) revealed a significant effect of time/AIH cycle number (P < 0.0001), but not of PVN treatment (P = 0.7321), and no time-PVN treatment interaction (P = 0.5598). One-factor, repeated-measures ANOVA indicated that splanchnic SNA was unchanged across episodes of normoxia in aCSF- and muscimol-injected rat. Analysis of MAP during hypoxic episodes revealed a significant effect of time/cycle number (P < 0.0001) with no effect of PVN treatment (P = 0.0920) and no time-treatment interaction (P = 0.9999). Within-group comparison by one-factor, repeated-measures ANOVA indicated that MAP in aCSF- and muscimol-injected rats (Fig. 4A, bottom, left) was significantly reduced relative to their Pre-AIH levels during all AIH cycles (P < 0.002), likely reflecting systemic hypoxic vasodilation. Analysis of MAP data during normoxic episodes (Fig. 4A, bottom, right) indicated a significant effect of time/AIH cycle number (P < 0.0001), but not of PVN treatment (P = 0.1145), and a significant time-treatment interaction (P = 0.0076). Within-group comparison by one-way ANOVA indicated that MAP during normoxic episodes rebounded in the aCSF-injected group to exceed baseline by 10–20 mmHg in all AIH cycles (P < 0.03). A similar response was observed in muscimol-injected rats, but the MAP overshoot was significant (P < 0.03) only during early AIH cycles (2–4). Overall, MAP responses during normoxia are consistent with lingering pressor effects of sympathetic activation and/or other vasoactive factors activated during prior bouts of hypoxia.
Relationship between arterial pressure and SNA: implications for AIH-induced S-LTF.
Because the level of splanchnic SNA during hypoxic and normoxic episodes of AIH almost certainly represents an integrated response to arterial chemoreceptor and baroreceptor activity, effects of PVN muscimol to blunt SNA responses could reflect PVN involvement in either or both of these reflexes. As shown in Fig. 4B, splanchnic SNA and MAP changes during each episode of hypoxia (left) and normoxia (right) were determined relative to their Pre-AIH values and expressed as a ratio normalized to the ratio of cycle 1 (see methods). Statistical analysis of data from hypoxic episodes by two-factor, repeated-measures ANOVA indicated a significant effect of time/cycle number (P < 0.0001), but not of PVN treatment (P = 0.3588), and no time-PVN treatment interaction (P = 0.5562). Within-group, one-factor ANOVA indicated that regardless of the magnitude of the reduction of MAP that occurred in aCSF-injected rats (open circles) during hypoxic episodes of AIH, the peak increase of splanchnic SNA was significantly increased (relative to cycle 1) during hypoxic episodes 3–10 (P < 0.05). In muscimol-injected rats (solid squares), by contrast, the SNA-MAP relationship remained constant throughout the hypoxic cycles of AIH. Statistical analysis of data from normoxic cycles (Fig. 4B, right) revealed a significant effect of time and cycle number only (P < 0.0001). One-factor ANOVA indicated that early normoxic episodes of AIH were accompanied by a below-baseline (<1.0) splanchnic SNA:MAP response ratio in both aCSF- and muscimol-injected groups, suggesting that recovery of MAP during early normoxic cycles of AIH involves factors in addition to ongoing splanchnic SNA. Residual adrenergic vasoconstriction from heightened SNA during the previous hypoxic episode seems a likely contributor, along with vascular recovery from hypoxic vasodilation (e.g., by nitric oxide). In both groups, the splanchnic SNA-MAP relation trended upward as AIH progressed, with effects reaching significance during later AIH cycles (7–10) in aCSF-injected rats only (P < 0.05). These findings suggest that the PVN is required to support enhanced SNA relative to MAP during repetitive bouts of hypoxia but that the gradual rise of splanchnic SNA in relation to blood pressure during normoxia progresses similarly with or without a functionally intact PVN.
Histology.
Injection sites marked with rhodamine beads were distributed within the histological boundary of the PVN, with no evidence of beads lining the third ventricle or in areas significantly dorsal or lateral to the PVN. As in previous studies (2, 15, 43), beads in aCSF- and muscimol-injected groups were distributed bilaterally within the dorsal cap (Fig. 5A), as well as in the central magnocellular and ventrolateral parvocellular areas throughout most of the rostrocaudal extent of the nucleus (Fig. 5B).
Fig. 5.
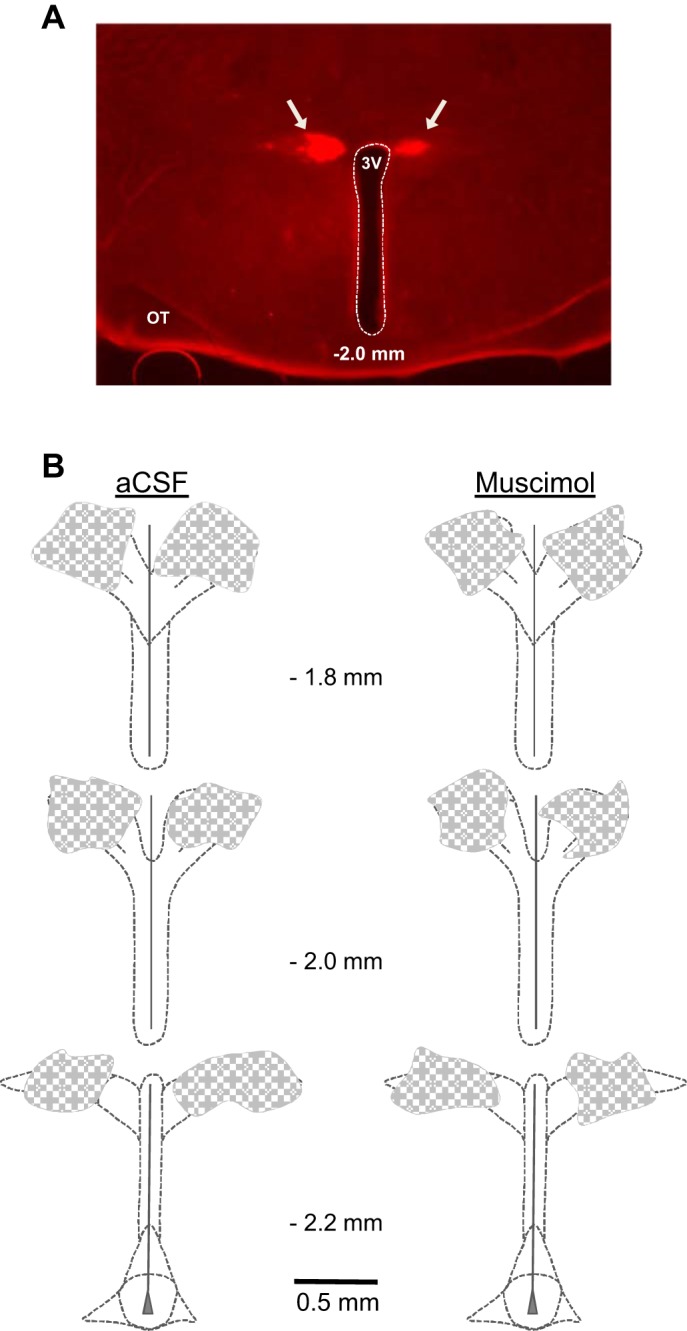
Location of PVN injection sites. A: histological section (50 µm thick) through the PVN at a rostral-mid plane showing representative distribution of rhodamine fluorescent nanospheres (arrows) marking sites of muscimol injection bilaterally into the PVN. B: summary representation of the location of aCSF (left; n = 5) and muscimol (right; n = 6) injections as indicated by the distribution of coinjected fluorescent nanospheres. Values in the center of each panel indicate the approximate rostral-caudal plane referenced to bregma. 3V, third cerebral ventricle; OT, optic tract.
DISCUSSION
After exposure to AIH and while undergoing continuous normoxic ventilation, a progressive increase of PNA and SNA can be observed in anesthetized, vagotomized, and paralyzed animals. These phenomena, referred to as phrenic and sympathetic long-term facilitation, rely on neuroplasticity mediated by incompletely understood mechanisms. Here, we extend current evidence of LTF processing by demonstrating that neuronal activity in the hypothalamic PVN contributes to AIH-induced LTF of integrated splanchnic SNA, but not of integrated PNA. We provide further evidence that PVN neuronal activity is required for full manifestation of putative arterial chemoreflex increases of splanchnic SNA while having little discernible impact on reflex increases of PNA.
Technical considerations and caveats to the interpretation of the results.
The majority of previous studies have quantified the magnitude of S-LTF and P-LTF developed over a period of 60 min or longer following exposure to AIH. Here, we chose to quantify LTF 30 min post-AIH to ensure maintenance of MAP in rats that underwent PVN inhibition because a decline of blood pressure is expected to increase SNA and PNA due to the unloading of arterial baroreceptors (48). Quantifying LTF under conditions where MAP had fallen would overestimate the contribution of LTF neuroplasticity to AIH-induced increases of SNA and PNA. As with previous studies of AIH-induced LTF in anesthetized animals (9, 17, 50), we did not observe recovery from S-LTF or P-LTF.
PVN involvement in hypoxic and normoxic episodes of AIH.
In the present study, we analyzed MAP, splanchnic SNA, and PNA responses during the individual episodes of hypoxic and hyperoxic ventilation of AIH. Consistent with arterial chemoreflex activation, hypoxic episodes were accompanied by increases of integrated splanchnic SNA and PNA (9, 13). Peak increases of SNA, but not those of PNA, were comparatively small in PVN-inhibited animals compared with their intact controls. During AIH, the frequency and amplitude of PNA bursts tended to be less in the PVN muscimol group, but the difference relative to the aCSF-injected group failed to reach statistical significance. This suggests that chemoreflex inspiratory drive is not dependent on PVN neuronal activity during repetitive episodes of hypoxia, which contrasts with evidence that the PVN contributes to increased respiratory activity in response to isolated chemoreflex activation with cyanide (37, 38) or by isolated bouts of acute hypoxia (5, 20, 21). A recent study by King et al. (21) further implicates catecholaminergic inputs to the PVN both in support of resting minute ventilation and in the increase of ventilation stimulated by a single prolonged bout of hypoxia. Collectively, data suggest that the PVN contribution to arterial chemoreflex responses could be masked in the setting of AIH by additional sympathoexcitatory and inspiratory drives.
It is noteworthy that PVN inhibition had no effect on the magnitude of MAP reductions during hypoxic episodes despite reducing increases of splanchnic SNA by nearly half. It would appear, therefore, that depressor effects of hypoxic episodes dominantly reflected direct vasodilatory responses and, moreover, that concurrent sympathetic activation was ineffective at buffering depressor responses even in PVN-intact animals. The latter could relate either to systemic hypoxia reducing sympathetic neurovascular coupling (10) or to the possibility that PVN-driven splanchnic SNA during episodes of hypoxia does not subserve vasomotor function (4, 14–16).
In the present study, during AIH, PNA declined toward baseline during episodes of normoxic ventilation consistent with an expected reduction of arterial chemoreceptor activation. Normoxia-associated recoveries of integrated PNA were largely due to reduced burst frequency, which likely reflected above-baseline arterial Po2 due to ventilation with 100% O2. This being the case, our observation that PNA burst amplitude remained relatively stable between hypoxic and normoxic episodes (see Fig. 2) is difficult to explain. It is tempting to speculate that below-baseline PNA frequency during normoxic episodes involves more than the relative silencing of arterial chemoreceptors but could also involve increased activity of arterial baroreceptors (29, 48). The latter is consistent with the fact that MAP increased during normoxic episodes to overshoot baseline. Another possible explanation for below-baseline PNA frequency during normoxic episodes is the activation of lateral pontine mechanisms mediating posthypoxia frequency decline (PHFD), as described by Coles and Dick (6). To the extent that PHFD mechanisms contribute to the reduction of PNA frequency during normoxic cycles, it is interesting that frequency reductions gradually decayed as AIH cycles progressed. This suggests that PHFD may have gradually failed with advancing cycles of AIH, suggesting a possible disruption of lateral pontine respiratory control. PVN muscimol largely prevented this decay, raising the possibility that PVN activation during AIH could actively interfere with pontine mechanisms that mediate PHFD.
Further analysis of PNA burst intervals indicated that PVN neuronal activity, despite being capable of eliciting tachypnea (18, 52), was largely inconsequential for maintaining the fidelity of rhythmic discharge among central inspiratory neurons. Thus, synaptic interplay among components of the respiratory rhythm-generating network appeared to be unaltered by our AIH protocol and relatively unaffected by the level of PVN neuronal activity before, during, or shortly after AIH exposure.
Upon returning to normoxic ventilation during AIH, we observed that elevated splanchnic SNA failed to return fully to baseline in aCSF-injected control animals. This may have contributed to MAP overshoots during these normoxic intervals. The degree to which SNA remained above baseline during normoxic episodes gradually increased during later cycles of AIH, giving the appearance that S-LTF began after only a few cycles of AIH. Rapid onset LTF has been previously reported (9, 34, 49, 51). PVN inhibition had little impact on recovery of PNA during normoxic episodes of AIH but tended to compromise concurrently recorded SNA and MAP responses. The latter indicates that whereas PVN neuronal activity is not the sole or dominant determinant of SNA or MAP during normoxic episodes of AIH, it, nevertheless, participates in the regulation of these variables, especially during later normoxic cycles of our AIH protocol.
Gradual recruitment of PVN by repeated cycles of intermittent hypoxia.
As noted, cycles of AIH in our study were accompanied by large and repeated reductions of MAP. Because of a trend for splanchnic SNA to progressively fail to return fully to baseline during normoxic episodes of AIH, a pattern not observed in PVN-inhibited animals, it appears that PVN activation may increase progressively with increasing cycles of intermittent hypoxia. A possible explanation for this could be that PVN neurons are gradually driven to higher and higher discharge by arterial chemoreceptor inputs due to gradually increasing hypoxemia, which seems a possibility since our vagotomized artificially ventilated rats were prevented from hyperventilating in proportion to their degree of hypoxemia. An alternative explanation is that increased SNA to the kidneys together with depressor responses during preceding bouts of hypoxia could have progressively stimulated the peripheral renin-angiotensin-aldosterone system (RAAS) (46). Increased RAAS activity is well known to activate PVN presympathetic neurons through descending inputs from the forebrain lamina terminalis (8, 27, 28, 40, 44). Therefore, it is tempting to speculate that progressive recruitment of the PVN during exposure to AIH could reflect gradually increasing RAAS activation of SNA, which could operate together with or instead of gradually increasing arterial chemoreceptor activation at later cycles of AIH. It should also be stressed that acute hypoxia and hypotension strongly stimulate the hypothalamo-pituitary-adrenal stress axis (5, 36), and the resulting increase of plasma corticosterone could have contributed to net increases of SNA by attenuating arterial baroreflex transmission through the NTS (41).
Whichever factors account for gradually increasing SNA during AIH, normalizing the recorded increases of SNA to concurrent reductions of MAP during hypoxic episodes revealed that the relationship between splanchnic SNA and MAP responses increased early during AIH and remain stable thereafter. This increase of SNA relative to MAP during hypoxia failed to develop in PVN-inhibited animals, suggesting that PVN neuronal reactivity to hypoxia increases after one or two cycles of AIH and becomes consistent thereafter. Accordingly, it appears that stably increased PVN activity during later cycles of hypoxia progressively failed to return to baseline during normoxic episodes of AIH, supporting increased splanchnic SNA relative to MAP. This indicates that in normal animals progressively greater PVN-driven SNA during later cycles of AIH aids MAP recovery (during normoxia) from repeated episodes of hypoxia.
PVN and post-AIH LTF.
Exposure to AIH was initially reported to consistently elicit both P-LTF and S-LTF (9), and as a result, AIH induced S-LTF was considered to arise secondarily to P-LTF, and to reflect increased inspiratory-sympathetic network coupling. This interpretation was reasonable in light of evidence that arterial chemoreflex sympathoexcitation normally results from enhancement of respiratory rhythmic bursting of SNA (23). It was subsequently reported that AIH can elicit S-LTF without concurrent P-LTF, indicating that S-LTF and P-LTF reflect separable neural mechanisms (49). Moreover, splanchnic S-LTF was found to reflect an increase of tonic activity rather than increased discharge during the inspiratory or expiratory phase of respiration. Of interest is that other studies have linked increased tonic SNA to activation of PVN neurons (14–16).
As noted, previous studies of AIH-induced S-LTF have failed to identify augmented inspiratory bursting as a driver of the net increase of SNA during the post-AIH LTF period (49–51). Recent data from unanesthetized rat preparations indicate that post-AIH LTF is associated with the emergence of active expiration and expiratory rhythmic bursting of SNA (24). The former response has been further linked to a serotonergic mechanism in the retrotrapezoid nucleus/parafacial respiratory group (25). In the present study, we did not record expiratory activity and did not address the extent to which similar post-AIH responses occurred in our anesthetized rats.
It was recently reported that intermittent optogenetic activation of the NTS, the CNS site where arterial chemoreceptor afferents initially synapse, is sufficient in the absence of intermittent hypoxia or arterial chemoreceptor activation to induce S-LTF and P-LTF (51). It was also determined that although S-LTF was accompanied by increased respiratory bursting of renal SNA, it did not strengthen respiratory-sympathetic coupling per se. These findings support the view that generation of S-LTF and P-LTF can utilize different mechanisms of neuroplasticity (34). Moreover, optogenetic activation of NTS did not cause reductions of MAP, affirming that S-LTF and P-LTF can be induced without intermittent baroreceptor unloading (50, 51). Since PVN and NTS are reciprocally connected (40, 47), the above findings raise the question of whether PVN neuronal activity represents a background synaptic substrate required for LTF neuroplasticity or whether PVN neuronal discharge increases progressively in pace with S-LTF, perhaps in response to noradrenergic NTS inputs (21), such that PVN presympathetic neurons themselves undergo LTF neuroplasticity. In vivo single neuron recording studies are needed to investigate these possibilities.
As noted, PVN neurons receive dense projections from forebrain circumventricular organs that sense and respond to circulating ANG II (27, 28, 40), which, as discussed, could be elevated during, and perhaps after, exposure to AIH (22, 44, 46). This is intriguing in light of a recent report in humans that S-LTF induced by acute intermittent breath holds is attenuated by systemic AT1R blockade (17). Interestingly, systemic AT1R blockade in the anesthetized rat failed to interfere with S-LTF induced by intermittent optogenetic activation of NTS (51), which might reflect failure to activate the RAAS since MAP increased rather than decreased as commonly occurs during hypoxic episodes of AIH. Additional studies are needed to determine the role of ANG II AT1R signaling in AIH-induced LTF neuroplasticity.
Summary.
The present findings indicate that PVN neuronal activity contributes significantly to the sympathetic limb of the arterial chemoreflex, but relatively little to the inspiratory limb during hypoxic episodes of AIH, and suggest that PVN neurons may undergo progressive recruitment with increasing cycles of AIH. Results further indicate that PVN neuronal activity is necessary for full development of post-AIH S-LTF, but plays only a minor role in the frequency component of P-LTF. Whether presympathetic PVN neurons themselves exhibit an LTF-like progressive increase in discharge post-AIH is unknown, which leaves open the possibility that their ongoing activity may simply be required for downstream sympathetic motor neurons to undergo LTF plasticity.
GRANTS
This work was supported by National Institutes of Health National Heart, Lung, and Blood Institute (NIH NHLBI) Grant HL-088052 and American Heart Association Grant 25710176 (to G. M. Toney). M. B. Blackburn received a stipend from NIH NHLBI T32 HL-07446.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
G.M.T. conceived and designed research; M.B.B. and M.A.A. performed experiments; M.B.B., M.A.A., and G.M.T. analyzed data; M.B.B. and G.M.T. interpreted results of experiments; M.B.B., M.A.A., and G.M.T. prepared figures; M.B.B. and G.M.T. drafted manuscript; M.B.B., M.A.A., and G.M.T. approved final version of manuscript; G.M.T. edited and revised manuscript.
ACKNOWLEDGMENTS
The authors thank Alfredo S. Calderon and Myrna Herrera-Rosales for excellent technical assistance and Drs. Steven Mifflin and J. Thomas Cunningham for comments and advice during preparation of the manuscript.
Present address: M. B. Blackburn, Tactical Combat Casualty Care Research, U.S. Army Institute of Surgical Research, 3698 Chambers Pass, JBSA Fort Sam Houston, TX 78234.
REFERENCES
- 1.Akine A, Montanaro M, Allen AM. Hypothalamic paraventricular nucleus inhibition decreases renal sympathetic nerve activity in hypertensive and normotensive rats. Auton Neurosci 108: 17–21, 2003. doi: 10.1016/j.autneu.2003.08.009. [DOI] [PubMed] [Google Scholar]
- 2.Bardgett ME, Holbein WW, Herrera-Rosales M, Toney GM. Ang II-salt hypertension depends on neuronal activity in the hypothalamic paraventricular nucleus but not on local actions of tumor necrosis factor-α. Hypertension 63: 527–534, 2014. doi: 10.1161/HYPERTENSIONAHA.113.02429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bardgett ME, Sharpe AL, Toney GM. Activation of corticotropin-releasing factor receptors in the rostral ventrolateral medulla is required for glucose-induced sympathoexcitation. Am J Physiol Endocrinol Metab 307: E944–E953, 2014. doi: 10.1152/ajpendo.00291.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chen QH, Toney GM. In vivo discharge properties of hypothalamic paraventricular nucleus neurons with axonal projections to the rostral ventrolateral medulla. J Neurophysiol 103: 4–15, 2010. doi: 10.1152/jn.00094.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Coldren KM, Li DP, Kline DD, Hasser EM, Heesch CM. Acute hypoxia activates neuroendocrine, but not presympathetic, neurons in the paraventricular nucleus of the hypothalamus: differential role of nitric oxide. Am J Physiol Regul Integr Comp Physiol 312: R982–R995, 2017. doi: 10.1152/ajpregu.00543.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Coles SK, Dick TE. Neurones in the ventrolateral pons are required for post-hypoxic frequency decline in rats. J Physiol 497: 79–94, 1996. doi: 10.1113/jphysiol.1996.sp021751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cutler MJ, Swift NM, Keller DM, Wasmund WL, Smith ML. Hypoxia-mediated prolonged elevation of sympathetic nerve activity after periods of intermittent hypoxic apnea. J Appl Physiol (1985) 96: 754–761, 2004. doi: 10.1152/japplphysiol.00506.2003. [DOI] [PubMed] [Google Scholar]
- 8.Denton DA, McKinley MJ, Weisinger RS. Hypothalamic integration of body fluid regulation. Proc Natl Acad Sci USA 93: 7397–7404, 1996. doi: 10.1073/pnas.93.14.7397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dick TE, Hsieh YH, Wang N, Prabhakar N. Acute intermittent hypoxia increases both phrenic and sympathetic nerve activities in the rat. Exp Physiol 92: 87–97, 2007. doi: 10.1113/expphysiol.2006.035758. [DOI] [PubMed] [Google Scholar]
- 10.Dinenno FA. Hypoxic regulation of blood flow in humans. α-adrenergic receptors and functional sympatholysis in skeletal muscle. Adv Exp Med Biol 543: 237–248, 2003. doi: 10.1007/978-1-4419-8997-0_17. [DOI] [PubMed] [Google Scholar]
- 11.Feldman JL, Mitchell GS, Nattie EE. Breathing: rhythmicity, plasticity, chemosensitivity. Annu Rev Neurosci 26: 239–266, 2003. doi: 10.1146/annurev.neuro.26.041002.131103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Grannell A, De Vito G. An investigation into the relationship between heart rate variability and the ventilatory threshold in healthy moderately trained males. Clin Physiol Funct Imaging In press. doi: 10.1111/cpf.12437. [DOI] [PubMed] [Google Scholar]
- 13.Guyenet PG, Darnall RA, Riley TA. Rostral ventrolateral medulla and sympathorespiratory integration in rats. Am J Physiol Regul Integr Comp Physiol 259: R1063–R1074, 1990. doi: 10.1152/ajpregu.1990.259.5.R1063. [DOI] [PubMed] [Google Scholar]
- 14.Holbein WW, Bardgett ME, Toney GM. Blood pressure is maintained during dehydration by hypothalamic paraventricular nucleus-driven tonic sympathetic nerve activity. J Physiol 592: 3783–3799, 2014. doi: 10.1113/jphysiol.2014.276261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Holbein WW, Toney GM. Activation of the hypothalamic paraventricular nucleus by forebrain hypertonicity selectively increases tonic vasomotor sympathetic nerve activity. Am J Physiol Regul Integr Comp Physiol 308: R351–R359, 2015. doi: 10.1152/ajpregu.00460.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Holbein WW, Toney GM. Sympathetic network drive during water deprivation does not increase respiratory or cardiac rhythmic sympathetic nerve activity. J Appl Physiol (1985) 114: 1689–1696, 2013. doi: 10.1152/japplphysiol.00078.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jouett NP, Moralez G, Raven PB, Smith ML. Losartan reduces the immediate and sustained increases in muscle sympathetic nerve activity after hyperacute intermittent hypoxia. J Appl Physiol (1985) 122: 884–892, 2017. doi: 10.1152/japplphysiol.00683.2016. [DOI] [PubMed] [Google Scholar]
- 18.Kc P, Balan KV, Tjoe SS, Martin RJ, Lamanna JC, Haxhiu MA, Dick TE. Increased vasopressin transmission from the paraventricular nucleus to the rostral medulla augments cardiorespiratory outflow in chronic intermittent hypoxia-conditioned rats. J Physiol 588: 725–740, 2010. doi: 10.1113/jphysiol.2009.184580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kc P, Haxhiu MA, Tolentino-Silva FP, Wu M, Trouth CO, Mack SO. Paraventricular vasopressin-containing neurons project to brain stem and spinal cord respiratory-related sites. Respir Physiol Neurobiol 133: 75–88, 2002. doi: 10.1016/S1569-9048(02)00131-3. [DOI] [PubMed] [Google Scholar]
- 20.King TL, Heesch CM, Clark CG, Kline DD, Hasser EM. Hypoxia activates nucleus tractus solitarii neurons projecting to the paraventricular nucleus of the hypothalamus. Am J Physiol Regul Integr Comp Physiol 302: R1219–R1232, 2012. doi: 10.1152/ajpregu.00028.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.King TL, Ruyle BC, Kline DD, Heesch CM, Hasser EM. Catecholaminergic neurons projecting to the paraventricular nucleus of the hypothalamus are essential for cardiorespiratory adjustments to hypoxia. Am J Physiol Regul Integr Comp Physiol 309: R721–R731, 2015. doi: 10.1152/ajpregu.00540.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Knight WD, Saxena A, Shell B, Nedungadi TP, Mifflin SW, Cunningham JT. Central losartan attenuates increases in arterial pressure and expression of FosB/ΔFosB along the autonomic axis associated with chronic intermittent hypoxia. Am J Physiol Regul Integr Comp Physiol 305: R1051–R1058, 2013. doi: 10.1152/ajpregu.00541.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Koshiya N, Guyenet PG. Tonic sympathetic chemoreflex after blockade of respiratory rhythmogenesis in the rat. J Physiol 491: 859–869, 1996. doi: 10.1113/jphysiol.1996.sp021263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lemes EV, Aiko S, Orbem CB, Formentin C, Bassi M, Colombari E, Zoccal DB. Long-term facilitation of expiratory and sympathetic activities following acute intermittent hypoxia in rats. Acta Physiol (Oxf) 217: 254–266, 2016. doi: 10.1111/apha.12661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lemes EV, Colombari E, Zoccal DB. Generation of active expiration by serotoninergic mechanisms of the ventral medulla of rats. J Appl Physiol (1985) 121: 1135–1144, 2016. doi: 10.1152/japplphysiol.00470.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li DP, Pan HL. Glutamatergic inputs in the hypothalamic paraventricular nucleus maintain sympathetic vasomotor tone in hypertension. Hypertension 49: 916–925, 2007. doi: 10.1161/01.HYP.0000259666.99449.74. [DOI] [PubMed] [Google Scholar]
- 27.McKinley MJ, Oldfield BJ. The brain as an endocrine target for peptide hormones. Trends Endocrinol Metab 9: 349–354, 1998. doi: 10.1016/S1043-2760(98)00092-7. [DOI] [PubMed] [Google Scholar]
- 28.McKinley MJ, Yao ST, Uschakov A, McAllen RM, Rundgren M, Martelli D. The median preoptic nucleus: front and centre for the regulation of body fluid, sodium, temperature, sleep and cardiovascular homeostasis. Acta Physiol (Oxf) 214: 8–32, 2015. doi: 10.1111/apha.12487. [DOI] [PubMed] [Google Scholar]
- 29.McMullan S, Dick TE, Farnham MM, Pilowsky PM. Effects of baroreceptor activation on respiratory variability in rat. Respir Physiol Neurobiol 166: 80–86, 2009. doi: 10.1016/j.resp.2009.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mifflin S, Cunningham JT, Toney GM. Neurogenic mechanisms underlying the rapid onset of sympathetic responses to intermittent hypoxia. J Appl Physiol (1985) 119: 1441–1448, 2015. doi: 10.1152/japplphysiol.00198.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mitchell GS, Baker TL, Nanda SA, Fuller DD, Zabka AG, Hodgeman BA, Bavis RW, Mack KJ, Olson EB Jr. Intermittent hypoxia and respiratory plasticity. J Appl Physiol (1985) 90: 2466–2475, 2001. doi: 10.1152/jappl.2001.90.6.2466. [DOI] [PubMed] [Google Scholar]
- 32.Moraes DJ, Machado BH, Paton JF. Specific respiratory neuron types have increased excitability that drive presympathetic neurones in neurogenic hypertension. Hypertension 63: 1309–1318, 2014. doi: 10.1161/HYPERTENSIONAHA.113.02283. [DOI] [PubMed] [Google Scholar]
- 33.Moraes DJ, Zoccal DB, Machado BH. Medullary respiratory network drives sympathetic overactivity and hypertension in rats submitted to chronic intermittent hypoxia. Hypertension 60: 1374–1380, 2012. doi: 10.1161/HYPERTENSIONAHA.111.189332. [DOI] [PubMed] [Google Scholar]
- 34.Pamenter ME, Powell FL. Signalling mechanisms of long-term facilitation of breathing with intermittent hypoxia. F1000Prime Rep 5: 23, 2013. doi: 10.12703/P5-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Prabhakar NR, Kumar GK, Nanduri J. Intermittent hypoxia augments acute hypoxic sensing via HIF-mediated ROS. Respir Physiol Neurobiol 174: 230–234, 2010. doi: 10.1016/j.resp.2010.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Przygodda F, Manfredi LH, Machado J, Gonçalves DAP, Zanon NM, Bonagamba LGH, Machado BH, Kettelhut IC, Navegantes LCC. Acute intermittent hypoxia in rats activates muscle proteolytic pathways through a glucocorticoid-dependent mechanism. J Appl Physiol (1985) 122: 1114–1124, 2017. doi: 10.1152/japplphysiol.00977.2015. [DOI] [PubMed] [Google Scholar]
- 37.Reddy MK, Patel KP, Schultz HD. Differential role of the paraventricular nucleus of the hypothalamus in modulating the sympathoexcitatory component of peripheral and central chemoreflexes. Am J Physiol Regul Integr Comp Physiol 289: R789–R797, 2005. doi: 10.1152/ajpregu.00222.2005. [DOI] [PubMed] [Google Scholar]
- 38.Reddy MK, Schultz HD, Zheng H, Patel KP. Altered nitric oxide mechanism within the paraventricular nucleus contributes to the augmented carotid body chemoreflex in heart failure. Am J Physiol Heart Circ Physiol 292: H149–H157, 2007. doi: 10.1152/ajpheart.00117.2006. [DOI] [PubMed] [Google Scholar]
- 39.Sawchenko PE, Swanson LW. The organization and biochemical specificity of afferent projections to the paraventricular and supraoptic nuclei. Prog Brain Res 60: 19–29, 1983. doi: 10.1016/S0079-6123(08)64371-X. [DOI] [PubMed] [Google Scholar]
- 40.Sawchenko PE, Swanson LW. The organization of forebrain afferents to the paraventricular and supraoptic nuclei of the rat. J Comp Neurol 218: 121–144, 1983. doi: 10.1002/cne.902180202. [DOI] [PubMed] [Google Scholar]
- 41.Scheuer DA, Mifflin SW. Glucocorticoids modulate baroreflex control of renal sympathetic nerve activity. Am J Physiol Regul Integr Comp Physiol 280: R1440–R1449, 2001. doi: 10.1152/ajpregu.2001.280.5.R1440. [DOI] [PubMed] [Google Scholar]
- 42.Sharpe AL, Andrade MA, Herrera-Rosales M, Britton SL, Koch LG, Toney GM. Rats selectively bred for differences in aerobic capacity have similar hypertensive responses to chronic intermittent hypoxia. Am J Physiol Heart Circ Physiol 305: H403–H409, 2013. doi: 10.1152/ajpheart.00317.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sharpe AL, Calderon AS, Andrade MA, Cunningham JT, Mifflin SW, Toney GM. Chronic intermittent hypoxia increases sympathetic control of blood pressure: role of neuronal activity in the hypothalamic paraventricular nucleus. Am J Physiol Heart Circ Physiol 305: H1772–H1780, 2013. doi: 10.1152/ajpheart.00592.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shell B, Faulk K, Cunningham JT. Neural control of blood pressure in chronic intermittent hypoxia. Curr Hypertens Rep 18: 19, 2016. doi: 10.1007/s11906-016-0627-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Simms AE, Paton JF, Pickering AE, Allen AM. Amplified respiratory-sympathetic coupling in the spontaneously hypertensive rat: does it contribute to hypertension? J Physiol 587: 597–610, 2009. doi: 10.1113/jphysiol.2008.165902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Skinner SL, McCubbin JW, Page IH. Renal baroceptor control of renin secretion. Science 141: 814–816, 1963. doi: 10.1126/science.141.3583.814. [DOI] [PubMed] [Google Scholar]
- 47.Warren Cottle MK, Calaresu FR. Projections from the nucleus and tractus solitarius in the cat. J Comp Neurol 161: 143–157, 1975. doi: 10.1002/cne.901610202. [DOI] [PubMed] [Google Scholar]
- 48.Wasicko MJ, Giering RW, Knuth SL, Leiter JC. Hypoglossal and phrenic nerve responses to carotid baroreceptor stimulation. J Appl Physiol (1985) 75: 1395–1403, 1993. doi: 10.1152/jappl.1993.75.3.1395. [DOI] [PubMed] [Google Scholar]
- 49.Xing T, Pilowsky PM. Acute intermittent hypoxia in rat in vivo elicits a robust increase in tonic sympathetic nerve activity that is independent of respiratory drive. J Physiol 588: 3075–3088, 2010. doi: 10.1113/jphysiol.2010.190454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Xing T, Pilowsky PM, Fong AY. Mechanism of sympathetic activation and blood pressure elevation in humans and animals following acute intermittent hypoxia. Prog Brain Res 209: 131–146, 2014. doi: 10.1016/B978-0-444-63274-6.00007-2. [DOI] [PubMed] [Google Scholar]
- 51.Yamamoto K, Lalley P, Mifflin S. Acute intermittent optogenetic stimulation of nucleus tractus solitarius neurons induces sympathetic long-term facilitation. Am J Physiol Regul Integr Comp Physiol 308: R266–R275, 2015. doi: 10.1152/ajpregu.00381.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yeh ER, Erokwu B, LaManna JC, Haxhiu MA. The paraventricular nucleus of the hypothalamus influences respiratory timing and activity in the rat. Neurosci Lett 232: 63–66, 1997. doi: 10.1016/S0304-3940(97)00579-X. [DOI] [PubMed] [Google Scholar]
- 53.Zoccal DB, Bonagamba LG, Paton JF, Machado BH. Sympathetic-mediated hypertension of awake juvenile rats submitted to chronic intermittent hypoxia is not linked to baroreflex dysfunction. Exp Physiol 94: 972–983, 2009. doi: 10.1113/expphysiol.2009.048306. [DOI] [PubMed] [Google Scholar]
- 54.Zoccal DB, Machado BH. Coupling between respiratory and sympathetic activities as a novel mechanism underpinning neurogenic hypertension. Curr Hypertens Rep 13: 229–236, 2011. doi: 10.1007/s11906-011-0198-7. [DOI] [PubMed] [Google Scholar]