

Abstract
We previously showed that the thioredoxin reductase-1 (TrxR1) inhibitor aurothioglucose (ATG) improves alveolarization in hyperoxia-exposed newborn C3H/HeN mice. Our data supported a mechanism by which the protective effects of ATG are mediated via sustained nuclear factor E2-related factor 2 (Nrf2) activation in hyperoxia-exposed C3H/HeN mice 72 h after ATG administration. Given that inbred mouse strains have differential sensitivity and endogenous Nrf2 activation by hyperoxia, the present studies utilized two C57BL/6 exposure models to evaluate the effects of ATG on lung development and Nrf2 activation. The first model (0–14 days) was used in our C3H/HeN studies and the 2nd model (4–14 days) is well characterized in C57BL/6 mice. ATG significantly inhibited lung TrxR1 activity in both models; however, there was no effect on parameters of alveolarization in C57BL/6 mice. In sharp contrast to C3H/HeN mice, there was no effect of ATG on pulmonary NADPH quinone oxidoreductase-1 (Nqo1) and heme oxygenase-1 (Hmox1) at 72 h in either C57BL/6 model. In conclusion, although ATG inhibited TrxR1 activity in the lungs of newborn C57BL/6 mice, effects on lung development and sustained Nrf2-dependent pulmonary responses were blunted. These findings also highlight the importance of strain-dependent hyperoxic sensitivity in evaluation of potential novel therapies.
Keywords: aurothioglucose, bronchopulmonary dysplasia, hyperoxia, nuclear factor (erythroid-derived-1)-like 2
INTRODUCTION
Bronchopulmonary dysplasia (BPD) remains the most common and costly long-term respiratory morbidity affecting prematurely born infants. Characterized by aberrant alveolarization, the pathogenesis includes O2 toxicity in the developing lung. Preterm neonates are more O2-sensitive due to developmental deficits in endogenous antioxidant defenses and impaired hyperoxia-induced antioxidant responses (2, 10, 12, 26, 27). Our group has used models of BPD and acute respiratory distress syndrome to identify thioredoxin reductase-1 (TrxR1), an NADPH-dependent oxidoreductase, as a novel therapeutic target to prevent O2-mediated lung injury (4, 13, 29). We recently demonstrated that a single injection of the TrxR1 inhibitor aurothioglucose (ATG) shortly after birth attenuated hyperoxia-induced alterations in alveolar development in newborn C3H/HeN mice (13). Overall, our studies support a model in which the effects of TrxR1 inhibition involve nuclear factor E2-related factor 2 (Nrf2) activation (4, 13, 14).
Nrf2 is a critical regulator of endogenous antioxidant responses, normal lung development, and protection from hyperoxia-mediated lung injury (9, 15). Nrf2 activation increases the transcription of antioxidant genes and enhances survival in many lung injury models (3, 8, 11, 18, 23, 31). Enhanced expression of the Nrf2-regulated genes heme oxygenase-1 (Hmox1) and NADPH quinone oxidoreductase-1 (Nqo1) at 72 h in ATG-treated hyperoxia-exposed mice suggested synergism between ATG and hyperoxia on Nrf2 activation (13). Genetic or pharmacological disruption of TrxR1 increases Nrf2 activation in vivo and in vitro, supporting the possible clinical utility of TrxR1 inhibitors to prevent and/or treat BPD (4, 29). We interpreted these findings to indicate that ATG pretreatment enhanced the ability of the newborn lung to mount rapid and sustained endogenous Nrf2-dependent antioxidant responses to hyperoxia resulting in preserved alveolar development.
Recently published studies in newborn C57BL/6 mice highlight the significance of timing, dose, and duration of hyperoxic exposure on lung morphological end points and therapeutic responses (16, 25). Given significant interstrain variation in the effects of hyperoxia on the newborn and adult lung, one cannot assume that findings generated in one strain will translate to similar findings in other strains (6, 7, 19, 32). In the present study we used two commonly employed neonatal hyperoxic exposure models to evaluate the effects of ATG on lung TrxR1 activity, lung morphometry, and indexes of Nrf2 activation in C57BL/6 mice. Our results indicate that although ATG significantly inhibited lung TrxR1 activity, similar to our findings in C3H/HeN newborn mice, ATG was not associated with sustained Nrf2 activation and did not alter hyperoxia-induced lung developmental deficits in either C57BL/6 model.
MATERIALS AND METHODS
Animal model.
Animal protocols were approved by the Institutional Animal Care and Use Committee at the University of Alabama at Birmingham. C57BL/6 mice were handled in accordance with National Institutes of Health guidelines and housed in a facility that is positive for murine norovirus and pathogenic Helicobacter. Mice were bred, and at least two dams were required to deliver within 12 h. Pups of both sexes were randomly assigned and equally distributed between two dams. For the 0- to 14-day-exposure studies, pups were injected intraperitoneally with saline or ATG [25 mg/kg (~20 μl); Research Diagnostics, Flanders, NJ; 1.25 mg/ml stock in saline] within 12 h of birth. For the 4- to 14-day-exposure studies, pups were injected intraperitoneally with saline or ATG [25 mg/kg (~40 μl)] immediately before exposure. One dam and litter were placed in a Plexiglas chamber containing 85% O2 [0.85 inspiratory O2 fraction () = 0.85 (hyperoxia)] with continuous O2 inflow and monitoring. The other dam and litter remained in room air ( = 0.21). The dams were switched every 24 h to prevent O2 toxicity. Pups were euthanized with a mixture of ketamine (100 mg/kg ip) and-xylazine (10 mg/kg ip) following exposure for 24 h, 72 h, or 14 days after birth. At 24 and 72 h, lungs were snap-frozen in liquid N2 and stored at −80°C.
Morphometric assessments.
Lungs were fixed, and morphometric analyses for alveolar number, alveolar area, and septal wall thickness were performed as previously described (30).
TrxR1 activity.
Frozen lungs were homogenized in lysis buffer [10 mM Tris buffer, pH 7.4, containing 0.1% Triton X-100, 100 µM diethylenetriamine pentaacetic acid, and proteinase inhibitor cocktail (Thermo Scientific, Rockford, IL)] on ice using a Dounce homogenizer. Supernatant was obtained by centrifugation at 20,000 g for 10 min. TrxR1 activity was determined by insulin disulfide reduction assay, as previously described (1, 5, 24).
Quantitative real-time PCR.
RNA was isolated from frozen lungs using the miRNeasy Mini Kit (Qiagen, Valencia, CA) and QIAcube (Qiagen). cDNA was synthesized using PrimeScript RT Master Mix (Takara/Clontech, Mountain View, CA). Quantitative real-time PCR was performed using Premix Ex Taq (Probe qPCR) (Takara/Clontech) and a real-time PCR cycler (Rotor-Gene Q, Qiagen). Primers for 18S (Hs99999901_s1), murine Hmox1 (Mm00516005_m1), and Nqo1 (01253561_m1) were obtained from Applied Biosystems/Thermo Fisher. Cycle threshold (CT) values were normalized to 18S (∆CT). Fold change relative to control was calculated using 2−∆∆CT.
Statistics.
Values are means ± SE. Data were analyzed using Prism 6.0 (GraphPad, La Jolla, CA). All data were tested for homogeneity of variances, logarithmically transformed when indicated, and analyzed by two-way ANOVA with Tukey’s post hoc test. Significance was accepted at P < 0.05.
RESULTS
ATG inhibits lung TrxR1 activity.
To confirm the TrxR1 inhibitory effects of ATG in C57BL/6 mice, we repeated the methods described in our previous study in C3H/HeN mice (13). Newborn pups were treated with a single intraperitoneal dose of 25 mg/kg ATG or saline within 12 h of birth and exposed to room air ( = 0.21) or hyperoxia ( = 0.85) for up to 14 days (Fig. 1A). In a separate set of experiments, we utilized another well-characterized model of hyperoxia-induced lung developmental deficits (21). In these studies, 4-day-old neonatal mice were treated with a single intraperitoneal dose of 25 mg/kg ATG or saline and exposed to room air or hyperoxia for up to 14 days (Fig. 1B). In each model, TrxR1 activity was determined in lung homogenates from tissues obtained 24 h after ATG administration.
Fig. 1.
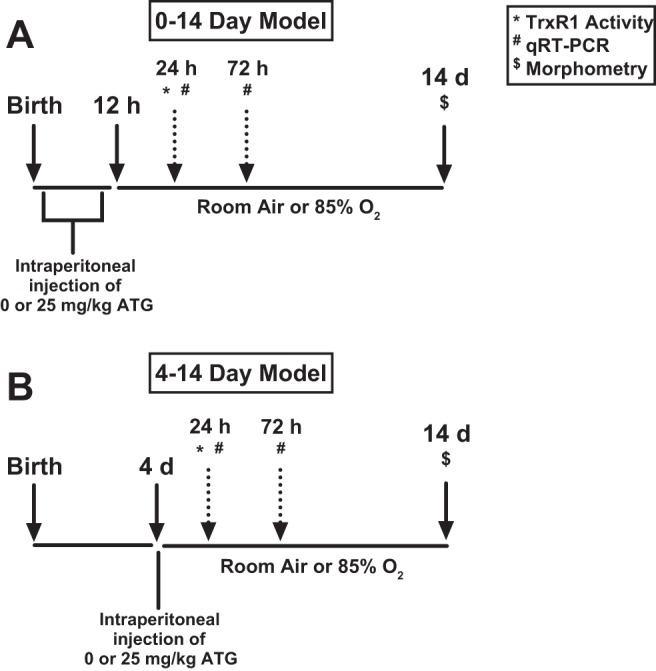
Exposure schemes for 0- to 14-day (A) and 4- to 14-day (B) models. A: newborn C57BL/6 mice were treated with a single intraperitoneal dose of 25 mg/kg aurothioglucose (ATG) or saline within 12 h of birth and subsequently exposed to room air [inspiratory O2 fraction () = 0.21] or hyperoxia ( = 0.85). B: 4-day-old C57BL/6 mice were treated with a single intraperitoneal dose of 25 mg/kg ATG or saline and subsequently exposed to room air (= 0.21) or hyperoxia ( = 0.85). TrxR1, thioredoxin reductase-1; qRT-PCR, quantitative real-time PCR.
Lung TrxR1 activity was similar in control-treated room air- and hyperoxia-exposed mice across both models. Two-way ANOVA indicated an independent effect of ATG on TrxR1 activity in both models (Fig. 2). ATG administered at birth decreased TrxR1 activity by 84% and 73% in room air- and hyperoxia-exposed newborn mice, respectively (Fig. 2A). ATG administered to 4-day-old mice decreased TrxR1 activity by 61% and 71% in room air- and hyperoxia-exposed groups, respectively (Fig. 2B).
Fig. 2.
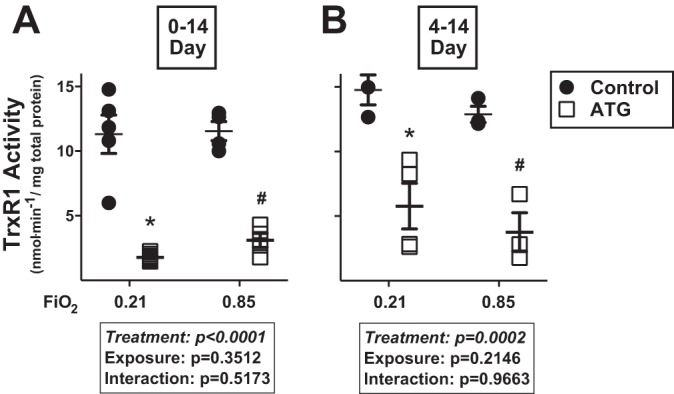
Lung TrxR1 activity in newborn mice exposed to room air ( = 0.21) or hyperoxia ( = 0.85) from 0 to 14 (A) or 4 to 14 (B) days of age. Neonatal C57BL/6 mice were treated and exposed as described in Fig. 1 legend. TrxR1 activity was determined by insulin disulfide reductase assay 24 h after ATG administration. Values are means ± SE; n = 3–8. Data were analyzed by 2-way ANOVA followed by Tukey’s post hoc test: *P < 0.007 vs. control + 0.21; #P < 0.0093 vs. control + 0.85.
ATG does not improve hyperoxia-induced alterations in alveolarization in C57BL/6 newborn mice.
Evaluation of lung morphology in both models demonstrated the well-described alveolar simplification phenotype in hyperoxia-exposed mice (Fig. 3). To quantitatively define the effects of hyperoxia and ATG on lung development, morphometric analyses were performed to assess alveolar number, individual alveolar area, and septal wall thickness using techniques previously described by our group (30).
Fig. 3.

Lung morphometry at 14 days of age. Neonatal C57BL/6 mice were treated and exposed as described in Fig. 1 legend. A and E: representative lung histology images (5 per lung) from hyperoxia-exposed control and ATG-treated mice. Scale bars = 100 μm. Alveolar number (B and F), alveolar area (C and G), and septal wall thickness (D and H) were determined as described in materials and methods. Values are means ± SE; n = 4–5 mice/group. Data were analyzed by 2-way ANOVA followed by Tukey’s post hoc test. Two-way ANOVA indicated an independent effect of exposure (P < 0.0001) on all 3 parameters. An independent effect of ATG on septal wall thickness (P = 0.0004) was detected in the 0- to 14-day, but not 4- to 14-day, model. *P < 0.0013 vs. control + 0.21; #P < 0.0052 vs. ATG + 0.21; $P < 0.006 vs. control + 0.85.
In general, hyperoxic exposure resulted in altered lung architecture, characterized by fewer and larger alveoli, and increased septal wall thickness. In both models, two-way ANOVA indicated an independent effect of hyperoxia on all three morphometric end points. In the 0- to 14-day model, hyperoxia decreased alveolar number by 61% (Fig. 3B), increased individual alveolar area by 2.4-fold (Fig. 3C), and increased septal wall thickness by 60% (Fig. 3D). In the 4- to 14-day model, hyperoxia decreased alveolar number by 51% (Fig. 3F), increased individual alveolar area by 2.3-fold (Fig. 3G), and increased septal wall thickness by 33% (Fig. 3H).
Alveolar number (Fig. 3, B and F) and area (Fig. 3, C and G) did not differ between hyperoxia-exposed control and hyperoxia-exposed ATG-treated mice. However, an independent effect of ATG on septal wall thickness was detected by two-way ANOVA in the 0- to 14-day (Fig. 3D), but not the 4- to 14-day (Fig. 3H), model. Septal wall thickness was 18% lower in ATG-treated hyperoxia-exposed than saline-treated control mice in the 0- to 14-day model (Fig. 3D). In the 4- to 14-day model, septal wall thickness was significantly lower (11%) in ATG-treated hyperoxia-exposed than saline-treated hyperoxia-exposed control mice (Fig. 3H).
ATG enhances Nrf2 activation at 24 h, but not 72 h, of hyperoxia.
Our overall working hypothesis, based on consistent findings in vitro and in vivo, is that sustained Nrf2 activation is a primary mechanism by which TrxR1 inhibitors attenuate lung injury (4, 13, 14, 28). Despite comparatively similar effects of ATG on TrxR1 activity in lungs from newborn C3H/HeN and C57BL/6 mice 24 h after administration, ATG modestly attenuated hyperoxia-induced increases in septal wall thickness and had no effects on alveolarization in C57BL/6 mice. Thus we next evaluated Nrf2 activation in the lungs of C57BL/6 mice by measuring Nqo1 and Hmox1 mRNA levels in both models after 24 or 72 h of hyperoxic exposure.
In our newborn C3H/HeN studies, we detected 1) no independent effect of ATG on Nqo1 and Hmox1 levels at 24 h and 2) an independent effect of ATG on Nqo1 and Hmox1 levels at 72 h (13). Our current data in the 0- to 14-day model revealed an independent effect of hyperoxia on Nqo1 and Hmox1 expression at 24 h (Fig. 4A) and 72 h (Fig. 4B). In addition, an independent effect of ATG on Hmox1, but not Nqo1, expression at 24 h was also detected by two-way ANOVA. There was no independent effect of ATG on Nqo1 or Hmox1 expression at 72 h. In the 4- to 14-day model (Fig. 4, C and D), an independent effect of hyperoxia on Nqo1 and Hmox1 expression was detected at 24 and 72 h. No independent effect of ATG on Nqo1 or Hmox1 was detected at either time point.
Fig. 4.

Lung NADPH quinone oxidoreductase-1 (Nqo1) and heme oxygenase-1 (Hmox1) expression. Newborn C57BL/6 mice were treated and exposed from birth (A and B) or 4 days of age (C and D) as described in Fig. 1 legend. Values are means ± SE; n = 3–5. Data were analyzed by 2-way ANOVA followed by Tukey’s post hoc test. A: *P < 0.002 vs. control + 0.21; #P < 0.0025 vs. ATG + 0.21; $P = 0.024 vs. control + 0.85. B: *P < 0.009 vs. control + 0.21; #P < 0.005 vs. ATG + 0.21. C: *P = 0.0458 vs. control + 0.21; #P < 0.05 vs. ATG + 0.21. D: *P < 0.002 vs. control + 0.21; #P < 0.009 vs. ATG + 0.21.
Our 72-h quantitative real-time PCR data suggest that Nrf2 responses to hyperoxia differ depending on the day on which the hyperoxic exposure was initiated. Nqo1, but not Hmox1, mRNA levels were modestly (2-fold) increased in saline-treated mice exposed to hyperoxia starting at birth (Fig. 4B). In contrast, compared with room air controls, lung Nqo1 and Hmox1 levels were increased 3.7- and 4.5-fold when hyperoxic exposure was initiated at 4 days of age (Fig. 4D).
DISCUSSION
The present studies indicate that although ATG significantly inhibits lung TrxR1 activity in two distinct C57BL/6 models of BPD, ATG did not improve alveolarization. While ATG modestly attenuated hyperoxia-induced increases in septal wall thickness in both C57BL/6 models, we did not observe sustained pulmonary Nrf2 activation in the lungs of hyperoxia-exposed ATG-treated neonatal pups. This is in contrast to our findings in C3H/HeN mice, in which TrxR1 inhibition was associated with persistent Nrf2 activation and improved alveolarization in hyperoxia-exposed mice. Our data also suggest that neonatal pulmonary Nrf2-dependent responses to hyperoxia differ not only by strain, but also by the day of life on which the hyperoxic exposure is initiated.
Based on our positive findings in C3H/HeN mice, we employed two C57BL/6 BPD models to test the efficacy of TrxR1 as a therapeutic target to improve lung development (13). Recent evidence suggests that the effects of therapeutic interventions, including cottonseed oil and caffeine, differ depending on the rodent BPD exposure model used (16, 22). Exposure of newborn C57BL/6 pups to 85% O2 from birth to 14 days of age recapitulates the scheme that we used in our previous C3H/HeN study. Compared with other exposure models, this approach is associated with concomitant decreases in total alveolar number and increased septal wall thickness, two hallmark features of BPD (17). We also utilized a model in which treatment and exposure were initiated at 4 days of age, when alveolarization starts, through 14 days of age. This study is the first to assess the effects of 85% O2 from 0 to 14 days compared with effects from 4 to 14 days in C57BL/6 mice with TrxR1 as the primary therapeutic target.
While C57BL/6 mice are among the most commonly utilized strains, BPD models utilize a wide variety of inbred strains. In a recent series of articles, Morty and colleagues highlight the challenges and complexities of using murine BPD models to study BPD pathogenesis and novel therapeutics (16, 17, 22, 25). With respect to C57BL/6 mice, this group elegantly demonstrated the differential effects of timing, concentration, and duration of O2 exposure on alveolar number and septal wall thickness in neonatal mice (16). Neonatal exposure to as little as 40% O2 from 0 to 14 days decreased alveolar number, while exposure to 85% O2 from 0 to 7 days altered alveolarization, but not septal wall thickness. Only 85% O2 from 0 to 14 days altered both alveolarization and septal wall thickness, which is consistent with our findings in the same model. We interpret our finding of increased septal wall thickness in the 4- to 14-day model to suggest that hyperoxia-induced increases in septal wall thickness are driven by exposure beyond 7 days of age.
Differences in the effects of hyperoxic exposure on lung injury in inbred mouse strains, including differential sensitivity to 100% O2 in adult C3H/HeJ and C57BL/6 mice (6, 7, 32), have been extensively characterized. Briefly, adult C3H/HeJ mice are considered “hyperoxia-resistant,” while C57BL/6 mice are considered “hyperoxia-sensitive.” In contrast, C3H/HeJ pups exhibited more extensive lung injury than C57BL/6 mice following exposure to 100% O2 for the first 3 days of life (19). It is difficult to interpret our current findings given differences in O2 concentrations (85% vs. 100%), the duration of hyperoxia exposure (0–14 days and 4–14 days vs. 0–3 days), and the lack of morphometric end points between C3H/HeJ and C57BL/6 mice in the earlier studies.
In both models, two-way ANOVA showed an independent effect of hyperoxia on Nqo1 and Hmox1 levels at both time points. This is consistent with our findings in C3H/HeN mice, indicating that hyperoxia induced endogenous Nrf2-dependent antioxidant responses in both strains (13). The robustness of Nrf2 activation in response to hyperoxia is strain-dependent in adult mice. Compared with C57BL/6 mice, C3H/HeJ mice exhibit enhanced Nrf2-dependent responses to 100% O2 exposure (6). We have historically used C3H/HeN mice in our hyperoxia studies, and we are unaware of any studies directly comparing the responses of neonatal C3H/HeN and C3H/HeJ mice in BPD models. The well-characterized Toll-like receptor 4 mutation in C3H/HeJ mice alters hyperoxia-induced inflammatory responses in adult mice compared with C3H/HeN mice, likely via NF κB-dependent mechanisms (20). In the present studies, hyperoxia more robustly enhanced Nrf2 activation in the 4- to 14-day model at 72 h than in the 0- to 14-day model. Thus our data indicate that neonatal pulmonary Nrf2-dependent responses to hyperoxia differ not only by strain, but also by the day of life on which the hyperoxic exposure is initiated in C57BL/6 mice.
Sex has been identified as a potentially important biological variable in neonatal hyperoxic responses. Male neonatal C57BL/6 mice exposed to 95% O2 from 1 to 5 days of age exhibit greater morphological abnormalities than female mice (16). Transcriptomic analyses in the same model suggest that sex-specific modulation of angiogenesis may, in part, be responsible for differences in hyperoxic responses (11). The present studies utilized both male and female pups, although sex was recorded only for animals used in morphometric analyses, and the present studies were not powered to test sex as an outcome.
Our data have consistently supported a role for sustained Nrf2 activation in pulmonary protection by TrxR1 inhibitors in vivo and in vitro (4, 13, 14, 29). The lack of sustained Nrf2 activation in ATG-treated hyperoxia-exposed C57BL/6 mice in the 0- to 14-day model, as evidenced by Nqo1 and Hmox1 levels at 72 h, is in contrast with our previous findings in C3H/HeN mice (13). In our previous studies we did not detect an effect of ATG on indexes of Nrf2 activation at 24 h; however, at 72 h, two-way ANOVA indicated an independent effect of ATG on Nqo1 and Hmox1 expression. The highest transcript levels were in ATG-treated hyperoxia-exposed C3H/HeN mice, suggesting synergism between ATG and hyperoxia on Nrf2 activation. In the current studies we detected an effect of ATG on Hmox1, but not Nqo1, expression at 24 h in the 0- to 14-day model, but no effect of ATG was detected at 72 h. We interpret the lack of an effect of ATG on alveolarization in the 0- to 14-day C57BL/6 BPD model, when combined with the lack of sustained Nrf2 activation, to support our overall hypothesis that the protective effects of ATG are likely to be Nrf2-mediated.
Our novel findings support the role of sustained Nrf2 activation as a mechanism by which TrxR1 inhibitors attenuate lung injury. Nonetheless, the lack of comparable efficacy in the C57BL/6 strain is emblematic of the inherent challenges of studying BPD by exposing neonatal mice to hyperoxia. In addition to recent suggestions regarding use of complementary animal models in BPD studies, our findings provide a rationale for the performance of mechanistic and/or therapeutic BPD studies in more than one murine strain (1). In conclusion, our findings highlight the need to exercise caution when attempting to extrapolate the benefits of candidate therapies between inbred strains of mice.
GRANTS
This work is supported by National Institutes of Health Grants R01-HL-119280 (to T. E. Tipple) and R01-HD-0888033 (to L. K. Rogers).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
Q.L., T.J., L.K.R., and T.E.T. conceived and designed research; Q.L., R.L., S.B.W., K.D., C.R., and L.K.R. performed experiments; Q.L., R.L., S.B.W., K.D., C.R., T.J., L.K.R., and T.E.T. analyzed data; Q.L., R.L., K.D., T.J., L.K.R., and T.E.T. interpreted results of experiments; Q.L. and T.E.T. prepared figures; Q.L. and T.E.T. drafted manuscript; Q.L. and T.E.T. edited and revised manuscript; Q.L., R.L., S.B.W., K.D., C.R., T.J., L.K.R., and T.E.T. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Morgan L. Locy, Cynthia L. Hill, Dr. Nelida Olave, and Mark MacEwen for advice on experimental design.
REFERENCES
- 1.Arnér ES, Zhong L, Holmgren A. Preparation and assay of mammalian thioredoxin and thioredoxin reductase. Methods Enzymol 300: 226–239, 1999. doi: 10.1016/S0076-6879(99)00129-9. [DOI] [PubMed] [Google Scholar]
- 2.Baydas G, Karatas F, Gursu MF, Bozkurt HA, Ilhan N, Yasar A, Canatan H. Antioxidant vitamin levels in term and preterm infants and their relation to maternal vitamin status. Arch Med Res 33: 276–280, 2002. doi: 10.1016/S0188-4409(02)00356-9. [DOI] [PubMed] [Google Scholar]
- 3.Boutten A, Goven D, Artaud-Macari E, Boczkowski J, Bonay M. NRF2 targeting: a promising therapeutic strategy in chronic obstructive pulmonary disease. Trends Mol Med 17: 363–371, 2011. doi: 10.1016/j.molmed.2011.02.006. [DOI] [PubMed] [Google Scholar]
- 4.Britt RD Jr, Velten M, Locy ML, Rogers LK, Tipple TE. The thioredoxin reductase-1 inhibitor aurothioglucose attenuates lung injury and improves survival in a murine model of acute respiratory distress syndrome. Antioxid Redox Signal 20: 2681–2691, 2014. doi: 10.1089/ars.2013.5332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cenas N, Prast S, Nivinskas H, Sarlauskas J, Arnér ES. Interactions of nitroaromatic compounds with the mammalian selenoprotein thioredoxin reductase and the relation to induction of apoptosis in human cancer cells. J Biol Chem 281: 5593–5603, 2006. doi: 10.1074/jbc.M511972200. [DOI] [PubMed] [Google Scholar]
- 6.Cho HY, Jedlicka AE, Gladwell W, Marzec J, McCaw ZR, Bienstock RJ, Kleeberger SR. Association of Nrf2 polymorphism haplotypes with acute lung injury phenotypes in inbred strains of mice. Antioxid Redox Signal 22: 325–338, 2015. doi: 10.1089/ars.2014.5942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cho HY, Jedlicka AE, Reddy SP, Zhang LY, Kensler TW, Kleeberger SR. Linkage analysis of susceptibility to hyperoxia. Nrf2 is a candidate gene. Am J Respir Cell Mol Biol 26: 42–51, 2002. doi: 10.1165/ajrcmb.26.1.4536. [DOI] [PubMed] [Google Scholar]
- 8.Cho HY, Kleeberger SR. Nrf2 protects against airway disorders. Toxicol Appl Pharmacol 244: 43–56, 2010. doi: 10.1016/j.taap.2009.07.024. [DOI] [PubMed] [Google Scholar]
- 9.Cho HY, van Houten B, Wang X, Miller-DeGraff L, Fostel J, Gladwell W, Perrow L, Panduri V, Kobzik L, Yamamoto M, Bell DA, Kleeberger SR. Targeted deletion of Nrf2 impairs lung development and oxidant injury in neonatal mice. Antioxid Redox Signal 17: 1066–1082, 2012. doi: 10.1089/ars.2011.4288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jain A, Mehta T, Auld PA, Rodrigues J, Ward RF, Schwartz MK, Mårtensson J. Glutathione metabolism in newborns: evidence for glutathione deficiency in plasma, bronchoalveolar lavage fluid, and lymphocytes in prematures. Pediatr Pulmonol 20: 160–166, 1995. doi: 10.1002/ppul.1950200306. [DOI] [PubMed] [Google Scholar]
- 11.Kensler TW, Wakabayashi N, Biswal S. Cell survival responses to environmental stresses via the Keap1-Nrf2-ARE pathway. Annu Rev Pharmacol Toxicol 47: 89–116, 2007. doi: 10.1146/annurev.pharmtox.46.120604.141046. [DOI] [PubMed] [Google Scholar]
- 12.Lee JW, Davis JM. Future applications of antioxidants in premature infants. Curr Opin Pediatr 23: 161–166, 2011. doi: 10.1097/MOP.0b013e3283423e51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li Q, Wall SB, Ren C, Velten M, Hill CL, Locy ML, Rogers LK, Tipple TE. Thioredoxin reductase inhibition attenuates neonatal hyperoxic lung injury and enhances Nrf2 activation. Am J Respir Cell Mol Biol 55: 419-428, 2016. doi: 10.1165/rcmb.2015-0228OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Locy ML, Rogers LK, Prigge JR, Schmidt EE, Arnér ES, Tipple TE. Thioredoxin reductase inhibition elicits Nrf2-mediated responses in Clara cells: implications for oxidant-induced lung injury. Antioxid Redox Signal 17: 1407–1416, 2012. doi: 10.1089/ars.2011.4377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.McGrath-Morrow S, Lauer T, Yee M, Neptune E, Podowski M, Thimmulappa RK, O’Reilly M, Biswal S. Nrf2 increases survival and attenuates alveolar growth inhibition in neonatal mice exposed to hyperoxia. Am J Physiol Lung Cell Mol Physiol 296: L565–L573, 2009. doi: 10.1152/ajplung.90487.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nardiello C, Mižíková I, Morty RE. Looking ahead: where to next for animal models of bronchopulmonary dysplasia? Cell Tissue Res 367: 457–468, 2017. doi: 10.1007/s00441-016-2534-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nardiello C, Mižíková I, Silva DM, Ruiz-Camp J, Mayer K, Vadász I, Herold S, Seeger W, Morty RE. Standardisation of oxygen exposure in the development of mouse models for bronchopulmonary dysplasia. Dis Model Mech 10: 185–196, 2017. doi: 10.1242/dmm.027086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nguyen T, Nioi P, Pickett CB. The Nrf2-antioxidant response element signaling pathway and its activation by oxidative stress. J Biol Chem 284: 13291–13295, 2009. doi: 10.1074/jbc.R900010200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nichols JL, Gladwell W, Verhein KC, Cho HY, Wess J, Suzuki O, Wiltshire T, Kleeberger SR. Genome-wide association mapping of acute lung injury in neonatal inbred mice. FASEB J 28: 2538–2550, 2014. doi: 10.1096/fj.13-247221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ogawa Y, Tasaka S, Yamada W, Saito F, Hasegawa N, Miyasho T, Ishizaka A. Role of Toll-like receptor 4 in hyperoxia-induced lung inflammation in mice. Inflamm Res 56: 334–338, 2007. doi: 10.1007/s00011-007-7052-z. [DOI] [PubMed] [Google Scholar]
- 21.Olave N, Lal CV, Halloran B, Pandit K, Cuna AC, Faye-Petersen OM, Kelly DR, Nicola T, Benos PV, Kaminski N, Ambalavanan N. Regulation of alveolar septation by microRNA-489. Am J Physiol Lung Cell Mol Physiol 310: L476–L487, 2016. doi: 10.1152/ajplung.00145.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rath P, Nardiello C, Surate Solaligue DE, Agius R, Mižíková I, Hühn S, Mayer K, Vadász I, Herold S, Runkel F, Seeger W, Morty RE. Caffeine administration modulates TGF-β signaling but does not attenuate blunted alveolarization in a hyperoxia-based mouse model of bronchopulmonary dysplasia. Pediatr Res 81: 795–805, 2017. doi: 10.1038/pr.2017.21. [DOI] [PubMed] [Google Scholar]
- 23.Reddy NM, Suryanaraya V, Yates MS, Kleeberger SR, Hassoun PM, Yamamoto M, Liby KT, Sporn MB, Kensler TW, Reddy SP. The triterpenoid CDDO-imidazolide confers potent protection against hyperoxic acute lung injury in mice. Am J Respir Crit Care Med 180: 867–874, 2009. doi: 10.1164/rccm.200905-0670OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rengby O, Cheng Q, Vahter M, Jörnvall H, Arnér ES. Highly active dimeric and low-activity tetrameric forms of selenium-containing rat thioredoxin reductase 1. Free Radic Biol Med 46: 893–904, 2009. doi: 10.1016/j.freeradbiomed.2008.12.017. [DOI] [PubMed] [Google Scholar]
- 25.Silva DM, Nardiello C, Pozarska A, Morty RE. Recent advances in the mechanisms of lung alveolarization and the pathogenesis of bronchopulmonary dysplasia. Am J Physiol Lung Cell Mol Physiol 309: L1239–L1272, 2015. doi: 10.1152/ajplung.00268.2015. [DOI] [PubMed] [Google Scholar]
- 26.Smith CV, Hansen TN, Martin NE, McMicken HW, Elliott SJ. Oxidant stress responses in premature infants during exposure to hyperoxia. Pediatr Res 34: 360–365, 1993. doi: 10.1203/00006450-199309000-00024. [DOI] [PubMed] [Google Scholar]
- 27.Stenmark KR, Abman SH. Lung vascular development: implications for the pathogenesis of bronchopulmonary dysplasia. Annu Rev Physiol 67: 623–661, 2005. doi: 10.1146/annurev.physiol.67.040403.102229. [DOI] [PubMed] [Google Scholar]
- 28.Tipple TE. The thioredoxin system in neonatal lung disease. Antioxid Redox Signal 21: 1916–1925, 2014. doi: 10.1089/ars.2013.5782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tipple TE, Welty SE, Rogers LK, Hansen TN, Choi YE, Kehrer JP, Smith CV. Thioredoxin-related mechanisms in hyperoxic lung injury in mice. Am J Respir Cell Mol Biol 37: 405–413, 2007. doi: 10.1165/rcmb.2006-0376OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Velten M, Heyob KM, Rogers LK, Welty SE. Deficits in lung alveolarization and function after systemic maternal inflammation and neonatal hyperoxia exposure. J Appl Physiol (1985) 108: 1347–1356, 2010. doi: 10.1152/japplphysiol.01392.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Walters DM, Cho HY, Kleeberger SR. Oxidative stress and antioxidants in the pathogenesis of pulmonary fibrosis: a potential role for Nrf2. Antioxid Redox Signal 10: 321–332, 2008. doi: 10.1089/ars.2007.1901. [DOI] [PubMed] [Google Scholar]
- 32.Whitehead GS, Burch LH, Berman KG, Piantadosi CA, Schwartz DA. Genetic basis of murine responses to hyperoxia-induced lung injury. Immunogenetics 58: 793–804, 2006. doi: 10.1007/s00251-006-0147-9. [DOI] [PMC free article] [PubMed] [Google Scholar]