

Abstract
Pulmonary arterial hypertension (PAH) is a lethal disease characterized by elevations in pulmonary arterial pressure, in part due to formation of occlusive lesions in the distal arterioles of the lung. These complex lesions may comprise multiple cell types, including endothelial cells (ECs). To better understand the molecular mechanisms underlying EC dysfunction in PAH, lung microvascular endothelial cells (MVECs) were isolated from normoxic rats (N-MVECs) and rats subjected to SU5416 plus hypoxia (SuHx), an experimental model of PAH. Compared with N-MVECs, MVECs isolated from SuHx rats (SuHx-MVECs) appeared larger and more spindle shaped morphologically and expressed canonical smooth muscle cell markers smooth muscle-specific α-actin and myosin heavy chain in addition to endothelial markers such as Griffonia simplicifolia and von Willebrand factor. SuHx-MVEC mitochondria were dysfunctional, as evidenced by increased fragmentation/fission, decreased oxidative phosphorylation, and increased reactive oxygen species (ROS) production. Functionally, SuHx-MVECs exhibited increased basal levels of intracellular calcium concentration ([Ca2+]i) and enhanced migratory and proliferative capacity. Treatment with global (TEMPOL) or mitochondria-specific (MitoQ) antioxidants decreased ROS levels and basal [Ca2]i in SuHx-MVECs. TEMPOL and MitoQ also decreased migration and proliferation in SuHx-MVECs. Additionally, inhibition of ROS-induced Ca2+ entry via pharmacologic blockade of transient receptor potential vanilloid-4 (TRPV4) attenuated [Ca2]i, migration, and proliferation. These findings suggest a role for mitochondrial ROS-induced Ca2+ influx via TRPV4 in promoting abnormal migration and proliferation in MVECs in this PAH model.
INTRODUCTION
Pulmonary arterial hypertension (PAH) is a lethal disease characterized by high pulmonary arterial pressure that progresses to right ventricular (RV) failure. In PAH, histologically complex occlusive lesions composed of various cell types contribute to increased pulmonary vascular resistance (7). Present in these lesions are abnormal, hypermigratory, and hyperproliferative endothelial cells (ECs) (7, 58), which are also 1) resistant to apoptosis (19), 2) thought to be microvascular in origin, and 3) display evidence of oxidant stress (7). However, the mechanisms underlying these abnormal features in human PAH ECs are not fully understood.
Several animal models have been developed to better understand the pathobiology of PAH. Although no animal model of PAH fully recapitulates the entirety of the histopathologic features of human PAH, the rat SU5416/hypoxia (SuHx) model is perhaps closest (49). In this model, rats are subcutaneously injected with one dose of SU5416, a vascular endothelial growth factor receptor antagonist, and subjected to hypoxia for 3 wk. After hypoxic exposure, rats return to normoxia, during which time remodeling progresses and rats continue to display robust increases in RV systolic pressure and hypertrophy (22). Histologically, vascular wall thickening and obliterative lesions similar to those seen in human PAH are observed (22, 36). However, little is known regarding whether ECs in SuHx rats retain their promigratory, proproliferative phenotype in vitro as has been shown with human PAH ECs (29) and, if so, the EC signaling underlying these changes.
Reactive oxygen species (ROS) are important for migration and proliferation in ECs from various vascular beds (2). The sources of ROS in ECs include various plasma membrane proteins, such as NAPDH oxidase, and the mitochondria (12). Mitochondrial ROS (mtROS) are generated as a side product of respiration via the electron transport chain (ETC) in the mitochondrial inner membrane. At baseline, low mtROS levels are maintained by tight regulation of electron supply (NADH, FADH2) and transport. However, under pathologic conditions, increased mtROS production can serve as a signaling mechanism for specific subcellular redox-sensitive targets. Although multiple sources of ROS production have been identified in ECs, the role of ROS in promoting EC migration and proliferation in the normal and PAH lung is not fully known.
To understand the molecular underpinnings of endothelial dysfunction in experimental PAH, we isolated lung microvascular ECs (MVECs) from normoxic rats (N-MVECs) and rats subjected to the SuHx protocol (SuHx-MVECs). To determine whether SuHx-MVECs recapitulated phenotypic and cell signaling abnormalities observed in human PAH ECs (29), we measured ROS (7, 57), markers of mitochondrial dysfunction (58), basal intracellular calcium concentration ([Ca2+]i), migration, and proliferation. We previously showed that exogenous ROS increased [Ca2+]i in MVECs by activating the transient receptor potential vanilloid-4 (TRPV4) calcium channel (45, 46). To determine whether this pathway could be activated by endogenously produced ROS in SuHx-MVECs, we also examined the effects of quenching ROS and TRPV4 inhibition on [Ca2+]i, migration, and proliferation in N- and SuHx-MVECs.
METHODS
All procedures were performed in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and were approved by the Animal Care and Use Committee of The Johns Hopkins University School of Medicine.
Drugs and reagents.
SU5416 was purchased from Tocris. Baculovirus constructs (MitoRFP, roGFP) were purchased from Life Technologies. MitoQ was provided by Antipodean Pharmaceuticals. Collagenase 1A was purchased from Worthington Laboratories. GSK2193874 was purchased from SelleckChem. HC-067047 was purchased from EMD Millipore. All other chemical reagents were purchased from Sigma-Aldrich.
SuHx animal model.
SU5416 was dissolved in a mixture of DMSO and carboxymethylcellulose solution containing 0.5% (wt/vol) carboxymethylcellulose sodium, 0.9% (wt/vol) sodium chloride, 0.4% (vol/vol) polysorbate, and 0.9% (vol/vol) benzyl alcohol in deionized water. Male Wistar rats (250–300 g) were subcutaneously injected with SU5416 (20 mg/kg) or vehicle and placed in sustained hypoxia ( = 10%) for 3 wk. The hypoxic chamber was continuously flushed with a mixture of room air and N2 to maintain CO2 concentrations at <0.5% and O2 at 10 ± 0.5%. Rats were briefly exposed to room air twice a week to clean cages and replenish food and water. Normoxic controls received vehicle injection and were kept in room air on racks next to the hypoxic chamber. Following 21 days of hypoxic exposure, SuHx rats were returned to normoxia for an additional 2 wk.
Hemodynamics.
Closed-chest RV pressure measurements were performed as previously described. Briefly, after anesthesia, a small midline abdominal incision was created and the diaphragm was visualized. A 23-gauge needle filled with heparinized saline and attached to a pressure transducer was inserted into the RV transdiaphragmatically, and the pressure waveform was recorded via Chart software. Only measurements from rats with stable tracings and adequate heart rate (>350 beats/min) were included in analysis. Following hemodynamic measurements, the chest was opened via sternotomy and the animal quickly killed via exsanguination before proceeding to MVEC harvest.
Isolation and culture of MVECs.
MVECs were obtained via a dual-selection method using beads conjugated with anti-CD31 (a pan endothelial cell marker) and Griffonia simplicifolia lectin 1 (GS), a cell surface marker more specific for microvascular ECs than pan-endothelial markers such as AcLDL or PECAM (44). Following hemodynamic measurements, the lungs were quickly removed by dissection and immersed in DMEM (GIBCO). Peripheral lung tissue was obtained by dissection, minced and digested in an incubator using collagenase (type 1A; 1 mg/ml) in EC growth media that contained the following: DMEM (GIBCO), 20% FBS (Hyclone), 1% nonessential amino acids (Sigma), 1% penicillin/streptomycin antibiotic solution, and 1 aliquot of endothelial growth supplement (Millipore). Following centrifugation and resuspension of the cell pellet, the cell suspension was incubated with CD31-conjugated beads (Invitrogen) for 30 min at room temperature. The beads were magnetically separated and then cultured on gelatin-coated (0.1%) cultureware using EC growth media. After reaching confluence, cells were trypsinized and incubated with beads conjugated with GS (Vector). Beads were again magnetically separated and cultured. Dual-selected cells were subsequently passaged and expanded. Repeat staining for endothelial markers CD31 (Abcam), von willebrand factor (vWF; BNL), and GS lectin (EY Laboratories) was performed at passage 3. All experiments were performed on cells incubated in DMEM media containing 5% FBS (Hyclone) at passage 3–4.
Isolation and culture of pulmonary arterial smooth muscle cells.
Pulmonary arteries measuring 200–600 µM were isolated, and the endothelium was removed by gentle rubbing. The denuded vessels were enzymatically digested, and smooth muscle cells were grown in culture media as previously described (22).
Western blot analysis.
Confluent flasks of N- and SuHx-MVECs were trypsinized, pelleted, and lysed with lysis buffer (T-PER) containing protease inhibitor cocktail tablet (Roche). Protein levels were quantified using a BCA kit (Pierce). Electrophoresis of equal amounts of protein was performed before transfer to PVDF membranes, blocking (5% BSA in TBS-T), incubation with primary antibody [TRPV4, Alomone, 1:2,000, overnight 4°C; endothelial nitric oxide synthase (eNOS), BD, 1:2,000, overnight 4°C; and phosphor and total dynamin-related protein 1 (DRP1), CST; 1:2,000, overnight 4°C] and appropriate secondary (goat anti-rabbit or anti-mouse; KPL; 1:10,000, 1 h room temperature), and ECL (Amersham; 3 min). Membranes were also probed for GAPDH (GAPDH-HRP; Bio-Rad; 1:10,000).
Immunofluorescence.
Cells were plated at ~50% confluence on glass coverslips for 24 h. Cells were fixed with formalin (4%), permeabilized with Triton-X (0.5%), and blocked in 2% BSA for 1 h. After blocking, samples were incubated with either FITC-conjugated GS Lectin (EY Laboratories; 1:100), anti-vWF (BNL; 1:1,000), FITC-conjugated smooth muscle-specific α-actin (FITC-SMA clone 1A4; Sigma; 1:2,000), or anti-myosin heavy chain (MHC; Cell Signaling; 1:2,000) primary antibodies, washed, and then incubated for 1 h with appropriate fluorescent secondary antibody (goat anti-mouse, goat-anti-rabbit or donkey anti-sheep; Alexa Fluor; 1:2,000) and for 2–3 min with DAPI nuclear stain (1:10,000 in PBS; Thermo). After being washed, coverslips were mounted with Prolong Anti-fade solution (Invitrogen). Images were obtained and analyzed using an Olympus IX51 inverted fluorescent microscope (×40 oil objective) and a Cooke digital camera connected to a computer running MicroManager. Percent positive cells were counted by a blinded investigator. For confocal images, mounted coverslips were imaged using a Zeiss LSM800 laser-scanning microscope (×40 oil objective). Confocal images were obtained and analyzed using Zen Blue (Zeiss) and Fiji (ImageJ) software. For each cell surface marker, slides were imaged at identical wide-field (magnification, exposure time, gain, and binning) and confocal (magnification, scan speed, laser power, detector gain, and pinhole diameter) settings.
Antibody validation.
The eNOS (BD Laboratories) and total DRP1 (Cell Signaling) antibodies have been previously validated using lysates from knockout mice (4, 52). The pDrp1Ser616 antibody was validated previously using phospho-mutant pDrp1 constructs (55). The serine phosphorylation sites used herein refer to human Drp1-isoform amino acid positions; Ser616 and Ser637 in human Drp-1 protein correspond to Ser635 and Ser656, respectively, in the rat (27). The TRPV4 antibody has also been validated previously, by us and others, with loss of the 75-kDa band for TRPV4 in TRPV4−/− lysates (46, 59). Full-length gel images have been included for all antibodies (Fig. 1). GS, vWF, SMA, and MHC immunofluorescence antibodies were validated by using appropriate negative controls (see Fig. 3A in results).
Fig. 1.

Antibody validation. Representative full-length gels of rat microvascular endothelial cell lysates probed for transient receptor potential vanilloid-4 (TRPV4; 75 kDa; A), endothelial nitric oxide synthase (eNOS; 140 kDa; B), pDrp1Ser616 (C), pDrp1Ser637 (D), and total dynamin-related protein 1 (Drp1; 75 kDa; E).
Fig. 3.

Cell surface marker expression in normoxic (N)-microvascular endothelial cells (MVECs) and SU5416 plus hypoxia (SuHx)-MVECs. A–D: representative photomicrographs showing N-MVECs, SuHx-MVECs and rat pulmonary arterial smooth muscle cells (RPASMCs) stained for Griffonia simplicifolia lectin (GS; green; A), von willebrand factor (vWF; red; B), smooth muscle α-actin (SMA; red; C), myosin heavy chain (MHC; red; D), and nuclear counterstain (DAPI; blue). Scatterplot (E) showing means ± SE percent positive cells in N- and SuHx-MVECs stained for GS, vWF, SMA, and MHC. *Significant difference from GS- and vWF-stained cells; n = 5–6 images per group from experiments performed on ECs isolated from at least 5 different animals.
Mitochondrial morphology.
Cells plated on coverslips were either 1) incubated with Mitotracker (100 nM) for 20 min before imaging, or 2) treated with a baculovirus vector containing MitoRFP (Life CellLight; 100 µl/2 ml media). To measure mitochondrial number, individual cells (at least 5 per experiment) in images of ECs isolated from at least five different animals were chosen by an investigator blinded to treatment/condition. Raw images were analyzed using CellProfiler software with a mitochondria-specific object segmentation workflow as described and validated previously (8). For mitochondrial morphologic measurements, it has recently been suggested that traditional measures of fragmentation such as aspect ratio and roundedness/form factor are not robust markers of changes in mitochondrial network connectivity (23, 30, 32). Thus we employed a newer morphology analytic algorithm that computationally “walks” the mitochondrial network within each cell and reconstructs a network map of connected mitochondria to determine fragmentation features such as changes in elements/cluster, junction/ends, and shifts in mitochondrial length/number distribution. This algorithm was recently validated using multiple cell lines treated with nonapopotic stimuli that stimulate either fusion or fission (32). For both mitochondrial number and morphology calculation, raw data files obtained from batch processing of confocal images were analyzed using R.
Mitochondrial metabolism measurements.
Equal numbers (20,000) of N- and SuHx-MVECs were seeded on a 96-well plate and incubated overnight in low-glucose DMEM (GIBCO) with 5% FBS (Hyclone). The cells were then washed with Seahorse XF base media (Agilent) supplemented with the following (in vol/vol): 1% 200 mM l-glutamine, 1% 100 mM pyruvate, and 0.4% 45% glucose (for a final concentration of 10 mM). This solution was pH adjusted to 7.4 and sterile filtered. One-hundred seventy-five microliters of the supplemented base media were added to each well. The injection ports for each well were loaded with 2 µM oligomycin, 0.5 µM FCCP, and 0.5 µM rotenone/antimycin A (all obtained from Agilent as part of the Mito Stress Kit pack). Metabolic flux analysis was conducted using the Seahorse Flux Analyzer (Model XFe96). The oxygen consumption rate was measured at baseline and following treatment with: oligomycin, FCCP, antimycin A, and rotenone. Values were normalized to cell number, measured using a CyQuant Kit (Thermo). The data were analyzed using Wave Software (Agilent) and GraphPad.
Intracellular Ca2+ measurements.
Cells were plated and grown to 50–60% confluence on glass coverslips. Three to four hours before measurements, the media were changed to DMEM with 5% serum. One h before experiments, cells were loaded with 5 μM fura-2 AM (Molecular Probes, Eugene, OR). Cells were placed in a temperature-controlled (37°C) laminar flow chamber in a live cell Ca2+ imaging system as described previously (45). Cells were perfused with a modified Krebs buffer containing the following (in mM): 118 NaCl, 4.7 KCl, 0.57 MgSO4, 1.18 KH2PO4, 25 NaHCO3, 2.5 CaCl2, and 10 glucose gassed with 16% O2 and 5% CO2. For Ca2+-free experiments, CaCl2 was replaced with 1 mM EGTA. Perfusate and chamber temperatures were maintained at 37°C. At the beginning of each experiment, cells were perfused for 15 min to allow for establishment of stable baseline before the start of measurements. [Ca2+]i was estimated from F340/F380 measured in calibration solutions with Ca2+ concentrations of 0–1350 nM (Molecular Probes).
Cell volume.
Confluent T75 flasks of N- and SuHx-MVECs were trypsinized, and cell volume was determined using a handheld automated cell counter (Scepter 2.0 Counter; 60-µM sensor; Millipore). Equal aliquots of media containing cell suspensions were loaded into the Scepter sensor, and the cell count and volume were measured as described by others (28, 40).
Intracellular ROS measurements.
MVECs were plated on coverslips at 60–80% confluence, and cells were infected with a baculovirus vector containing a roGFP-GRX1 plasmid (Premo ROS sensor; Life Biotechnologies) at 80 multiplicity of infection. At 48 h, cells were imaged using fluorescent microscopy. A subset of F400/F490 measurements were made using the Ca2+ imaging system described earlier and analyzed using Incytim2 Software (Intracellular Imaging). To reproduce and validate our findings, roGFP infected MVECs were also imaged using 1) an Olympus 1X51 inverted microscope fitted with appropriate excitation and emission filters, and 2) a Zeiss LSM800 laser scanning confocal microscope fitted with a heated live-cell imaging chamber. As all three measurement techniques yielded similar roGFP ratios, the data are reported together. For all three methods, individual cells were selected and F400/F490 was calculated after background subtraction. The frame rates for the three methods were five images/min (Intracellular Imaging and Olympus fluorescence microscopes) or three images/minute (Zeiss confocal microscope). The individual cell ratio values were averaged to yield a single roGFP ratio value for each experiment. For time traces of roGFP fluorescence, coverslips were incubated in heated HEPES buffer containing the following (in mmol/l): 130 NaCl, 5 KCl, 1.2 MgCl2, 1.5 CaCl2, 10 HEPES, and 10 glucose, with pH adjusted to 7.2 with 5 mol/l NaOH, and a single field of cells was serially imaged at baseline and following addition of 1 µM MitoQ, 1 mM DTT, or 1 mM H2O2. Each SuHx-MVEC experiment was performed immediately after a corresponding N-MVEC control experiment using all the same imaging settings, and the SuHx F400/F490 ratios were normalized to normoxic controls. Photomicrographs of F400/F490 were obtained using the Ratio Plus plug-in (ImageJ).
Migration.
Transwell filters (Corning) were immersed in cultureware containing DMEM with 5% serum. After 2 h, 100,000 cells were added to the top chamber of each filter. Equal numbers of cells were used for all experiments. Cells were incubated for 24 h, after which cells were fixed with ice cold ethanol (100%; 10 min) and stained with Brilliant Blue (5 min). To determine total and migrated cell number, images were obtained before (total cells) and after removal of cells on the top portion of the Transwell filter by scraping (migrated cells). Percent migration was calculated by dividing the number of migrated cells by the total number of cells and multiplying by 100.
Proliferation.
Equal numbers of N- and SuHx-MVECs were seeded in triplicate in a 96-well plate containing 200 µl of DMEM with 5% serum per well and incubated overnight before addition of bromodeoxyuridine (1:100). At 18 h, cells were fixed, blocked, and stained with anti-bromodeoxyuridine per kit instructions (Biotrak; GE HealthCare). Triplicates were averaged, and N- and SuHx-MVECs proliferation measurements were made pairwise. SuHx-MVEC proliferation measurements were normalized to normoxic controls. For each experiment, one set (triplicates) of N-MVECs also received 20% FBS as a positive control.
Data analysis/statistics.
All values are expressed as means ± SE. For [Ca2+]i and roGFP measurements, data were collected from up to 30 cells, and the values were averaged to obtain a single value for each experiment. Basal [Ca2+]i was calculated by averaging values over 3–5 min after stabilization. All experiments were performed using cells isolated from at least five different rats. Data were compared using unpaired Student’s t-test or by one-way ANOVA with a Holm-Sidak post hoc test to determine differences between groups. The earthmover distance metric was used to compare differences between the length/number distributions between N- and SuHx-MVEC mitochondria. As described previously (31, 32), this statistic can be used to evaluate changes in the distribution of no-parametric histogram data. Earthmover distance was calculated using the Momito program written by Ouellet et al. (32). Inter- and intragroup differences in EMD distance between normoxic and SuHx mitochondria were compared using ANOVA. P < 0.05 was accepted as statistically significant for all experiments.
RESULTS
Validation of antibodies.
To improve transparency and rigor, we have provided full-length gels for antibodies used in this article (Fig. 1). As discussed in methods, the antibodies used in our current study have been validated by us previously or by others using lysates from genetically deficient animals and/or genetically silenced cells.
Validation of SuHx model.
As shown in Fig. 2, compared with controls, SuHx rats exhibited significantly higher RV systolic pressure (Fig. 2A). Markers of RV hypertrophy, including the ratio of RV weight to left ventricular (LV) and septal (S) weight (RV/LV + S; Fig. 2B) and RV weight normalized to body weight (Fig. 2C) were also significantly increased in SuHx rats, indicating development of pulmonary hypertension.
Fig. 2.

Phenotyping of normoxic (N)-microvascular endothelial cells (MVECs) and SU5416 plus hypoxia (SuHx)-MVECs. A–C: hemodynamics in normoxic and SuHx rats used for MVEC isolation. Scatterplots showing means ± SE values for right ventricular systolic pressure (RVSP; A), Fulton index [right ventricular to left ventricular + septal weight (RV/LV + S); B], and RV/body weight (C) in normoxic (N) and SuHx rats. *Significant difference from N rats; n = 5–7 animals/group. D: representative phase contrast photomicrographs of N-MVECs and SuHx-MVECs showing cell morphology. E and F: representative immunoblot (E) and densitometry (F) showing means ± SE endothelial nitric oxide synthase (eNOS) expression in N- and SuHx-MVECs. G: scatterplot showing means ± SE cell volume in N- and SuHx-MVECs. *Significant difference from N-MVEC control; n = 6–10 per group.
MVEC phenotype in ECs isolated from rats following SuHx.
MVECs from control and SuHx rats were initially characterized using phase contrast and immunofluorescence microscopy. As shown in Fig. 2D, unlike the characteristic “cobblestone” appearance of N-MVECs, SuHx-MVECs appeared larger and more spindle shaped. As eNOS expression is a common marker for ECs, we evaluated eNOS expression (Fig. 2E) and found similar expression of eNOS (140 kDa) in N- and SuHx-MVECs (Fig. 2F). Interestingly, compared with N-MVECs, SuHx-MVECs expressed a strong eNOS band at a higher (~150 kDa) molecular mass; this band likely corresponds to phosphorylated eNOS. Lastly, since the SuHx cells appeared larger on bright-field microscopy, we measured cell volume in trypsinized N- and SuHx-MVECs. As shown in Fig. 2G, cell volume was significantly higher in SuHx-MVECs, suggesting cell hypertrophy.
Next, we assessed cell surface marker expression in N- and SuHx-MVECs. Immunofluorescence experiments (Fig. 3, A–D) revealed that N-MVECs stained positive for GS, a MVEC marker, and vWF, a pan-EC marker, but did not show positivity for the smooth muscle cell markers SMA and MHC. In contrast, rat pulmonary arterial smooth muscle cells stained negative for both EC markers but were positive for SMA and MHC. Interestingly, while SuHx-MVECs stained positive for EC markers, these cells also exhibited reactivity to SMA and MHC (summarized in Fig. 3E). Confocal images of SuHx-MVECs (Fig. 4) showed that SuHx-MVECs coexpress EC and smooth muscle cell markers.
Fig. 4.

Confocal imaging of surface marker coexpression. Representative images showing individual channel and merged photomicrographs obtained by confocal microscopy of normoxic (N)-microvascular endothelial cells (MVECs) (A) and SU5416 plus hypoxia (SuHx)-MVECs (B) costained with EC markers Griffonia simplicifolia (GS) and von willebrand factor (vWF), SuHx-MVECs costained with EC marker vWF and smooth muscle cell (SMC) marker smooth muscle α-actin SMA (C), and SuHx-MVECs costained with SMC markers SMA and myosin heavy chain (MHC) (D).
Altered mitochondrial morphology and increased intracellular ROS in SuHx MVECs.
Mitochondrial morphology was characterized in N- and SuHx-MVECs using Mitotracker and MitoRFP. We initially measured mitochondrial number using MVECs stained with MitoTracker (Fig. 5A). We noted a small but significant increase in total mitochondrial number in SuHx-MVECs (Fig. 5B). To validate these findings, we measured both mitochondrial number and morphology using confocal microscopy in MVECs following infection with MitoRFP-containing baculovirus. Similar to our Mitotracker data, SuHx MVEC mitochondria were increased in number when measured using MitoRFP (Fig. 5, A and B). To quantitatively characterize changes in mitochondrial connectivity/fragmentation, we employed an unsupervised algorithm to map mitochondrial networks in cells transfected with mitoRFP. As shown in Fig. 6A, this validated algorithm (32) utilizes changes in network architecture that occur with fragmentation to evaluate for differences in mitochondrial connectivity. As summarized in Fig. 6B, it has been shown that as mitochondria become fragmented, the increase in shorter mitochondrial fragments leads to a leftward shift in the mitochondrial length/number distribution and a decrease in number of elements per cluster as well as junctions/ends (i.e., fragmentation, a larger number of mitochondrial clusters, each with a more simplified network structure) (32). As shown in Fig. 6, C–E, in SuHx-MVECs, we observed a left shift of the mitochondrial length/number distribution, decreased elements/cluster, and decreased junctions/ends. To confirm that the observed morphologic differences were in fact due to increased fission, we probed MVEC lysates for phosphorylated and total protein levels of DRP1 since DRP1 phosphorylation has been shown to be strongly correlated with mitochondrial fission (9). As shown in Fig. 7, A–C, phosphorylated Drp1 (pDrp1Ser616 and pDrp1Ser637) was increased in SuHx-MVECs, whereas there was no difference in total Drp1.
Fig. 5.
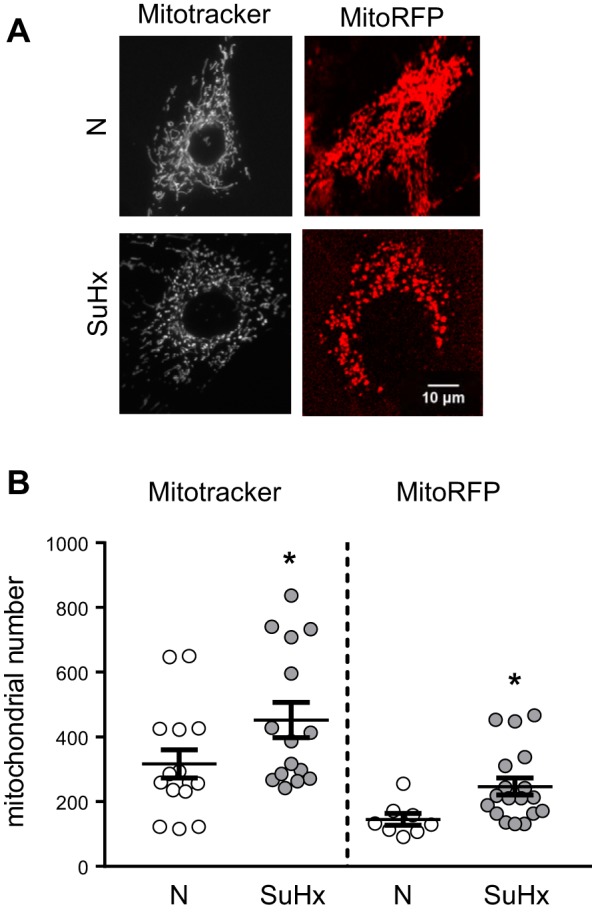
Altered mitochondrial morphology in SU5416 plus hypoxia (SuHx)-microvascular endothelial cells (MVECs). A: representative single cell images of mitochondria in normoxic (N)-MVECs and SuHx-MVECs stained with the mitochondrial dye Mitotracker (100 nM; 30 min) or transfected with MitoRFP. B: scatterplot showing means ± SE mitochondrial number in N- and SuHx-MVECs following mitochondrial tagging with either MitoTracker or MitoRFP. *Significant difference from N-MVEC control, n = 15–20 images from at least 5 different animals per group.
Fig. 6.

Computational analysis of mitochondrial network in SU5416 plus hypoxia (SuHx)-microvascular endothelial cells (MVECs). A: represented images of normoxic (N)- and SuHx- mitochondrial networks extracted from images of N- and SuHx-MVECs transfected with MitoRFP. B: table showing expected results of computational analysis of the mitochondrial network under conditions of increased fission or fusion. C: mitochondrial length/number distribution curves for N- and SuHx-MVEC mitochondria showing left shift in SuHx-MVEC mitochondria. D: scatterplot showing means ± SE elements/cluster (Ec) for N- and SuHx-MVEC mitochondria computationally extracted from images of MitoRFP-transfected cells. E: scatterplot showing means ± SE junctions/ends ratio (J/E) for N- and SuHx-MVEC mitochondria computational extracted from images of MitoRFP-transfected cells. *Significant difference from N-MVEC control; n = 10 per group.
Fig. 7.
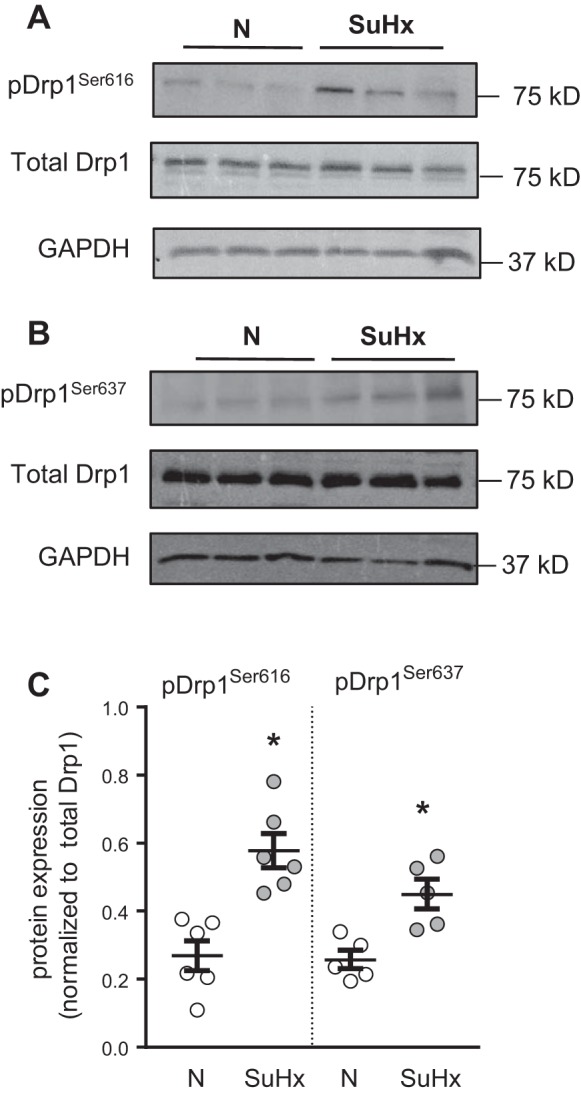
Dynamin-related protein 1 (Drp1) phosphorylation in SU5416 plus hypoxia (SuHx)-microvascular endothelial cells (MVECs). A and B: representative immunoblots demonstrating pDrp1Ser616 (A) and pDrp1Ser636 (B) protein levels in relation to total Drp1 and GAPDH expression in normoxic (N)- and SuHx-MVECs. C: scatterplot showing means ± SE densitometry values for pDrp1Ser616 and pDrp1Ser636, normalized to total Drp1. *Significant difference from N-MVEC control; n = 5–6 per group.
We next measured mitochondrial respiration in N- and SuHx-MVECs. As shown in Fig. 8, basal and maximal oxygen consumption was significantly lower in SuHx-MVECs (Fig. 8, A–C), whereas basal and maximal extracellular acidification rate were slightly, but significantly, increased in SuHx-MVECs (Fig. 8, D–F), suggesting increased H+ ion export across the cell membrane. These changes are suggestive of glycolytic shift. To determine whether changes in mitochondrial morphology and respiration were accompanied by increased mtROS levels, we measured total cytosolic ROS levels using roGFP in N- and SuHx-MVECs with and without mtROS quenching with the mitochondria-targeted antioxidant MitoQ (MQ; 1 µM). roGFP is a ratiometric ROS dye, which controls for potential differences in the amount of roGFP expression both within and between samples. As shown in Fig. 9A ratiometric ROS measurements were stable over time and responsive to maximal reduction with DTT followed by oxidation with exogenous ROS (H2O2). ROS levels were increased in SuHx-MVECs at baseline compared with N-MVECs (Fig. 9, B and C) and decreased to N-MVECs levels when treated with MitoQ (Fig. 9, C and D).
Fig. 8.

Decreased oxidative phosphorylation in SU5416 plus hypoxia (SuHx)-microvascular endothelial cells (MVECs). A: representative traces of oxygen consumption rate (OCR) at baseline and following treatment with oligomycin, FCCP, and rotenone/antimycin in Normoxic (N) (dashed lines)- and SuHx-MVECs (solid lines). B and C: scatterplots showing means ± SE OCR values for basal respiration (B) and maximal respiration (C) in N- and SuHx-MVECs. D: representative traces of extracellular acidification rate (ECAR) at baseline and with treatment with oligomycin, FCCP, and rotenone/entimycin in N (dashed lines)- and SuHx-MVECs (solid lines). E and F: scatterplots showing means ± SE ECAR values for basal ECAR (E) and maximal ECAR (F) in N- and SuHx-MVECs. *Significant difference from N-MVECs control; n = 18–20 experiments using cells isolated from 5 different animals per group.
Fig. 9.

Reactive oxygen species (ROS) levels in SU5416 plus hypoxia (SuHx)-microvascular endothelial cells (MVECs). A: representative trace of fluorescence ratio (F380/F490) in SuHx-MVECs following infection with roGFP plasmid showing baseline roGFP ratio and decrease in ratio following reduction with DTT(1 mM) followed by a rise in roGFP ratio with addition of exogenous oxidant (1 mM H2O2). B: confocal images of individual excitation/emission images (400/510 and 490/510) as well as 400/490 ratio in normoxic (N)-MVECs and SuHx-MVECs following infection with roGFP-containing plasmid. C: scatterplot showing means ± SE roGFP fluorescence ratio in N- and SuHx-MVECs with and without MitoQ treatment. D: representative traces of roGFP ratio in N-MVECs (dashed line) and SuHx-MVECs normalized to normoxic control (solid line) at baseline and following acute treatment with MitoQ (1 µM). *Significant difference from N-MVEC control; **significant difference from untreated SuHx-MVECs; n = 6–15 individual experiments using cells isolated from 5 different animals.
Basal [Ca2+]i, migration, and proliferation in SuHx MVECs.
Given our prior work implicating ROS in activation of Ca2+ entry via the calcium channel TRPV4 in MVECs (45, 46), we measured basal [Ca2+]i in N- and SuHx-MVECs at baseline and with nonspecific and mitochondrial-specific quenching of ROS with TEMPOL and MitoQ, respectively. Basal [Ca2+]i was significantly higher in SuHx-MVECs (Fig. 10, A and F). While removal of extracellular Ca2+ had no significant effect on baseline [Ca2+]i in N-MVECs, the elevated basal [Ca2+]i observed in SuHx-MVECs was rapidly normalized when extracellular Ca2+ was removed (Fig. 10A). Treatment with MitoQ (Fig. 10B) also reversed baseline elevations in SuHx-MVECs [Ca2+]i. Since our prior work showed that ROS activated TRPV4 to increase [Ca2+]i in MVECs (45), we measured [Ca2+]i levels following TRPV4 blockade in N- and SuHx-MVECs. Inhibition of TRPV4 with HC-067047 (10 µM) attenuated [Ca2+]i in SuHx-MVECs (Fig. 10, C and E) to a similar extent as removal of extracellular Ca2+ or treatment with antioxidants. To confirm these findings, we also measured [Ca2+]i in N- and SuHx-MVECs before and after treatment with GSK2193874 (30 nM), another specific inhibitor of TRPV4 (Fig. 10, D and E) (50). Interestingly, TRPV4 expression was similar between N- and SuHx-MVECs (Fig. 11, A and B). To determine whether baseline activation of TRPV4 affected ROS levels, we measured ROS levels in N- and SuHx-MVECs with and without TRPV4 inhibition (using HC-067047; 10 µM). Unlike MitoQ, HC-067047 treatment did not normalize elevated ROS levels in SuHx-MVECs (Fig. 11C). Finally, we measured migration and proliferation in N- and SuHx-MVECs. Consistent with reports in human pulmonary arterial smooth muscle cells from PAH patients (34, 62), baseline migration (Fig. 12A) and proliferation (Fig. 12B) were both higher in SuHx-MVECs than N-MVECs. Similar to our Ca2+ data, quenching ROS with either TEMPOL or MitoQ, or inhibiting TRPV4 with HC-067047, reduced migration and proliferation in SuHx-MVECs to near control levels (Fig. 13).
Fig. 10.

Abnormalities in intracellular calcium concentration ([Ca2+]i) in SU5416 plus hypoxia (SuHx)-microvascular endothelial cells (MVECs). A–D: representative tracings showing [Ca2+]i in normoxic (N)-MVECs and SuHx-MVECs before and after removal of extracellular Ca2+ (A), treatment with MitoQ (B), treatment with HC-067047 (HC; C), and treatment with GSK2193874 (GSK2; D). E: scatterplot showing means ± SE [Ca2+]i in N- and SuHx-MVECs at baseline and following removal of extracellular Ca2+ via perfusion with Ca2+-free buffer (CF) or treatment with HC (10 µM), GSK2193874 (30 nM; GSK2), TEMPOL (0.5 mM; Tem), and MitoQ (1 µM; MQ). TRPV4, transient receptor potential vanilloid-4; ROS, reactive oxygen species. *Significant difference from N-MVEC control; **significant difference from untreated SuHx-MVECs; n = 5–12/group.
Fig. 11.
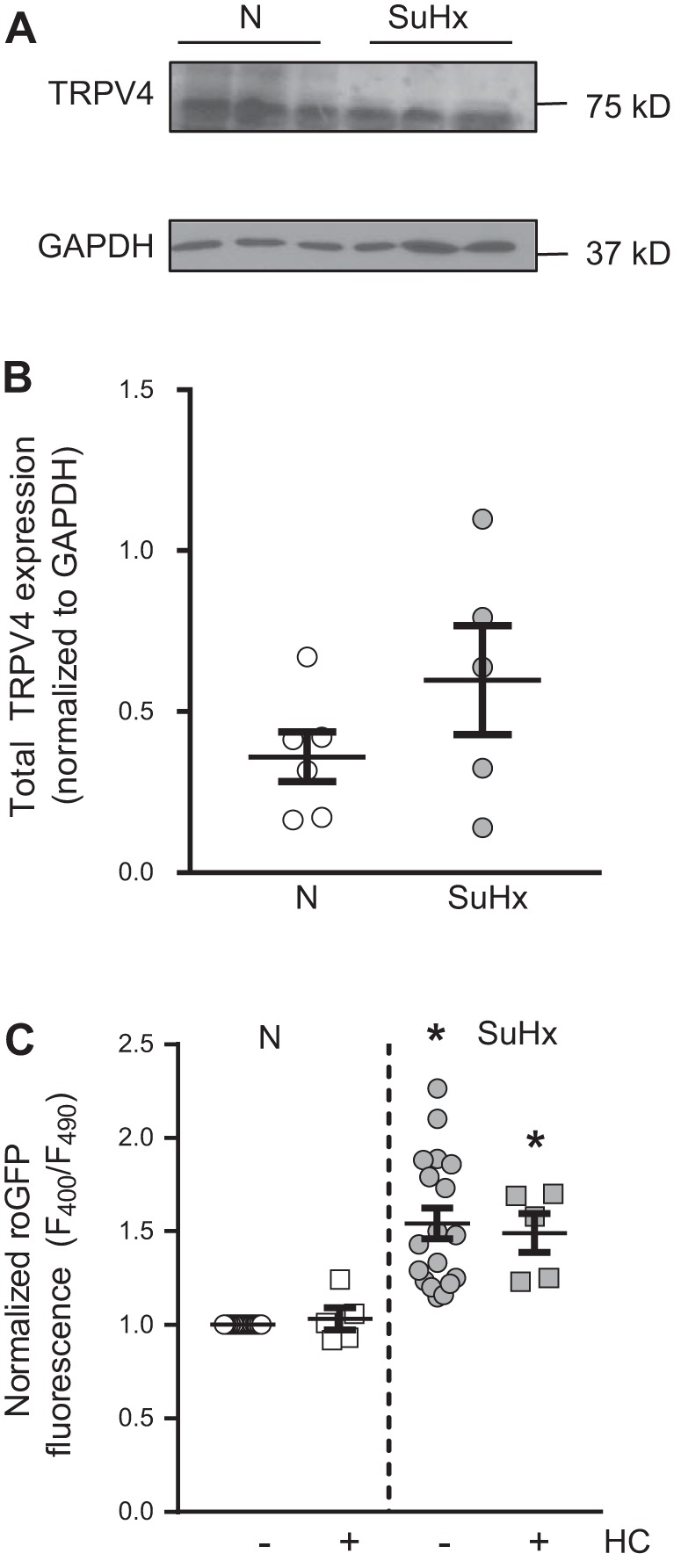
Transient receptor potential vanilloid-4 (TRPV4) expression and effect of TRPV4 inhibition of reactive oxygen species (ROS) levels in SU5416 plus hypoxia (SuHx)-microvascular endothelial cells (MVECs). A: representative immunoblot showing total TRPV4 expression in normoxic (N)- and SuHx-MVECs. B: scatterplot of means ± SE TRPV4 densitometry values normalized to GAPDH. C: scatterplot showing means ± SE roGFP fluorescence levels in N- and SuHx-MVECs with and without treatment with the TRPV4 inhibitor HC-067047 (10 µM; HC). *Significant difference from N-MVEC control; n = 5–15 individual experiments using cells isolated from 5 different animals.
Fig. 12.
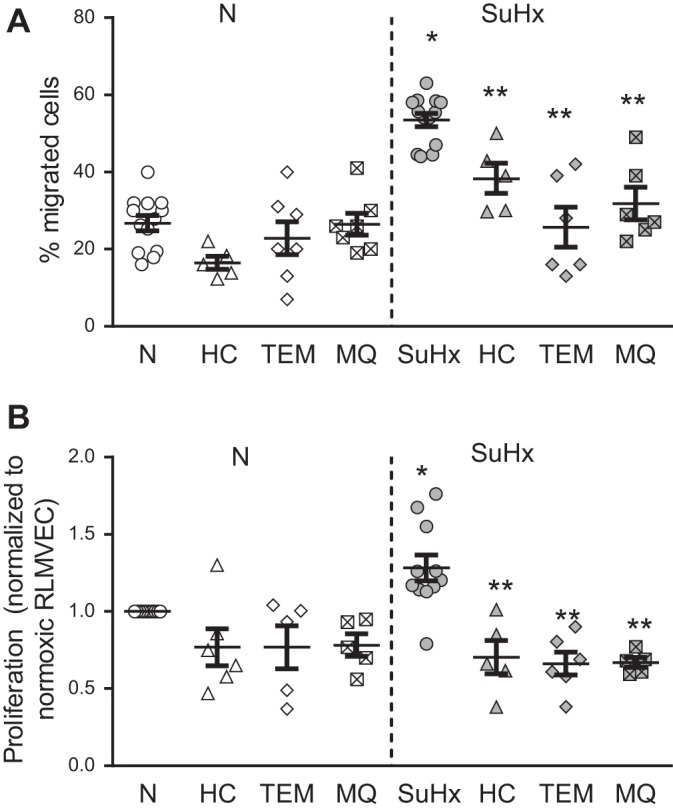
Increased migration and proliferation in SU5416 plus hypoxia (SuHx)-microvascular endothelial cells (MVECs). Scatterplots showing migration (A) and proliferation normoxic (N)-MVECs and SuHx-MVECs (B) with and without treatment with TEMPOL (0.5 mM; Tem), HC-067047 (10 µM; HC), and MitoQ (1 µM; MQ). *Significant difference from N-MVECs control; **significant difference from untreated SuHx MVECs; n = 5–12/group.
Fig. 13.

A: table showing observed molecular, structural, and functional changes in SU5416 plus hypoxia (SuHx)-microvascular endothelial cells (MVECs). B: proposed mechanisms linking mitochondrail reactive oxygen species (ROS) in intracellular calcium concentration ([Ca2+]i), migration, and proliferation in SuHx-MVECs. Mitochondrial dysfunction (i.e., increased fission, increased ROS production, and decreased oxidative phosphorylation) leads to increased cytosolic ROS levels, which activate transient receptor potential vanilloid-4 (TRPV4), increase [Ca2+]i, and promote migration and proliferation in SuHx-MVECs; thus inhibition of this pathway could be achieved with selective TRPV4 blocker (HC-067047) quenching of cytosolic ROS with TEMPOL quenching of mitochondria-specific ROS with MitoQ. eNOS, endothelial nitric oxide synthase; GS, Griffonia simplicifolia; vWF, von willebrand factor; SMA, smooth muscle α-actin; MHC, myosin heavy chain.
DISCUSSION
In this study, we show that MVECs isolated from SuHx rats exhibit several phenotypic and functional abnormalities that contribute to increased migration and proliferation in vitro. In addition to abnormal morphology, increased basal [Ca2+]i, and expression of smooth muscle cell markers, SuHx-MVECs exhibit evidence of mitochondrial dysfunction characterized by increased fission, decreased oxidative phosphorylation, and increased ROS production. These cellular abnormalities (i.e., evidence of mesenchymal transition, mitochondrial dysfunction, increased [Ca2+]i, increased migration, and proliferation) are also seen in ECs isolated from patients with PAH (15, 29, 57, 58), suggesting that abnormalities in PAH ECs, possibly triggered by extrinsic factors like soluble mediators in serum or inflammatory cells, persist in culture and may be playing a role in the pathobiology of experimental and human PAH. In addition, our data show that inhibition of ROS either globally (i.e., TEMPOL) or in a mitochondrial-specific fashion (i.e., MitoQ) decreases [Ca2+]i migration and proliferation in SuHx-MVECs to an extent similar to what was observed when inhibiting TRPV4-mediated Ca2+ entry. Taken together, our data suggest a role for elevated production of mitochondrial-derived ROS in inducing Ca2+ influx via TRPV4 to promote enhanced migratory and proliferative capacity in SuHx-MVECs.
Our method for isolating MVECs is similar to previous descriptions of flow-cytometry and magnet-based EC isolation (6, 10, 13); however, we used a sequential selection process to exclude non-ECs (by selecting CD31+ cells) followed by exclusion of nonmicrovascular cells (by selecting for GS+ cells). At the time of experiments, cells from normoxic and SuHx rats had been isolated in culture under identical conditions for the same amount of time, suggesting that the observed changes in morphology and positive staining for smooth muscle markers represent a true phenotypic difference between N- and SuHx-MVECs rather than an artifact due to differential time spent in vitro. One explanation for these findings is SuHx-MVECs may be undergoing differentiation to a phenotype more similar to smooth muscle cells, while retaining EC cell marker expression [i.e., endothelial to mesenchymal transdifferentiation (EndMT)], similar to what is seen in human PAH (21, 38).
Since our method involves passaging the cells, it is difficult estimate the exact number of MVECs exhibiting these characteristics in vivo. However, Suzuki et al. (47) recently published in vivo evidence of EndMT in the mouse SuHx model, suggesting that the EndMT phenomenon may be occurring in vivo in both human and rodent models of PAH. In our studies, while we observed phenotypic changes in nearly all the cells studied, we cannot exclude the possibility that ECs with greater proliferative potential could outpace the slower growing more normal cells, and lead to overestimation of the number of cells that undergo these changes in vivo. We attempted to visualize VE-cadherin expression, loss of which is a hallmark of EndMT, in our cells using several commercially available antibodies. However, significant nonspecific binding was observed in rat lung MVECs despite repeated attempts at optimization. Moreover, while VE-cadherin staining has been repeatedly reported in human ECs (14, 26), and VE-cadherin expression has been assessed using Western blotting in rat MVECs (1, 33), to our knowledge, few studies have reported immunofluorescence staining using VE-cadherin antibodies in rat MVECs.
Our subsequent mitochondrial, ROS, and Ca2+ data support the hypothesis that EndMT accompanies the cellular signaling and metabolic abnormalities that contribute to increased migration and proliferation. In addition to possible EndMT, SuHx-MVECs exhibit increased volume, and on our confocal images, the nucleus of SuHx-MVECs appears larger than normoxic controls. It is possible that cellular hypertrophy may occur in SuHx-MVECs, raising the interesting possibility that in addition to increased migration and proliferation, increased EC cell size itself may also be contributing to vascular obliteration in PAH. This initial observation requires further validation with additional robust measures of cell size and hypertrophic markers.
Mitochondria are a prominent source of ROS in ECs. At baseline, mitochondria are highly dynamic organelles that spontaneously merge (fusion) and separate (fission) based on alterations in cellular energy demand and other factors (24). Increased fission occurs naturally as part of mitochondrial quality control (i.e., to dispose of mitochondria with heavily damaged DNA), as a part of cell division, and as a response to alterations in nutrient availability. For instance, high glucose concentration and increased fatty acid oxidation induce mitochondrial fission (61). Moreover, fission is associated with increased mtROS production and decreased oxidative phosphorylation (36), abnormalities that constitute a mitochondrial phenotype associated with increased cellular stress. In SuHx-MVECs, we found evidence of increased fission, reduced oxidative phosphorylation, and increased ROS that is likely mitochondrial in origin. While the number of mitochondria per cell in N-MVECs (~200) is similar to the number of mitochondria reported in control human pulmonary artery endothelial cells (56), unlike the reduction in mitochondria observed in human PAH pulmonary artery endothelial cells, our rat SuHx-MVECs displayed a small but significant increase in mitochondrial number compared with control cells. This difference could be due to differences in cell type (conduit vs. microvascular), differences in species, or the effect of fixation, serum, and/or media. Another possible explanation is that human PAH ECs are obtained from patients with end-stage disease, at which point sustained mitochondrial stress may have progressed to frank mitophagy. A third possibility is that increased mitochondrial numbers reflect a higher energetic need of SuHx-MVECs, which are larger than N-MVECs. Due to these considerations, the mitochondrial number per se cannot specifically identify which of the processes governing mitochondrial dynamics (increased fission, decreased mitophagy, or increased biogenesis) is responsible. However, our observations of fragmented mitochondrial morphology on confocal imaging and our Drp1 phosphorylation results are consistent with a role for fission. As reviewed recently (9), phosphorylation of Drp1 at the Ser616 residue has been associated with translocation of Drp1 to the mitochondria from the cytoplasm and initiation of mitochondrial fission. Our data suggest that in addition to Ser616, Ser637 phosphorylation is also increased in SuHx-MVECs. The differing effects of Ser637 phosphorylation on mitochondrial fission have been discussed in detail elsewhere (9); relevant to the cell type used in our studies, Wang et al. (53) recently demonstrated that Ser637 phosphorylation was critical in Drp1 recruitment to the mitochondria and initiation of fission in kidney microvascular endothelial cells. Whether processes other than fission (such as decrease in defective mitochondrial removal or increased mitochondrial production) are also playing a role is currently unknown. Finally, our findings of decreased oxidative phosphorylation in SuHx-MVECs are also consistent with findings in human ECs (39, 41, 56), and coupled with our extracellular acidification rate data, suggest glycolytic shift, a metabolic adaptation seen in cancer cells (51), although additional metabolic experiments examining activity of TCA and glycolytic cycle enzymes will be needed to confirm these initial findings.
In addition to mitochondrial dysfunction, our data indicate that basal levels of cytosolic ROS are elevated in SuHx-MVECs compared with controls. Since the roGFP fluorescence ratio was attenuated with application of MitoQ at a dose previously shown to abolish acute hypoxia-induced ROS production at complex III (5), it is likely that mitochondria are a main source of increased ROS in SuHx-MVECs. Maximal reduction with DTT decreased ROS levels to below those seen in N-MVECs, suggesting that MitoQ quenching did not simply completely reduce the intracellular compartment but rather normalized ROS levels to those seen in N-MVECs. It is important to note that we cannot definitively rule out contributions from nonmitochondrial ROS sources as the roGFP construct used in these studies was not targeted to mitochondria, and non-mtROS sources such as NAPDH oxidase (25) and uncoupled eNOS (18, 58) have also been suggested as sources of increased EC ROS in PAH. Interestingly, our eNOS immunoblots revealed additional higher molecular weight bands in SuHx-MVECs but not N-MVECs, potentially reflecting increased phosphorylation status. eNOS phosphorylation has been shown to be dysregulated in human PAH ECs, with increased phosphorylation at certain sites associated with eNOS uncoupling (57), raising the interesting possibility that eNOS may be uncoupled in SuHx-MVECs, contributing to elevated ROS levels. Moreover, these experiments were performed in the absence of exogenous growth factors, and it is possible that additional nonmitochondrial sources of ROS may be contributing to MVEC function in vivo due to exposure to circulating and paracrine signaling molecules.
Basal [Ca2+]i was increased in SuHx-MVECs, with near normalization of [Ca2+]i when cells were perfused with Ca2+-free buffer, suggesting Ca2+ influx as the primary source. We have shown that application of exogenous ROS increased [Ca2+]i in normal MVECs via influx through the Ca2+-permeable channel TRPV4 (45). Our current data show similar attenuation of SuHx-MVECs [Ca2+]i with 1) TEMPOL, 2) MitoQ, and 3) HC-067047, suggesting a link between increased endogenous ROS production and TRPV4 activation. That treatment with HC-067047, MitoQ, or TEMPOL acutely decreased basal [Ca2+]i suggests that ROS-induced activation of TRPV4 occurs at baseline in SuHx-MVECs. A role for TRPV4 in regulating hypoxic vasoconstriction has been previously reported (20), and TRPV4 has been implicated in migration and proliferation of tumor endothelial cells (17), but, to our knowledge, a role for a mitochondrial ROS-TRPV4 axis in regulating lung MVEC migration and proliferation has not been previously reported. Interestingly, a recent report noted that mitochondrial ROS activated TRPV4 and contributed to vasorelaxation in isolated cerebral arteries (48). Although our data similarly suggest a link between mtROS and TRPV4 and our prior work showed that exogenous H2O2 activated TRPV4 in MVECs (45), further studies are needed to determine the mechanism by which increased ROS (mitochondrial or otherwise) activate the TRPV4 channel. Lastly, based on our data showing attenuation of basal Ca2+ with TRPV4 inhibition, we hypothesized that TRPV4 expression would be increased in SuHx-MVECs. However, our immunoblot data suggests that total TRPV4 expression is not different between N- and SuHx-MVECs. One explanation for this finding is that, in SuHx-MVECs, membrane-bound TRPV4 may be elevated due to increased translocation of TRPV4 to the cell surface. Such a mechanism of TRPV4 regulation was recently reported in macrophages in the context of LPS-induced lung injury (43). We are currently undertaking studies aimed at determining the relationship between mtROS and TRPV4 translocation/activation in SuHx-MVECs.
Reduction of basal [Ca2+]i with both MitoQ and HC-067047, as well as lack of attenuation of roGFP fluorescence with HC-067047 treatment, suggest that mtROS production is upstream of increased [Ca2+]i in SuHx-MVECs. Since acute hypoxia induces similar mitochondrial ROS production (3), one possibility is that in the SuHx model, mitochondria dysfunction is initiated by hypoxia and continues unabated despite return to normoxia. In acute hypoxia, hypoxia-inducible factors (HIFs) play a prominent role in mediating cell signaling (60), with mtROS production being critical for stabilization of the α-subunit of HIF-1 (54) in pulmonary arterial smooth muscle cells. HIF signaling is activated in both animal and human pulmonary hypertension (16, 37, 60) and upregulates TRPC channels responsible for elevated basal [Ca2+]i in pulmonary arterial smooth muscle cells from mice and rats with hypoxia-induced pulmonary hypertension (15). Whether continued mtROS production in SuHx-MVECs leads to “pseudohypoxic” stabilization of HIFs and/or changes in the activity of HIF family members warrants further investigation.
Increased migration and proliferation have been reported in ECs isolated from patients with PAH (29), similar to our findings in MVECs isolated from SuHx rats. There are several limitations and caveats to our migration/proliferation results. We did not synchronize cells with a period of exposure to serum-free media in part because serum starvation may have induced quiescence (42) and because, in our experience, human and rodent MVECs exhibit significant cell stress and death after even a few hours in serum-free media. The increase in proliferation measured in SuHx-MVECs, while significant, is modest; stimulation with factors known to be elevated in PAH (such as PDGF or VEGF) may lead to larger differences in MVEC proliferation. Our experiments were also performed in the absence of coculture with smooth muscle, fibroblast, or inflammatory cells. Thus the changes observed reflect the contributions to increased PAH EC migratory and proliferative capacity due solely to abnormalities intrinsic to ECs and may differ in magnitude from what occurs in vivo, where increased production of growth factors might augment these responses.
In conclusion, our data suggest that MVECs isolated from SuHx rats exhibit a variety of phenotypic and functional abnormalities similar to those observed in ECs isolated from humans with PAH. Both ROS and [Ca2+]i promote migration and proliferation in ECs (11, 35); thus one explanation for our findings is that augmented mobility and growth in SuHx-MVECs may be driven, in part, by the intrinsic abnormalities in SuHx-MVECs that cause elevated mtROS production, which in turn activates TRPV4 to increase basal [Ca2+]i (Fig. 13). Further studies will be required to determine whether EndMT and possibly cellular hypertrophy occurs before, or as a consequence of, increases in [Ca2+]i and ROS. Given the similar abnormalities observed in human PAH ECs and SuHx-MVECs, the limited supply of human EC samples, and the technical difficulty in performing transfections and other genetic manipulations in human primary cells, N- and SuHx-MVECs may serve as a novel platform for studies exploring the signaling and functional abnormalities that contribute to vascular remodeling and MVEC dysfunction in PAH.
GRANTS
This study was supported by National Heart, Lung and Blood Institute Grants F32-HL-124930 and K08-HL-132055 (to K. Suresh); R01-HL-073859, R25-HL-084762, and R01-HL-126514 (to L. A. Shimoda); F32-HL-124727 and K08-HL-133475 (to J. Huestch); and T32-HL-007534 (to K. Suresh and J. Huetsch). Additional support includes a Gilead PAH Research Scholar Award (to K. Suresh) and National Insitutes of Health Grant S10-OD-016374 (to S. Kuo, Johns Hopkins University School of Medicine Microscope Facility).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
K.S., M.D., and L.A.S. conceived and designed research; K.S., L.S., H.J., Z.B., X.Y., C.K., and J.C.H. performed experiments; K.S. and Z.B. analyzed data; K.S., M.D., and L.A.S. interpreted results of experiments; K.S. and J.C.H. prepared figures; K.S. drafted manuscript; M.D. and L.A.S. approved final version of manuscript; L.A.S. edited and revised manuscript.
ACKNOWLEDGMENTS
We thank Drs. Jimmie Sylvester and David Pearse for thoughtful comments and guidance on experimental design and data analysis. We also thank Dr. Michael Caterina for providing expertise regarding TRPV4 and the Johns Hopkins School of Medicine Microscope Facility for assistance with fluorescence confocal imaging.
REFERENCES
- 1.Adkison JB, Miller GT, Weber DS, Miyahara T, Ballard ST, Frost JR, Parker JC. Differential responses of pulmonary endothelial phenotypes to cyclical stretch. Microvasc Res 71: 175–184, 2006. doi: 10.1016/j.mvr.2006.02.007. [DOI] [PubMed] [Google Scholar]
- 2.Aggarwal S, Gross CM, Sharma S, Fineman JR, Black SM. Reactive oxygen species in pulmonary vascular remodeling. Compr Physiol 3: 1011–1034, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Al-Mehdi AB, Pastukh VM, Swiger BM, Reed DJ, Patel MR, Bardwell GC, Pastukh VV, Alexeyev MF, Gillespie MN. Perinuclear mitochondrial clustering creates an oxidant-rich nuclear domain required for hypoxia-induced transcription. Sci Signal 5: ra47, 2012. doi: 10.1126/scisignal.2002712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Austin SA, Katusic ZS. Loss of endothelial nitric oxide synthase promotes p25 generation and tau phosphorylation in a murine model of Alzheimer’s disease. Circ Res 119: 1128–1134, 2016. doi: 10.1161/CIRCRESAHA.116.309686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bell EL, Klimova TA, Eisenbart J, Moraes CT, Murphy MP, Budinger GR, Chandel NS. The Qo site of the mitochondrial complex III is required for the transduction of hypoxic signaling via reactive oxygen species production. J Cell Biol 177: 1029–1036, 2007. doi: 10.1083/jcb.200609074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bonnet S, Provencher S, Guignabert C, Perros F, Boucherat O, Schermuly RT, Hassoun PM, Rabinovitch M, Nicolls MR, Humbert M. Translating research into improved patient care in pulmonary arterial hypertension. Am J Respir Crit Care Med 195: 583–595, 2017. doi: 10.1164/rccm.201607-1515PP. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bowers R, Cool C, Murphy RC, Tuder RM, Hopken MW, Flores SC, Voelkel NF. Oxidative stress in severe pulmonary hypertension. Am J Respir Crit Care Med 169: 764–769, 2004. doi: 10.1164/rccm.200301-147OC. [DOI] [PubMed] [Google Scholar]
- 8.Cataldo AM, McPhie DL, Lange NT, Punzell S, Elmiligy S, Ye NZ, Froimowitz MP, Hassinger LC, Menesale EB, Sargent LW, Logan DJ, Carpenter AE, Cohen BM. Abnormalities in mitochondrial structure in cells from patients with bipolar disorder. Am J Pathol 177: 575–585, 2010. doi: 10.2353/ajpath.2010.081068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cho B, Choi SY, Cho HM, Kim HJ, Sun W. Physiological and pathological significance of dynamin-related protein 1 (drp1)-dependent mitochondrial fission in the nervous system. Exp Neurobiol 22: 149–157, 2013. doi: 10.5607/en.2013.22.3.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dagda RK, Cherra SJ 3rd, Kulich SM, Tandon A, Park D, Chu CT. Loss of PINK1 function promotes mitophagy through effects on oxidative stress and mitochondrial fission. J Biol Chem 284: 13843–13855, 2009. doi: 10.1074/jbc.M808515200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Datla SR, Peshavariya H, Dusting GJ, Mahadev K, Goldstein BJ, Jiang F. Important role of Nox4 type NADPH oxidase in angiogenic responses in human microvascular endothelial cells in vitro. Arterioscler Thromb Vasc Biol 27: 2319–2324, 2007. doi: 10.1161/ATVBAHA.107.149450. [DOI] [PubMed] [Google Scholar]
- 12.Dröge W. Free radicals in the physiological control of cell function. Physiol Rev 82: 47–95, 2002. doi: 10.1152/physrev.00018.2001. [DOI] [PubMed] [Google Scholar]
- 13.Duthu GS, Smith JR. In vitro proliferation and lifespan of bovine aorta endothelial cells: effect of culture conditions and fibroblast growth factor. J Cell Physiol 103: 385–392, 1980. doi: 10.1002/jcp.1041030303. [DOI] [PubMed] [Google Scholar]
- 14.Evrard SM, Lecce L, Michelis KC, Nomura-Kitabayashi A, Pandey G, Purushothaman KR, d’Escamard V, Li JR, Hadri L, Fujitani K, Moreno PR, Benard L, Rimmele P, Cohain A, Mecham B, Randolph GJ, Nabel EG, Hajjar R, Fuster V, Boehm M, Kovacic JC. Endothelial to mesenchymal transition is common in atherosclerotic lesions and is associated with plaque instability. Nat Commun 7: 11853, 2016. doi: 10.1038/ncomms11853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fantozzi I, Zhang S, Platoshyn O, Remillard CV, Cowling RT, Yuan JX. Hypoxia increases AP-1 binding activity by enhancing capacitative Ca2+ entry in human pulmonary artery endothelial cells. Am J Physiol Lung Cell Mol Physiol 285: L1233–L1245, 2003. doi: 10.1152/ajplung.00445.2002. [DOI] [PubMed] [Google Scholar]
- 16.Fijalkowska I, Xu W, Comhair SA, Janocha AJ, Mavrakis LA, Krishnamachary B, Zhen L, Mao T, Richter A, Erzurum SC, Tuder RM. Hypoxia inducible-factor1alpha regulates the metabolic shift of pulmonary hypertensive endothelial cells. Am J Pathol 176: 1130–1138, 2010. doi: 10.2353/ajpath.2010.090832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fiorio Pla A, Ong HL, Cheng KT, Brossa A, Bussolati B, Lockwich T, Paria B, Munaron L, Ambudkar IS. TRPV4 mediates tumor-derived endothelial cell migration via arachidonic acid-activated actin remodeling. Oncogene 31: 200–212, 2012. doi: 10.1038/onc.2011.231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ghosh S, Gupta M, Xu W, Mavrakis DA, Janocha AJ, Comhair SA, Haque MM, Stuehr DJ, Yu J, Polgar P, Naga Prasad SV, Erzurum SC. Phosphorylation inactivation of endothelial nitric oxide synthesis in pulmonary arterial hypertension. Am J Physiol Lung Cell Mol Physiol 310: L1199–L1205, 2016. doi: 10.1152/ajplung.00092.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Giedt RJ, Yang C, Zweier JL, Matzavinos A, Alevriadou BR. Mitochondrial fission in endothelial cells after simulated ischemia/reperfusion: role of nitric oxide and reactive oxygen species. Free Radic Biol Med 52: 348–356, 2012. doi: 10.1016/j.freeradbiomed.2011.10.491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Goldenberg NM, Wang L, Ranke H, Liedtke W, Tabuchi A, Kuebler WM. TRPV4 is required for hypoxic pulmonary vasoconstriction. Anesthesiology 122: 1338–1348, 2015. doi: 10.1097/ALN.0000000000000647. [DOI] [PubMed] [Google Scholar]
- 21.Hopper RK, Moonen JR, Diebold I, Cao A, Rhodes CJ, Tojais NF, Hennigs JK, Gu M, Wang L, Rabinovitch M. In pulmonary arterial hypertension, reduced BMPR2 promotes endothelial-to-mesenchymal transition via hmga1 and its target slug. Circulation 133: 1783–1794, 2016. doi: 10.1161/CIRCULATIONAHA.115.020617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Huetsch JC, Jiang H, Larrain C, Shimoda LA. The Na+/H+ exchanger contributes to increased smooth muscle proliferation and migration in a rat model of pulmonary arterial hypertension. Physiol Rep 4: e12729, 2016. doi: 10.14814/phy2.12729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Koopman WJ, Visch HJ, Smeitink JA, Willems PH. Simultaneous quantitative measurement and automated analysis of mitochondrial morphology, mass, potential, and motility in living human skin fibroblasts. Cytometry A 69: 1–12, 2006. doi: 10.1002/cyto.a.20198. [DOI] [PubMed] [Google Scholar]
- 24.Liesa M, Shirihai OS. Mitochondrial dynamics in the regulation of nutrient utilization and energy expenditure. Cell Metab 17: 491–506, 2013. doi: 10.1016/j.cmet.2013.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liu JQ, Zelko IN, Erbynn EM, Sham JS, Folz RJ. Hypoxic pulmonary hypertension: role of superoxide and NADPH oxidase (gp91phox). Am J Physiol Lung Cell Mol Physiol 290: L2–L10, 2006. doi: 10.1152/ajplung.00135.2005. [DOI] [PubMed] [Google Scholar]
- 26.Long L, Ormiston ML, Yang X, Southwood M, Gräf S, Machado RD, Mueller M, Kinzel B, Yung LM, Wilkinson JM, Moore SD, Drake KM, Aldred MA, Yu PB, Upton PD, Morrell NW. Selective enhancement of endothelial BMPR-II with BMP9 reverses pulmonary arterial hypertension. Nat Med 21: 777–785, 2015. doi: 10.1038/nm.3877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lundby A, Secher A, Lage K, Nordsborg NB, Dmytriyev A, Lundby C, Olsen JV. Quantitative maps of protein phosphorylation sites across 14 different rat organs and tissues. Nat Commun 3: 876, 2012. doi: 10.1038/ncomms1871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lyons SM, Alizadeh E, Mannheimer J, Schuamberg K, Castle J, Schroder B, Turk P, Thamm D, Prasad A. Changes in cell shape are correlated with metastatic potential in murine and human osteosarcomas. Biol Open 5: 289–299, 2016. doi: 10.1242/bio.013409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Masri FA, Xu W, Comhair SA, Asosingh K, Koo M, Vasanji A, Drazba J, Anand-Apte B, Erzurum SC. Hyperproliferative apoptosis-resistant endothelial cells in idiopathic pulmonary arterial hypertension. Am J Physiol Lung Cell Mol Physiol 293: L548–L554, 2007. doi: 10.1152/ajplung.00428.2006. [DOI] [PubMed] [Google Scholar]
- 30.Nikolaisen J, Nilsson LI, Pettersen IK, Willems PH, Lorens JB, Koopman WJ, Tronstad KJ. Automated quantification and integrative analysis of 2D and 3D mitochondrial shape and network properties. PLoS One 9: e101365, 2014. doi: 10.1371/journal.pone.0101365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Orlova DY, Zimmerman N, Meehan S, Meehan C, Waters J, Ghosn EE, Filatenkov A, Kolyagin GA, Gernez Y, Tsuda S, Moore W, Moss RB, Herzenberg LA, Walther G. Earth mover’s distance (EMD): a true metric for comparing biomarker expression levels in cell populations. PLoS One 11: e0151859, 2016. doi: 10.1371/journal.pone.0151859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ouellet M, Guillebaud G, Gervais V, Lupien St-Pierre D, Germain M. A novel algorithm identifies stress-induced alterations in mitochondrial connectivity and inner membrane structure from confocal images. PLOS Comput Biol 13: e1005612, 2017. doi: 10.1371/journal.pcbi.1005612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Parker JC, Stevens T, Randall J, Weber DS, King JA. Hydraulic conductance of pulmonary microvascular and macrovascular endothelial cell monolayers. Am J Physiol Lung Cell Mol Physiol 291: L30–L37, 2006. doi: 10.1152/ajplung.00317.2005. [DOI] [PubMed] [Google Scholar]
- 34.Paulin R, Meloche J, Courboulin A, Lambert C, Haromy A, Courchesne A, Bonnet P, Provencher S, Michelakis ED, Bonnet S. Targeting cell motility in pulmonary arterial hypertension. Eur Respir J 43: 531–544, 2014. doi: 10.1183/09031936.00181312. [DOI] [PubMed] [Google Scholar]
- 35.Pendyala S, Gorshkova IA, Usatyuk PV, He D, Pennathur A, Lambeth JD, Thannickal VJ, Natarajan V. Role of Nox4 and Nox2 in hyperoxia-induced reactive oxygen species generation and migration of human lung endothelial cells. Antioxid Redox Signal 11: 747–764, 2009. doi: 10.1089/ars.2008.2203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Picard M, Shirihai OS, Gentil BJ, Burelle Y. Mitochondrial morphology transitions and functions: implications for retrograde signaling? Am J Physiol Regul Integr Comp Physiol 304: R393–R406, 2013. doi: 10.1152/ajpregu.00584.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pisarcik S, Maylor J, Lu W, Yun X, Undem C, Sylvester JT, Semenza GL, Shimoda LA. Activation of hypoxia-inducible factor-1 in pulmonary arterial smooth muscle cells by endothelin-1. Am J Physiol Lung Cell Mol Physiol 304: L549–L561, 2013. doi: 10.1152/ajplung.00081.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ranchoux B, Antigny F, Rucker-Martin C, Hautefort A, Péchoux C, Bogaard HJ, Dorfmüller P, Remy S, Lecerf F, Planté S, Chat S, Fadel E, Houssaini A, Anegon I, Adnot S, Simonneau G, Humbert M, Cohen-Kaminsky S, Perros F. Endothelial-to-mesenchymal transition in pulmonary hypertension. Circulation 131: 1006–1018, 2015. doi: 10.1161/CIRCULATIONAHA.114.008750. [DOI] [PubMed] [Google Scholar]
- 39.Rehman J, Archer SL. A proposed mitochondrial-metabolic mechanism for initiation and maintenance of pulmonary arterial hypertension in fawn-hooded rats: the Warburg model of pulmonary arterial hypertension. Adv Exp Med Biol 661: 171–185, 2010. doi: 10.1007/978-1-60761-500-2_11. [DOI] [PubMed] [Google Scholar]
- 40.Rotondo F, Romero MD, Ho-Palma AC, Remesar X, Fernández-López JA, Alemany M. Quantitative analysis of rat adipose tissue cell recovery, and non-fat cell volume, in primary cell cultures. PeerJ 4: e2725, 2016. doi: 10.7717/peerj.2725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ryan J, Dasgupta A, Huston J, Chen KH, Archer SL. Mitochondrial dynamics in pulmonary arterial hypertension. J Mol Med (Berl) 93: 229–242, 2015. doi: 10.1007/s00109-015-1263-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ryan JM. Effect of different fetal bovine serum concentrations on the replicative life span of cultured chick cells. In Vitro 15: 895–899, 1979. doi: 10.1007/BF02618046. [DOI] [PubMed] [Google Scholar]
- 43.Scheraga RG, Abraham S, Niese KA, Southern BD, Grove LM, Hite RD, McDonald C, Hamilton TA, Olman MA. TRPV4 mechanosensitive ion channel regulates lipopolysaccharide-stimulated macrophage phagocytosis. J Immunol 196: 428–436, 2016. doi: 10.4049/jimmunol.1501688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Stevens T. Functional and molecular heterogeneity of pulmonary endothelial cells. Proc Am Thorac Soc 8: 453–457, 2011. doi: 10.1513/pats.201101-004MW. [DOI] [PubMed] [Google Scholar]
- 45.Suresh K, Servinsky L, Reyes J, Baksh S, Undem C, Caterina M, Pearse DB, Shimoda LA. Hydrogen peroxide-induced calcium influx in lung microvascular endothelial cells involves TRPV4. Am J Physiol Lung Cell Mol Physiol 309: L1467–L1477, 2015. doi: 10.1152/ajplung.00275.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Suresh K, Servinsky L, Reyes J, Undem C, Zaldumbide J, Rentsendorj O, Modekurty S, Dodd OJ, Scott AL, Pearse DB, Shimoda LA. CD36 mediates H2O2-induced calcium influx in lung microvascular endothelial cells. Am J Physiol Lung Cell Mol Physiol 312: L143–L153, 2017. doi: 10.1152/ajplung.00361.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Suzuki T, Carrier EJ, Talati MH, Rathinasabapathy A, Chen X, Nishimura R, Tada Y, Tatsumi K, West JD. Isolation and characterization of endothelial-to-mesenchymal transition-cells in pulmonary arterial hypertension. Am J Physiol Lung Cell Mol Physiol 314: L118–L126, 2018. doi: 10.1152/ajplung.00296.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Szarka N, Pabbidi MR, Amrein K, Czeiter E, Berta G, Pohoczky K, Helyes Z, Ungvari Z, Koller A, Buki A, Toth P. Traumatic brain injury impairs myogenic constriction of cerebral arteries: role of mitochondria-derived H2O2 and TRPV4-dependent activation of BKCa channels. J Neurotrauma 2018 Jan 12. [Epub ahead of print]. doi: 10.1089/neu.2017.5056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Taraseviciene-Stewart L, Kasahara Y, Alger L, Hirth P, Mc Mahon G, Waltenberger J, Voelkel NF, Tuder RM. Inhibition of the VEGF receptor 2 combined with chronic hypoxia causes cell death-dependent pulmonary endothelial cell proliferation and severe pulmonary hypertension. FASEB J 15: 427–438, 2001. doi: 10.1096/fj.00-0343com. [DOI] [PubMed] [Google Scholar]
- 50.Thorneloe KS, Cheung M, Bao W, Alsaid H, Lenhard S, Jian MY, Costell M, Maniscalco-Hauk K, Krawiec JA, Olzinski A, Gordon E, Lozinskaya I, Elefante L, Qin P, Matasic DS, James C, Tunstead J, Donovan B, Kallal L, Waszkiewicz A, Vaidya K, Davenport EA, Larkin J, Burgert M, Casillas LN, Marquis RW, Ye G, Eidam HS, Goodman KB, Toomey JR, Roethke TJ, Jucker BM, Schnackenberg CG, Townsley MI, Lepore JJ, Willette RN. An orally active TRPV4 channel blocker prevents and resolves pulmonary edema induced by heart failure. Sci Transl Med 4: 159ra148, 2012. doi: 10.1126/scitranslmed.3004276. [DOI] [PubMed] [Google Scholar]
- 51.Tuder RM, Davis LA, Graham BB. Targeting energetic metabolism: a new frontier in the pathogenesis and treatment of pulmonary hypertension. Am J Respir Crit Care Med 185: 260–266, 2012. doi: 10.1164/rccm.201108-1536PP. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wakabayashi J, Zhang Z, Wakabayashi N, Tamura Y, Fukaya M, Kensler TW, Iijima M, Sesaki H. The dynamin-related GTPase Drp1 is required for embryonic and brain development in mice. J Cell Biol 186: 805–816, 2009. doi: 10.1083/jcb.200903065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wang W, Wang Y, Long J, Wang J, Haudek SB, Overbeek P, Chang BH, Schumacker PT, Danesh FR. Mitochondrial fission triggered by hyperglycemia is mediated by ROCK1 activation in podocytes and endothelial cells. Cell Metab 15: 186–200, 2012. doi: 10.1016/j.cmet.2012.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Waypa GB, Smith KA, Schumacker PT. O2 sensing, mitochondria and ROS signaling: The fog is lifting. Mol Aspects Med 47-48: 76–89, 2016. doi: 10.1016/j.mam.2016.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Xie Q, Wu Q, Horbinski CM, Flavahan WA, Yang K, Zhou W, Dombrowski SM, Huang Z, Fang X, Shi Y, Ferguson AN, Kashatus DF, Bao S, Rich JN. Mitochondrial control by DRP1 in brain tumor initiating cells. Nat Neurosci 18: 501–510, 2015. doi: 10.1038/nn.3960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Xu W, Erzurum SC. Endothelial cell energy metabolism, proliferation, and apoptosis in pulmonary hypertension. Compr Physiol 1: 357–372, 2011. doi: 10.1002/cphy.c090005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Xu W, Kaneko FT, Zheng S, Comhair SAA, Janocha AJ, Goggans T, Thunnissen FBJM, Farver C, Hazen SL, Jennings C, Dweik RA, Arroliga AC, Erzurum SC. Increased arginase II and decreased NO synthesis in endothelial cells of patients with pulmonary arterial hypertension. FASEB J 18: 1746–1748, 2004. doi: 10.1096/fj.04-2317fje. [DOI] [PubMed] [Google Scholar]
- 58.Xu W, Koeck T, Lara AR, Neumann D, DiFilippo FP, Koo M, Janocha AJ, Masri FA, Arroliga AC, Jennings C, Dweik RA, Tuder RM, Stuehr DJ, Erzurum SC. Alterations of cellular bioenergetics in pulmonary artery endothelial cells. Proc Natl Acad Sci USA 104: 1342–1347, 2007. doi: 10.1073/pnas.0605080104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ye L, Kleiner S, Wu J, Sah R, Gupta RK, Banks AS, Cohen P, Khandekar MJ, Boström P, Mepani RJ, Laznik D, Kamenecka TM, Song X, Liedtke W, Mootha VK, Puigserver P, Griffin PR, Clapham DE, Spiegelman BM. TRPV4 is a regulator of adipose oxidative metabolism, inflammation, and energy homeostasis. Cell 151: 96–110, 2012. doi: 10.1016/j.cell.2012.08.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yu AY, Frid MG, Shimoda LA, Wiener CM, Stenmark K, Semenza GL. Temporal, spatial, and oxygen-regulated expression of hypoxia-inducible factor-1 in the lung. Am J Physiol Lung Mol Cell Physiol 275: L818–L826, 1998. [DOI] [PubMed] [Google Scholar]
- 61.Yu T, Robotham JL, Yoon Y. Increased production of reactive oxygen species in hyperglycemic conditions requires dynamic change of mitochondrial morphology. Proc Natl Acad Sci USA 103: 2653–2658, 2006. doi: 10.1073/pnas.0511154103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Yu Y, Fantozzi I, Remillard CV, Landsberg JW, Kunichika N, Platoshyn O, Tigno DD, Thistlethwaite PA, Rubin LJ, Yuan JX. Enhanced expression of transient receptor potential channels in idiopathic pulmonary arterial hypertension. Proc Natl Acad Sci USA 101: 13861–13866, 2004. doi: 10.1073/pnas.0405908101. [DOI] [PMC free article] [PubMed] [Google Scholar]