

Abstract
Critically ill patients are commonly treated with high levels of oxygen, hyperoxia, for prolonged periods of time. Unfortunately, extended exposure to hyperoxia can exacerbate respiratory failure and lead to a high mortality rate. Mitochondrial A-kinase anchoring protein (Akap) has been shown to regulate mitochondrial function. It has been reported that, under hypoxic conditions, Akap121 undergoes proteolytic degradation and promotes cardiac injury. However, the role of Akap1 in hyperoxia-induced acute lung injury (ALI) is largely unknown. To address this gap in our understanding of Akap1, we exposed wild-type (wt) and Akap1−/− mice to 100% oxygen for 48 h, a time point associated with lung damage in the murine model of ALI. We found that under hyperoxia, Akap1−/− mice display increased levels of proinflammatory cytokines, immune cell infiltration, and protein leakage in lungs, as well as increased alveolar capillary permeability compared with wt controls. Further analysis revealed that Akap1 deletion enhances lung NF-κB p65 activity as assessed by immunoblotting and DNA-binding assay and mitochondrial autophagy-related markers, PINK1 and Parkin. Ultrastructural analysis using electron microscopy revealed that Akap1 deletion was associated with remarkably aberrant mitochondria and lamellar bodies in type II alveolar epithelial cells. Taken together, these results demonstrate that Akap1 genetic deletion increases the severity of hyperoxia-induced acute lung injury in mice.
Keywords: acute lung injury, AKAP121, Akap1, autophagy, hyperoxia, mitochondria, oxygen treatment
INTRODUCTION
Hyperoxic treatment is a first-line therapy used in acute respiratory failure patients. However, as a down side, prolonged hyperoxia ( > 0.8) may lead to hyperoxia-induced acute lung injury (HALI), a condition characterized by severe alveolar damage (23). Furthermore, hyperoxia has been shown to cause mortality in small animal and higher-order primate models (23). HALI triggers an acute inflammatory response that leads to proinflammatory cytokine production, neutrophil and macrophage infiltration, as well as edema and damage to the alveolar epithelium (13, 14, 29). Thus using hyperoxia as a form of oxygen toxicity is clinically relevant to study the pathophysiology of ALI. HALI is known to exacerbate respiratory failure, for which there is no effective pharmacotherapy available (11). Thus identifying the molecular mechanisms and pathways involved in the promotion and resolution of hyperoxia is essential for the design of effective treatments against oxidant lung injury.
Mitochondria play a central role in several cellular processes, as they are a source for both energy and reactive oxygen species (ROS). Consequently, mitochondria are critical regulators of cell survival in the lung and in other inflammatory diseases (4, 28). Our previous studies indicate that ROS, as well as the intermediate 4-hydroxy-2-nonenal (4-HNE), are produced during HALI and heavily impact mitochondrial integrity (16, 26, 44). Based on numerous studies of ROS involvement in ALI, many trials have targeted ROS for the treatment of ALI and acute respiratory distress syndrome (27, 35). Unfortunately, molecular instability makes ROS a challenging therapeutic target, and antioxidants (which target ROS) do not rectify mitochondrial damage (5, 18, 19). Recent studies in murine models have shown that targeting the proteins involved in mitochondrial function confers greater protection than antioxidant treatment (1, 3, 22).
One of the major mitochondrial proteins, A-kinase anchoring proteins (AKAPs) have been recently implicated in the transmission of cAMP signals to the outer membrane of mitochondria (2, 10). Mitochondrial AKAP121, AKAP100, and AKAP84 are products of the Akap1 gene, and are generated by alternative splicing (20). Downregulation of Akap121 was observed under cardiac pressure overload and was found to be associated with mitochondrial abnormalities and contributes to the development of cardiac dysfunction (33). In a recent study, it was reported that Akap1 deletion promotes mitochondrial aberrations, autophagy, and increases the severity of cardiac injury in a murine model of myocardial ischemia (39). The importance of Akap121 has been recently established in hypoxic cardiac disease models, but its role in lung disease has not yet been studied. In the present study, we hypothesize that Akap1 genetic deletion might affect mitochondrial function and facilitate a change in the severity of HALI.
METHODS
Experimental animals.
All animal experiments were approved by the Institution of Animal Care and Use Committee of the University of South Florida. Akap1−/− mice have been previously generated by Dr. Stanley McKnight (Univ. of Washington) (32) and were donated by Dr. Stefan Strack (Univ. of Iowa). All animals were housed in isolated cages, maintained under identical conditions of temperature (22 ± 1°C), humidity (60 ± 5%), and dark-light cycle. Mice were fed normal diet ad libitum.
In vivo hyperoxia exposure.
Akap1−/− mice and their littermates (C57BL/6 background, 7–9 wk of age) were placed in cages in an airtight chamber (75 × 50 × 50 cm) and exposed to room air or 100% O2 (hyperoxia, HO) for 48 h. Oxygen concentration was controlled with a proOx P100 sensor (BioSpherix), as previously described (25). All the experiments were repeated two to three times with n = 5 mice per group. Mice were maintained in a specific-pathogen-free animal facility at the University of South Florida.
Mitochondria isolation.
Mitochondria were isolated from lung tissue using a Mitochondrial Isolation Kit (cat. no: 89801, Thermo Fisher Scientifics), according to manufacturer’s instructions. The mitochondrial fraction was verified based on high expression of VDAC.
Western blotting.
Wild type (wt) and Akap1−/− mouse lungs were perfused with 5 ml of PBS and snap-frozen in liquid nitrogen for further use. Frozen lung tissue was pulverized and lysed in protein extraction buffer (20 mM Tris·HCl, pH 7.4, 150 mM NaCl, and 0.5% Triton X-100), and the supernatant, containing soluble protein, was collected by high-speed centrifugation (14,000 g) for 15 min at 4°C. Protein was estimated by BCA Protein Assay kit (Pierce, Rockford, IL). Equal amounts of protein (20 µg) were subjected SDS-PAGE by using 4–20% Tris-glycine gels (Bio-Rad), followed by electrotransfer to PVDF membrane. Membranes were blocked in 5% skim milk with TBS-T followed by incubation with appropriate primary and secondary antibodies. Antibodies to AKAP1, phospho p65, total p65, anti-p62, LC3B, VDAC, β-actin (Cell-signaling Technology), PINK1 (Millipore), and Parkin (Abcam) were used. The proteins were visualized using Pierce ECL Western blotting substrate (Thermo Fisher Scientifics). Protein bands were scanned, and densitometry was analyzed with NIH ImageJ software. Ratios of concerned protein with control loading protein were recorded, and results were expressed as percent controls.
NF-kB p65 DNA-binding activity assay.
Lung nuclear proteins were extracted using a nuclear extraction kit (Active Motif, Carlsbad, CA). Protein concentrations were determined using the BCA protein assay kit (Pierce). Equal amounts of nuclear extracts were used to quantify the DNA binding activity of the NF-kB p65 subunit using ELISA-based TransAM kit (40096; Active Motif) according to the manufacturer’s instructions and as described previously (34).
Bronchoalveolar lavage fluid (BALF) analysis.
BALF was collected by flushing 1 ml PBS into the lung via a tracheal cannula three times. The pooled BALF was centrifuged at 400 g in 4°C for 10 min. Immediately, BALF supernatant was collected in a fresh tube and stored at −80°C until further use. Pelleted cells were then resuspended in 1 ml PBS. Total cell number was counted using a hemocytometer, and a differential cell count was performed using cytospin preparations stained with Diff-Quik (Fisher Scientifics) as described previously (41). The total protein concentration in the supernatant, following BALF centrifugation, was determined using the BCA protein assay kit (Pierce).
Levels of IL-6 (cat. no. EM2IL6; sensitivity < 7 pg/ml) and TNFα (cat. no. KMC3011; sensitivity < 3 pg/ml) in BALF were measured using mouse ELISA kits (Thermo Fisher Scientifics) according to the manufacturer’s instructions, and they do not cross-react with any other cytokines.
Assessment of lung capillary leakage.
To assess lung permeability, 50 mg/kg of Evans blue dye (EBD; Sigma-Aldrich) dissolved in 200 µl of PBS was injected into the tail veins of mice following 48 h of hyperoxia. After 30 min, the animals were euthanized and the lungs perfused with 5 ml PBS, followed by BAL fluid collection in 2 ml of PBS as described above. The lungs were photographed, excised, and snap-frozen in liquid nitrogen for further analysis. BALF EBD was measured at 630 nm by spectrophotometry. Frozen lung EBD dye content was measured as described previously (34) and expressed as micrograms per gram of lung.
Myeloperoxidase assay.
As an index of neutrophil infiltration, lung tissue-associated myeloperoxidase (MPO) activity was measured as described previously (34) using a commercially available fluorometric assay kit (Cayman Chemicals) according to the manufacturer’s instructions.
Lung histology.
Formalin-fixed paraffin-embedded lung tissue sections were stained with hematoxylin and eosin. The extent of lung injury was evaluated as described previously (14).
Transmission electron microscopy.
Lung pieces from wt and Akap1−/− mice were fixed in 2% glutaraldehyde containing 0.5% tannic acid. Samples were further fixed in osmium tetroxide for 30 min at 37°C. All fixed samples were then dehydrated in graded ethanol and acetone. The dehydrated lung samples were processed for epoxy resin embedding using EMbed-812 (Electron Microscopy Sciences, Hatfield, PA). Thin sections (90–100 nm) were cut using an ultramicrotome (UCT; Leica, Wetzlar, Germany), treated with uranyl acetate, and analyzed as described previously (15). The number of abnormal mitochondria (mitochondrial aberrations) in type II AECs were counted in electron microscopy images as described previously (39), and results are expressed as percentage.
Statistical analysis.
All data are expressed as means ± SE. Significance of difference was assessed by Student’s t-test (single comparisons) or by one-way ANOVA with post hoc Tukey test (multiple comparisons) by using GraphPad Prism 7.0 (GraphPad Software, San Diego, CA). P < 0.05 was considered statistically significant.
RESULTS
Akap1 genetic deletion increases inflammatory cell infiltration into the airspace after hyperoxia.
To investigate the functional role of mitochondrial AKAP in the lung, we studied mice with global genetic deletion of the Akap1 gene (Akap1−/−) along with their wt littermates. To know the basal expression levels of Akap1 protein in the lung, we measured lung mitochondrial Akap1 protein in wt littermates and Akap1−/− mice by immunoblotting. Akap1 protein is constitutively expressed in lung mitochondrial extract, which suggests that it plays an important role in lung physiology. We first verified the efficacy of our global Akap1 knockout mice by analyzing Akap1 expression in the mitochondrial lysate. We observed no Akap1 expression in Akap1−/− mice relative to their littermate wt control (Fig. 1). To study the functional role of Akap1 in hyperoxic acute lung injury, wt and Akap1−/− mice were exposed to hyperoxia for 48 h and BAL fluid was collected as described in methods. Since infiltration of inflammatory cells into the injured area is an established hallmark of HALI (37), we analyzed cells in BAL fluid by total and differential cell counts. After 48 h of hyperoxia, wt mice displayed a significant increase in total cell numbers (P < 0.01) compared with room air-exposed wt mice (Fig. 2A). Analysis of Akap1−/− mice under hyperoxia displayed a significant increase (P < 0.01) in the total cell number (Fig. 2A), as well as neutrophil and macrophage levels (data not shown), compared with the wt littermate controls. However, there was no significant difference in total cell count between room air exposed BAL fluid samples in wt and Akap1−/− mice (Fig. 2A). Increased capillary permeability results in lung edema, a driving force for hypoxemia that is observed in ALI and causes escape of plasma protein into the alveolar space. Analysis of BAL fluid for protein by BCA assay shows increased levels of protein (P < 0.05) in the Akap1−/− mice compared with wt mice under hyperoxia (Fig. 2B). These data suggest that Akap1 deletion promotes alveolar leakage into the airspace. A prominent aspect of acute lung inflammation is infiltration of inflammatory cells into the lungs and thus to the BAL fluid. BAL fluid cells were cytospun for neutrophil cell counts after Diff-Quick staining. Our results revealed an increase in the neutrophil count in hyperoxia-exposed Akap1−/− mice compared with wt mice (Fig. 2C). However, MPO activity was significantly higher (P < 0.05) after hyperoxia in Akap1−/− mice lung compared with wt mice (Fig. 2D). MPO expression in the pulmonary system is an indicator for the presence of neutrophils. These results clearly indicate that Akap1 gene deletion increases the pulmonary infiltration of inflammatory cells following hyperoxic lung injury.
Fig. 1.
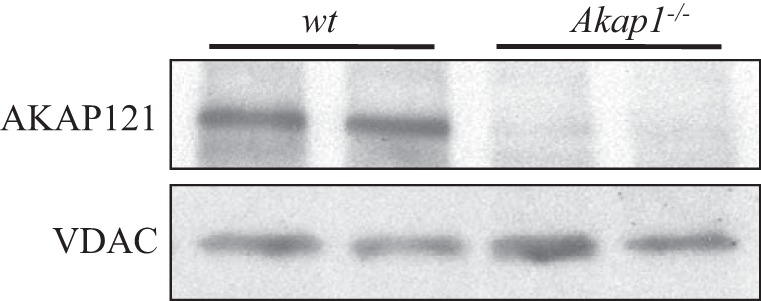
Western blot analysis of AKAP121 protein in wild-type (wt) and Akap1−/− mice lung. Representative immunoblot of mitochondrial protein isolated from the lungs. VDAC protein was used as loading control.
Fig. 2.

Akap1 genetic deletion increases inflammatory cell infiltration into the airspace (BAL fluid) after hyperoxia. Wild-type (wt) and Akap1−/− mice were exposed to room air or hyperoxia for 48 h, then BAL fluid was collected and a total cell count was conducted using a hemocytometer (A); total protein in BAL fluids was measured by BCA protein assay (B) (**P < 0.01 vs. wt room air; #P < 0.01 vs. wt hyperoxia; n = 5 mice/group). C: microscopic examination following cytospin and Diff-Quik–stained cells from BAL fluid collected from hyperoxia-exposed mice from wt and Akap1−/−; arrows denote neutrophils. Magnification: × 200. D: myeloperoxidase activity (% control) in lung tissue of hyperoxia exposed mice from wt and Akap1−/−. Data are shown as means ± SE (*P < 0.05 vs. wt hyperoxia; n = 5 mice/group).
Akap1 genetic deletion increases lung injury and alveolar permeability after hyperoxia.
To evaluate the effect of Akap1 genetic deletion on histopathological findings of mouse lungs exposed to hyperoxia, lungs were examined histologically and then subjected to H&E staining. Wild-type (wt) lungs exposed to 48 h of hyperoxia demonstrated an increased infiltration of immune cells, damage in alveoli, and thickening of the bronchial epithelium (Fig. 3A). This hyperoxia-induced morphological alteration was very high in Akap1−/− lungs relative to control lungs (Fig. 3A). To evaluate the lung capillary permeability following Evans blue dye (EBD) injection, BAL fluid and lungs from wt and Akap1−/− mice were analyzed for EBD content. Extravasation of EBD was photographed. Akap1−/− mice lungs exposed to hyperoxia show intense blue coloration compared with wt mice (Fig. 3B, top). Significant increase in EBD content was observed in Akap1−/− BAL fluid (Fig. 3B, bottom left) and in lung tissue (Fig. 3B, bottom right) compared with wt mice exposed to hyperoxia. Histological and EBD data suggest that Akap1−/− mice have increased lung damage, alveolar leakage, and exacerbation of HALI compared with the wt mice.
Fig. 3.

Akap1 genetic deletion increases lung injury and alveolar permeability after hyperoxia. Wild-type (wt) and Akap1−/− mice were exposed to hyperoxia (HO) for 48 h. A: Akap1−/− mice show increased damage against hyperoxia-induced ALI. Representative hematoxylin and eosin (H&E) image has been shown from a total of 5 mice/group. The extent of lung injury was evaluated from H&E-stained lung tissue sections. Original magnification, ×40 (insets, × 200). B: extravasation of Evans blue dye (EBD) into the lungs following intravenous injection was photographed (top; scale bar = 1 cm) and quantified by spectrophotometry (bottom panel). BAL fluid (B, bottom left) and lung tissue (B, bottom right) EBD concentration. Data are shown as means ± SE (*P < 0.05 vs. wt hyperoxia; n = 5 mice/group).
Akap1 genetic deletion increases inflammatory cytokine secretion into the BAL fluid and phosphorylation and DNA binding activity of p65 subunit of NF-κB in lung tissue after hyperoxia.
Inflammation in HALI is associated with release of proinflammatory cytokines by neutrophils, macrophages, and other cells. Secretion of inflammatory mediators disrupts the immune-repressed alveolar state of resident macrophages and epithelial cells (6). We measured the proinflammatory cytokines IL-6 and TNFα in the BAL fluid. Levels of these two markers were significantly increased in hyperoxic conditions compared with room air-exposed wt mice. However, the levels were further increased in BAL fluid of Akap1−/− mice compared with wt under hyperoxia (P < 0.01) (Fig. 4, A and B). Further, we analyzed NF-κB transcription factor by immunoblotting, which is associated with the production of these inflammatory cytokines. Lung protein analysis reveals an increase in both phospho-p65 and total p65 subunits of NF-κB in response to hyperoxia in wt and Akap1−/− mice lungs compared with room air-exposed mice (Fig. 4C). Densitometry analysis reveals that the levels of phospho-p65 subunit in hyperoxia-exposed Akap1−/− mice were significantly higher compared with wt controls (Fig. 4C); however, no difference was observed in the levels of total p65 subunit of NF-κB between two groups after hyperoxia. To further validate NF-κB p65 transcription factor binding activity to its DNA response elements, we isolated nuclear fractions from lungs of wt and Akap1−/− mice exposed to either room air or hyperoxia for 48 h, and analyzed p65 DNA-binding activity. The data show that exposure to hyperoxia significantly increases p65 binding activity in both wt and Akap1−/− mice compared with their respective room air-exposed controls (Fig. 4D). Notably, p65 binding activity was also significantly increased in Akap1−/− mice exposed to hyperoxia compared with wt mice exposed to hyperoxia, which is consistent with the Western blot results (P < 0.05) (Fig. 4D). The data conclusively demonstrate that Akap1−/− mice have exacerbated HALI as a result of p65 subunit activation-mediated proinflammatory cytokine production.
Fig. 4.

Akap1 genetic deletion increases inflammatory cytokine secretion into the BAL fluid, and increases phosphorylation and binding activity of NF-kB p65 in lung tissue after hyperoxia. Wild-type (wt) and Akap1−/− mice were exposed to room air or hyperoxia for 48 h, then BAL fluid was collected from each group and analyzed by ELISA for IL-6 (A) and TNFα (B) cytokine measurement. Data are shown as means ± SE (**P < 0.01 vs. wt room air; #P < 0.01 vs. wt hyperoxia; n = 5 mice/group). C: in separate experiments, total lung protein was collected and subjected to SDS-PAGE and Western blotting for phospho (P) and total (T) NF-kB p65 subunit. β-Actin served as loading control. Densitometry of phosphorylated NF-kB p65 shown below. Data are shown as means ± SE (**P < 0.01 vs. wt room air; *P < 0.05 vs. wt hyperoxia; n = 5 mice/group). D: analysis of DNA-binding activities of NF-kB p65 in lung nuclear extracts by TransAM NF-kB p65 activity assay kit. Data are shown as means ± SE (*P < 0.05 vs. wt room air; #P < 0.05 vs. wt hyperoxia; n = 5 mice/group).
Akap1 genetic deletion promotes mitochondrial mitophagy associated marker expression.
Previous reports have shown that under the conditions of pressure overload and myocardial infarction, the downregulation of cardiac Akap121 is associated with mitochondrial abnormalities (33, 39). To determine whether Akap1 genetic deletion directly affects mitochondrial response to oxidative stress, we investigated mitophagy-associated markers in the lung by immunoblotting. Basal expression of PINK1, an initiator of mitophagy, is slightly elevated in room air-exposed Akap1−/− mice compared with wt mice. We found that Akap1−/− lungs had higher PINK1 expression under hyperoxic conditions compared with wt, which is related to mitophagy induction in Akap1-deficient conditions (Fig. 5A). Under normal physiological conditions, PINK1 recruits its downstream target, Parkin, to the mitochondria and eliminates abnormal mitochondria through mitophagy (38). Our results show that there is no significant difference in the basal expression of Parkin in either group (wt and Akap1−/−) exposed to room air (Fig. 5A). Parkin expression in hyperoxia lungs of wt mice shows significant increase over room air-exposed wt mice lungs, and this increase is further elevated in Akap1−/− lungs (Fig. 5A). We also analyzed another autophagy-associated protein marker, p62/SQSTM1, which plays an important role in recognizing and eliminating toxic cellular waste (24). Our data show that basal p62 expression in wt mice is very low, whereas it is elevated in Akap1−/− lungs under room air (Fig. 5B). Conversely, hyperoxia induces high levels of p62/SQSTM1 expression in both wt and Akap1−/− lungs compared with room air-exposed samples (Fig. 5B). Densitometric scans revealed a significant increase in p62 expression in hyperoxia-exposed Akap1−/− lungs compared with hyperoxia-exposed wt lungs. The ratio of another autophagy marker, LC3B II/I, was significantly decreased under hyperoxia in wt mice; however, densitometry analysis of LC3B II/I shows no change between wt and Akap1−/− mice under hyperoxia (Fig. 5C). However, we observed a low basal LCBII/I ratio in Akap1−/− mice compared with wt mice exposed to room air (Fig. 5C). This suggests that there is no link between p62 and LC3B expression patterns between wt and Akap1−/− mice under hyperoxia. Taken together, we conclude that the PINK1-Parkin and p62 pathways are actively involved in Akap1−/− lungs to promote mitophagy in response to oxidative stress.
Fig. 5.

Akap1 genetic deletion promotes mitochondrial mitophagy associated markers after hyperoxia. Wild-type (wt) and Akap1−/− mice were exposed to room air or hyperoxia for 48 h, then lung protein was collected and subjected to SDS-PAGE followed by Western blotting. A: representative Western blot and densitometric analysis of PINK1 and Parkin. Data are shown as means ± SE (*P < 0.05 vs. wt room air; #P < 0.05 vs. wt hyperoxia; n = 5 mice/group). B: representative Western blot and densitometric analysis of p62. Data are shown as means ± SE (*P < 0.05 vs. wt room air; #P < 0.05 vs. wt hyperoxia; n = 5 mice/group). C: representative Western blot and densitometric analysis of LC3BII/I ratio to β-actin. Data are shown as means ± SE (**P < 0.01 vs. room air; *P < 0.01 vs. room air; **P < 0.05 vs. room air; ns, no significance; n = 5 mice/group).
Akap1 genetic deletion leads to morphological alteration in both mitochondria and lamellar bodies of type II AECs following hyperoxic challenge.
To determine whether Akap1 deletion affects mitochondrial structure, we next investigated mitochondrial morphology in the lung by electron microscopy. In room air-exposed wt mice, type II AECs display intact lamellar bodies and normal mitochondria in cytoplasm (oval shape) (Fig. 6, A and C). Conversely, type II AECs from Akap1−/− mice display relatively electron-lucent nuclei and cytoplasm compared with wt mice (Fig. 6, B and D). Type II AECs of wt mice under hyperoxia display disorganized lamellar bodies. The shape and size of mitochondria also varied depending on each cell (Fig. 6, E and G). Small circular mitochondria are apparent, but sparse (Fig. 6E). Type II AECs of Akap1−/− mice display under hyperoxia diverse morphology. Many type II AECs show vacuolated cytoplasm, which is reminiscent of necrotic or autophagic cell death (circled by red dashed line in Fig. 6F). Type II AECs that still look viable display disorganized lamellar bodies and mitochondria with different sizes and morphologies (Fig. 6H). The number of abnormal mitochondria were found to be significantly higher in Akap1−/− mice compared with wt mice under hyperoxia (P < 0.05) (Fig. 6I). Overall Akap1−/− mice exposed to hyperoxia display disorganized abnormal mitochondria, especially in type II AECs. These findings are consistent with mitophagy associated PINK1 and Parkin protein expression in the lungs.
Fig. 6.

Akap1 genetic deletion leads to morphological alteration in mitochondria and lamellar bodies of type II AECs after hyperoxia. Representative transmission electron microscopy images of resin-embedded lung sections from wild-type (wt) (A, C, E, and G) and Akap1−/− (n = 5 mice/group) (B, D, F, and H) mice under room air (A–D) or 48 h of hyperoxia (E–H). Blue asterisks denote mitochondria. Lamellar bodies are circled by yellow dashed lines. A and C: type II AECs from wt mice display apparently intact lamellar bodies and mitochondria in cytoplasm. B and D: type II AECs from Akap1−/− mice under room air display relatively electron-lucent nuclei and cytoplasm compared with type II AECs of wt mice under room air. E and G: type II AECs of wt mice under hyperoxia display disorganized lamellar bodies. F and H: type II AECs of Akap1−/− mice display diverse morphology. Many type II AECs show vacuolated cytoplasm, which is reminiscent of necrotic or autophagic cell death (type II AEC is circled by red dashed line in F). Type II AECs that still look viable, display disorganized lamellar bodies and mitochondria with varying size and morphology (H). Nuc, nucleus; AS, airspace. Original magnifications: ×15,000 (A, B, D, E, F); ×30,000 (C, H); ×60,000 (G). I: numerical counting on abnormal mitochondria was performed on the electron microscopic images of wt and Akap1−/− mice exposed to hyperoxia. Data are shown as means ± SE (*P < 0.05 vs. wt hyperoxia; n = 5 mice/group).
DISCUSSION
Here, for the first time, we demonstrate that Akap1 genetic deletion increases the severity of inflammation, causes mitochondrial abnormalities, and enhances mitophagy in the lung under hyperoxia. Akap1 has been recently identified as a key protein which mediates transmission of extracellular signals into the mitochondria and is responsible for the proper functioning of mitochondrial dynamics (8). In addition, recent studies indicate ROS and 4-HNE produced during HALI as the key contributors of mitochondrial damage (16, 26, 44). The purpose of this study is to elucidate the role of Akap1 in HALI and its involvement in many of the mitochondrial functions. In this study we show that 1) Akap1 deletion increases the severity of hyperoxia-induced lung inflammation in murine lungs, 2) Akap1 deletion exacerbates hyperoxia-induced infiltration of inflammatory cells and protein leakage, 3) production of proinflammatory cytokines and activation of its NF-κB transcription factor is elevated in Akap1−/− mice exposed to hyperoxia, and 4) Akap1 deletion promotes inflammation by inducing autophagy and altering the morphology of mitochondria and lamellar bodies of type II AECs following hyperoxic challenge.
Stress-induced mitochondrial damage increases ROS production and can cause damage to nearby mitochondria, which leads to the release of proapoptotic signals and the amplification of cellular damage (43). Recently, it was shown using a rat model of cardiac hypertrophy that Akap1 is an important regulator of mitochondrial function and cell survival (33). In the current study, we exposed Akap1 deficient mice to hyperoxia and studied its effects on inflammatory cell infiltration and protein leakage into the BAL fluid. We revealed that Akap1 deficiency leads to increased infiltration of total cell counts and protein leakage into the BAL fluid, suggesting that Akap1 plays an important role in recruiting inflammatory mediators. Even though we did not see a significant difference in BAL fluid neutrophil counts between wt and Akap1−/− following hyperoxia, we could see significant differences in the MPO activity, an index for the presence of neutrophils.
The alveolar capillary barrier plays an important homeostatic role in the regulation of immune cell infiltration, and hyperoxia is known to deteriorate this barrier function (23, 37). To understand the role of Akap1 genetic deletion in alveolar capillary permeability, we injected mice with Evans blue dye through the tail vein and measured dye content in BAL fluid and in the lung. Our results reveal that, following hyperoxia, Akap1−/− mice have increased dye content in both BAL fluid and lung tissue, indicating that these mice are more susceptible to the hyperoxic-induced lung injury.
Hyperoxic lung injury results in increased inflammation and permeability (6, 17). To determine whether Akap1 participates in inflammatory cytokine production and lung permeability, we exposed Akap1−/− mice to hyperoxia for 48 h and then analyzed BAL fluid for proinflammatory cytokines IL-6 and TNFα by ELISA. The results reveal that Akap1−/− mice have a significant increase of both cytokines compared with wt mice under hyperoxic conditions. Taken together, our research demonstrates for the first time that Akap1−/− mice are more prone to produce inflammatory cytokines and increase the severity of lung injury. Based on our current results presented, we surmise that Akap1 deficiency compromises the functional integrity of mitochondria under hyperoxic as well as normoxic conditions. We posit that in hyperoxic conditions this Akap1 deficiency-related mitochondrial dysfunction culminates in cytokine burst, cytokine-dependent inflammation, and ultimately epithelial cell apoptosis most commonly triggered by epithelial permeability and barrier dysfunction (23, 29). In future studies, it would be prudent to examine the long-term effects of Akap1 depletion on parameters such as survival after hyperoxia. Understanding the full biological role of this anchoring protein would greatly benefit the field.
Proinflammatory transcription factor NF-κB signaling pathway is involved in inflammatory diseases (42) and is associated with the production of inflammatory cytokines, chemokines (6). NF-κB can have both beneficial (46) and detrimental effects in HALI (12, 47). Phosphorylation of NF-κB is a key signaling event involved in its transcriptional activation. To assess the effects of Akap1 genetic deletion on NF-κB activation, we analyzed room air and hyperoxia-exposed mouse lungs for the p65 subunit of NF-κB. Following hyperoxia, Akap1−/− mice exhibited elevated phospho-p65 subunit expression compared with wt mice. However, we did not see a difference in total p65 expression between these groups, suggesting that Akap1−/− increases the severity of inflammation and is associated with the activation of NF-κB signaling. Although we found elevated NF-κB p65 phosphorylation and DNA binding activity from Akap1−/− mice compared with wt controls, additional studies should be conducted to confirm how the NF-κB pathway is directly involved in Akap1-mediated HALI.
Autophagy involves the formation of autophagosomes that target and engulf cytosolic material, including damaged organelles and nonfunctional proteins. Hyperoxia induces mitochondrial oxidant generation and inhibition of autophagy in mouse lungs and lung endothelial cells (48). PINK1 and its downstream signaling protein Parkin mediate autophagy of damaged mitochondria in a process of mitophagy (31). Mitophagy is a more selective degradation and engulfment pathway for autophagy of mitochondria. We found that Akap1 genetic deletion increased PINK1 and Parkin and p62 protein expression by hyperoxia compared with wt mice and low basal levels in both wt and Akap1−/− in room air-exposed mice. This is relevant due to the fact that the PINK1-Parkin pathway promotes mitochondrial fission and inhibits its fusion (21, 45). Notably, the LC3B II/I ratio was lower in wt hyperoxia-exposed lungs, which is compatible with previous reports (49); conversely, the ratio between room air and hyperoxia-exposed Akap1−/− mice was not significantly different, suggesting that LC3B may not be involved in the exacerbation of lung injury.
In this study, our results also unveil the function of Akap1 in the control of mitophagy during hyperoxia and suggest that mitoAKAPs may play a crucial role in the pathogenesis of ALI. Type II AECs exhibit the highest level of pulmonary oxygen utilization and contain 50% of mitochondria in the lung (7, 30). Profound defects in cellular oxygen consumption in ALI are associated with damage of type II AECs. To determine the direct effects of Akap1 deletion on type II AECs mitochondria, following hyperoxia, we analyzed lungs by electron microscopy. In the absence of Akap1, under hyperoxia, type II AECs exhibit diverse morphology of mitochondria and lamellar bodies (LB). Vacuolated cytoplasm, an indication of autophagic cells, is found more frequently in Akap1−/− mice under hyperoxia. The LB of the lung epithelium secrete lung surfactant on the surface of lung alveoli to lower surface tension necessary for optimal gas exchange and act as a protective lining against environmental influences (40). In Akap−/− mice, type II cells had uncharacteristically small LB whereas wt cells had predominantly large LB under hyperoxia. Wt and Akap−/− mice type II AECs exhibited structural abnormalities in LB that could lead to surfactant deficiency and thus respiratory distress syndrome (9). These results suggest that balance in regulating mitochondrial dynamics is lost in the absence of Akap1 during hyperoxia. Similar results of mitochondrial abnormalities were observed in cardiomyocytes exposed to hypoxic conditions (39). It was found that inhibition of mitophagy can provide protection against cardiac dysfunction in Akap1−/− mice (39).
The increased severity in inflammation, alveolar leakage, and mitochondrial abnormalities in type II AECs observed in Akap1−/− lungs are likely attributable to the reduced targeting of protein kinase A and associated complex on the outer mitochondrial membrane. The precise molecular signaling pathways involved in the mitophagy and mitochondrial abnormalities in Akap1 deficient mice remain to be investigated. Recently, a study indicated that high levels of Akap1 were found in advanced stage cancers. In lung cancer, Akap1 expression correlates with high levels of Myc, mTOR phosphorylation, and patient survival (36). These findings suggest a contrasting role for Akap1 in the pathology of cancers.
In conclusion, our findings highlight the critical role of mitochondrial AKAPs (Akap1) in HALI. Here, for the first time, we shed light on the role of Akap1 as a new molecular target in ALI pathogenesis, reveal its biological significance in regulating ALI, and decipher its connection to mitochondrial destabilization. We demonstrated that Akap1 loss can increase the severity of lung inflammation, alveolar capillary permeability, and autophagy-associated markers, as well as cause aberrant change in the mitochondria and lamellar bodies of type II AECs. Therefore, Akap1 might represent a novel therapeutic target in inflammatory lung diseases, especially HALI. Expanding our knowledge of Akap1-mediated HALI may also lead to the development of clinical interventions capable of protecting countless patients that require supplemental oxygen.
GRANTS
N. Kolliputi was funded by American Heart Association (AHA) National Scientist Development Grant 09SDG2260957; National Heart, Lung, and Blood Institute Grant R01-HL-105932,; and the Joy McCann Culverhouse Endowment to the Division of Allergy and Immunology. J. Fukumoto was funded by AHA Postdoctoral Fellowship Award 14POST18200004. L. Galam was funded by AHA National Scientist Development Grant 17SDG32780002. V. R. Narala was supported by the University Grants Commission, Raman Postdoctoral Fellowship Award in USA [No. F-90/2016 (IC)], New Delhi, India.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
V.R.N., J.F., R.F.L., and N.K. conceived and designed research; V.R.N., J.F., H.H.-C., S.S.P., S.K., and L.G. performed experiments; V.R.N., J.F., and R.S. analyzed data; V.R.N. interpreted results of experiments; V.R.N., J.F., and M.B. prepared figures; V.R.N. and M.B. drafted manuscript; V.R.N. and M.B. edited and revised manuscript; N.K. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Profs. S. Strack and S. McKnight for graciously providing us with the Akap1−/− mice. We also thank A. Garces at the Lisa Muma Weitz Advanced Microscopy Core Laboratory for technical assistance in the use of transmission electron microscopy.
REFERENCES
- 1.Adlam VJ, Harrison JC, Porteous CM, James AM, Smith RA, Murphy MP, Sammut IA. Targeting an antioxidant to mitochondria decreases cardiac ischemia-reperfusion injury. FASEB J 19: 1088–1095, 2005. doi: 10.1096/fj.05-3718com. [DOI] [PubMed] [Google Scholar]
- 2.Carlucci A, Lignitto L, Feliciello A. Control of mitochondria dynamics and oxidative metabolism by cAMP, AKAPs and the proteasome. Trends Cell Biol 18: 604–613, 2008. doi: 10.1016/j.tcb.2008.09.006. [DOI] [PubMed] [Google Scholar]
- 3.Chacko BK, Reily C, Srivastava A, Johnson MS, Ye Y, Ulasova E, Agarwal A, Zinn KR, Murphy MP, Kalyanaraman B, Darley-Usmar V. Prevention of diabetic nephropathy in Ins2(+/)−(AkitaJ) mice by the mitochondria-targeted therapy MitoQ. Biochem J 432: 9–19, 2010. doi: 10.1042/BJ20100308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cloonan SM, Choi AM. Mitochondria in lung disease. J Clin Invest 126: 809–820, 2016. doi: 10.1172/JCI81113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cochemé HM, Murphy MP. Can antioxidants be effective therapeutics? Curr Opin Investig Drugs 11: 426–431, 2010. [PubMed] [Google Scholar]
- 6.Cox R Jr, Phillips O, Fukumoto J, Fukumoto I, Parthasarathy PT, Arias S, Cho Y, Lockey RF, Kolliputi N. Enhanced resolution of hyperoxic acute lung injury as a result of aspirin triggered resolvin D1 treatment. Am J Respir Cell Mol Biol 53: 422–435, 2015. doi: 10.1165/rcmb.2014-0339OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Crapo JD, Barry BE, Foscue HA, Shelburne J. Structural and biochemical changes in rat lungs occurring during exposures to lethal and adaptive doses of oxygen. Am Rev Respir Dis 122: 123–143, 1980. doi: 10.1164/arrd.1980.122.1.123. [DOI] [PubMed] [Google Scholar]
- 8.Czachor A, Failla A, Lockey R, Kolliputi N. Pivotal role of AKAP121 in mitochondrial physiology. Am J Physiol Cell Physiol 310: C625–C628, 2016. doi: 10.1152/ajpcell.00292.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Edwards V, Cutz E, Viero S, Moore AM, Nogee L. Ultrastructure of lamellar bodies in congenital surfactant deficiency. Ultrastruct Pathol 29: 503–509, 2005. doi: 10.1080/01913120500323480. [DOI] [PubMed] [Google Scholar]
- 10.Feliciello A, Gottesman ME, Avvedimento EV. The biological functions of A-kinase anchor proteins. J Mol Biol 308: 99–114, 2001. doi: 10.1006/jmbi.2001.4585. [DOI] [PubMed] [Google Scholar]
- 11.Fisher AB. Oxygen therapy. Side effects and toxicity. Am Rev Respir Dis 122: 61–69, 1980. doi: 10.1164/arrd.1980.122.5P2.61. [DOI] [PubMed] [Google Scholar]
- 12.Franek WR, Morrow DM, Zhu H, Vancurova I, Miskolci V, Darley-Usmar K, Simms HH, Mantell LL. NF-kappaB protects lung epithelium against hyperoxia-induced nonapoptotic cell death-oncosis. Free Radic Biol Med 37: 1670–1679, 2004. doi: 10.1016/j.freeradbiomed.2004.08.007. [DOI] [PubMed] [Google Scholar]
- 13.Fukumoto J, Cox R Jr, Fukumoto I, Cho Y, Parthasarathy PT, Galam L, Lockey RF, Kolliputi N. Deletion of ASK1 protects against hyperoxia-induced acute lung injury. PLoS One 11: e0147652, 2016. doi: 10.1371/journal.pone.0147652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fukumoto J, Fukumoto I, Parthasarathy PT, Cox R, Huynh B, Ramanathan GK, Venugopal RB, Allen-Gipson DS, Lockey RF, Kolliputi N. NLRP3 deletion protects from hyperoxia-induced acute lung injury. Am J Physiol Cell Physiol 305: C182–C189, 2013. doi: 10.1152/ajpcell.00086.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fukumoto J, Soundararajan R, Leung J, Cox R, Mahendrasah S, Muthavarapu N, Herrin T, Czachor A, Tan LC, Hosseinian N, Patel P, Gone J, Breitzig MT, Cho Y, Cooke AJ, Galam L, Narala VR, Pathak Y, Lockey RF, Kolliputi N. The role of club cell phenoconversion and migration in idiopathic pulmonary fibrosis. Aging (Albany NY) 8: 3091–3109, 2016. doi: 10.18632/aging.101115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Galam L, Failla A, Soundararajan R, Lockey RF, Kolliputi N. 4-hydroxynonenal regulates mitochondrial function in human small airway epithelial cells. Oncotarget 6: 41508–41521, 2015. doi: 10.18632/oncotarget.6131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Galam L, Parthasarathy PT, Cho Y, Cho SH, Lee YC, Lockey RF, Kolliputi N. Adenovirus-mediated transfer of the SOCS-1 gene to mouse lung confers protection against hyperoxic acute lung injury. Free Radic Biol Med 84: 196–205, 2015. doi: 10.1016/j.freeradbiomed.2015.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gomes KM, Campos JC, Bechara LR, Queliconi B, Lima VM, Disatnik MH, Magno P, Chen CH, Brum PC, Kowaltowski AJ, Mochly-Rosen D, Ferreira JC. Aldehyde dehydrogenase 2 activation in heart failure restores mitochondrial function and improves ventricular function and remodelling. Cardiovasc Res 103: 498–508, 2014. doi: 10.1093/cvr/cvu125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Graham D, Huynh NN, Hamilton CA, Beattie E, Smith RA, Cochemé HM, Murphy MP, Dominiczak AF. Mitochondria-targeted antioxidant MitoQ10 improves endothelial function and attenuates cardiac hypertrophy. Hypertension 54: 322–328, 2009. doi: 10.1161/HYPERTENSIONAHA.109.130351. [DOI] [PubMed] [Google Scholar]
- 20.Huang LJ, Durick K, Weiner JA, Chun J, Taylor SS. Identification of a novel protein kinase A anchoring protein that binds both type I and type II regulatory subunits. J Biol Chem 272: 8057–8064, 1997. doi: 10.1074/jbc.272.12.8057. [DOI] [PubMed] [Google Scholar]
- 21.Huang P, Galloway CA, Yoon Y. Control of mitochondrial morphology through differential interactions of mitochondrial fusion and fission proteins. PLoS One 6: e20655, 2011. doi: 10.1371/journal.pone.0020655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Islam MN, Das SR, Emin MT, Wei M, Sun L, Westphalen K, Rowlands DJ, Quadri SK, Bhattacharya S, Bhattacharya J. Mitochondrial transfer from bone-marrow-derived stromal cells to pulmonary alveoli protects against acute lung injury. Nat Med 18: 759–765, 2012. doi: 10.1038/nm.2736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kallet RH, Matthay MA. Hyperoxic acute lung injury. Respir Care 58: 123–141, 2013. doi: 10.4187/respcare.01963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Katsuragi Y, Ichimura Y, Komatsu M. p62/SQSTM1 functions as a signaling hub and an autophagy adaptor. FEBS J 282: 4672–4678, 2015. doi: 10.1111/febs.13540. [DOI] [PubMed] [Google Scholar]
- 25.Kolliputi N, Shaik RS, Waxman AB. The inflammasome mediates hyperoxia-induced alveolar cell permeability. J Immunol 184: 5819–5826, 2010. doi: 10.4049/jimmunol.0902766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kolliputi N, Waxman AB. IL-6 cytoprotection in hyperoxic acute lung injury occurs via PI3K/Akt-mediated Bax phosphorylation. Am J Physiol Lung Cell Mol Physiol 297: L6–L16, 2009. doi: 10.1152/ajplung.90381.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li C, Bo L, Liu W, Lu X, Jin F. Enteral immunomodulatory diet (omega-3 fatty acid, γ-linolenic acid and antioxidant supplementation) for acute lung injury and acute respiratory distress syndrome: an updated systematic review and meta-analysis. Nutrients 7: 5572–5585, 2015. doi: 10.3390/nu7075239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li X, Fang P, Yang WY, Chan K, Lavallee M, Xu K, Gao T, Wang H, Yang X. Mitochondrial ROS, uncoupled from ATP synthesis, determine endothelial activation for both physiological recruitment of patrolling cells and pathological recruitment of inflammatory cells. Can J Physiol Pharmacol 95: 247–252, 2017. doi: 10.1139/cjpp-2016-0515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mach WJ, Thimmesch AR, Pierce JT, Pierce JD. Consequences of hyperoxia and the toxicity of oxygen in the lung. Nurs Res Pract 2011: 260482, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Massaro GD, Gail DB, Massaro D. Lung oxygen consumption and mitochondria of alveolar epithelial and endothelial cells. J Appl Physiol 38: 588–592, 1975. doi: 10.1152/jappl.1975.38.4.588. [DOI] [PubMed] [Google Scholar]
- 31.McWilliams TG, Muqit MMK. PINK1 and Parkin: emerging themes in mitochondrial homeostasis. Curr Opin Cell Biol 45: 83–91, 2017. doi: 10.1016/j.ceb.2017.03.013. [DOI] [PubMed] [Google Scholar]
- 32.Newhall KJ, Criniti AR, Cheah CS, Smith KC, Kafer KE, Burkart AD, McKnight GS. Dynamic anchoring of PKA is essential during oocyte maturation. Curr Biol 16: 321–327, 2006. doi: 10.1016/j.cub.2005.12.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Perrino C, Feliciello A, Schiattarella GG, Esposito G, Guerriero R, Zaccaro L, Del Gatto A, Saviano M, Garbi C, Carangi R, Di Lorenzo E, Donato G, Indolfi C, Avvedimento VE, Chiariello M. AKAP121 downregulation impairs protective cAMP signals, promotes mitochondrial dysfunction, and increases oxidative stress. Cardiovasc Res 88: 101–110, 2010. doi: 10.1093/cvr/cvq155. [DOI] [PubMed] [Google Scholar]
- 34.Reddy AT, Lakshmi SP, Kleinhenz JM, Sutliff RL, Hart CM, Reddy RC. Endothelial cell peroxisome proliferator-activated receptor γ reduces endotoxemic pulmonary inflammation and injury. J Immunol 189: 5411–5420, 2012. doi: 10.4049/jimmunol.1201487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rice TW, Wheeler AP, Thompson BT, deBoisblanc BP, Steingrub J, Rock P; NIH NHLBI Acute Respiratory Distress Syndrome Network of Investigators . Enteral omega-3 fatty acid, gamma-linolenic acid, and antioxidant supplementation in acute lung injury. JAMA 306: 1574–1581, 2011. doi: 10.1001/jama.2011.1435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rinaldi L, Sepe M, Delle Donne R, Conte K, Arcella A, Borzacchiello D, Amente S, De Vita F, Porpora M, Garbi C, Oliva MA, Procaccini C, Faicchia D, Matarese G, Zito Marino F, Rocco G, Pignatiello S, Franco R, Insabato L, Majello B, Feliciello A. Mitochondrial AKAP1 supports mTOR pathway and tumor growth. Cell Death Dis 8: e2842, 2017. doi: 10.1038/cddis.2017.241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rubenfeld GD, Caldwell E, Peabody E, Weaver J, Martin DP, Neff M, Stern EJ, Hudson LD. Incidence and outcomes of acute lung injury. N Engl J Med 353: 1685–1693, 2005. doi: 10.1056/NEJMoa050333. [DOI] [PubMed] [Google Scholar]
- 38.Scarffe LA, Stevens DA, Dawson VL, Dawson TM. Parkin and PINK1: much more than mitophagy. Trends Neurosci 37: 315–324, 2014. doi: 10.1016/j.tins.2014.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schiattarella GG, Cattaneo F, Pironti G, Magliulo F, Carotenuto G, Pirozzi M, Polishchuk R, Borzacchiello D, Paolillo R, Oliveti M, Boccella N, Avvedimento M, Sepe M, Lombardi A, Busiello RA, Trimarco B, Esposito G, Feliciello A, Perrino C. Akap1 deficiency promotes mitochondrial aberrations and exacerbates cardiac injury following permanent coronary ligation via enhanced mitophagy and apoptosis. PLoS One 11: e0154076, 2016. doi: 10.1371/journal.pone.0154076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schmitz G, Müller G. Structure and function of lamellar bodies, lipid-protein complexes involved in storage and secretion of cellular lipids. J Lipid Res 32: 1539–1570, 1991. [PubMed] [Google Scholar]
- 41.Shaik FB, Panati K, Narasimha VR, Narala VR. Chenodeoxycholic acid attenuates ovalbumin-induced airway inflammation in murine model of asthma by inhibiting the T(H)2 cytokines. Biochem Biophys Res Commun 463: 600–605, 2015. doi: 10.1016/j.bbrc.2015.05.104. [DOI] [PubMed] [Google Scholar]
- 42.Shih VF, Tsui R, Caldwell A, Hoffmann A. A single NFκB system for both canonical and non-canonical signaling. Cell Res 21: 86–102, 2011. doi: 10.1038/cr.2010.161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Song M, Chen Y, Gong G, Murphy E, Rabinovitch PS, Dorn GW II. Super-suppression of mitochondrial reactive oxygen species signaling impairs compensatory autophagy in primary mitophagic cardiomyopathy. Circ Res 115: 348–353, 2014. doi: 10.1161/CIRCRESAHA.115.304384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Waxman AB, Kolliputi N. IL-6 protects against hyperoxia-induced mitochondrial damage via Bcl-2-induced Bak interactions with mitofusins. Am J Respir Cell Mol Biol 41: 385–396, 2009. doi: 10.1165/rcmb.2008-0302OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Whitworth AJ, Pallanck LJ. The PINK1/Parkin pathway: a mitochondrial quality control system? J Bioenerg Biomembr 41: 499–503, 2009. doi: 10.1007/s10863-009-9253-3. [DOI] [PubMed] [Google Scholar]
- 46.Yang G, Abate A, George AG, Weng YH, Dennery PA. Maturational differences in lung NF-kappaB activation and their role in tolerance to hyperoxia. J Clin Invest 114: 669–678, 2004. doi: 10.1172/JCI200419300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zaher TE, Miller EJ, Morrow DM, Javdan M, Mantell LL. Hyperoxia-induced signal transduction pathways in pulmonary epithelial cells. Free Radic Biol Med 42: 897–908, 2007. doi: 10.1016/j.freeradbiomed.2007.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhang Y, Jiang G, Sauler M, Lee PJ. Lung endothelial HO-1 targeting in vivo using lentiviral miRNA regulates apoptosis and autophagy during oxidant injury. FASEB J 27: 4041–4058, 2013. doi: 10.1096/fj.13-231225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhang Y, Sauler M, Shinn AS, Gong H, Haslip M, Shan P, Mannam P, Lee PJ. Endothelial PINK1 mediates the protective effects of NLRP3 deficiency during lethal oxidant injury. J Immunol 192: 5296–5304, 2014. doi: 10.4049/jimmunol.1400653. [DOI] [PMC free article] [PubMed] [Google Scholar]