

Abstract
Cigarette smoking is the leading cause of preventable disease and death in the United States. Cardiovascular comorbidities associated with both active and secondhand cigarette smoking indicate the vascular toxicity of smoke exposure. Growing evidence supports the injurious effect of cigarette smoke on pulmonary endothelial cells and the roles of endothelial cell injury in development of acute respiratory distress syndrome (ARDS), emphysema, and pulmonary hypertension. This review summarizes results from studies of humans, preclinical animal models, and cultured endothelial cells that document toxicities of cigarette smoke exposure on pulmonary endothelial cell functions, including barrier dysfunction, endothelial activation and inflammation, apoptosis, and vasoactive mediator production. The discussion is focused on effects of cigarette smoke-induced endothelial injury in the development of ARDS, emphysema, and vascular remodeling in chronic obstructive pulmonary disease.
Keywords: acute respiratory distress syndrome, cigarette smoke, emphysema, endothelial cells, pulmonary hypertension
INTRODUCTION
Cigarette smoking is the leading cause of preventable disease and death in the US, causing >480,000 deaths per year in the US, according to the 2014 Surgeon General’s report (136a). Worldwide, the World Health Organization estimates that tobacco use kills >7 million people each year, 6 million of whom are smokers while 890,000 die from the effects of secondhand smoke exposure [World Health Organization Tobacco Fact Sheet (150a)]. Although the incidence of tobacco use in the US has decreased, 20.1% of adults still continued to use tobacco products in 2015 (106). Thus cigarette smoking remains a significant cause of morbidity and mortality.
Cigarette smoking has systemic effects, in addition to altering lung function. Causes of smoking-related deaths include cancers, cardiovascular and metabolic diseases, pulmonary diseases, and conditions related to pregnancy and birth [2014 Surgeon General’s report (136a)]. Chronic lung diseases caused by cigarette smoking include chronic obstructive pulmonary disease (COPD), idiopathic pulmonary fibrosis, and asthma. COPD in particular is associated with systemic comorbidities (“the comorbidome”; 35). Among the systemic comorbidities, cardiovascular diseases, including pulmonary hypertension, are the most prevalent and carry a high risk of mortality (32). Furthermore, secondhand smoke exposure is also associated with increased risk of coronary artery disease and other cardiovascular diseases (13). These cardiovascular comorbidities of both mainstream and secondhand smoke exposure suggest that toxic cigarette smoke constituents are absorbed from the airways into the blood and that the inhalation of cigarette smoke causes systemic toxicity.
A growing body of evidence indicates that cigarette smoke alters vascular function, in addition to airways and immune function (127). Voelkel and colleagues were among the first to demonstrate lung endothelial cell apoptosis in human emphysema [66; Imai et al. (59)] and in animal models of cigarette smoke exposure [Marwick et al. (88)] and suggested the “vascular hypothesis” for pathogenesis of emphysema (140). Endothelial progenitor cell dysfunction has been recognized in acute exacerbation of COPD, suggesting that cigarette smoke (CS)-induced inadequate repair of the endothelium may also contribute to COPD mortality (80).
CS is complex and includes ~4,500 components. Among these, CO, nicotine, oxidants, fine particulate matter, aryl hydrocarbons, and aldehydes such as formaldehyde and acrolein have potential for causing endothelial cell injury. Acrolein is of particular interest since increased levels have been found in blood and urine of smokers (7). Intraperitoneal administration of acrolein causes emphysema in rats (71), and intratracheal instillation increases lung vascular permeability in mice (82). A recent review summarized the known effects on lung function of nicotine administered by electronic cigarettes (30).
It is now recognized that the lung endothelium is not a passive barrier to gas exchange, but that it is metabolically active and is a key mediator of inflammation. Among the functions of lung endothelium, barrier function is critical in preventing extravasation of water, protein, and cells from the circulation, thus maintaining normal gas exchange. A consequence of injury to the lung endothelium is loss of barrier function and increased-permeability pulmonary edema, the pathophysiologic hallmark of acute respiratory distress syndrome (ARDS). ARDS is an important cause of mortality that does not have specific treatment, despite great improvements in supportive care. In this review we consider growing epidemiologic evidence that cigarette smokers are at increased risk for ARDS and are more susceptible to loss of lung endothelial barrier function from other events associated with development of ARDS (Table 1). In addition, we summarize evidence for cigarette smoke effects on alveolar loss in emphysema and on vascular remodeling and reactivity in pulmonary hypertension associated with COPD. We review studies using cell and animal models of lung endothelial toxicity caused by cigarette smoke exposure, resulting in abnormal barrier function, increased permeability pulmonary edema, endothelial cell apoptosis, endothelial activation and inflammation, and endothelial-dependent vascular remodeling.
Table 1.
Human studies on effects of cigarette smoke on development of ARDS
| Authors | Year | Cohort | Smoking Status | Association |
|---|---|---|---|---|
| Iribarren et al. (60) | 2000 | HMO database, extracted ARDS admissions | Patient report | Dose-response effect between cigarette smoking and risk of ARDS |
| Tandon et al. (132) | 2001 | Esophagectomy patients in the United Kingdom | Patient report | Smoking was associated with postesophagectomy ARDS |
| Calfee et al. (26) | 2011 | Emergency department visits for blunt trauma | Cotinine levels measured at admission | Cotinine level correlated with risk of ARDS |
| Hsieh et al. (57) | 2014 | ARDSNET cohort (2 studies) | 4-(Methylnitrosamino)-1-(3-pyridyl)-1-butanol levels | Similar risk of ARDS despite being younger and healthier |
| Ware et al. (146) | 2014 | Brain-dead donors for Beta-Agonists for Oxygenation in Lung Donors (BOLD) study | Medical record and interviews with next of kin | Current smokers had higher lung weights than ever smokers, and ever smokers had higher lung weights than lifelong nonsmokers (measure of edema) |
| Ware et al. (147) | 2016 | Validating Acute Lung Injury Biomarkers for Diagnosis (VALID) study cohort | Medical record/patient report | Ozone exposure significantly associated with ARDS risk only in current smokers |
| Wacharasint et al. (141) | 2015 (poster) | Thai-Surgical Intensive Care Unit (Thai-SICU) study databases | Medical record/patient report | Smokers with higher number of pack years and heavier smokers had higher incidence of ARDS and longer ICU length of stay |
| Moazed et al. (94) | 2016 | Volunteer smokers and nonsmokers | Self-report | Greater increase in total protein in smokers (measure of permeability) after exposure to LPS, multiple biomarkers |
| García-Lucio et al. (43) | 2016 | Lobectomy/pneumonectomy patients for solitary pulmonary nodule | Patient report | Elevated ANGPT-2 in smokers |
| Gajic et al. (42) | 2011 | Admitted patients with risk factors for ARDS | Patient report | No association between smoking and ARDS |
| Ferro et al. (38) | 2010 | Admitted trauma patients | Patient report | No difference in adverse outcomes when stratified by smoking status |
ANGPT-2, angiopoietin-2; ARDS, acute respiratory distress syndrome; ARDSNET, ARDS Network; HMO, health maintenance organization; ICU, intensive care unit.
EFFECT OF CIGARETTE SMOKING ON ENDOTHELIAL PERMEABILITY AND ACTIVATION IN ARDS
CS Is a Priming Factor for ARDS in Human Subjects
CS increases susceptibility to lung infections and development of ARDS in human subjects.
Respiratory tract infections and complications are common causes of morbidity and mortality among smokers (136a). Smokers are more susceptible to infections by human immunodeficiency virus (HIV; 107, 113), Mycobacterium tuberculosis (27), Streptococcus pneumoniae (15), and influenza A (150). Current smokers with community-acquired pneumonia often progress to severe sepsis (15). In addition to increased risk of infections, recent work has implicated primary and secondhand smoke exposure as a risk factor for the development of ARDS. Iribarren et al. extracted ARDS cases from a Kaiser Permanente database of hospitalizations (60). They found that current heavy smokers (>20 cigarettes per day) had 5.7 times the risk of developing ARDS, with a dose-response association in a multivariate regression. They estimated that 50% of the risk of ARDS is attributable to cigarette smoking. In a study of esophagectomy patients, a history of smoking was one of two preoperative factors associated with the later development of ARDS (132). Wacharasint et al. found that in a cohort of patients admitted to a surgical intensive care unit in Thailand, those who were active smokers had a higher incidence of ARDS than former smokers, who in turn had a higher incidence than never smokers (141). This study also demonstrated a dose-response association in which pack years correlated with risk of ARDS. Ware et al. evaluated the cohort from the Validating Acute Lung Injury Biomarkers for Diagnosis (VALID) study (147). Although they were testing for an association between ambient ozone levels and risk of ARDS, they found that in this critically ill population, ozone exposure was associated with ARDS only in patients who smoked (147). Furthermore, when ARDS survivors were followed as in the ARDS Network (ARDSNET) Long-Term Outcomes Study (ALTOS), those who smoked had a poorer health-related quality of life (22).
There are a few studies that have not shown smoking to be a significant factor. Gajic et al. examined a variety of variables in a prospective study to determine which were associated with the development of mild and severe ARDS (42). While characteristics including alcohol abuse, obesity, and male sex were significant, smoking was not associated with higher rates of ARDS (42). In a retrospective study of trauma patients, Ferro et al. showed no association between smoking and various adverse outcomes including ARDS, as well as sepsis, septic shock, and mortality (38). Smoking also did not impact the number of days of mechanical ventilation or intensive care unit length of stay (38).
Notably, all the aforementioned studies relied on self-report and/or chart review to determine smoking status. To better avoid bias inherent in self-report and to capture secondhand smoke exposure, a few studies have used biomarkers such as cotinine and NNAL [4-(methylnitrosamino)-1-(3-pyridyl)-1-butanol] to assess smoking status. Calfee et al. measured levels of plasma cotinine and tracked the subsequent incidence of ARDS (26). They found that patients in the highest quartile of cotinine concentrations were 3.25 times as likely as those in the lowest quartile to develop ARDS (26). Hsieh et al. performed a secondary analysis of the ARDSNET Albuterol for the Treatment of Acute Lung Injury (ALTA) and Omega-3 Fatty Acid/Antioxidant Supplementation for Treating People with Acute Lung Injury or Acute Respiratory Distress Syndrome (Omega) studies. Smoking status was determined on the basis of urine NNAL concentrations at the time of randomization in the primary study (57). Smokers tended to be younger and healthier and have fewer comorbidities and lower Acute Physiology and Chronic Health Evaluation (APACHE) scores than patients with undetectable NNAL, but they nonetheless had equally severe lung injury. In these patients, smoking nullified the protective benefit of overall better health when they developed ARDS. Smokers were also overrepresented in this sample of patients with ARDS compared with national smoking rates (57).
CS increases alveolar-capillary barrier permeability and inflammation in humans.
The poor outcomes of ARDS among smokers are likely due to inflammation, alveolar epithelial and endothelial injury, and increased alveolar-capillary permeability, all of which predispose to development of pulmonary edema. In a study of donor lungs harvested with the intention of transplant, Ware et al. (146) showed that lungs from current smokers weighed more than lungs of former smokers and that these were heavier than the lungs of never smokers, all presumably because of edema. They found the same pattern for efficiency of oxygenation [arterial partial pressure of O2 ()/inspired O2 fraction ()] before harvest. Additionally, they found that a more extensive smoking history was associated with worse results in a laboratory test of alveolar fluid clearance (146), a measure of alveolar epithelial cell function. Relative to the more permeable pulmonary capillary endothelial barrier (pore radius of intercellular junctions is ~40–80 Å), the alveolar epithelial barrier is less permeable because of tight junctions of an estimated pore radius of 4–10 Å (133). Pulmonary clearance of inhaled 99mtechnetium-labeled diethylenetriamine pentaacetic acid (99mTc-DTPA) has been used to noninvasively assess alveolar epithelial barrier permeability in humans (14). Using this technique, Jones et al. have documented increased alveolar epithelial barrier permeability in cigarette smokers (63). Taken together, CS increases alveolar epithelial barrier permeability.
Whether CS exposure increases pulmonary endothelial barrier permeability in humans has been controversial. A pilot study on four smokers suggests that smokers may have decreased pulmonary capillary barrier permeability to urea (145). In contrast, pulmonary endothelial denudation has been documented in smokers with COPD (9). Altered endothelial function has also been suggested by biomarkers in human subjects. García-Lucio et al. (43) obtained pneumonectomy specimens from a sample of patients with solitary nodules, who either had COPD, were smokers with normal lung function, or were nonsmokers with normal lung function. In these patients, expression of angiopoietin-2 (ANGPT-2), as measured by immunohistochemistry, was associated with increased pulmonary artery vessel wall remodeling. In another cohort, they found that plasma levels of ANGPT-2 were higher in current smokers than in former smokers and that COPD patients overall had higher levels of ANGPT-2 than nonsmokers. Again, they demonstrated a dose-response association with pack years (43). A similar association was demonstrated by Petta et al. in healthy smokers with asthma (105). Since ANGPT-2 is secreted by vascular endothelial cells in Weibel-Palade bodies, these findings support the notion that cigarette smoking alters endothelial cell function in humans.
Moazed et al. (94) demonstrated experimentally that after exposure to inhaled lipopolysaccharide (LPS), smokers had a greater increase in plasma IL-8, a known mediator of inflammation in ARDS, compared with nonsmokers. These smokers had increased bronchoalveolar lavage (BAL) protein, suggesting increased alveolar-capillary permeability. They also had elevated plasma matrix metalloproteinase-8 (MMP-8), a potential cause of lung matrix breakdown. MMP-8 is secreted by neutrophils, which were elevated in total numbers and percentages in BAL fluid. LPS inhalation caused an exaggerated decrease in BAL VEGF, which has been shown to be decreased in ARDS patients and to increase with recovery (1). Smokers had increased levels of soluble VEGF receptor 1 (VEGFR1), which inhibits VEGF and may contribute to increased permeability (94). A recent study of mechanically ventilated trauma patients also showed differences in the lung microbiota of smokers and nonsmokers, at admission and after 48 h, which in turn affected expression of inflammatory markers and was associated with differences in incidence and severity of ARDS (101).
In summary, with some exceptions, these clinical studies present significant evidence that cigarette smoking potentiates ARDS and worsens outcomes. Cigarette smoke exposure increases both epithelial and endothelial barrier permeability at baseline and upregulates inflammatory pathways in humans. The role of inflammation in ARDS may differ among cases; however, evidence from several ARDSNET trials suggests that there are at least two subphenotypes of ARDS with distinct biochemical and inflammatory profiles (25). This is clinically significant because these subphenotypes respond differently to liberal vs. conservative fluid strategies and to high vs. low positive end-expiratory pressure (25, 36, 122). In clinical practice, subphenotype determination is limited by the biochemical assays available at the bedside. Future studies should incorporate smoking history as a factor in the characterization of ARDS subphenotypes and determine whether it can be used to identify the subphenotype and influence treatment strategies (25, 36, 122).
Both Brief and Subacute CS Exposures Increase Alveolar-Capillary Barrier Permeability in Animal Models
Loss of alveolar-capillary barrier function is a hallmark of ARDS. By assessing pulmonary clearance of fluorescein isothiocyanate-dextran (FITC-D; molecular radius at 22.2 Å), Burns et al. reported that CS exposure increases alveolar epithelial barrier permeability in guinea pigs (23). When the alveolar epithelial barrier is compromised, components of CS may pass through the leaky epithelial barrier and potentially result in endothelial cell injury and increased permeability. By assessing blood concentrations of intratracheally instilled iodine-125 bovine serum albumin (125I-BSA), which has dimensions of 140 × 40 × 40 Å and is impermeable to a healthy capillary endothelial barrier, Li et al. reported that intratracheal instillation of cigarette smoke condensate increases pulmonary capillary barrier permeability in rats (78). They also suggested that CS-induced increase in alveolar-capillary barrier permeability was due to oxidative stress but not a consequence of lung inflammation (78). We have demonstrated that lung microvascular endothelial cells (LMVEC) isolated from mice exposed to CS had a greater permeability and incomplete recovery after challenges by LPS or thrombin (Fig. 1, A and B; 20). Our results suggest that CS exposure may cause epigenetic changes in endothelial cells, which make these cells more vulnerable to a second injury. Both brief (hours) and subacute (4 wk) CS exposures increased BAL protein levels in guinea pigs (74). We have shown that acute CS preexposure followed by saline (control) caused lung edema; this effect was augmented in response to a second injury (LPS or Pseudomonas) in mice (Fig. 2; 20, 84). To our surprise, CS exposure alone without saline did not increase BAL protein levels or lung wet-to-dry weight ratio (data not shown). We suspect that the increased alveolar-capillary permeability by CS exposure alone is so covert in mice that it is not detectable by BAL protein content or wet-to-dry lung weight ratio. We speculate that more sensitive methods may be necessary to assess the effect of CS alone on alveolar-capillary permeability in mice. Nevertheless, there is strong evidence suggesting that CS exposure increases lung alveolar-capillary barrier permeability both in humans and in other animal models.
Fig. 1.
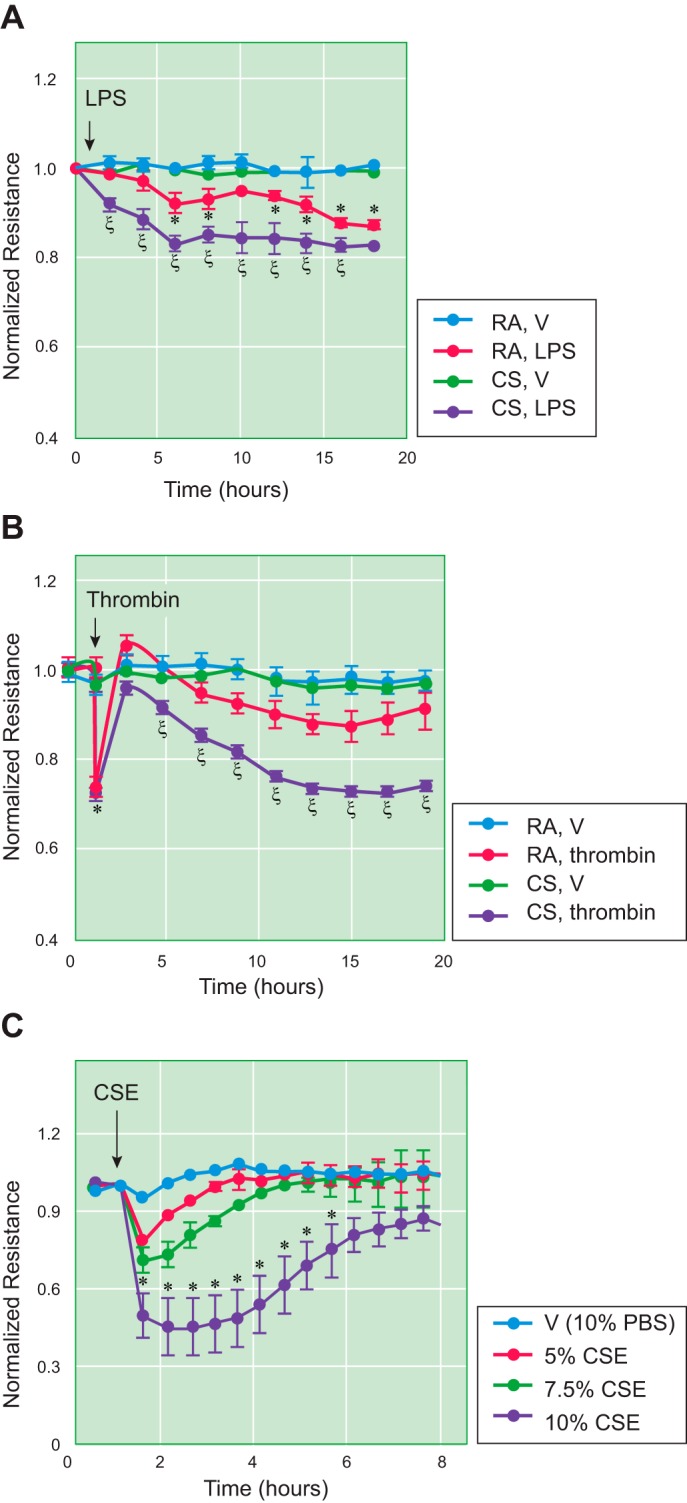
Cigarette smoke (CS) increased lung microvascular endothelial cell (LMVEC) permeability in vitro. A and B: 6-wk-old male C57BL/6 mice were exposed to room air (RA) or CS for 6 h. After overnight rest, LMVEC were isolated from cortical lung tissues. Isolated and characterized LMVEC at passage 1 were plated onto electric cell-substrate impedance sensing (ECIS) arrays at equal numbers of cells (2.5 × 105 cells per well). After overnight attachment, cells were treated with vehicle (V), LPS (1 µg/ml), or thrombin (2 U/ml) for 20 h, and monolayer permeability was assessed by measuring electric resistance across the monolayers. Four independent experiments with duplicated ECIS wells for each condition at each time were conducted. C: primary human LMVEC were treated with vehicle (10% PBS) or varying concentrations of cigarette smoke extract (CSE) for indicated times, and monolayer permeability was assessed by ECIS. Three independent experiments with duplicated ECIS wells for each condition at each time were conducted. Data are presented as the means ± SE of the normalized electrical resistance at the selected time points relative to their initial resistance. ANOVA and Tukey-Kramer post hoc test were used to determine statistically significant difference across means among groups. *P < 0.05 vs. RA+V; ξP < 0.05 vs. RA+LPS (A) or RA+thrombin (B). Arrows indicate the time for addition of treatments. [Reprinted with permission of the American Journal of Respiratory Cell and Molecular Biology (20).]
Fig. 2.
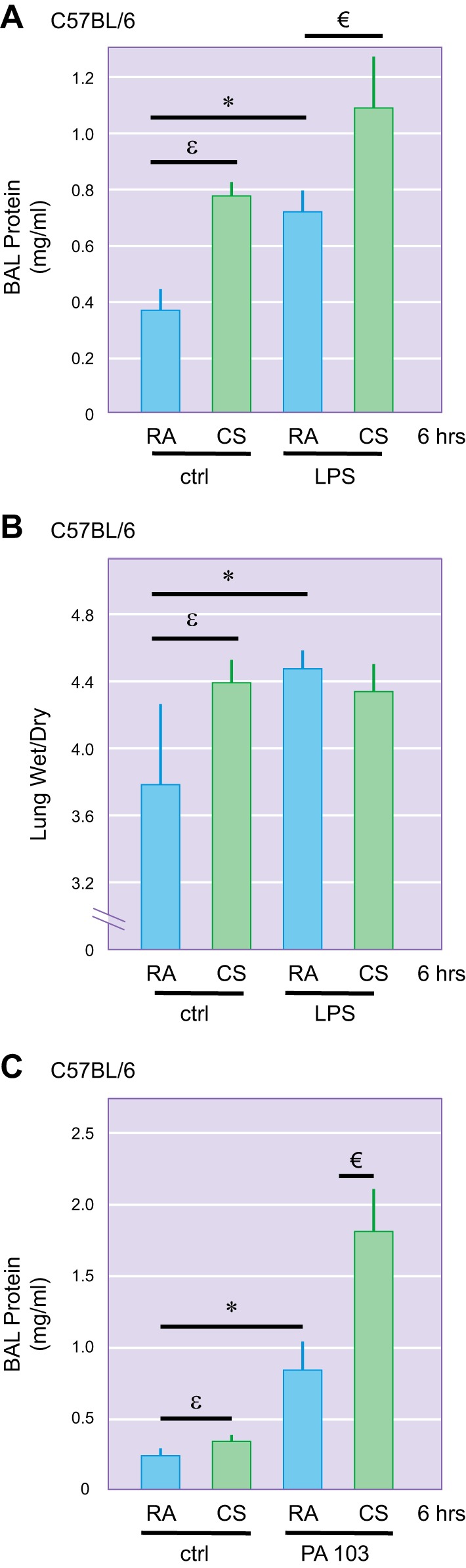
Effects of a brief cigarette smoke (CS) exposure on LPS- and Pseudomonas-induced lung edema. Male 6-wk-old C57BL/6 were exposed to room air (RA) or CS for 6 h. One hour after CS exposure, mice were intratracheally administered with 2.5 mg/kg of LPS or 5 × 103 colony-forming units of P. aeruginosa (strain PA103) or equal volume of saline as a control (ctrl). After 18 h, bronchoalveolar lavage (BAL) protein content (A and C) and lung wet-to-dry weight ratio (B) were assessed. Four to six mice per group were used. εP < 0.05 CS/ctrl vs. RA/ctrl; *P < 0.05 RA/LPS vs. RA/ctrl; €P < 0.05 CS/LPS vs. RA/LPS or RA/PA103. [Reprinted with permission of the American Journal of Physiology Lung Cellular and Molecular Physiology (84) and the American Journal of Respiratory Cell and Molecular Biology (20).]
Both Brief and Subacute CS Exposures Increase Susceptibility to Acute Lung Injury in Animal Models
Similarly to the human studies described above, CS preexposure delayed bacterial clearance and promoted lung inflammation induced by Pseudomonas aeruginosa infection in mice (33). Two weeks of CS exposure also enhanced alveolar inflammation and apoptosis induced by poly(I:C), a viral pathogen-associated molecular pattern (PAMP) in mice (65). Four weeks of CS exposure exacerbated lung inflammation and compromised adaptive immunity to infection by Hemophilus influenzae in mice (86). Two weeks of CS exposure did not change basal BAL protein levels or neutrophil count but significantly exacerbated mechanical ventilation-induced increase in BAL protein content and neutrophil influx in rats (54). Similarly, CS exposure for 3–4 wk aggravated blunt chest trauma-induced lung inflammation and lung cell apoptosis in mice (142).
Using two strains of inbred mice with different susceptibility to CS-induced emphysema (48), we found that as little as 3–6 h of exposure to CS potentiated LPS-induced lung edema in both C57BL/6 (Fig. 2A) and AKR mice (116). Similar exacerbating effects were observed after 3 wk of preexposure to CS (Fig. 3A). We also noted a strain difference in susceptibility to CS priming for acute lung injury, with a greater effect in AKR mice (Fig. 3A; 116).
Fig. 3.
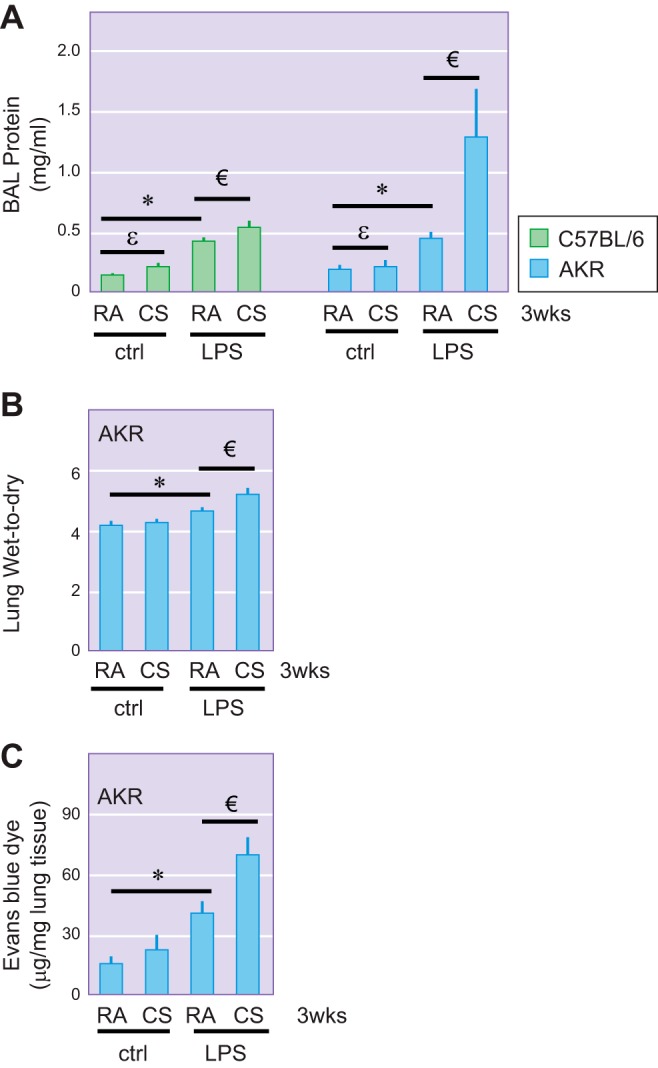
Effects of prolonged cigarette smoke (CS) exposure on LPS-induced lung edema. Male 6-wk-old C57BL/6 and AKR mice were exposed to room air (RA) or CS for 3 wk. One hour after the last CS exposure, mice were intratracheally administered with 2.5 mg/kg of LPS or equal volume of saline as a control (ctrl). After 18 h, bronchoalveolar lavage (BAL) protein content (A), lung wet-to-dry weight ratio (B), and lung extravasation of albumin-conjugated Evans blue dye (C) were assessed. Three to four C57BL/6 mice per group and 4–6 AKR mice per group were used. εP < 0.05 CS/ctrl vs. RA/ctrl; *P < 0.05 RA/LPS vs. RA/ctrl; €P < 0.05 CS/LPS vs. RA/LPS. [Reprinted with permission of the American Journal of Physiology Lung Cellular and Molecular Physiology (116).]
Taken together, studies of both human and preclinical animal models indicate that increased susceptibility to and severity of acute lung injury/ARDS are important adverse health consequences of CS exposure that need to be taken into consideration when treating critically ill individuals. In addition, there is evidence from humans and from animal studies that CS exposure increases lung barrier permeability, through effects on both epithelial and endothelial cells. In this review we focus on mechanisms of CS effects on lung endothelial cell permeability.
Cigarette Smoke Extract Increases Endothelial Monolayer Permeability in Vitro
Although the nature and concentrations of smoke components may differ between CS inhalation in vivo and aqueous cigarette smoke extract (CSE) exposure of culture cells in vitro, CSE exposure is a widely used and acceptable in vitro system to address effects of CS on specific cells and underlying mechanisms (92, 155). As has been shown in human and animal studies, exposure to CSE in vitro increases pulmonary macrovascular and microvascular endothelial monolayer permeability (Fig. 1C; 20, 55). The underlying mechanism involves inhibition of RhoA and focal adhesion kinase (FAK) signaling (84), microtubule depolymerization (16), histone deacetylase-6 (HDAC6) activation (20), p38 activation (81), ceramide upregulation (120), and disruption of intercellular adhesion molecules, as depicted in Fig. 4.
Fig. 4.

Proposed model of cigarette smoke-induced pulmonary endothelial cell injury. Cigarette smoke (CS) exposure causes lung endothelial cell (EC) activation, resulting in inflammation, via activation of NF-κB signaling; CS exposure directly increases EC permeability likely through multiple signaling pathways, including inhibition of RhoA, focal adhesion kinase (FAK), and intercellular adhesion molecules (IAM) and activation/upregulation of histone deacetylase-6 (HDAC6), p38, and ceramide; CS exposure also causes EC apoptosis through signaling pathways involving downregulation of VEGF, FAK, α1-antitrypsin (AAT), and unfolded protein response (UPR) and upregulation of ceramide, adenosine, p38, and p53. Excessive EC apoptosis can lead to necrosis and autoimmunity because of release of mitochondrial damage-associated molecular patterns (DAMPs), endothelial microparticles (eMPs), and other cellular contents. CS-induced increases in EC activation, permeability, apoptosis, and necrosis collectively contribute to increased risk of development of acute respiratory distress syndrome (ARDS). CS-induced lung inflammation, EC apoptosis and necrosis, and autoimmunity cause emphysema. CS exposure increases EC endothelin (ET)-1 levels, leading to increased smooth muscle cell (SMC) proliferation and vasoconstriction. CS exposure also decreases vasodilation because of reduction in EC NO. All these changes, in combination with increased blood flow in remaining vessels due to EC apoptosis and loss of vessels, cause vascular remodeling and pulmonary hypertension (PH) in chronic obstructive pulmonary disease (COPD) patients.
Endothelial barrier integrity is regulated by adherens junctions (AJs), F-actin cytoskeleton, microtubules (102), and focal adhesion complexes (FAC; 138). It has been well documented that RhoA is required for coordinated assembly of F-actin fibers and that AJs and FAC are critical for establishment of cell-cell contacts (21). RhoA activation also mediates increased endothelial permeability induced by various edemagenic agents, such as thrombin and histamine, by increasing contractile F-actin stress fiber formation (136). The role of RhoA signaling in CSE-induced endothelial monolayer permeability has been controversial. Inhibition of Rho kinase has been reported to attenuate CSE-induced increases in lung microvascular endothelial monolayer permeability (120). In contrast, we have shown that CSE inactivated RhoA via oxidative stress in a dose- and time-dependent manner (84). In support of our results, reactive oxygen species (ROS) have been shown to inhibit RhoA, leading to structural disruption of FAC and F-actin fibers in other cells (24). We further demonstrated that overexpression of constitutively active RhoA blunted CSE-induced disassembly of AJs, FAC, and cortical F-actin fibers in pulmonary endothelial cells (84). Our data suggest that inhibition of RhoA contributes to CSE-induced endothelial barrier dysfunction. The apparent discrepancy of these results may be due to different preparations and concentrations of CSE used. RhoA contains a redox-sensitive motif [GXXXCGK(S/T)C]. It has been suggested that physiological levels of ROS and high reduction potential may directly oxidize and activate RhoA through direct oxidation of two cysteine residues located in the redox-sensitive motif (2). Conversely, high levels of oxidants inhibit RhoA through formation of an intramolecular disulfide bridge that prevents GTP binding (53). Whether CSE inhibits RhoA via intramolecular disulfide bridge formation needs further investigation.
FAK is a key component of FAC and an essential survival factor for anchorage-dependent cells (83). Although FAK activation has been implicated in endothelial barrier dysfunction in some settings (49, 153), FAK activity is also required for maintenance of endothelial barrier function (72). Endothelial cells isolated from FAK knockout mouse embryos or embryos expressing kinase-defective FAK displayed increased monolayer permeability (160). Knockdown of FAK by small interfering RNA (siRNA) also caused disruption of tight junctions in the testis (126). FAK activity is also required for resealing of endothelial adhesion junctions after exposure to H2O2 (112) and thrombin (56). We have shown that CSE dose and time dependently inhibits FAK activity via oxidative stress and that overexpression of FAK attenuates CSE-induced disruption of AJs, FAC, and F-actin fibers (84). The reactive aldehyde, 4-hydroxy-2-nonenal (4-HNE), a component of CSE, also decreases FAK activity and endothelial barrier function via oxidative stress (137). Taken together, oxidative stress-induced FAK inhibition contributes to CSE-induced endothelial barrier dysfunction.
Intact microtubules are critical for maintenance of endothelial barrier integrity (17, 91, 139). α-Tubulin is a building block of microtubules, and acetylation of α-tubulin is characteristic of stable microtubules (89, 159). CSE has been shown to cause microtubule depolymerization in human umbilical vein endothelial cells (HUVEC; 16). We have shown that CSE decreases α-tubulin acetylation and causes microtubule depolymerization in pulmonary endothelial cells (20). The microtubule stabilizer paclitaxel elevates α-tubulin acetylation and prevents CSE-induced pulmonary endothelial barrier dysfunction (20). These results indicate that microtubule instability contributes to CSE-induced endothelial barrier dysfunction.
Acetylated α-tubulin is deacetylated by HDAC6 (158). HDAC6 is a microtubule-associated protein deacetylase that is ubiquitously expressed and predominantly located in the cytoplasm (58). HDAC6 knockout mice do not have a visible abnormal phenotype (158) but have better survival and reduced lung injury after LPS challenge (28, 144). Histone deacetylase inhibitors attenuate lung injury induced by LPS (96), P. aeruginosa (20), mechanical ventilation (29), and cecal ligation and puncture (157). HDAC6 inhibitors also blunt increased permeability of lung endothelial monolayers induced by thrombin (114) and LPS (28). In addition, inhibition of HDAC6 has been proposed as a novel promising therapeutic option for a variety of disorders, including cardiac dysfunction, depression, Alzheimer’s disease, COPD-associated cilia dysfunction, and diabetes (31, 41, 46, 62, 75, 149, 154).
The role of HDAC6 in CS-induced lung injury is less well understood. We have recently shown that HDAC6 activity is enhanced in cultured lung endothelial cells exposed to CSE likely via oxidative stress-mediated Akt inactivation and subsequent GSK-3β-mediated phosphorylation of HDAC6 at serine-22 (20). Tubacin, a specific HDAC6 inhibitor (50), and HDAC6 siRNA abolished CSE-induced barrier dysfunction in primary human LMVEC in vitro (20). Importantly, tubacin prevented CS priming for lung injury after infection with P. aeruginosa in mice (20). We speculated that HDAC6 inhibitors may become a novel therapeutic option for CS-associated lung injury.
Endothelial intercellular adhesion molecule CD146, also known as cell surface glycoprotein MUC18, is decreased in lung tissue of smokers with COPD and of rats exposed to CS, as well as in cultured lung macrovascular and microvascular endothelial cells exposed to CSE (73). CD146 knockout mice exhibit perivascular edema and lung inflammation (73). However, it remains unknown whether loss of CD146 plays a role in CS-induced endothelial barrier dysfunction and increased vascular permeability in vivo. Notably, soluble sCD146, which lacks transmembrane and intracellular domains of CD146, is increased in plasma and BAL of COPD patients as well as in BAL of rats exposed to CS (73). Further investigation is needed to determine whether circulating or BAL sCD146 can be used as biomarkers of CS-induced lung injury.
Platelet endothelial cell adhesion molecule-1 (PECAM-1) regulates endothelial barrier permeability by facilitating dephosphorylation of β-catenin (18). PECAM-1 null mice exhibit prolonged and increased permeability after inflammatory insults (18). CSE causes a 10-fold increase in tyrosine phosphorylation of PECAM-1 in HUVEC (124). However, the role of PECAM-1 in CS-induced lung endothelial injury remains unknown.
It is likely that multiple components of cigarette smoke are responsible for increased endothelial permeability. We have demonstrated that acrolein increased endothelial monolayer and lung microvascular permeability in vivo (82). Schweitzer et al. reported that nicotine, e-cigarette solution, and condensed e-cigarette vapor increased endothelial monolayer permeability (119). In addition, they demonstrated nicotine-independent effects of e-cigarette solutions on endothelial permeability. Acrolein is among the components of combustion of e-cigarette solutions.
CS Causes Lung Endothelial Cell Activation and Inflammation
CS activates the lung endothelium and causes inflammatory cell accumulation. Sharma et al. showed in human LMVEC that this mechanism involves the inhibition of platelet aggregating factor acetylhydrolase (PAF-AH), which degrades platelet aggregating factor (PAF), leading to increased levels of PAF (123). Increased PAF is in turn associated with neutrophil [polymorphonuclear neutrophil (PMN)] adherence to the endothelium. They also showed that CSE stimulates expression of other endothelium-activating molecules, including P-selectin, E-selectin, intercellular adhesion molecule-1 (ICAM-1), and vascular cell adhesion molecule-1 (VCAM-1; 123). The same group subsequently showed that increased PAF is associated with increased adherence of metastatic breast cancer cells to the lung endothelium and that CSE stimulates the upregulation of the PAF receptor on the breast cancer cells as well (70).
Acute CS exposure also activates xanthine oxidase and elicits the rolling and adhesion of leukocytes to endothelium of arterioles and postcapillary venules of striated muscle microcirculation and aortic endothelium in hamsters in vivo; these effects were prevented by superoxide dismutase and water-soluble antioxidants but not by lipid-soluble antioxidants (76, 77). CSE increases the surface expression of adhesion molecules, including ICAM-1, endothelial leukocyte adhesion molecule-1 (ELAM-1), VCAM-1, and E-selectin, as well as cytokines and chemokines, including tumor necrosis factor-α, IL-6, and IL-1β, via NADPH oxidase-dependent NF-κB transcriptional activation in endothelial cells (98) CSE also increases adherence of monocytes to the endothelium and transendothelial migration (124), as well as neutrophil transmigration across HUVEC (99). CS-induced upregulation of the C-X-C motif chemokine receptor 3 (CXCR3) receptor in endothelial cells may mediate endothelial cell apoptosis (47). CSE synergizes with the inflammatory cytokine IL-1β to increase vascular permeability and endothelial dysfunction via ROS/p38/phosphatase and tensin homolog (PTEN)-mediated tyrosine phosphorylation of vascular endothelial cadherin and β-catenin, as well as subsequent β-catenin nuclear translocation and expression of inflammatory genes, such as cyclooxygenase-2 (COX-2) in cardiac endothelial cells (11, 12). Taken together, CS causes lung inflammation via its direct effect on endothelial cell activation, leading to enhanced endothelial cell apoptosis, increased barrier permeability, and endothelial dysfunction.
Damage-associated molecular patterns (DAMPs) enhance PMN adherence to endothelial cells by increasing surface expression of adhesion molecules in both PMN and human pulmonary artery endothelial cells in vitro (130). CS exposure induces necrosis of bronchial epithelial cells and neutrophils, leading to DAMP release and proinflammatory responses (51, 111). Levels of DAMPs, including high-mobility group box-1 (HMGB1), are increased in extracellular lung fluids of COPD patients (109), and increased DAMPs have been implicated in inflammation in COPD (110). Future studies are needed to address the sources and roles of DAMPs in CS-induced endothelial activation and related lung diseases.
EFFECT OF CIGARETTE SMOKE ON ENDOTHELIAL APOPTOSIS IN COPD
Robust pulmonary endothelial cell apoptosis has been observed in patients with severe ARDS (1) and in mice with mild ARDS induced by LPS (40). Pulmonary microvascular endothelial cell apoptosis has been suggested as a cause of barrier dysfunction and edema in septic mice (44, 45). Inhibition of apoptosis by a broad-spectrum caspase inhibitor prolonged survival of mice exposed to LPS (67). These studies suggest that lung endothelial cell apoptosis contributes to the pathogenesis of ARDS.
Recent clinical studies also implicate vascular endothelial damage in the pathogenesis of COPD. This has been observed outside the lungs as well, most notably with the loss of glomerular integrity in patients with COPD. Polverino et al. (108) showed that COPD patients had lower estimated glomerular filtration rates than smokers without COPD and nonsmoking controls and that lower forced expiratory volume in 1 s (FEV1) %predicted was associated with double contouring of the glomerular basement membrane. Most smokers had albuminuria as well, but the quantity did not correlate with FEV1. In mouse studies, cigarette smoke also increased albuminuria. This group also showed that levels of oxidative stress-advanced glycation end products (AGEs) and their receptors (RAGEs) increased in mice with cigarette smoke exposure. Treatment with enalapril blunted these effects in mice, suggesting a role of angiotensin I-converting enzyme (ACE) inhibitors for renal protection in patients with COPD (108). Agrawal et al. also demonstrated increased albuminuria in patients with acute exacerbations of COPD (3).
Lung tissue from patients with emphysema displays increased apoptosis of alveolar epithelial and endothelial cells (59, 66). We also observed an increase in lung endothelial cell apoptosis in AKR mice exposed to CS for 3 wk (115). Circulating endothelial microparticles (eMPs) are thought to be shed into the bloodstream from activated, apoptotic, or necrotic endothelial cells. Blood eMPs are significantly elevated in healthy smokers (128) and patients with COPD (131, 134). Circulating eMP levels remain elevated in COPD patients despite smoking cessation but return to normal low levels in healthy former smokers, suggesting that pulmonary endothelial cell injury is reversible only in healthy smokers who quit smoking (128). Circulating eMPs are also increased in rats exposed to CS for 2–6 mo (79). Further studies are needed to determine whether circulating eMPs are markers of vascular endothelial injury or part of the pathogenesis of diseases linked to endothelial injury and inflammation in smokers (121).
CSE triggers apoptosis of pulmonary endothelial cells (115, 135) and induces necrosis and inhibits apoptosis of alveolar epithelial cells and HUVEC (148). The potential mechanisms underlying CS-induced endothelial apoptosis include inhibition of VEGF, FAK, and α1-antitrypsin (AAT) signaling and upregulation of ceramide and adenosine, as well as impairment of unfolded protein response (UPR), as depicted in Fig. 4.
VEGF/VEGFR2 signaling is reduced in the lungs of smokers with and without COPD (59, 66) and of rodents exposed to CS (88). CSE decreases expression of VEGF in pulmonary endothelial cells in vitro (135). We observed that FAK activity is reduced in AKR mice exposed to CS for 3 wk and in lung endothelial cells exposed to CSE; these effects were associated with enhanced pulmonary endothelial cell apoptosis in vivo and in vitro (115). Overexpression of FAK prevented CSE-induced endothelial cell apoptosis, suggesting that reduced FAK activity may contribute to CSE-induced endothelial cell apoptosis (115). Inhibition of FAK also causes emphysema-like changes in rat lungs (93). Further studies are necessary to address whether reduced FAK activity contributes to CS-induced lung endothelial cell apoptosis in vivo. AAT inhibits apoptosis of cultured lung endothelial cells by blocking interaction of caspases-3, -6, and -7 with their substrates (103). Overexpression of AAT inhibits apoptosis of cultured lung endothelial cells and attenuates emphysema caused by active caspase-3 and blockade of VEGF signaling in vivo (103). Exogenous AAT protects cultured pulmonary endothelial cells from CSE-induced apoptosis in vitro (6). Whether exogenous AAT or overexpression of AAT prevents or reverses CS-induced lung endothelial cell apoptosis and emphysema in vivo remains to be determined. Ceramide is upregulated in emphysematous lungs of patients and animal models, as well as in cultured pulmonary endothelial cells exposed to CSE (104). This increase in ceramide enhances alveolar cell apoptosis (104).
CS exposure increases lung tissue adenosine levels in mice, an effect associated with lung endothelial cell apoptosis and early emphysema (85). Sustained increased adenosine in adenosine deaminase (ADA)-deficient mice enhances alveolar cell apoptosis and causes emphysema-like changes in mice (161). ADA expression and activity are reduced in lungs of smokers with COPD (162). Whether chronically elevated adenosine contributes to CS-induced lung endothelial cell apoptosis and development of emphysema remains to be investigated. Other investigators have reported that CS increased xanthine oxidoreductase (XOR) expression and activity and that this increase was sufficient and necessary for p53 induction and subsequent endothelial cell apoptosis (69).
The UPR is an important mechanism of elimination of endoplasmic reticulum stress and enhanced cell survival (118). The UPR is activated in lung tissue of smokers without emphysema (68). UPR is also activated by CSE in cultured human bronchial epithelial cells and 3T3 fibroblasts (52, 64) and in cultured pulmonary endothelial cells (115). Using mouse models of CS exposure, we have observed a strong link between impairment of eukaryotic initiation factor 2α (eIF2α) signaling and lung endothelial cell apoptosis in a mouse strain susceptible to CS-induced emphysema (115). Future studies are necessary to determine whether impaired eIF2α signaling contributes to lung endothelial cell apoptosis and emphysema.
In summary, CS exposure causes lung endothelial cell apoptosis. The underlying mechanism is rather complicated, as summarized in Fig. 4. This may be due to the complexity of toxins in smoke. Therefore it would be difficult to prevent and treat cigarette smoke-induced lung diseases by using a single approach/drug. It may be important to target multiple pathways.
EFFECT OF CIGARETTE SMOKE ON ENDOTHELIAL DYSFUNCTION IN PULMONARY HYPERTENSION ASSOCIATED WITH COPD
Pulmonary hypertension is a complication of smoking-induced COPD with a prevalence of ~19% in COPD patients (39). The causes of pulmonary hypertension in COPD are complex and include vasoconstriction caused by acute hypoxia and acidosis, loss of microcirculation in emphysema, increased blood viscosity caused by polycythemia, and vascular remodeling caused by chronic hypoxia.
Pulmonary vasoconstriction and vascular remodeling have been recognized in smokers with normal lung function and in patients with mild COPD without hypoxemia (117). Pulmonary endothelial dysfunction has also been reported in otherwise healthy young smokers (100). In addition, pulmonary endothelial dysfunction and pulmonary vascular smooth muscle cell proliferation precede emphysema in guinea pigs exposed to cigarette smoke (37). These results suggest that cigarette smoke directly alters the structure and function of pulmonary vessels at early stage COPD. CS-induced pulmonary endothelial cell dysfunction might predispose patients with COPD to further smooth muscle hypertrophy, thus contributing to development of pulmonary hypertension in COPD. Furthermore, changes in endogenous pulmonary vasoconstrictors and vasodilators produced by lung endothelium can cause vasoconstriction. The potential role of endothelial dysfunction in the pathophysiology of pulmonary hypertension in COPD suggests that therapies targeting the endothelium may be effective in treating these patients.
Endothelin (ET)-1 and nitric oxide (NO) are endothelium-derived mediators with opposite effects on vascular tone and cell growth. ET-1 is a potent vasoconstrictor and mitogenic agent (19). Plasma ET-1 levels are elevated in smokers, and patients with both COPD and pulmonary hypertension have increased expression of ET-1 in pulmonary endothelial cells (156). ET-1 levels are also elevated in hypertensive intrapulmonary arteries of guinea pigs exposed to CS for 6 mo (151). CSE promotes ET receptor expression in pulmonary artery endothelial cells (90). Future studies are needed to address the role of ET-1 in pulmonary vascular remodeling in COPD patients with pulmonary hypertension.
NO, a major vasodilator, is produced by endothelial NO synthase (eNOS). Expression of eNOS is reduced in pulmonary arteries of healthy smokers (10). Pulmonary arterial relaxation was also reduced in otherwise healthy smokers; this effect was associated with decreased eNOS activity and reduced NO in exhaled air (34). Diminished lung eNOS levels, lower NO, and reduced endothelium-dependent vasodilatation in pulmonary arteries precede the development of emphysema in guinea pigs (37) and mice (125). After 6 mo of cigarette smoke exposure, eNOS knockout mice had enhanced vascular remodeling and pulmonary hypertension (152). CSE irreversibly decreased eNOS mRNA and protein levels and inhibited eNOS enzymatic activity in pulmonary endothelial cells (129, 143). CSE also increases eNOS acetylation via oxidative stress-dependent downregulation of sirtuin 1, leading to blunted eNOS activity (8). In addition, CSE-induced ROS may interact with NO to produce peroxynitrite (ONOO−), thereby reducing NO bioactivity (61). In fact, reactive nitrogen species are increased in lung vessels of smokers with COPD and further increased in smokers with combined COPD and pulmonary hypertension (152). Taken together, oxidative stress-mediated reduction in eNOS-NO signaling is a significant contributor to CS-induced vascular endothelial dysfunction.
Nana-Sinkam et al. demonstrated decreased expression of prostacyclin synthase (PGI2S) and lower levels of prostacyclin, which prevents apoptosis, in lung tissue of patients with COPD (95). In cultured human pulmonary microvascular endothelial cells, treatment with CSE directly decreased expression of COX-1 and increased expression of COX-2 and cytosolic phospholipase A2 (CPLA2), potential causes of oxidative stress. This was prevented by pretreatment with the antioxidant N-acetylcysteine (95). These results suggest that decreased prostacyclin and increased oxidative stress also contribute to CS-induced pulmonary endothelial dysfunction and pulmonary hypertension in patients with COPD.
PERSPECTIVES AND FUTURE DIRECTIONS
Growing evidence from clinical studies supports an adverse effect of cigarette smoke exposure as a risk factor for ARDS and poor outcome. Numerous studies of preclinical animal models and cultured cells have demonstrated that cigarette smoke exposure increases pulmonary endothelial barrier permeability and causes endothelial activation resulting in enhanced lung inflammation. Cigarette smoke exposure also causes pulmonary endothelial cell apoptosis; this may initiate and promote development of the lung destruction characteristic of emphysema. Cigarette smoke exposure directly causes endothelial dysfunction associated with vascular remodeling and vasoconstriction in smokers.
We hypothesize that the normal alveolar epithelial barrier protects endothelial cells from exposure to toxins in cigarette smoke. However, as illustrated in Fig. 4, after smoke-induced injury to and disruption of the tight alveolar epithelial barrier, components of smoke and/or DAMPs released from injured airway epithelial cells can reach and cause injury to endothelial cells and subsequently enter the circulation via a more permeable endothelial barrier. Future studies should include developing strategies to protect endothelial cells in order to prevent and treat cigarette smoking-associated lung diseases, such as ARDS, emphysema, and pulmonary hypertension associated with COPD. Since components of cigarette smoke and/or DAMPs released from injured epithelial and endothelial cells can cross the injured endothelial barrier and enter the systemic circulation, it would be reasonable to strengthen the pulmonary endothelial barrier as a measure to prevent cardiovascular and other cigarette smoke-induced systemic diseases.
GRANTS
This study was supported using facilities at the Providence Veterans Affairs Medical Center and by Veterans Affairs Merit Review (S. Rounds), National Heart, Lung, and Blood Institute Grant R01-HL-130230 (Q. Lu), a Brown University Dean’s Emerging Areas of New Science (DEANS) award (S. Rounds), National Institute of General Medical Sciences (NIGMS) Grant U54-GM-115677 (S. Rounds), and an Institutional Development award from NIGMS under Grant P20-GM-103652 (S. Rounds; project 1 to Q. Lu).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
Q.L. and E.G. prepared figures; Q.L., E.G., and S.R. drafted manuscript; Q.L., E.G., and S.R. edited and revised manuscript; Q.L., E.G., and S.R. approved final version of manuscript.
REFERENCES
- 1.Abadie Y, Bregeon F, Papazian L, Lange F, Chailley-Heu B, Thomas P, Duvaldestin P, Adnot S, Maitre B, Delclaux C. Decreased VEGF concentration in lung tissue and vascular injury during ARDS. Eur Respir J 25: 139–146, 2005. doi: 10.1183/09031936.04.00065504. [DOI] [PubMed] [Google Scholar]
- 2.Aghajanian A, Wittchen ES, Campbell SL, Burridge K. Direct activation of RhoA by reactive oxygen species requires a redox-sensitive motif. PLoS One 4: e8045, 2009. doi: 10.1371/journal.pone.0008045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Agrawal A, Garg R, Sahu D, Kumar M. Study the association of chronic obstructive pulmonary disease with early endothelial dysfunction and its impact on cardiovascular system by estimating urinary albumin creatinine ratio. Lung India 34: 138–143, 2017. doi: 10.4103/0970-2113.201299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Aldonyte R, Hutchinson TE, Jin B, Brantly M, Block E, Patel J, Zhang J. Endothelial alpha-1-antitrypsin attenuates cigarette smoke induced apoptosis in vitro. COPD 5: 153–162, 2008. [Erratum in COPD 5: 405, 2008.] doi: 10.1080/15412550802092936. [DOI] [PubMed] [Google Scholar]
- 7.Alwis KU, deCastro BR, Morrow JC, Blount BC. Acrolein exposure in U.S. tobacco smokers and non-tobacco users: NHANES 2005-2006. Environ Health Perspect 123: 1302–1308, 2015. doi: 10.1289/ehp.1409251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Arunachalam G, Yao H, Sundar IK, Caito S, Rahman I. SIRT1 regulates oxidant- and cigarette smoke-induced eNOS acetylation in endothelial cells: role of resveratrol. Biochem Biophys Res Commun 393: 66–72, 2010. doi: 10.1016/j.bbrc.2010.01.080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Barberà JA. Mechanisms of development of chronic obstructive pulmonary disease-associated pulmonary hypertension. Pulm Circ 3: 160–164, 2013. doi: 10.4103/2045-8932.109949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Barberà JA, Peinado VI, Santos S, Ramirez J, Roca J, Rodriguez-Roisin R. Reduced expression of endothelial nitric oxide synthase in pulmonary arteries of smokers. Am J Respir Crit Care Med 164: 709–713, 2001. doi: 10.1164/ajrccm.164.4.2101023. [DOI] [PubMed] [Google Scholar]
- 11.Barbieri SS, Ruggiero L, Tremoli E, Weksler BB. Suppressing PTEN activity by tobacco smoke plus interleukin-1β modulates dissociation of VE-cadherin/β-catenin complexes in endothelium. Arterioscler Thromb Vasc Biol 28: 732–738, 2008. doi: 10.1161/ATVBAHA.107.159434. [DOI] [PubMed] [Google Scholar]
- 12.Barbieri SS, Weksler BB. Tobacco smoke cooperates with interleukin-1β to alter β-catenin trafficking in vascular endothelium resulting in increased permeability and induction of cyclooxygenase-2 expression in vitro and in vivo. FASEB J 21: 1831–1843, 2007. doi: 10.1096/fj.06-7557com. [DOI] [PubMed] [Google Scholar]
- 13.Barnoya J, Glantz SA. Cardiovascular effects of secondhand smoke: nearly as large as smoking. Circulation 111: 2684–2698, 2005. doi: 10.1161/CIRCULATIONAHA.104.492215. [DOI] [PubMed] [Google Scholar]
- 14.Barrowcliffe MP, Jones JG. Pulmonary clearance of 99mTc-DTPA in the diagnosis and evolution of increased permeability pulmonary oedema. Anaesth Intensive Care 17: 422–432, 1989. [DOI] [PubMed] [Google Scholar]
- 15.Bello S, Menéndez R, Antoni T, Reyes S, Zalacain R, Capelastegui A, Aspa J, Borderías L, Martin-Villasclaras JJ, Alfageme I, Rodríguez de Castro F, Rello J, Luis M, Ruiz-Manzano J. Tobacco smoking increases the risk for death from pneumococcal pneumonia. Chest 146: 1029–1037, 2014. doi: 10.1378/chest.13-2853. [DOI] [PubMed] [Google Scholar]
- 16.Bernhard D, Csordas A, Henderson B, Rossmann A, Kind M, Wick G. Cigarette smoke metal-catalyzed protein oxidation leads to vascular endothelial cell contraction by depolymerization of microtubules. FASEB J 19: 1096–1107, 2005. doi: 10.1096/fj.04-3192com. [DOI] [PubMed] [Google Scholar]
- 17.Birukova AA, Birukov KG, Smurova K, Adyshev D, Kaibuchi K, Alieva I, Garcia JG, Verin AD. Novel role of microtubules in thrombin-induced endothelial barrier dysfunction. FASEB J 18: 1879–1890, 2004. doi: 10.1096/fj.04-2328com. [DOI] [PubMed] [Google Scholar]
- 18.Biswas P, Canosa S, Schoenfeld D, Schoenfeld J, Li P, Cheas LC, Zhang J, Cordova A, Sumpio B, Madri JA. PECAM-1 affects GSK-3β-mediated β-catenin phosphorylation and degradation. Am J Pathol 169: 314–324, 2006. doi: 10.2353/ajpath.2006.051112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Böhm F, Pernow J. The importance of endothelin-1 for vascular dysfunction in cardiovascular disease. Cardiovasc Res 76: 8–18, 2007. doi: 10.1016/j.cardiores.2007.06.004. [DOI] [PubMed] [Google Scholar]
- 20.Borgas D, Chambers E, Newton J, Ko J, Rivera S, Rounds S, Lu Q. Cigarette smoke disrupted lung endothelial barrier integrity and increased susceptibility to acute lung injury via histone deacetylase 6. Am J Respir Cell Mol Biol 54: 683–696, 2016. doi: 10.1165/rcmb.2015-0149OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Braga VM, Machesky LM, Hall A, Hotchin NA. The small GTPases Rho and Rac are required for the establishment of cadherin-dependent cell-cell contacts. J Cell Biol 137: 1421–1431, 1997. doi: 10.1083/jcb.137.6.1421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Brown SM, Wilson E, Presson AP, Zhang C, Dinglas VD, Greene T, Hopkins RO, Needham DM; National Institutes of Health NHLBI ARDS Network . Predictors of 6-month health utility outcomes in survivors of acute respiratory distress syndrome. Thorax 72: 311–317, 2017. doi: 10.1136/thoraxjnl-2016-208560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Burns AR, Hosford SP, Dunn LA, Walker DC, Hogg JC. Respiratory epithelial permeability after cigarette smoke exposure in guinea pigs. J Appl Physiol (1985) 66: 2109–2116, 1989. doi: 10.1152/jappl.1989.66.5.2109. [DOI] [PubMed] [Google Scholar]
- 24.Cai J, Niu X, Chen Y, Hu Q, Shi G, Wu H, Wang J, Yi J. Emodin-induced generation of reactive oxygen species inhibits RhoA activation to sensitize gastric carcinoma cells to anoikis. Neoplasia 10: 41–51, 2008. doi: 10.1593/neo.07754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Calfee CS, Delucchi K, Parsons PE, Thompson BT, Ware LB, Matthay MA; NHLBI ARDS Network . Subphenotypes in acute respiratory distress syndrome: latent class analysis of data from two randomised controlled trials. Lancet Respir Med 2: 611–620, 2014. doi: 10.1016/S2213-2600(14)70097-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Calfee CS, Matthay MA, Eisner MD, Benowitz N, Call M, Pittet JF, Cohen MJ. Active and passive cigarette smoking and acute lung injury after severe blunt trauma. Am J Respir Crit Care Med 183: 1660–1665, 2011. doi: 10.1164/rccm.201011-1802OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chan ED, Kinney WH, Honda JR, Bishwakarma R, Gangavelli A, Mya J, Bai X, Ordway DJ. Tobacco exposure and susceptibility to tuberculosis: is there a smoking gun? Tuberculosis (Edinb) 94: 544–550, 2014. doi: 10.1016/j.tube.2014.08.010. [DOI] [PubMed] [Google Scholar]
- 28.Chattopadhyay S, Fensterl V, Zhang Y, Veleeparambil M, Wetzel JL, Sen GC. Inhibition of viral pathogenesis and promotion of the septic shock response to bacterial infection by IRF-3 are regulated by the acetylation and phosphorylation of its coactivators. MBio 4: e00636-12, 2013. doi: 10.1128/mBio.00636-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chen HY, Li L, Fu ZJ. Histone deacetylase inhibitors trichostatin A and suberoylanilide hydroxamic acid attenuate ventilator-induced lung injury. Pharmazie 69: 55–59, 2014. doi: 10.1691/ph.2014.3716. [DOI] [PubMed] [Google Scholar]
- 30.Chun LF, Moazed F, Calfee CS, Matthay MA, Gotts JE. Pulmonary toxicity of e-cigarettes. Am J Physiol Lung Cell Mol Physiol 313: L193–L206, 2017. doi: 10.1152/ajplung.00071.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Demos-Davies KM, Ferguson BS, Cavasin MA, Mahaffey JH, Williams SM, Spiltoir JI, Schuetze KB, Horn TR, Chen B, Ferrara C, Scellini B, Piroddi N, Tesi C, Poggesi C, Jeong MY, McKinsey TA. HDAC6 contributes to pathological responses of heart and skeletal muscle to chronic angiotensin-II signaling. Am J Physiol Heart Circ Physiol 307: H252–H258, 2014. doi: 10.1152/ajpheart.00149.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Divo M. COPD, co-morbidities and health-related quality of life (HRQOL): more is less. COPD 10: 275–276, 2013. doi: 10.3109/15412555.2013.795409. [DOI] [PubMed] [Google Scholar]
- 33.Drannik AG, Pouladi MA, Robbins CS, Goncharova SI, Kianpour S, Stämpfli MR. Impact of cigarette smoke on clearance and inflammation after Pseudomonas aeruginosa infection. Am J Respir Crit Care Med 170: 1164–1171, 2004. doi: 10.1164/rccm.200311-1521OC. [DOI] [PubMed] [Google Scholar]
- 34.Duong-Quy S, Dao P, Hua-Huy T, Guilluy C, Pacaud P, Dinh-Xuan AT. Increased Rho-kinase expression and activity and pulmonary endothelial dysfunction in smokers with normal lung function. Eur Respir J 37: 349–355, 2011. doi: 10.1183/09031936.00056610. [DOI] [PubMed] [Google Scholar]
- 35.Fabbri LM, Beghé B, Agustí A. COPD and the solar system: introducing the chronic obstructive pulmonary disease comorbidome. Am J Respir Crit Care Med 186: 117–119, 2012. doi: 10.1164/rccm.201205-0906ED. [DOI] [PubMed] [Google Scholar]
- 36.Famous KR, Delucchi K, Ware LB, Kangelaris KN, Liu KD, Thompson BT, Calfee CS; ARDS Network . Acute respiratory distress syndrome subphenotypes respond differently to randomized fluid management strategy. Am J Respir Crit Care Med 195: 331–338, 2017. doi: 10.1164/rccm.201603-0645OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ferrer E, Peinado VI, Díez M, Carrasco JL, Musri MM, Martínez A, Rodríguez-Roisin R, Barberà JA. Effects of cigarette smoke on endothelial function of pulmonary arteries in the guinea pig. Respir Res 10: 76, 2009. doi: 10.1186/1465-9921-10-76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ferro TN, Goslar PW, Romanovsky AA, Petersen SR. Smoking in trauma patients: the effects on the incidence of sepsis, respiratory failure, organ failure, and mortality. J Trauma 69: 308–312, 2010. doi: 10.1097/TA.0b013e3181e1761e. [DOI] [PubMed] [Google Scholar]
- 39.Freixa X, Portillo K, Paré C, Garcia-Aymerich J, Gomez FP, Benet M, Roca J, Farrero E, Ferrer J, Fernandez-Palomeque C, Antó JM, Barberà JA; PAC-COPD Study Investigators . Echocardiographic abnormalities in patients with COPD at their first hospital admission. Eur Respir J 41: 784–791, 2013. doi: 10.1183/09031936.00222511. [DOI] [PubMed] [Google Scholar]
- 40.Fujita M, Kuwano K, Kunitake R, Hagimoto N, Miyazaki H, Kaneko Y, Kawasaki M, Maeyama T, Hara N. Endothelial cell apoptosis in lipopolysaccharide-induced lung injury in mice. Int Arch Allergy Immunol 117: 202–208, 1998. doi: 10.1159/000024011. [DOI] [PubMed] [Google Scholar]
- 41.Fukada M, Hanai A, Nakayama A, Suzuki T, Miyata N, Rodriguiz RM, Wetsel WC, Yao TP, Kawaguchi Y. Loss of deacetylation activity of Hdac6 affects emotional behavior in mice. PLoS One 7: e30924, 2012. doi: 10.1371/journal.pone.0030924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gajic O, Dabbagh O, Park PK, Adesanya A, Chang SY, Hou P, Anderson H III, Hoth JJ, Mikkelsen ME, Gentile NT, Gong MN, Talmor D, Bajwa E, Watkins TR, Festic E, Yilmaz M, Iscimen R, Kaufman DA, Esper AM, Sadikot R, Douglas I, Sevransky J, Malinchoc M; U.S. Critical Illness and Injury Trials Group: Lung Injury Prevention Study Investigators (USCIITG-LIPS) . Early identification of patients at risk of acute lung injury: evaluation of lung injury prediction score in a multicenter cohort study. Am J Respir Crit Care Med 183: 462–470, 2011. doi: 10.1164/rccm.201004-0549OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.García-Lucio J, Argemi G, Tura-Ceide O, Diez M, Paul T, Bonjoch C, Coll-Bonfill N, Blanco I, Barberà JA, Musri MM, Peinado VI. Gene expression profile of angiogenic factors in pulmonary arteries in COPD: relationship with vascular remodeling. Am J Physiol Lung Cell Mol Physiol 310: L583–L592, 2016. doi: 10.1152/ajplung.00261.2015. [DOI] [PubMed] [Google Scholar]
- 44.Gill SE, Rohan M, Mehta S. Role of pulmonary microvascular endothelial cell apoptosis in murine sepsis-induced lung injury in vivo. Respir Res 16: 109, 2015. doi: 10.1186/s12931-015-0266-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gill SE, Taneja R, Rohan M, Wang L, Mehta S. Pulmonary microvascular albumin leak is associated with endothelial cell death in murine sepsis-induced lung injury in vivo. PLoS One 9: e88501, 2014. doi: 10.1371/journal.pone.0088501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Govindarajan N, Rao P, Burkhardt S, Sananbenesi F, Schlüter OM, Bradke F, Lu J, Fischer A. Reducing HDAC6 ameliorates cognitive deficits in a mouse model for Alzheimer’s disease. EMBO Mol Med 5: 52–63, 2013. doi: 10.1002/emmm.201201923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Green LA, Petrusca D, Rajashekhar G, Gianaris T, Schweitzer KS, Wang L, Justice MJ, Petrache I, Clauss M. Cigarette smoke-induced CXCR3 receptor up-regulation mediates endothelial apoptosis. Am J Respir Cell Mol Biol 47: 807–814, 2012. doi: 10.1165/rcmb.2012-0132OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Guerassimov A, Hoshino Y, Takubo Y, Turcotte A, Yamamoto M, Ghezzo H, Triantafillopoulos A, Whittaker K, Hoidal JR, Cosio MG. The development of emphysema in cigarette smoke-exposed mice is strain dependent. Am J Respir Crit Care Med 170: 974–980, 2004. doi: 10.1164/rccm.200309-1270OC. [DOI] [PubMed] [Google Scholar]
- 49.Guo M, Wu MH, Granger HJ, Yuan SY. Focal adhesion kinase in neutrophil-induced microvascular hyperpermeability. Microcirculation 12: 223–232, 2005. doi: 10.1080/10739680590905251. [DOI] [PubMed] [Google Scholar]
- 50.Haggarty SJ, Koeller KM, Wong JC, Grozinger CM, Schreiber SL. Domain-selective small-molecule inhibitor of histone deacetylase 6 (HDAC6)-mediated tubulin deacetylation. Proc Natl Acad Sci USA 100: 4389–4394, 2003. doi: 10.1073/pnas.0430973100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Heijink IH, Pouwels SD, Leijendekker C, de Bruin HG, Zijlstra GJ, van der Vaart H, Ten Hacken NH, van Oosterhout AJ, Nawijn MC, van der Toorn M. Cigarette smoke induced damage-associated molecular pattern release from necrotic neutrophils triggers proinflammatory mediator release. Am J Respir Cell Mol Biol 52: 554–562, 2015. doi: 10.1165/rcmb.2013-0505OC. [DOI] [PubMed] [Google Scholar]
- 52.Hengstermann A, Müller T. Endoplasmic reticulum stress induced by aqueous extracts of cigarette smoke in 3T3 cells activates the unfolded-protein-response-dependent PERK pathway of cell survival. Free Radic Biol Med 44: 1097–1107, 2008. doi: 10.1016/j.freeradbiomed.2007.12.009. [DOI] [PubMed] [Google Scholar]
- 53.Heo J, Raines KW, Mocanu V, Campbell SL. Redox regulation of RhoA. Biochemistry 45: 14481–14489, 2006. doi: 10.1021/bi0610101. [DOI] [PubMed] [Google Scholar]
- 54.Hirsch J, Chalkley RJ, Bentley T, Burlingame AL, Frank JA. Double impact of cigarette smoke and mechanical ventilation on the alveolar epithelial type II cell. Crit Care 18: R50, 2014. doi: 10.1186/cc13795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Holden WE, Maier JM, Malinow MR. Cigarette smoke extract increases albumin flux across pulmonary endothelium in vitro. J Appl Physiol (1985) 66: 443–449, 1989. doi: 10.1152/jappl.1989.66.1.443. [DOI] [PubMed] [Google Scholar]
- 56.Holinstat M, Knezevic N, Broman M, Samarel AM, Malik AB, Mehta D. Suppression of RhoA activity by focal adhesion kinase-induced activation of p190RhoGAP: role in regulation of endothelial permeability. J Biol Chem 281: 2296–2305, 2006. doi: 10.1074/jbc.M511248200. [DOI] [PubMed] [Google Scholar]
- 57.Hsieh SJ, Zhuo H, Benowitz NL, Thompson BT, Liu KD, Matthay MA, Calfee CS; National Heart, Lung, and Blood Institute Acute Respiratory Distress Syndrome Network; National Heart Lung and Blood Institute Acute Respiratory Distress Syndrome Network . Prevalence and impact of active and passive cigarette smoking in acute respiratory distress syndrome. Crit Care Med 42: 2058–2068, 2014. doi: 10.1097/CCM.0000000000000418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hubbert C, Guardiola A, Shao R, Kawaguchi Y, Ito A, Nixon A, Yoshida M, Wang XF, Yao TP. HDAC6 is a microtubule-associated deacetylase. Nature 417: 455–458, 2002. doi: 10.1038/417455a. [DOI] [PubMed] [Google Scholar]
- 59.Imai K, Mercer BA, Schulman LL, Sonett JR, D’Armiento JM. Correlation of lung surface area to apoptosis and proliferation in human emphysema. Eur Respir J 25: 250–258, 2005. doi: 10.1183/09031936.05.00023704. [DOI] [PubMed] [Google Scholar]
- 60.Iribarren C, Jacobs DR Jr, Sidney S, Gross MD, Eisner MD. Cigarette smoking, alcohol consumption, and risk of ARDS: a 15-year cohort study in a managed care setting. Chest 117: 163–168, 2000. doi: 10.1378/chest.117.1.163. [DOI] [PubMed] [Google Scholar]
- 61.Jaimes EA, DeMaster EG, Tian RX, Raij L. Stable compounds of cigarette smoke induce endothelial superoxide anion production via NADPH oxidase activation. Arterioscler Thromb Vasc Biol 24: 1031–1036, 2004. doi: 10.1161/01.ATV.0000127083.88549.58. [DOI] [PubMed] [Google Scholar]
- 62.Jochems J, Boulden J, Lee BG, Blendy JA, Jarpe M, Mazitschek R, Van Duzer JH, Jones S, Berton O. Antidepressant-like properties of novel HDAC6-selective inhibitors with improved brain bioavailability. Neuropsychopharmacology 39: 389–400, 2014. doi: 10.1038/npp.2013.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Jones JG, Lawler P, Crawley JC, Minty BD, Hulands G, Veall N. Increased alveolar epithelial permeability in cigarette smokers. Lancet 315: 66–68, 1980. doi: 10.1016/S0140-6736(80)90493-6. [DOI] [PubMed] [Google Scholar]
- 64.Jorgensen E, Stinson A, Shan L, Yang J, Gietl D, Albino AP. Cigarette smoke induces endoplasmic reticulum stress and the unfolded protein response in normal and malignant human lung cells. BMC Cancer 8: 229, 2008. doi: 10.1186/1471-2407-8-229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kang MJ, Lee CG, Lee JY, Dela Cruz CS, Chen ZJ, Enelow R, Elias JA. Cigarette smoke selectively enhances viral PAMP- and virus-induced pulmonary innate immune and remodeling responses in mice. J Clin Invest 118: 2771–2784, 2008. doi: 10.1172/JCI32709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kasahara Y, Tuder RM, Cool CD, Lynch DA, Flores SC, Voelkel NF. Endothelial cell death and decreased expression of vascular endothelial growth factor and vascular endothelial growth factor receptor 2 in emphysema. Am J Respir Crit Care Med 163: 737–744, 2001. doi: 10.1164/ajrccm.163.3.2002117. [DOI] [PubMed] [Google Scholar]
- 67.Kawasaki M, Kuwano K, Hagimoto N, Matsuba T, Kunitake R, Tanaka T, Maeyama T, Hara N. Protection from lethal apoptosis in lipopolysaccharide-induced acute lung injury in mice by a caspase inhibitor. Am J Pathol 157: 597–603, 2000. doi: 10.1016/S0002-9440(10)64570-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kelsen SG, Duan X, Ji R, Perez O, Liu C, Merali S. Cigarette smoke induces an unfolded protein response in the human lung: a proteomic approach. Am J Respir Cell Mol Biol 38: 541–550, 2008. doi: 10.1165/rcmb.2007-0221OC. [DOI] [PubMed] [Google Scholar]
- 69.Kim BS, Serebreni L, Hamdan O, Wang L, Parniani A, Sussan T, Scott Stephens R, Boyer L, Damarla M, Hassoun PM, Damico R. Xanthine oxidoreductase is a critical mediator of cigarette smoke-induced endothelial cell DNA damage and apoptosis. Free Radic Biol Med 60: 336–346, 2013. doi: 10.1016/j.freeradbiomed.2013.01.023. [DOI] [PubMed] [Google Scholar]
- 70.Kispert SE, Marentette JO, McHowat J. Enhanced breast cancer cell adherence to the lung endothelium via PAF acetylhydrolase inhibition: a potential mechanism for enhanced metastasis in smokers. Am J Physiol Cell Physiol 307: C951–C956, 2014. doi: 10.1152/ajpcell.00218.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kitaguchi Y, Taraseviciene-Stewart L, Hanaoka M, Natarajan R, Kraskauskas D, Voelkel NF. Acrolein induces endoplasmic reticulum stress and causes airspace enlargement. PLoS One 7: e38038, 2012. doi: 10.1371/journal.pone.0038038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Knezevic N, Tauseef M, Thennes T, Mehta D. The G protein βγ subunit mediates reannealing of adherens junctions to reverse endothelial permeability increase by thrombin. J Exp Med 206: 2761–2777, 2009. doi: 10.1084/jem.20090652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kratzer A, Chu HW, Salys J, Moumen Z, Leberl M, Bowler R, Cool C, Zamora M, Taraseviciene-Stewart L. Endothelial cell adhesion molecule CD146: implications for its role in the pathogenesis of COPD. J Pathol 230: 388–398, 2013. doi: 10.1002/path.4197. [DOI] [PubMed] [Google Scholar]
- 74.Kubo S, Kobayashi M, Masunaga Y, Ishii H, Hirano Y, Takahashi K, Shimizu Y. Cytokine and chemokine expression in cigarette smoke-induced lung injury in guinea pigs. Eur Respir J 26: 993–1001, 2005. doi: 10.1183/09031936.05.00042405. [DOI] [PubMed] [Google Scholar]
- 75.Lam HC, Cloonan SM, Bhashyam AR, Haspel JA, Singh A, Sathirapongsasuti JF, Cervo M, Yao H, Chung AL, Mizumura K, An CH, Shan B, Franks JM, Haley KJ, Owen CA, Tesfaigzi Y, Washko GR, Quackenbush J, Silverman EK, Rahman I, Kim HP, Mahmood A, Biswal SS, Ryter SW, Choi AM. Histone deacetylase 6-mediated selective autophagy regulates COPD-associated cilia dysfunction. J Clin Invest 123: 5212–5230, 2013. doi: 10.1172/JCI69636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Lehr HA, Frei B, Arfors KE. Vitamin C prevents cigarette smoke-induced leukocyte aggregation and adhesion to endothelium in vivo. Proc Natl Acad Sci USA 91: 7688–7692, 1994. doi: 10.1073/pnas.91.16.7688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Lehr HA, Kress E, Menger MD, Friedl HP, Hübner C, Arfors KE, Messmer K. Cigarette smoke elicits leukocyte adhesion to endothelium in hamsters: inhibition by CuZn-SOD. Free Radic Biol Med 14: 573–581, 1993. doi: 10.1016/0891-5849(93)90138-K. [DOI] [PubMed] [Google Scholar]
- 78.Li XY, Rahman I, Donaldson K, MacNee W. Mechanisms of cigarette smoke induced increased airspace permeability. Thorax 51: 465–471, 1996. doi: 10.1136/thx.51.5.465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Liu H, Ding L, Zhang Y, Ni S. Circulating endothelial microparticles involved in lung function decline in a rat exposed in cigarette smoke maybe from apoptotic pulmonary capillary endothelial cells. J Thorac Dis 6: 649–655, 2014. doi: 10.3978/j.issn.2072-1439.2014.06.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Liu X, Liu Y, Huang X, Lin G, Xie C. Endothelial progenitor cell dysfunction in acute exacerbation of chronic obstructive pulmonary disease. Mol Med Rep 16: 5294–5302, 2017. doi: 10.3892/mmr.2017.7260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Low B, Liang M, Fu J. p38 mitogen-activated protein kinase mediates sidestream cigarette smoke-induced endothelial permeability. J Pharmacol Sci 104: 225–231, 2007. doi: 10.1254/jphs.FP0070385. [DOI] [PubMed] [Google Scholar]
- 82.Lu Q, Mundy M, Chambers E, Lange T, Newton J, Borgas D, Yao H, Choudhary G, Basak R, Oldham M, Rounds S. Alda-1 protects against acrolein-induced acute lung injury and endothelial barrier dysfunction. Am J Respir Cell Mol Biol 57: 662–673, 2017. doi: 10.1165/rcmb.2016-0342OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Lu Q, Rounds S. Focal adhesion kinase and endothelial cell apoptosis. Microvasc Res 83: 56–63, 2012. doi: 10.1016/j.mvr.2011.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Lu Q, Sakhatskyy P, Grinnell K, Newton J, Ortiz M, Wang Y, Sanchez-Esteban J, Harrington EO, Rounds S. Cigarette smoke causes lung vascular barrier dysfunction via oxidative stress-mediated inhibition of RhoA and focal adhesion kinase. Am J Physiol Lung Cell Mol Physiol 301: L847–L857, 2011. doi: 10.1152/ajplung.00178.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Lu Q, Sakhatskyy P, Newton J, Shamirian P, Hsiao V, Curren S, Gabino Miranda GA, Pedroza M, Blackburn MR, Rounds S. Sustained adenosine exposure causes lung endothelial apoptosis: a possible contributor to cigarette smoke-induced endothelial apoptosis and lung injury. Am J Physiol Lung Cell Mol Physiol 304: L361–L370, 2013. doi: 10.1152/ajplung.00161.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Lugade AA, Bogner PN, Thatcher TH, Sime PJ, Phipps RP, Thanavala Y. Cigarette smoke exposure exacerbates lung inflammation and compromises immunity to bacterial infection. J Immunol 192: 5226–5235, 2014. doi: 10.4049/jimmunol.1302584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Marwick JA, Stevenson CS, Giddings J, MacNee W, Butler K, Rahman I, Kirkham PA. Cigarette smoke disrupts VEGF165-VEGFR-2 receptor signaling complex in rat lungs and patients with COPD: morphological impact of VEGFR-2 inhibition. Am J Physiol Lung Cell Mol Physiol 290: L897–L908, 2006. doi: 10.1152/ajplung.00116.2005. [DOI] [PubMed] [Google Scholar]
- 89.Matsuyama A, Shimazu T, Sumida Y, Saito A, Yoshimatsu Y, Seigneurin-Berny D, Osada H, Komatsu Y, Nishino N, Khochbin S, Horinouchi S, Yoshida M. In vivo destabilization of dynamic microtubules by HDAC6-mediated deacetylation. EMBO J 21: 6820–6831, 2002. doi: 10.1093/emboj/cdf682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Milara J, Ortiz JL, Juan G, Guijarro R, Almudever P, Martorell M, Morcillo EJ, Cortijo J. Cigarette smoke exposure up-regulates endothelin receptor B in human pulmonary artery endothelial cells: molecular and functional consequences. Br J Pharmacol 161: 1599–1615, 2010. doi: 10.1111/j.1476-5381.2010.00979.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Mirzapoiazova T, Kolosova IA, Moreno L, Sammani S, Garcia JG, Verin AD. Suppression of endotoxin-induced inflammation by taxol. Eur Respir J 30: 429–435, 2007. doi: 10.1183/09031936.00154206. [DOI] [PubMed] [Google Scholar]
- 92.Mizumura K, Cloonan SM, Nakahira K, Bhashyam AR, Cervo M, Kitada T, Glass K, Owen CA, Mahmood A, Washko GR, Hashimoto S, Ryter SW, Choi AM. Mitophagy-dependent necroptosis contributes to the pathogenesis of COPD. J Clin Invest 124: 3987–4003, 2014. doi: 10.1172/JCI74985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Mizuno S, Yasuo M, Bogaard HJ, Kraskauskas D, Alhussaini A, Gomez-Arroyo J, Farkas D, Farkas L, Voelkel NF. Copper deficiency induced emphysema is associated with focal adhesion kinase inactivation. PLoS One 7: e30678, 2012. doi: 10.1371/journal.pone.0030678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Moazed F, Burnham EL, Vandivier RW, O’Kane CM, Shyamsundar M, Hamid U, Abbott J, Thickett DR, Matthay MA, McAuley DF, Calfee CS. Cigarette smokers have exaggerated alveolar barrier disruption in response to lipopolysaccharide inhalation. Thorax 71: 1130–1136, 2016. doi: 10.1136/thoraxjnl-2015-207886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Nana-Sinkam SP, Lee JD, Sotto-Santiago S, Stearman RS, Keith RL, Choudhury Q, Cool C, Parr J, Moore MD, Bull TM, Voelkel NF, Geraci MW. Prostacyclin prevents pulmonary endothelial cell apoptosis induced by cigarette smoke. Am J Respir Crit Care Med 175: 676–685, 2007. doi: 10.1164/rccm.200605-724OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Ni YF, Wang J, Yan XL, Tian F, Zhao JB, Wang YJ, Jiang T. Histone deacetylase inhibitor, butyrate, attenuates lipopolysaccharide-induced acute lung injury in mice. Respir Res 11: 33, 2010. doi: 10.1186/1465-9921-11-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Orosz Z, Csiszar A, Labinskyy N, Smith K, Kaminski PM, Ferdinandy P, Wolin MS, Rivera A, Ungvari Z. Cigarette smoke-induced proinflammatory alterations in the endothelial phenotype: role of NAD(P)H oxidase activation. Am J Physiol Heart Circ Physiol 292: H130–H139, 2007. doi: 10.1152/ajpheart.00599.2006. [DOI] [PubMed] [Google Scholar]
- 99.Overbeek SA, Braber S, Henricks PA, Kleinjan M, Kamp VM, Georgiou NA, Garssen J, Kraneveld AD, Folkerts G. Cigarette smoke induces β2-integrin-dependent neutrophil migration across human endothelium. Respir Res 12: 75, 2011. doi: 10.1186/1465-9921-12-75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Ozaki K, Hori T, Ishibashi T, Nishio M, Aizawa Y. Effects of chronic cigarette smoking on endothelial function in young men. J Cardiol 56: 307–313, 2010. doi: 10.1016/j.jjcc.2010.07.003. [DOI] [PubMed] [Google Scholar]
- 101.Panzer AR, Lynch SV, Langelier C, Christie JD, McCauley K, Nelson M, Cheung CK, Benowitz NL, Cohen MJ, Calfee CS. Lung microbiota is related to smoking status and to development of acute respiratory distress syndrome in critically ill trauma patients. Am J Respir Crit Care Med 197: 621–631, 2018. doi: 10.1164/rccm.201702-0441OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Petrache I, Birukova A, Ramirez SI, Garcia JG, Verin AD. The role of the microtubules in tumor necrosis factor-α-induced endothelial cell permeability. Am J Respir Cell Mol Biol 28: 574–581, 2003. doi: 10.1165/rcmb.2002-0075OC. [DOI] [PubMed] [Google Scholar]
- 103.Petrache I, Fijalkowska I, Medler TR, Skirball J, Cruz P, Zhen L, Petrache HI, Flotte TR, Tuder RM. α-1 antitrypsin inhibits caspase-3 activity, preventing lung endothelial cell apoptosis. Am J Pathol 169: 1155–1166, 2006. doi: 10.2353/ajpath.2006.060058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Petrache I, Natarajan V, Zhen L, Medler TR, Richter AT, Cho C, Hubbard WC, Berdyshev EV, Tuder RM. Ceramide upregulation causes pulmonary cell apoptosis and emphysema-like disease in mice. Nat Med 11: 491–498, 2005. doi: 10.1038/nm1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Petta V, Bakakos P, Tseliou E, Kostikas K, Simoes DC, Konstantellou E, Hillas G, Koulouris NG, Papiris S, Loukides S. Angiopoietins 1 and 2 in sputum supernatant of optimally treated asthmatics: the effect of smoking. Eur J Clin Invest 45: 56–62, 2015. doi: 10.1111/eci.12379. [DOI] [PubMed] [Google Scholar]
- 106.Phillips E, Wang TW, Husten CG, Corey CG, Apelberg BJ, Jamal A, Homa DM, King BA. Tobacco product use among adults: United States, 2015. MMWR Morb Mortal Wkly Rep 66: 1209–1215, 2017. doi: 10.15585/mmwr.mm6644a2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Pines H, Koutsky L, Buskin S. Cigarette smoking and mortality among HIV-infected individuals in Seattle, Washington (1996–2008). AIDS Behav 15: 243–251, 2011. doi: 10.1007/s10461-010-9682-3. [DOI] [PubMed] [Google Scholar]
- 108.Polverino F, Laucho-Contreras ME, Petersen H, Bijol V, Sholl LM, Choi ME, Divo M, Pinto-Plata V, Chetta A, Tesfaigzi Y, Celli BR, Owen CA. A pilot study linking endothelial injury in lungs and kidneys in chronic obstructive pulmonary disease. Am J Respir Crit Care Med 195: 1464–1476, 2017. doi: 10.1164/rccm.201609-1765OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Pouwels SD, Heijink IH, ten Hacken NH, Vandenabeele P, Krysko DV, Nawijn MC, van Oosterhout AJ. DAMPs activating innate and adaptive immune responses in COPD. Mucosal Immunol 7: 215–226, 2014. doi: 10.1038/mi.2013.77. [DOI] [PubMed] [Google Scholar]
- 110.Pouwels SD, Hesse L, Faiz A, Lubbers J, Bodha PK, Ten Hacken NH, van Oosterhout AJ, Nawijn MC, Heijink IH. Susceptibility for cigarette smoke-induced DAMP release and DAMP-induced inflammation in COPD. Am J Physiol Lung Cell Mol Physiol 311: L881–L892, 2016. doi: 10.1152/ajplung.00135.2016. [DOI] [PubMed] [Google Scholar]
- 111.Pouwels SD, Zijlstra GJ, van der Toorn M, Hesse L, Gras R, Ten Hacken NH, Krysko DV, Vandenabeele P, de Vries M, van Oosterhout AJ, Heijink IH, Nawijn MC. Cigarette smoke-induced necroptosis and DAMP release trigger neutrophilic airway inflammation in mice. Am J Physiol Lung Cell Mol Physiol 310: L377–L386, 2016. doi: 10.1152/ajplung.00174.2015. [DOI] [PubMed] [Google Scholar]
- 112.Quadri SK, Bhattacharya J. Resealing of endothelial junctions by focal adhesion kinase. Am J Physiol Lung Cell Mol Physiol 292: L334–L342, 2007. doi: 10.1152/ajplung.00228.2006. [DOI] [PubMed] [Google Scholar]
- 113.Reddy KP, Parker RA, Losina E, Baggett TP, Paltiel AD, Rigotti NA, Weinstein MC, Freedberg KA, Walensky RP. Impact of cigarette smoking and smoking cessation on life expectancy among people with HIV: a US-based modeling study. J Infect Dis 214: 1672–1681, 2016. doi: 10.1093/infdis/jiw430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Saito S, Lasky JA, Guo W, Nguyen H, Mai A, Danchuk S, Sullivan DE, Shan B. Pharmacological inhibition of HDAC6 attenuates endothelial barrier dysfunction induced by thrombin. Biochem Biophys Res Commun 408: 630–634, 2011. doi: 10.1016/j.bbrc.2011.04.075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Sakhatskyy P, Gabino Miranda GA, Newton J, Lee CG, Choudhary G, Vang A, Rounds S, Lu Q. Cigarette smoke-induced lung endothelial apoptosis and emphysema are associated with impairment of FAK and eIF2α. Microvasc Res 94: 80–89, 2014. doi: 10.1016/j.mvr.2014.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Sakhatskyy P, Wang Z, Borgas D, Lomas-Neira J, Chen Y, Ayala A, Rounds S, Lu Q. Double-hit mouse model of cigarette smoke priming for acute lung injury. Am J Physiol Lung Cell Mol Physiol 312: L56–L67, 2017. doi: 10.1152/ajplung.00436.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Santos S, Peinado VI, Ramírez J, Melgosa T, Roca J, Rodriguez-Roisin R, Barberà JA. Characterization of pulmonary vascular remodelling in smokers and patients with mild COPD. Eur Respir J 19: 632–638, 2002. doi: 10.1183/09031936.02.00245902. [DOI] [PubMed] [Google Scholar]
- 118.Schröder M, Kaufman RJ. The mammalian unfolded protein response. Annu Rev Biochem 74: 739–789, 2005. doi: 10.1146/annurev.biochem.73.011303.074134. [DOI] [PubMed] [Google Scholar]
- 119.Schweitzer KS, Chen SX, Law S, Van Demark M, Poirier C, Justice MJ, Hubbard WC, Kim ES, Lai X, Wang M, Kranz WD, Carroll CJ, Ray BD, Bittman R, Goodpaster J, Petrache I. Endothelial disruptive proinflammatory effects of nicotine and e-cigarette vapor exposures. Am J Physiol Lung Cell Mol Physiol 309: L175–L187, 2015. doi: 10.1152/ajplung.00411.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Schweitzer KS, Hatoum H, Brown MB, Gupta M, Justice MJ, Beteck B, Van Demark M, Gu Y, Presson RG Jr, Hubbard WC, Petrache I. Mechanisms of lung endothelial barrier disruption induced by cigarette smoke: role of oxidative stress and ceramides. Am J Physiol Lung Cell Mol Physiol 301: L836–L846, 2011. doi: 10.1152/ajplung.00385.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Serban KA, Rezania S, Petrusca DN, Poirier C, Cao D, Justice MJ, Patel M, Tsvetkova I, Kamocki K, Mikosz A, Schweitzer KS, Jacobson S, Cardoso A, Carlesso N, Hubbard WC, Kechris K, Dragnea B, Berdyshev EV, McClintock J, Petrache I. Structural and functional characterization of endothelial microparticles released by cigarette smoke. Sci Rep 6: 31596, 2016. doi: 10.1038/srep31596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Shankar-Hari M, McAuley DF. Acute respiratory distress syndrome phenotypes and identifying treatable traits. The dawn of personalized medicine for ARDS. Am J Respir Crit Care Med 195: 280–281, 2017. doi: 10.1164/rccm.201608-1729ED. [DOI] [PubMed] [Google Scholar]
- 123.Sharma J, Young DM, Marentette JO, Rastogi P, Turk J, McHowat J. Lung endothelial cell platelet-activating factor production and inflammatory cell adherence are increased in response to cigarette smoke component exposure. Am J Physiol Lung Cell Mol Physiol 302: L47–L55, 2012. doi: 10.1152/ajplung.00179.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Shen Y, Rattan V, Sultana C, Kalra VK. Cigarette smoke condensate-induced adhesion molecule expression and transendothelial migration of monocytes. Am J Physiol Heart Circ Physiol 270: H1624–H1633, 1996. doi: 10.1152/ajpheart.1996.270.5.H1624. [DOI] [PubMed] [Google Scholar]
- 125.Sikka G, Pandey D, Bhuniya AK, Steppan J, Armstrong D, Santhanam L, Nyhan D, Berkowitz DE. Contribution of arginase activation to vascular dysfunction in cigarette smoking. Atherosclerosis 231: 91–94, 2013. doi: 10.1016/j.atherosclerosis.2013.08.026. [DOI] [PubMed] [Google Scholar]
- 126.Siu ER, Wong EW, Mruk DD, Porto CS, Cheng CY. Focal adhesion kinase is a blood-testis barrier regulator. Proc Natl Acad Sci USA 106: 9298–9303, 2009. doi: 10.1073/pnas.0813113106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Stämpfli MR, Anderson GP. How cigarette smoke skews immune responses to promote infection, lung disease and cancer. Nat Rev Immunol 9: 377–384, 2009. doi: 10.1038/nri2530. [DOI] [PubMed] [Google Scholar]
- 128.Strulovici-Barel Y, Staudt MR, Krause A, Gordon C, Tilley AE, Harvey BG, Kaner RJ, Hollmann C, Mezey JG, Bitter H, Pillai SG, Hilton H, Wolff G, Stevenson CS, Visvanathan S, Fine JS, Crystal RG. Persistence of circulating endothelial microparticles in COPD despite smoking cessation. Thorax 71: 1137–1144, 2016. doi: 10.1136/thoraxjnl-2015-208274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Su Y, Han W, Giraldo C, De Li Y, Block ER. Effect of cigarette smoke extract on nitric oxide synthase in pulmonary artery endothelial cells. Am J Respir Cell Mol Biol 19: 819–825, 1998. doi: 10.1165/ajrcmb.19.5.3091. [DOI] [PubMed] [Google Scholar]
- 130.Sun S, Sursal T, Adibnia Y, Zhao C, Zheng Y, Li H, Otterbein LE, Hauser CJ, Itagaki K. Mitochondrial DAMPs increase endothelial permeability through neutrophil dependent and independent pathways. PLoS One 8: e59989, 2013. doi: 10.1371/journal.pone.0059989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Takahashi T, Kobayashi S, Fujino N, Suzuki T, Ota C, He M, Yamada M, Suzuki S, Yanai M, Kurosawa S, Yamaya M, Kubo H. Increased circulating endothelial microparticles in COPD patients: a potential biomarker for COPD exacerbation susceptibility. Thorax 67: 1067–1074, 2012. doi: 10.1136/thoraxjnl-2011-201395. [DOI] [PubMed] [Google Scholar]
- 132.Tandon S, Batchelor A, Bullock R, Gascoigne A, Griffin M, Hayes N, Hing J, Shaw I, Warnell I, Baudouin SV. Peri-operative risk factors for acute lung injury after elective oesophagectomy. Br J Anaesth 86: 633–638, 2001. doi: 10.1093/bja/86.5.633. [DOI] [PubMed] [Google Scholar]
- 133.Taylor AE, Gaar KA Jr. Estimation of equivalent pore radii of pulmonary capillary and alveolar membranes. Am J Physiol 218: 1133–1140, 1970. doi: 10.1152/ajplegacy.1970.218.4.1133. [DOI] [PubMed] [Google Scholar]
- 134.Thomashow MA, Shimbo D, Parikh MA, Hoffman EA, Vogel-Claussen J, Hueper K, Fu J, Liu CY, Bluemke DA, Ventetuolo CE, Doyle MF, Barr RG. Endothelial microparticles in mild chronic obstructive pulmonary disease and emphysema. The Multi-Ethnic Study of Atherosclerosis Chronic Obstructive Pulmonary Disease study. Am J Respir Crit Care Med 188: 60–68, 2013. doi: 10.1164/rccm.201209-1697OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Tuder RM, Wood K, Taraseviciene L, Flores SC, Voekel NF. Cigarette smoke extract decreases the expression of vascular endothelial growth factor by cultured cells and triggers apoptosis of pulmonary endothelial cells. Chest 117, Suppl 1: 241S–242S, 2000. doi: 10.1378/chest.117.5_suppl_1.241S. [DOI] [PubMed] [Google Scholar]
- 136.Tzima E. Role of small GTPases in endothelial cytoskeletal dynamics and the shear stress response. Circ Res 98: 176–185, 2006. doi: 10.1161/01.RES.0000200162.94463.d7. [DOI] [PubMed] [Google Scholar]
- 136a.United States. Department of Health and Human Services The Health Consequences of Smoking: 50 Years of Progress. A Report of the Surgeon General. Atlanta, GA: U.S. Dept. of Health and Human Services, Centers for Disease Control and Prevention, National Center for Chronic Disease Prevention and Health Promotion, Office on Smoking and Health, 2014. [Google Scholar]
- 137.Usatyuk PV, Parinandi NL, Natarajan V. Redox regulation of 4-hydroxy-2-nonenal-mediated endothelial barrier dysfunction by focal adhesion, adherens, and tight junction proteins. J Biol Chem 281: 35554–35566, 2006. doi: 10.1074/jbc.M607305200. [DOI] [PubMed] [Google Scholar]
- 138.Vandenbroucke E, Mehta D, Minshall R, Malik AB. Regulation of endothelial junctional permeability. Ann N Y Acad Sci 1123: 134–145, 2008. doi: 10.1196/annals.1420.016. [DOI] [PubMed] [Google Scholar]
- 139.Verin AD, Birukova A, Wang P, Liu F, Becker P, Birukov K, Garcia JG. Microtubule disassembly increases endothelial cell barrier dysfunction: role of MLC phosphorylation. Am J Physiol Lung Cell Mol Physiol 281: L565–L574, 2001. doi: 10.1152/ajplung.2001.281.3.L565. [DOI] [PubMed] [Google Scholar]
- 140.Voelkel NF. Cigarette smoke is an endothelial cell toxin. Am J Respir Crit Care Med 197: 274, 2018. doi: 10.1164/rccm.201706-1123LE. [DOI] [PubMed] [Google Scholar]
- 141.Wacharasint P, Fuengfoo P, Sukcharoen N, Rangsin R; THAI-SICU Trial Group . Smoking increased risk of ARDS in surgical critically ill patients: results from the multicenter THAI-SICU study (Abstract). Crit Care 19, Suppl 1: 238, 2015. doi: 10.1186/cc14318. 26036415 [DOI] [Google Scholar]
- 142.Wagner K, Gröger M, McCook O, Scheuerle A, Asfar P, Stahl B, Huber-Lang M, Ignatius A, Jung B, Duechs M, Möller P, Georgieff M, Calzia E, Radermacher P, Wagner F. Blunt chest trauma in mice after cigarette smoke-exposure: effects of mechanical ventilation with 100% O2. PLoS One 10: e0132810, 2015. doi: 10.1371/journal.pone.0132810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Wagner L, Laczy B, Tamaskó M, Mazák I, Markó L, Molnár GA, Wagner Z, Mohás M, Cseh J, Fekete A, Wittmann I. Cigarette smoke-induced alterations in endothelial nitric oxide synthase phosphorylation: role of protein kinase C. Endothelium 14: 245–255, 2007. doi: 10.1080/10623320701606707. [DOI] [PubMed] [Google Scholar]
- 144.Wang B, Rao YH, Inoue M, Hao R, Lai CH, Chen D, McDonald SL, Choi MC, Wang Q, Shinohara ML, Yao TP. Microtubule acetylation amplifies p38 kinase signalling and anti-inflammatory IL-10 production. Nat Commun 5: 3479, 2014. doi: 10.1038/ncomms4479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Ward C, Thien F, Secombe J, Gollant S, Walters EH. Bronchoalveolar lavage fluid urea as a measure of pulmonary permeability in healthy smokers. Eur Respir J 15: 285–290, 2000. doi: 10.1034/j.1399-3003.2000.15b11.x. [DOI] [PubMed] [Google Scholar]
- 146.Ware LB, Lee JW, Wickersham N, Nguyen J, Matthay MA, Calfee CS; California Transplant Donor Network . Donor smoking is associated with pulmonary edema, inflammation and epithelial dysfunction in ex vivo human donor lungs. Am J Transplant 14: 2295–2302, 2014. doi: 10.1111/ajt.12853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Ware LB, Zhao Z, Koyama T, May AK, Matthay MA, Lurmann FW, Balmes JR, Calfee CS. Long-term ozone exposure increases the risk of developing the acute respiratory distress syndrome. Am J Respir Crit Care Med 193: 1143–1150, 2016. doi: 10.1164/rccm.201507-1418OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Wickenden JA, Clarke MC, Rossi AG, Rahman I, Faux SP, Donaldson K, MacNee W. Cigarette smoke prevents apoptosis through inhibition of caspase activation and induces necrosis. Am J Respir Cell Mol Biol 29: 562–570, 2003. doi: 10.1165/rcmb.2002-0235OC. [DOI] [PubMed] [Google Scholar]
- 149.Winkler R, Benz V, Clemenz M, Bloch M, Foryst-Ludwig A, Wardat S, Witte N, Trappiel M, Namsolleck P, Mai K, Spranger J, Matthias G, Roloff T, Truee O, Kappert K, Schupp M, Matthias P, Kintscher U. Histone deacetylase 6 (HDAC6) is an essential modifier of glucocorticoid-induced hepatic gluconeogenesis. Diabetes 61: 513–523, 2012. doi: 10.2337/db11-0313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Wong CM, Yang L, Chan KP, Chan WM, Song L, Lai HK, Thach TQ, Ho LM, Chan KH, Lam TH, Peiris JS. Cigarette smoking as a risk factor for influenza-associated mortality: evidence from an elderly cohort. Influenza Other Respi Viruses 7: 531–539, 2013. doi: 10.1111/j.1750-2659.2012.00411.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150a.World Health Organization Tobacco Fact Sheet [Online]. Geneva: WHO; http://www.who.int/mediacentre/factsheets/fs339/en/ [ March 2018]. [Google Scholar]
- 151.Wright JL, Tai H, Churg A. Vasoactive mediators and pulmonary hypertension after cigarette smoke exposure in the guinea pig. J Appl Physiol (1985) 100: 672–678, 2006. doi: 10.1152/japplphysiol.00274.2005. [DOI] [PubMed] [Google Scholar]
- 152.Wright JL, Zhou S, Churg A. Pulmonary hypertension and vascular oxidative damage in cigarette smoke exposed eNOS−/− mice and human smokers. Inhal Toxicol 24: 732–740, 2012. doi: 10.3109/08958378.2012.715698. [DOI] [PubMed] [Google Scholar]
- 153.Wu MH. Endothelial focal adhesions and barrier function. J Physiol 569: 359–366, 2005. doi: 10.1113/jphysiol.2005.096537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Xiong Y, Zhao K, Wu J, Xu Z, Jin S, Zhang YQ. HDAC6 mutations rescue human tau-induced microtubule defects in Drosophila. Proc Natl Acad Sci USA 110: 4604–4609, 2013. doi: 10.1073/pnas.1207586110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Yao H, Rahman I. Role of histone deacetylase 2 in epigenetics and cellular senescence: implications in lung inflammaging and COPD. Am J Physiol Lung Cell Mol Physiol 303: L557–L566, 2012. doi: 10.1152/ajplung.00175.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Yildiz L, Akçay F, Kaynar H, Bakan N. Increased plasma endothelin-1 in heavy and light smokers. Clin Chem 42: 483–484, 1996. [PubMed] [Google Scholar]
- 157.Zhang L, Jin S, Wang C, Jiang R, Wan J. Histone deacetylase inhibitors attenuate acute lung injury during cecal ligation and puncture-induced polymicrobial sepsis. World J Surg 34: 1676–1683, 2010. doi: 10.1007/s00268-010-0493-5. [DOI] [PubMed] [Google Scholar]
- 158.Zhang Y, Kwon S, Yamaguchi T, Cubizolles F, Rousseaux S, Kneissel M, Cao C, Li N, Cheng HL, Chua K, Lombard D, Mizeracki A, Matthias G, Alt FW, Khochbin S, Matthias P. Mice lacking histone deacetylase 6 have hyperacetylated tubulin but are viable and develop normally. Mol Cell Biol 28: 1688–1701, 2008. doi: 10.1128/MCB.01154-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Zhang Y, Li N, Caron C, Matthias G, Hess D, Khochbin S, Matthias P. HDAC-6 interacts with and deacetylates tubulin and microtubules in vivo. EMBO J 22: 1168–1179, 2003. doi: 10.1093/emboj/cdg115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Zhao X, Peng X, Sun S, Park AY, Guan JL. Role of kinase-independent and -dependent functions of FAK in endothelial cell survival and barrier function during embryonic development. J Cell Biol 189: 955–965, 2010. doi: 10.1083/jcb.200912094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Zhou Y, Mohsenin A, Morschl E, Young HW, Molina JG, Ma W, Sun CX, Martinez-Valdez H, Blackburn MR. Enhanced airway inflammation and remodeling in adenosine deaminase-deficient mice lacking the A2B adenosine receptor. J Immunol 182: 8037–8046, 2009. doi: 10.4049/jimmunol.0900515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Zhou Y, Murthy JN, Zeng D, Belardinelli L, Blackburn MR. Alterations in adenosine metabolism and signaling in patients with chronic obstructive pulmonary disease and idiopathic pulmonary fibrosis. PLoS One 5: e9224, 2010. doi: 10.1371/journal.pone.0009224. [DOI] [PMC free article] [PubMed] [Google Scholar]