

Abstract
Hypoxia-induced pulmonary vasoconstriction (HPV) is attributed to an increase in intracellular Ca2+ concentration ([Ca2+]i) in pulmonary artery smooth muscle cells (PASMCs). We have reported that phospholipase C-γ1 (PLCγ1) plays a significant role in the hypoxia-induced increase in [Ca2+]i in PASMCs and attendant HPV. In this study, we intended to determine molecular mechanisms for hypoxic Ca2+ and contractile responses in PASMCs. Our data reveal that hypoxic vasoconstriction occurs in pulmonary arteries, but not in mesenteric arteries. Hypoxia caused a large increase in [Ca2+]i in PASMCs, which is diminished by the PLC inhibitor U73122 and not by its inactive analog U73433. Hypoxia augments PLCγ1-dependent inositol 1,4,5-trisphosphate (IP3) generation. Exogenous ROS, hydrogen peroxide (H2O2), increases PLCγ1 phosphorylation at tyrosine-783 and IP3 production. IP3 receptor-1 (IP3R1) knock-down remarkably diminishes hypoxia- or H2O2-induced increase in [Ca2+]i. Hypoxia or H2O2 increases the activity of IP3Rs, which is significantly reduced in protein kinase C-ε (PKCε) knockout PASMCs. A higher PLCγ1 expression, activity, and basal [Ca2+]i are found in PASMCs, but not in mesenteric artery smooth muscle cells from mice exposed to chronic hypoxia (CH) for 21 days. CH enhances H2O2- and ATP-induced increase in [Ca2+]i in PASMCs and PLC-dependent, norepinephrine-evoked pulmonary vasoconstriction. In conclusion, acute hypoxia uniquely causes ROS-dependent PLCγ1 activation, IP3 production, PKCε activation, IP3R1 opening, Ca2+ release, and contraction in mouse PASMCs; CH enhances PASM PLCγ1 expression, activity, and function, playing an essential role in pulmonary hypertension in mice.
Keywords: calcium; hypoxia; hypoxia-induced pulmonary vasoconstriction; inositol 1,4,5-trisphosphate receptor-1; phospholipase C-γ1; protein kinase C-ε; reactive oxygen species
INTRODUCTION
Hypoxia-induced pulmonary vasoconstriction (HPV) increases pulmonary vascular resistance to direct blood to well-ventilated alveoli of the lungs to match ventilation perfusion, with the ultimate aim of proper oxygen delivery to the systemic circulation. Importantly, persistent HPV commonly results in fatal pulmonary hypertension and right ventricular heart failure (7, 10, 34, 38, 54).
HPV occurs mainly because of an increase in [Ca2+]i in pulmonary artery smooth muscle cells (PASMCs). The hypoxia-induced initial increase in [Ca2+]i is due to Ca2+ release from the sarcoplasmic reticulum (SR), and the subsequent sustained Ca2+ response results from Ca2+ influx from the extracellular source (8, 13, 15, 34, 58). Intracellular Ca2+ release channels, ryanodine receptors (RyRs), are activated by Ca2+, whereas inositol 1,4,5-trisphosphate receptors (IP3Rs) are activated upon IP3 binding, produced by the activity of phospholipase C (PLC). RyRs are involved in the hypoxia-induced increase in [Ca2+]i in PASMCs (3, 13, 26, 63–65). In addition, hypoxia can increase IP3 generation in pulmonary artery fibroblasts (60), which then release Ca2+ from the SR via IP3Rs. Recently, we have found that PLCγ1 is activated, playing an important role in the hypoxia-induced increase in [Ca2+]i in PASMCs and HPV (61). Importantly, Ca2+ released from IP3Rs activates RyRs in PASMCs (62, 65), indicating a cross talk between these two Ca2+ release channels.
Reactive oxygen species (ROS) are important second messengers, which mediate hypoxia-induced increase in [Ca2+]i in PASMCs (1, 2, 9, 23, 30, 36, 37, 41, 42, 43, 52, 55, 56) and are determinant of the heterogeneity of hypoxic responses in pulmonary artery (PA) and systemic artery (SA) smooth muscle cells (SMCs) (41, 52, 54). Mitochondria and NADPH oxidase (NOX) are the two major sources that generate ROS in PASMCs. Interestingly, hypoxia-induced mitochondrial ROS induce NOX-mediated ROS generation in PASMCs, ROS-induced ROS generation (RIRG) (41, 42). The hypoxia-induced mitochondrial ROS generation is primarily mediated by the mitochondrial electron train chain molecules before the complex III ubisemiquinone site, in which Rieske iron-sulfur protein (RISP) in the complex III is an indispensable element (25, 52). Hypoxia-induced ROS can activate protein kinase C-ε (PKCε) and, thus, increase [Ca2+]i in PASMCs. Activation of PKCε induces NOX-mediated ROS generation in PASMCs (41, 42, 54). Importantly, hypoxia-induced activation of PLCγ1 is downstream to the complex III-mediated, RISP-dependent intracellular ROS generation in PASMCs (61). Thus, PLCγ1 and PKCε mediate ROS-dependent hypoxia-induced increase in [Ca2+]i in PASMCs.
Various external stimuli cause an increase in [Ca2+]i in a variety of cell types by producing IP3 through activation of PLC. IP3 then binds to IP3Rs and causes the opening of the channel and release of Ca2+ from the SR (4, 5). Application of hypoxia-induced mitogenic factor results in a U73122-sensitive increase in [Ca2+]i in cultured human PASMCs and IP3 generation in cultured human PASMCs, as well as in pulmonary artery fibroblasts (14, 60). Importantly, hypoxia causes an increase in IP3 generation that is fully abolished by pretreatment with the specific PLC inhibitor U73122, and IP3R inhibition diminishes the hypoxic increase in [Ca2+]i in PASMCs (61).
IP3Rs are classified into three subtypes (IP3R1, IP3R2, and IP3R3), encoded by the three separate genes. The functional unit of IP3Rs is either present as homotetramers or heterotetramers. The three subtypes of IP3Rs share 60–80% similarity in sequence (4, 5, 32, 39, 46, 47). Almost all tissues express IP3R1, IP3R2, and IP3R3 to various extents; however, IP3R1 predominates in adult vascular SMCs (47). In support, IP3R1 mRNA is highly expressed in all arteries. It is also noted that IP3R1 is expressed in vascular SMCs and endothelial cells, while IP3R2 and IP3R3 are present in endothelial cells; furthermore, weak expression of IP3R2 is present in basilar artery SMCs (17). All three subtypes of IP3Rs were expressed in primary cultured rat PASMCs, but their relative contributions to intracellular Ca2+ release are unknown (62). Thus, IP3R1 may play a significant role in hypoxia-induced increase in [Ca2+]i in PASMCs.
The opening of IP3Rs requires the binding of IP3; generally, each subunit of IP3Rs binds to one IP3 molecule. The binding of IP3 to IP3Rs is regulated by Ca2+, phosphorylation of IP3Rs by various kinases, ATP, and various accessory proteins (4, 5, 32, 46, 47). Phosphorylation by protein kinase A (PKA), cyclic GMP dependent protein kinase G (PKG), PKC, and Ca2+ calmodulin-dependent protein kinase II (CaMKII) alters the channel functions in various tissues. Phosphorylation of IP3Rs by PKA is found to enhance IP3-induced Ca2+ release in hepatocytes, while inhibiting IP3R functions in cerebellar cells. Phosphorylation of IP3Rs by PKG, PKC, and CaMKII enhances the channel function by facilitating IP3 binding to IP3Rs (39). Importantly, PKC can phosphorylate IP3Rs and enhance the binding of IP3 to IP3Rs, contributing to increased [Ca2+]i. The phosphorylation of IP3R1 is eight times more than IP3R3 (49). Thus, hypoxia-induced activation of PKCε may regulate IP3R function and, thereby, increase hypoxic responses in PASMCs.
Thus, in the current study, we sought to determine the molecular mechanisms for hypoxia-induced, PLCγ1-dependent increase in [Ca2+]i in PASMCs. Further, we also investigated the possible involvement of PLCγ1 in chronic hypoxia (CH)-induced vascular remodeling, which plays an important role in the development of pulmonary hypertension.
MATERIALS AND METHODS
Reagents.
Fura-2 AM was purchased from Molecular Probes (Eugene, OR); phospho-PLCγ1 (Tyr-783) antibody was obtained from Cell Signaling Technologies; PLCγ1 antibody, smooth muscle actin antibody, and GAPDH were obtained from Santa Cruz Biotechnology (Santa Cruz, CA). The specificity and effectiveness of the antibodies against PLCγ1 and phospho-PLCγ1 have been validated in our previous studies (61). U73122, U73433, and H2O2 were obtained from Sigma Aldrich (St. Louis, MO); IP-One HTRF assay kit from Cisbio Bioassays (Bedford, MA). A set of mouse pLKO.1 lentiviral PLCγ1 shRNAs (TRC-Mm1.0; cat. no.: RMM4534), IP3R1 shRNA (RMM4431-200348227), and lentiviral scrambled (nonsilencing, NS) shRNAs (cat. no. RHS4346) from Open Biosystems (Lafayette, CO). IP3R1 (sc6093), IP3R2 (sc7278), and IP3R3 (sc7277) antibodies were obtained from Santa Cruz Biotechnology).
Animals.
C57/BALBc and PKCɛ−/− mice were purchased from the Jackson Laboratory (Bar Harbor, ME); Swiss-Webster mice were purchased from Taconic (Germantown, NY). All animal experiments were approved by the Institutional Animal Care and Use Committee of Albany Medical College.
Preparation of isolated PAs and PASMCs.
PAs and PASMCs were prepared from C57BalbC and Swiss-Webster, as we described previously (25–27, 41, 42, 52, 61, 64, 65). Briefly, the heart and lungs were carefully removed from mice and placed in ice-cold physiological saline solution (PSS) containing (in mM): 130 NaCl, 1 MgSO4, 1.8 CaCl2, 5.4 KCl, 10 HEPES [4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid], and 10 glucose (pH 7.4). Resistance pulmonary and mesenteric arteries were then carefully dissected. To obtain PASMCs, PAs were enzymatically digested using the two-step digestion method: first in 2-ml PSS containing (in mg): 2.4 papain (Sigma; P4762) 0.4 dithioerythritol (Sigma; D8255) and 2 BSA (Sigma; A7888) for 20 min at 37°C (prewarmed for 20 min at 37°C), and then in 1.5 ml PSS (prewarmed at 37°C for 7–8 min) containing (in mg): 1.5 collagenase II (Sigma; C6885), 1.2 collagenase F (Sigma; 7926) 1.2 dithiothreitol (Sigma; D9779), and 1.5 BSA for 15 min at 37°C. The digested arteries were then incubated in ice-cold PSS containing 1% BSA on ice for 15 min with washing every 5 min. Single cells were harvested by gentle triturating using Pasteur pipettes. To examine the effects of pharmacological reagents, control experiments were carried out in the same mouse cells.
Hypoxia.
In in vitro experiments, acute hypoxia-induced responses were obtained by switching perfusion of PAs or PASMCs from a normoxic to hypoxia-inducing Krebs PSS (25–27, 41, 42, 52, 61, 64, 65). To test the effect of hypoxia on the activity of PLC, vascular smooth muscle tissues and cells were first incubated with a normoxic Krebs PSS (that was continuously aerated with a normoxic gas) for 10 min and then with a hypoxia-inducing Krebs PSS (continuously bubbled with a hypoxia-inducing gas) for 5 min. In control experiments, vascular smooth muscle tissues or cells were incubated with the normoxic Krebs PSS for 10 min, followed by continuous incubation with the normoxic Krebs PSS for 5 min. The composition of Krebs PSS was (in mM): 110 NaCl, 3.4 KCl, 2.4 CaCl2, 0.8 MgSO4, 25.8 NaHCO3, 1.2 KH2PO4, and 5.6 glucose (pH 7.4). The normoxic PSS was made by bubbling 20% O2, 5% CO2, and 75% N2, while hypoxia-inducing PSS was made by switching the gas to 1% O2, 5% CO2, and 94% N2.
In the determination of the effect of hypoxia on [Ca2+]i in PASMCs, single cells were first perfused with the normoxic PSS for 10 min and then with the hypoxia-inducing PSS for 5 min. As a control, cells were treated identically but only perfused with normoxic PSS.
To scrutinize the effect of CH in animals, mice were placed for 21 days in a BioSpherix chamber that was continuously flushed with hypoxic air (10% O2) generated from a mixture of room air and N2 by a BioSpherix ProOx oxygen controller (24, 66). As control mice were exposed to normoxia (21% O2) for 21 days. PAs and PASMCs were prepared from mice exposed to hypoxia or normoxia and used for various experiments.
Total PLC and PLCγ1 activity assay.
Total PLC activity was determined using a [3H]-phosphatidylinositol bisphosphate (PIP2) assay (51). Twenty microliters of total lysates from PAs were mixed and incubated with 20 µl of PLC activity assay buffer (containing in mM): 100 HEPES, 6 EGTA, 160 KCl, and 1 CaCl2 (pH 7.2) with [3H]-PIP2 (10,000 cpm counts/reaction) for 30 min at 37°C. The reaction was stopped using 200 µl 10% TCA and 100 µl of 1% BSA. The reaction content was vortexed and centrifuged at 8,600 g for 5 min. Three hundred microliters of the supernatant was mixed with 5 ml of Formula-989 liquid scintillation cocktail (PerkinElmer) and then subjected to scintillation counting to reading [3H]-IP3 production from [3H]-PIP2, which reflects PLC activity.
PLCγ1 activity was assessed by examining phosphorylation of PLCγ1 at tyrosine-783 using Western blot analysis, as described previously (51), as well as in our previous publications (25, 42, 64). Fifteen-microgram proteins of cell lysates were loaded onto 8% SDS-PAGE and subsequently transferred to PVDF membranes. The membranes were incubated with primary antibodies for PLCγ1 and PLCγ1 phosphorylated on tyrosine-783 at a 1:250 dilution, and then secondary horseradish peroxidase (HRP)-conjugated antibodies at a 1:1,500 dilution. Blots were developed using an enhanced chemiluminescence detection kit (Santa Cruz Biotechnology). The intensity of the bands was quantified using multigauge software (Fujifilm).
Western blot analysis.
Western blot analysis was performed as described above (25, 42, 51, 64). For PLCγ1, 15 µg of sample proteins were loaded on 8% SDS-PAGE. Primary antibodies against PLCγ1 at a 1:250 dilution and secondary HRP-conjugated antibodies at a 1:1,500 dilution were used, respectively. For IP3R1, 200 μg of sample proteins were loaded on 8% SDS-PAGE. Primary antibodies against IP3R1 at a 1:200 dilution and secondary HRP-conjugated antibodies at a 1:1,000 dilution were used, respectively. Blots were developed and quantified.
Measurement of [Ca2+]i.
Measurement of [Ca2+]i was made using a Till fluorescence imaging system (Till Photonics, Gräfelfing, Germany) (25–27, 41, 42, 52, 64, 65). PASMCs were incubated with the fluorescent dye fura-2 AM (10 µM) for 30 min. Fura-2 was excited at 340 and 380 nm, respectively. The emitted fluorescence was detected at 510 nm. [Ca2+]i was calculated using the equation: [Ca2+]i = β·Kd·(R − Rmin)/(Rmax − R), in which Rmin represents 340/380 fluorescence ratio obtained in cells exposed to 0 Ca2+ with ionomycin (10 µM) and EGTA (70 mM), Rmax 340/380 fluorescence ratio obtained in cells exposed to high Ca2+ (10 mM) with ionomycin (10 µM), R experimental values of 340/380 ratio, β R380max/R380min value, and Kd (a dissociation constant of fura-2) 386 nM.
Muscle tension measurement.
Muscle tension measurement was performed using a PowerLab data acquisition system (AD Instruments), as described earlier (25, 26, 64, 65). Endothelium-denuded resistant PA rings were placed in organ bath chambers that contained PSS aerated with a normoxic gas mixture (20% O2, 5% CO2, and 75% N2), given a preload of 800 mg, and equilibrated for 90 min. During an equilibration period, the α-adrenergic receptor agonist phenylephrine (10 µM) was applied to induce contraction for the verification of the responsiveness of PAs. Following phenylephrine-induced contraction, ACh (10 µM) was added to confirm no relaxation (i.e., the complete removal of the endothelium). A difference between the basal tension and maximal tension following norepinephrine (NE) stimulation was calculated and normalized to tissue weight (mg/mg), which was presented as NE-induced muscle tension.
Lentiviral shRNA preparation and infection.
Lentiviral particles containing mouse pLKO.1 shRNAs specific for PLCγ1 (TRCN0000024974-75), shRNAs specific for IP3R1 (RMM4431-200348227), and NS shRNA were produced using a classic Ca2+ phosphate transfection protocol modified from Icardi et al. (20) and as described earlier (61). Lentiviral packaging pCMV-dR8.2 dvpr and pVSV-G constructs were kindly provided by Dr. Peter Vincent (Center for Cardiovascular Sciences, Albany Medical College, Albany, NY). In brief, 11.1 μg of the plasmid shRNAmir vectors were cotransfected with 8.33 μg of pCMV-dR8.2 dvpr, and 5.55 μg of pVSV-G in HEK-293FT cells were seeded in a 10-cm dish. The supernatant containing the viral particles was harvested 48 and 72 h after transfection and concentrated by ultracentrifugation for 2 h at 22,000 rotations per minute in an SW28 rotor at 4°C. Target cells were then transduced accordingly with the concentrated supernatant lentiviral particles. Primary cultured PASMCs in culture medium (DMEM, 1×) with 4.5 g/l glucose, l-glutamine and sodium pyruvate medium, 10% FBS, and 1% penicillin-streptomycin) were infected with lentiviral particles for 24 h, followed by incubation with the medium in the absence of lentiviral particles for 48 h. Then, cells were incubated with puromycin (4 µM) for 48 h to kill the noninfected cells and ensure near 100% infection of cells. The infected cells were incubated with 0.3% FBS containing culture medium for 48 h. The KD efficiency of each clone was determined using Western blot analysis. The clones showing maximum KD efficiency were selected for further experiments.
IP3 production assay.
Production of IP3 was measured by analyzing the production of inositol monophosphate (IP1, downstream metabolite of IP3, which is stable in the presence of LiCl) using IP-One HTRF assay (48), according to the manufacturer’s protocol. Primary cultured PASMCs were incubated in assay buffer containing LiCl to inhibit IP1 degradation for 1.5 h at 37°C. Eu3+-labeled anti-IP1 antibody (acceptor) was added, followed by IP1-d2 conjugate (donor). After incubation for 1 h at room temperature, the reaction plate wells were excited at 343 nm on a FlexStation-III Microplate Reader (Molecular Devices), and emitted light was measured at 620 nm and 665 nm. The time-resolved fluorescence resonance energy transfer (ratio of 665 nm/620 nm), which is inversely proportional to the IP1 amount, was used to determine IP3 production.
[3H]-IP3 binding assay.
The [3H]-IP3 binding assay was performed, modified as previously described (27, 31). Briefly, cell lysate (90μg protein) was incubated with [3H]-IP3 at 5–25nM at 37°C for 3h in a binding assay solution (300μl) containing 25mM Tris, 50mM HEPES, a protease inhibitor mix, and 100μM CaCl2 (pH 7.4). The binding mixture was diluted with ice-cold washing buffer (3ml) containing 25mM Tris (pH 8.0) and 250mM KCl and immediately filtered through Millipore Membrane filters presoaked with washing buffer. The filters were washed three times with ice-cold washing buffer (5ml). The radioactivity associated with the filters was determined by liquid scintillation counting. Nonspecific binding was determined in the presence of 50μM unlabeled IP3. All binding assays were executed in duplicate.
Statistical analysis.
All of the data was presented as box and whisker plots with a vertical line to mark the median vertical. The comparison between two groups was made using Student’s t-test. Experiments comprising three or more groups were compared using one-way ANOVA followed by Tukey’s multiple-comparison post hoc analysis. Values at P < 0.05 were considered statistically significant. Wherever used, numbers in parentheses indicate the number of cells isolated and tested from four or more animals.
RESULTS
PLCγ1 may play a role in the hypoxia-induced increase in [Ca2+]i in PASMCs.
It is generally agreed that hypoxia causes vasoconstriction in PAs, but not SAs; thus, we first used a muscle tension measurement method to examine the effect of hypoxia on contraction in isolated PA and MA rings. As shown in Fig. 1, A and B, we observed that hypoxia produced vasoconstriction in PA rings, whereas there was no contraction in MA rings.
Fig. 1.

Phospholipase C-γ1 (PLCγ1) play an important role in hypoxia-induced pulmonary vasoconstriction (HPV). A: pulmonary artery (PA) and mesenteric artery (MA) rings were exposed to a physiological saline solution (PSS) bubbled with a normoxic gas (21% O2, 5% CO2, and the balance with N2) for 10 min and then a hypoxic gas (1% O2, 5% CO2, and the balance with N2) for 30 min. B: graph summarizes the tension generated in response to hypoxia in PAs and MAs (n = 4; *P < 0.05 compared with PA). C: representative Western blots of PLCγ1 expression in pulmonary artery smooth muscle cells (PASMCs) and mesenteric artery smooth muscle cells (MASMCs). D: graph summarizes the quantification of Western blots of PLCγ1 from three independent experiments. *P < 0.05 compared with MASMCs. E: representative traces show a hypoxia-induced increase in [Ca2+]i in PASMCs preincubated without or with U73122 (1 µM) or U73433 (1 µM) for 30 min. F: quantification of the hypoxic increase in [Ca2+]i in PASMCs untreated and treated with U73122 or U73433. Data were obtained from at least four different animals. *P < 0.05 compared with control.
The aforementioned results, together with our previous report that PLCγ1 plays an important role in HPV (61), inspired us to assess the potential role of PLCγ1 in the heterogeneity of hypoxic responses. The results indicated that expression of PLCγ1 was significantly higher in PASMCs compared with that in mesenteric artery smooth muscle cells (MASMCs) (Fig. 1, C and D). We also found a hypoxia-induced increase in [Ca2+]i in PASMCs was inhibited by the PLC inhibitor U73122 (1 µM) and was unaffected by its inactive analog U73433 (1 µM, Fig. 1, E and F).
Exogenous ROS cause PLCγ1 activation and IP3 production in PASMCs.
We have reported that hypoxia-induced increase in [Ca2+]i is associated with ROS-dependent increase in the activity of PLCγ1 in PASMCs (61). In view of this previous finding, we investigated whether application of exogenous ROS, H2O2, may cause an increase in the activity of PLCγ1 in PASMCs, assessed by determining phosphorylation of PLCγ1 at tyrosine-783, as described previously (16, 18, 53). The results revealed that hypoxia and H2O2 both significantly increased the activity of PLCγ1 (Fig. 2, A and B).
Fig. 2.
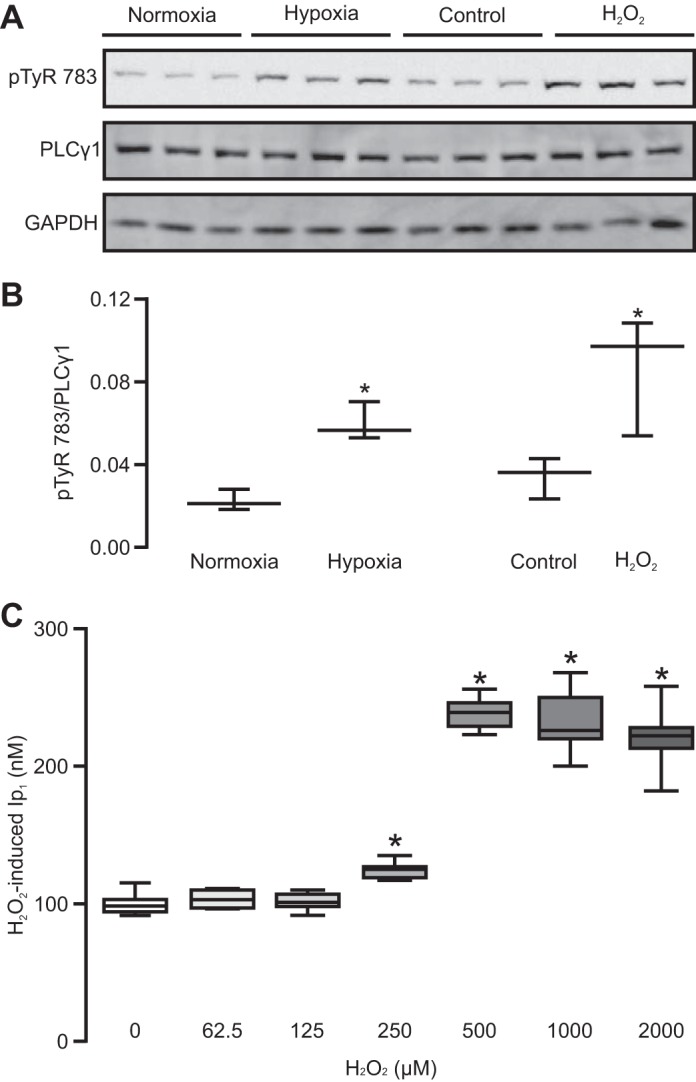
Hypoxia and H2O2 both increase PLCγ1 activity and inositol monophosphate-1 (IP1) production in PASMCs. A: Western blots of PLCγ1 phosphorylated at tyrosine 783 (pTyr-783) and total PLCγ1 in PASMCs exposed to normoxia, hypoxia, untreated control, and H2O2 (500 µM, 10 min). B: bar graph summarizes the ratio of pTyr-783 expression level relative to total PLCγ1 protein expression level from three separate experiments. *P < 0.05 compared with respective control. C: IP1 production in cells untreated and treated with H2O2 in various concentrations for 10 min. Data were obtained from three separate experiments. *P < 0.05 compared with untreated group.
Furthermore, our data indicated that H2O2 produced a concentration-dependent increase in IP3 production, as determined by measuring its downstream metabolite IP1 production, in PASMCs (Fig. 2C).
PLCγ1 mediates hypoxia- and H2O2-induced increase in IP3 production in PASMCs.
Our previous report has shown that hypoxia leads to PLC-dependent IP3 production in PASMCs, and PLCγ1 regulates the hypoxia-induced increase in [Ca2+]i in PASMCs (61). This suggests a possible role of PLCγ1 in hypoxia-induced IP3 production. To address this possibility, we infected PASMCs with lentiviral particles, enclosing specific PLCγ1 or NS shRNAs, and the shRNA-infected PASMCs were used to assay the effect of PLCγ1 knockdown (KD) on hypoxic IP3 production. Unsurprisingly, corresponding to its effect on PLCγ1 expression, PLCγ1 KD significantly attenuated the hypoxia-induced increase in IP1 production (Fig. 3), demonstrating the inhibitory effect of PLCγ1 KD on IP3 production in PASMCs. In contrast, NS shRNA did not affect either PLCγ1 expression or hypoxic IP1 production. In addition, the basal IP1 levels were also reduced by PLCγ1 KD.
Fig. 3.
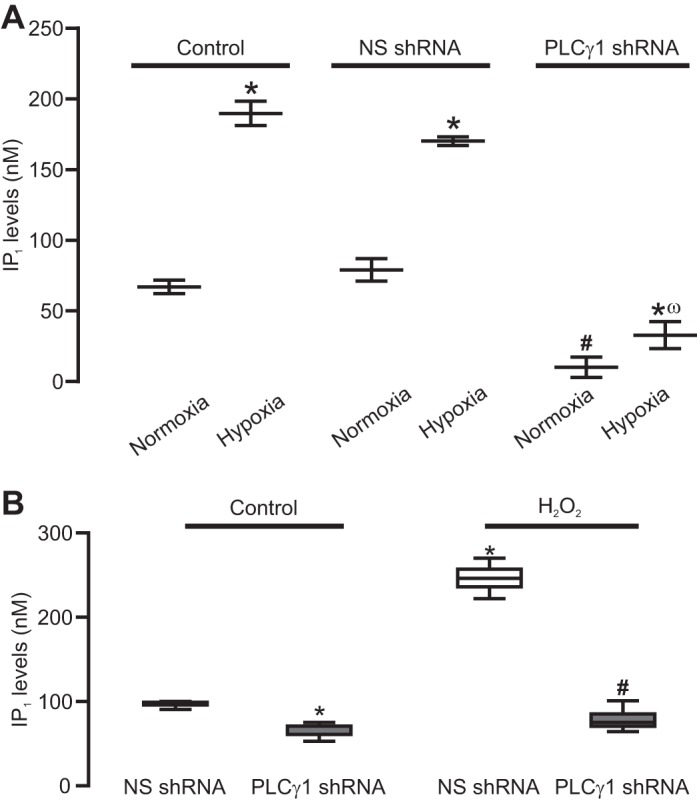
PLCγ1 knock-down (KD) abolishes hypoxia- and H2O2-induced increase in IP1 production in pulmonary artery smooth muscle cells PASMCs. A: summary of the hypoxia-induced IP1 production in PASMCs. Cells were uninfected (control) or infected with lentiviral particles containing nonsilencing (NS) short hairpin (sh)RNAs or shRNAs for PLCγ1. Data were obtained from three different experiments. *P < 0.05 compared with respective normoxia group. #P < 0.05 compared with normoxia. ωP < 0.05 compared with normoxia. B: H2O2 (500 µM) induced IP1 production in PASMCs infected with NS shRNA or PLCγ1 shRNAs. Data were obtained from three different experiments *P < 0.05 compared with NS shRNA. #P < 0.05 compared with NS shRNA.
We have also shown that H2O2-induced increase in IP1 production was significantly reduced in PLCγ1 KD cells. Thus, PLCγ1 mediates the basal and hypoxia- as well as H2O2-induced increase in IP3 production in PASMCs.
IP3R1 serves as an essential downstream molecule in hypoxia-induced increase in [Ca2+]i in PASMCs.
Acute hypoxia enhances IP3 production in PASMCs; IP3 can activate IP3Rs and release Ca2+ from the SR. Among all three isoforms of IP3Rs, IP3R1 is predominantly present in PASMCs (45, 47). Thus, we thought that IP3R1 might be involved in the hypoxia-induced Ca2+ responses as well. To investigate our hypothesis, we created lentiviral particle containing shRNAs targeting IP3R1. These shRNAs were able to knock down IP3R1 protein levels in PASMCs (Fig. 4, A and B). To substantiate the specificity of IP3R1 KD, we measured the levels of IP3R2 and IP3R3 in IP3R1 KD cells. Our data revealed that levels of IP3R2 and IP3R3 were unaffected in PASCMCs infected with shRNA targeting IP3R1 (Fig. 4, C–E). All of these findings also validate the specificity and effectiveness of IP3R1 antibodies.
Fig. 4.

Ca2+ release from IP3 receptor 1 (R1) is involved in hypoxia- and H2O2-induced increase in [Ca2+]i in PASMCs. A: Western blot of IP3R1 in PASMCs infected with lentiviral particles containing shRNAs targeting IP3R1 and NS shRNAs. B: graph summarizes the quantification of Western blots of IP3R1 from three independent experiments. C: Western blots of the expression of IP3R2 and IP3R3 in PASMCs infected with NS shRNA and IP3R1 shRNA. D: summary of the quantification of IP3R2 protein expression. E: graph summarizes the average IP3R3 expression. F: effect of IP3R1 gene KD on hypoxia-induced induced increase in [Ca2+]i. G: effect of IP3R1 KD on H2O2 (500 µM)-induced increase in [Ca2+]i. Cells were uninfected (control) or infected with lentiviral particles containing NS shRNAs and shRNAs specific for IP3R1. *P < 0.05 compared with control.
Importantly, the hypoxia-produced increase in [Ca2+]i was significantly attenuated in IP3R1 KD PASMCs, whereas cells infected with the NS shRNAs showed a similar increase in [Ca2+]i to that in control (uninfected) cells (Fig. 4F). In agreement with the hypoxia-induced response, H2O2 (500 µM) induced a much smaller increase in [Ca2+]i in IP3R1 KD PASMCs (Fig. 4G). These data clearly point toward the involvement of IP3R1 in the hypoxia- and H2O2-induced increase in [Ca2+]i in PASMCs.
PKCε regulates hypoxia- and H2O2-induced binding affinity or activity of IP3Rs in PASMCs.
We have shown that mitochondrial ROS cause PKCε activation, which is involved in the hypoxia-induced increase in [Ca2+]i and NOX-dependent ROS production in PASMCs (41, 42). It is also known that PKC can phosphorylate IP3Rs to enhance their activity, and the PKC-mediated phosphorylation of IP3R1 is eight times higher than that of IP3R3 (49). Thus, we have speculated that hypoxia-induced activation of PKCε might phosphorylate IP3Rs, thereby increasing the binding of IP3 to its receptors and inducing Ca2+ release in PASMCs. To investigate this possibility, we compared the IP3 binding in PASMCs from control (wild-type) and PKCε gene knockout (PKCε−/−) mice. As expected, we have found that hypoxia caused a large increase in the binding of IP3 to IP3Rs in PASMCs from control mice, but a much smaller increase in cells from PKCε−/− mice (Fig. 5A).
Fig. 5.
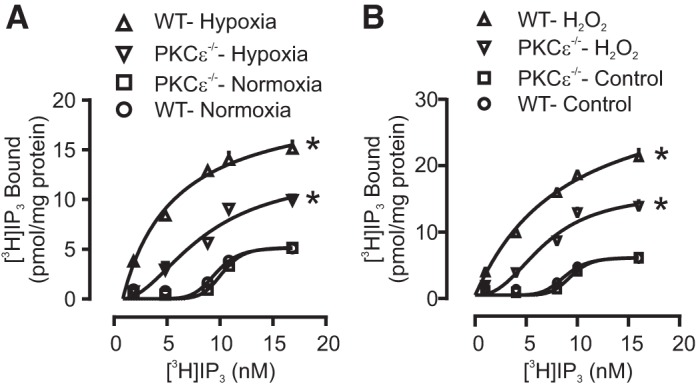
PKCε regulates hypoxia- and H2O2-induced increase in IP3R binding affinity or activity in PASMCs. Radiolabeled [3H] IP3 binding assay was conducted in lysates of PAMSCs to test the activity of IP3Rs. A: effect of acute hypoxia on the binding of IP3Rs in control (wild-type, WT) and PKCε−/− PASMCs. B: effect of H2O2 (500 μM) on [3H] IP3 binding of IP3Rs in control and PKCε−/− PASMCs. Data were obtained from three independent experiments. *P < 0.05 compared with control.
In addition, H2O2 (500 µM) increased the binding of IP3 in control PASMCs, and the effect of H2O2 was significantly reduced in PKCε−/− cells (Fig. 5B). Thus, the hypoxia-induced activation of PKCε may phosphorylate IP3Rs, thereby increasing IP3 binding, IP3R activity, Ca2+ release, and ultimately [Ca2+]i in PASMCs.
Mice exposed to CH show an increase in PLCγ1 activity in PASMCs and a decrease in MASMCs.
Acute hypoxia activates PLCγ1 through the mitochondrial ROS-dependent signaling mechanism (61) and, thus, increases [Ca2+]i via the IP3 production-mediated, IP3R1-based Ca2+ release process (Figs. 3 and 4C). The increase in [Ca2+]i in PASMCs may make a significant contribution to the development of HPV (Fig. 1). This unique response, if persisted, together with pulmonary arterial remodeling, ultimately leads to pulmonary hypertension (19, 50). Accordingly, we wanted to study the role of PLCγ1 in hypoxic pulmonary hypertension. In these experiments, mice were exposed to 10% or 21% O2 for 21 days. As shown in Fig. 6A, PLC activity was significantly higher in PASM tissues from mice exposed to CH than from control (normoxic) animals; in contrast, PLC activity was lower in MASM tissues from hypoxic mice.
Fig. 6.
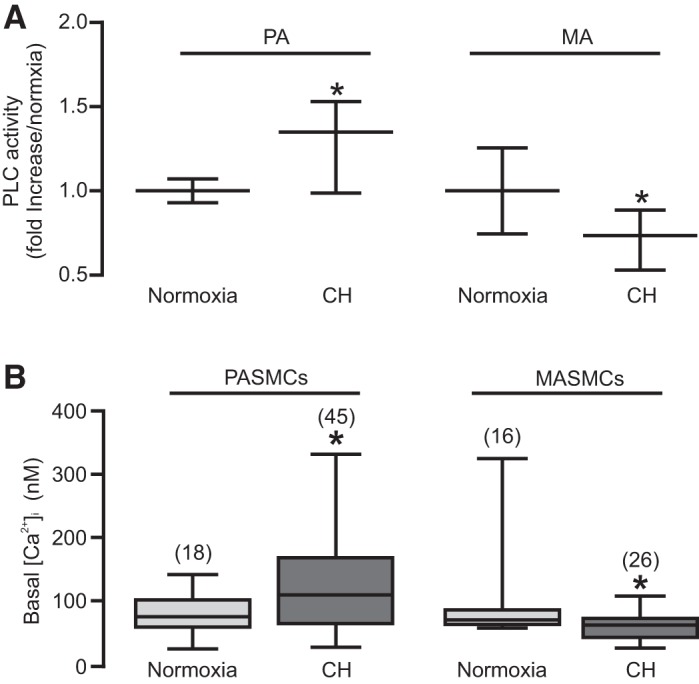
Mice exposed to chronic hypoxia (CH) show higher basal PLC activity and [Ca2+]i in PASMCs than that in MASMCs. A: lysates from PAs and MAs from mice exposed to CH for 3 wk were subjected to PLC activity assay. Data were obtained from four independent experiments. The data were obtained by taking the average of the test values in the normoxia group, dividing the individual value from each test in the normoxia and hypoxia groups by the averaged value from the normoxia group, and finally taking the average of the calculated values in the normoxia and hypoxia groups (B) to provide basal intracellular [Ca2+]i in PASMCs and MASMCs. Data were obtained from four animals. *P < 0.05 compared with normoxia.
In accordance with the altered PLC activity, the resting [Ca2+]i was higher in PASMCs from hypoxic mice, while it was lower in MASMCs from hypoxic mice (Fig. 6B).
To further determine the role of PLCγ1 in hypoxic pulmonary hypertension, we isolated PASMCs from normoxic and hypoxic mice. The cells were cultured and then subjected to Western blot analysis. The results indicated that PLCγ1 expression levels were higher in PASMCs from hypoxic mice compared with normoxic mice (Fig. 7A).
Fig. 7.
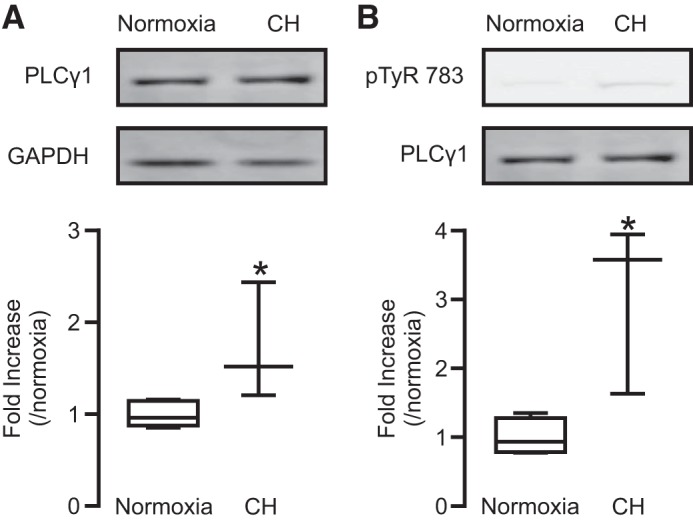
Mice exposed CH showed higher expression and basal activity of PLCγ1 in PASMCs than in MASMCs. A: lysates from PASMCs of mice exposed to normoxia and CH were subjected to Western blot analysis. A, top: representative blots of PLCγ1 and GAPDH. A, bottom: quantification of PLCγ1 expression levels in PASMCs from five animals exposed to normoxia and three animals to CH. B, top: representative Western blots of protein expression of PLCγ1 phosphorylated at tyrosine 783 (pTyr-783) and total PLCγ1 in PASMCs from normoxia and CH mice. B, bottom: summary of the fold increase in the ratio of pTyr-783 expression level relative to total PLCγ1 protein expression level in PASMCs. Data were obtained from four mice exposed to normoxia and three mice exposed to CH. *P < 0.05 compared with normoxia.
Further, we examined PLCγ1 activation by assessing its Tyr-783 phosphorylation. The data revealed that the basal activity of PLCγ1 was higher in PASMCs from hypoxic mice; thus, Tyr-783 phosphorylation was significantly increased. (Fig. 7B). The higher expression and activity of PLCγ1 in PASMCs point toward its possible involvement in hypoxic pulmonary hypertension.
PLC-dependent agonist-induced pulmonary vasoconstriction is largely augmented in mice exposed to CH.
Hypoxic pulmonary hypertension is well characterized by pulmonary vasoconstriction and remodeling; these two major cellular responses are primarily mediated by the increased [Ca2+]i in PASMCs (28, 44, 58, 59). Concordantly, the purinergic receptor agonist ATP (10 µM) induced a larger increase in [Ca2+]i in PASMCs from mice exposed to CH relative to normoxic mice (Fig. 8A). Similarly, H2O2 (500 µM) produced a similar result.
Fig. 8.
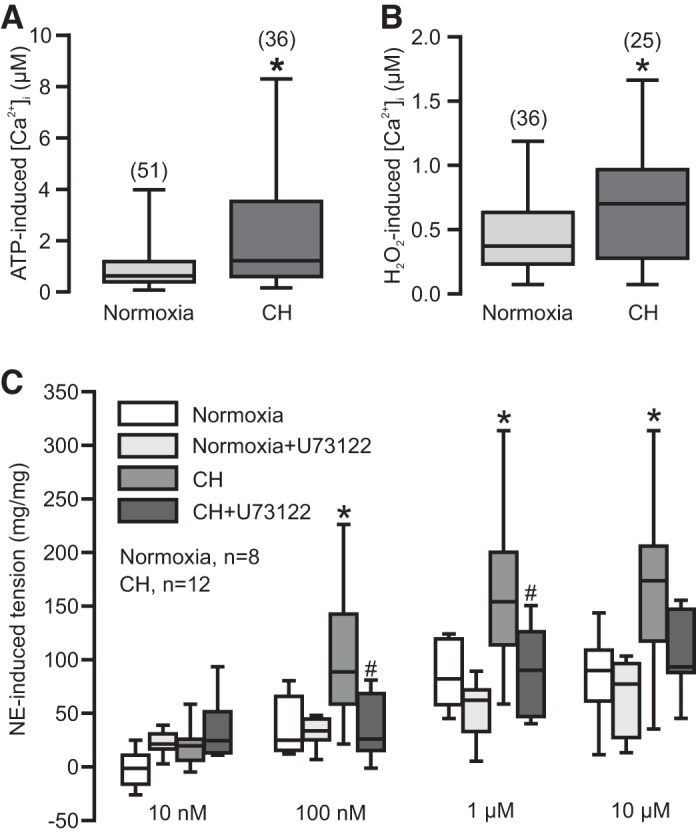
ATP and H2O2-induced increase in [Ca2+]i in PASMCs and PLC-dependent norepinephrine (NE)-induced contraction in PAs are greatly enhanced in mice exposed to CH. A: effect of ATP (10 µM) on [Ca2+]i in PASMCs from normoxic and CH mice. B: effect of H2O2 (500 µM) on [Ca2+]i in PASMCs from normoxic and CH mice. Cells were obtained from at least four different animals. *P < 0.05 compared with normoxia. C: summary of NE-induced contraction in PA rings from normoxia and CH mice treated with and without U73122 (10 µM). Data were obtained from three normoxic and four CH mice. *P < 0.05 compared with normoxia, and #P < 0.05 compared with CH.
Application of the α-adrenergic receptor agonist NE induced a contraction-dependent vasoconstriction in PAs (Fig. 8C). This agonist-induced pulmonary vasoconstriction was remarkably augmented in mice exposed to CH. More interestingly, the hypoxic augmentation of agonist-evoked pulmonary vasoconstriction was almost eliminated by pretreatment with the PLC inhibitor U73122 (10 µM) for 10 min. Notably, treatment with U73122 did not affect agonist-induced pulmonary vasoconstriction in PAs from normoxic mice. Thus, PLC, particularly PLCγ1, may mediate the enhanced pulmonary vasoconstriction in mice following CH.
DISCUSSION
HPV and associated pulmonary hypertension are mainly associated with the increased [Ca2+]i in PASMCs, and the underlying signaling mechanisms are still being energetically explored. Recently, we have reported that acute hypoxia causes a significant increase in the activity of PLCγ1, which plays an important role in the hypoxia-induced increase in [Ca2+]i and attendant contraction in PASMCs. The hypoxia-induced increase in the activity of PLCγ1 is secondary to the increased mitochondrial ROS production primarily through RISP-based ROS generation system in the complex III. The role of PLCγ1 in mediating hypoxia-induced Ca2+ and contractile responses in PASMCs are a result of the increased PLC-reliant IP3 production, IP3Rs opening, and SR Ca2+ release (61). However, the downstream molecular players that are involved in the mechanisms for PLCγ1-dependent hypoxic increase in [Ca2+]i in PASMCs are undetermined. In the present study, we have confirmed that PLCγ1 mediates the hypoxia-induced increase in IP3 production in PASMCs. Importantly, we have found that IP3R1 is involved in the hypoxia-induced increase in [Ca2+]i in PASMCs. Notably, PKCε regulates the hypoxia-induced increase in the activity of IP3Rs in PASMCs. In addition, our data also indicate that PLCγ1 may play a vital role in the CH-induced pulmonary hypertension.
Hypoxia causes vasoconstriction in PAs and produces relaxation in SAs (1, 2, 12). We have observed HPV in isolated PAs from mice, while MAs show no contractile responses to hypoxia (Fig. 1, A and B). The heterogeneity of these hypoxic responses in PAs and SAs has been attributed primarily to a large increase in [Ca2+]i in PASMCs, but not in mesenteric and other SA SMCs. Importantly, the hypoxia-induced increase in [Ca2+]i in PASMCs is dependent on the production of mitochondrial superoxide (O2·−), H2O2, and other ROS molecules in PASMCs. The hypoxia-induced mitochondrial ROS-dependent activation of PKCε has been observed in PASMCs. Overall, the generation of ROS in response to hypoxia is an important factor that mediates the heterogeneity of hypoxia responses in the pulmonary and systemic vasculature (41, 52, 54). In accordance, acute hypoxia results in a significant increase in the activity of total PLC in PAs and not in MAs (61). To test the potential role of PLCγ1 in the heterogeneity of hypoxia responses in PAs and Mas, we determined the expression levels of PLCγ1 in PASMCs and MASMCs. Out data reveal that PLCγ1 expression in PASMCs is significantly higher compared with that in MASMCs (Fig. 1, C and D). Interestingly, the hypoxia-induced increase in the activity of PLCγ1 is dependent on RISP-dependent mitochondrial ROS generation in PASMCs (61). Thus, the differential ROS-dependent activation of PLCγ1 in PAs during hypoxia exposure may play an essential role in the heterogeneity of hypoxia responses in PAs and SAs. Consistent with our previous report (61), our current study has discovered that hypoxia stimulation increases [Ca2+]i in PASMCs. This increase in [Ca2+]i is diminished upon PLC inhibition with U3122 and is unaffected by the inactive U73122 analog U73433 (Fig. 1, E and F).
Hypoxia-induced increase in [Ca2+]i in PASMCs is attributed to an increase in intracellular ROS produced by mitochondria and NOX (54). Mitochondrial O2·− is generated through the electron transport chain (ETC) and then rapidly converted to H2O2 by manganese superoxide dismutase. H2O2 is a more diffusible and active ROS. H2O2, with O2·− and other ROS, activates various intracellular signaling molecules and mediators. The hypoxic generation of mitochondrial ROS is primarily mediated by the ETC molecules before the complex III ubisemiquinone site, in which RISP in the complex III is an indispensable element (25, 52). Interestingly, the hypoxic increase in RISP-dependent mitochondrial ROS may cause activation of PKCε and subsequently NOX, leading to further ROS generation, termed RIRG (41, 42). In addition, RISP-dependent mitochondrial ROS activate PLCγ1 and, thereby, regulate the hypoxic Ca2+ response in PASMCs (61). Thus, the newly discovered RIRG and PLCγ1 activation may play a significant role in hypoxic Ca2+ and contractile responses in PASMCs. Furthermore, we have reported that the H2O2-induced increase in [Ca2+]i in PASMCs is dependent on PLCγ1 (61). In agreement with this previous report, the current study has revealed that hypoxia and H2O2 both cause PLCγ1 activation (Fig. 2, A and B). H2O2 also increases IP1 production (Fig. 2C). All of the current data support the report that the blockade of IP3Rs inhibits the H2O2-induced increase in [Ca2+]i in PASMCs and contraction in PAs (61).
IP3, a product of PLC, opens IP3Rs on the SR and subsequently induces Ca2+ release, which mediates various cellular responses in almost all types of cells. Hypoxia causes a large increase in the activity of PLCγ1, and PLCγ1 KD brings about a large reduction in the hypoxia-induced increase in [Ca2+]i in PASMCs (61). In accord with our expectation, the data obtained in this study demonstrate that PLCγ1 mediates the production of IP3 in response to acute hypoxia in PASMCs and also maintains the basal IP3 production in PASMCs (Fig. 3A). In addition, H2O2-induced IP1 production is also dependent on the activity of PLCγ1 in PASMCs (Fig. 3B).
We have reported that inhibition of IP3Rs reduces the hypoxia-induced increase in [Ca2+]i in PASMCs (61). Three subtypes of IP3Rs (IP3R1, IP3R2, and IP3R3) are expressed in mammalian cells and encoded by three separate genes. IP3R1, IP3R2, and IP3R3 are present to various extents in every type of cells, but IP3R1 predominates in vascular SMCs (17, 45–47). Considering this fact, we determined the role of IP3R1 in the hypoxia-induced increase in [Ca2+]i in PASMCs using the gene KD approach. Transfection of lentiviral shRNAs specific for IP3R1, but not NS shRNAs, greatly knocked down its protein expression in PASMCs (Fig. 4, A and B). Importantly, our data reveal that IP3R1 KD significantly diminishes the hypoxia-induced increase in [Ca2+]i in PASMCs (Fig. 4F) and also H2O2-induced response (Fig. 4G). These findings are consistent with the general notion that IP3R1 expression and activity predominate in vascular SMCs (17, 45–47) and also demonstrate that IP3R1 plays an important role in the hypoxia- and H2O2-induced increase in [Ca2+]i in PASMCs. It should also be noted that further investigations are needed to exclude the potential role of IP3R2 and/or IP3R3 in the hypoxia-induced [Ca2+]i response in PASMCs.
IP3Rs are opened as a result of IP3 binding; on the other hand, phosphorylation of IP3Rs can regulate its binding by IP3, opening, and function (46). For instance, PKC phosphorylates IP3Rs, enhances IP3 binding, and increases Ca2+ release. Interestingly, PKC causes the phosphorylation of IP3R1 eight times more than that of IP3R3 (49). We and other scientists have disclosed that PKCε is involved in the hypoxia-induced increase in [Ca2+]i and contraction in PASMCs (41), as well as HPV in isolated lungs (29). In the current study, we have found that IP3R1 mediates the hypoxia-induced increase in [Ca2+]i in PASMCs (Fig. 4). Considering the aforementioned facts, we speculate that PKCε may regulate IP3Rs channel activation and, thus, contribute to the hypoxia-induced increase in [Ca2+]i in PASMCs. Indeed, we have found that hypoxia enhances the binding of IP3 to IP3Rs (Fig. 5A). Furthermore, the hypoxia-induced increase in IP3 binding is significantly reduced in PASMCs from PKCε−/− mice. In addition, H2O2 also enhances IP3 binding, which is also significantly inhibited in PASMCs from PKCε−/− mice (Fig. 5B). Thus, the hypoxia-induced activation of PKCε may increase the activity of IP3R1, increasing IP3 binding and subsequently [Ca2+]i in PASMCs.
Our previous report (61) and the current findings reveal the importance of PLCγ1 in the hypoxia-induced increase in [Ca2+]i in PASMCs. The increase in [Ca2+]i in PASMCs is important for HPV, which serves as a vital physiological process in the lungs (8, 11, 34, 54, 58). Instead, persistent HPV, together with pulmonary vascular remodeling, leads to the development of pulmonary hypertension and right ventricular heart failure (10, 31). We have shown that hypoxia activates PLC to increase [Ca2+]i in PASMCs and to cause HPV (61). As a natural and important extension of our previous findings, the current study has found that the basal PLC activity is much higher in PAs from mice exposed to CH for 21 days (Fig. 6A). The higher PLC activity is related very well to the higher basal [Ca2+]i in PASMCs (Fig. 6B) and higher vascular tone (Fig. 8C). Similarly, the correlation of the higher pulmonary artery pressure with higher vascular tone in PA has been reported (35, 57). Thus, the higher PLC activity corresponds well to the higher basal tone. In addition, we have found that the basal PLC activity and [Ca2+]i are lower in MASMCs from CH mice (Fig. 6, A and B). In accordance, we have also observed that PLCγ1 expression and activity are significantly higher in PASMCs from CH mice (Fig. 7, A and B). CH is known to be associated with variable degrees of the proliferation of SMCs or smooth muscle-like cells (33), and the hypoxia-induced increase in [Ca2+]i functions as an important signaling player to cause pulmonary vasoconstriction and remodeling (40). In particular, pulmonary hypertension is characterized by the increased PA tone due to the elevated [Ca2+]i in PASMCs (21, 22, 28, 44, 58, 59). Thus, pulmonary vasoconstriction and remodeling may use the overlapping signaling processes in mediating their vasoconstrictive and structural responses. Consistent with the higher PLCγ1 activity, we have further observed that PAs from CH mice show the hypercontractility to the major vascular neurotransmitter NE, and this hypercontraction is blocked by the PLC inhibitor U73122 at 100 nM and 1 µM concentrations (Fig. 8C). Equally intriguingly, H2O2 and ATP both induce a larger increase in [Ca2+]i in PASMCs from CH mice. Application of ATP stimulates purinergic P2X and P2Y receptors. The former receptors are the Ca2+-permeable ligand-gated ion channels and can allow Ca2+ influx upon stimulation. Activation of P2Y receptors can activate PLC and produce IP3 and DAG (6). Conceivably, CH may perhaps affect these two receptor-dependent signaling processes in coordination with PLCγ1 to mediate the development of pulmonary hypertension.
On the basis of the current findings with our previous report (61), we present a schematic diagram, as shown in Fig. 9, to conclude that hypoxia increases RISP-dependent mitochondrial ROS production, activates PLCγ1, causes IP3 production, opens IP3R, and induces Ca2+ release in PASMCs. On the other hand, hypoxia-induced mitochondrial ROS activate PKCε, enhance IP3R1 activity, IP3 binding, and induce further Ca2+ release. Altogether, these two signaling pathways contribute toward the hypoxia-induced increase in [Ca2+]i in PASMCs, HPV, and pulmonary hypertension.
Fig. 9.
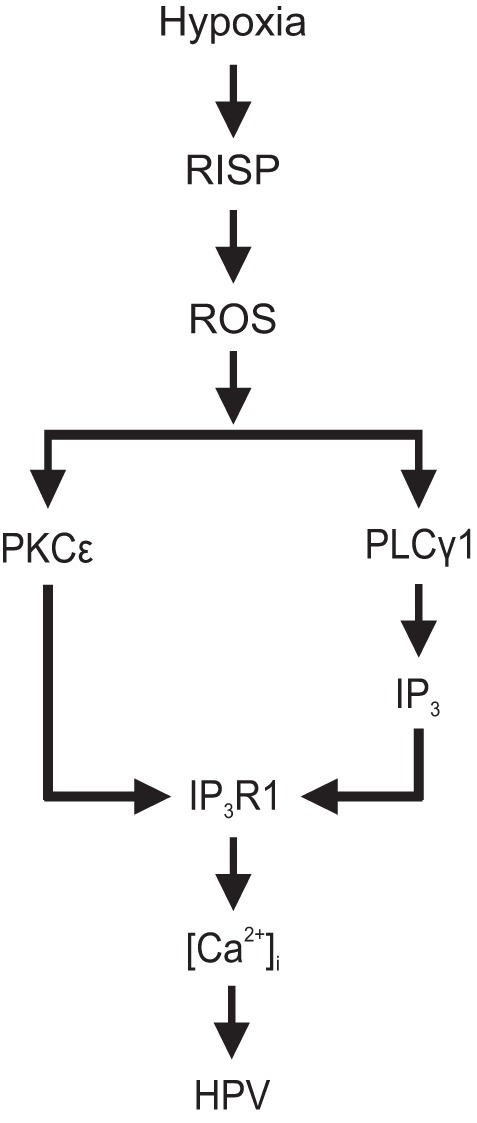
A schematic diagram of the signaling mechanisms for hypoxia-induced PLCγ1- and PKCε-dependent increase in [Ca2+]i in PASMCs and HPV. Hypoxia causes an increase in RISP-dependent mitochondrial ROS production, which causes PLCγ1 activation, IP3 production, IP3R1 opening, and Ca2+ release. RISP-dependent mitochondrial ROS may also activate PKCε, phosphorylate IP3R1, enhance IP3 binding, induce Ca2+ release in PASMCs. Taken together, PLCγ1 and PKCε can synergistically contribute to the hypoxic increase in [Ca2+]i in PASMCs, HPV, and pulmonary hypertension.
GRANTS
This work was supported by an American Heart Association Established Investigator Award 0340160N (to Y.-X. Wang) and Scientist Development Grant 0630236N (to Y.-M. Zheng) and National Institutes of Health Grants R01HL64043, HL064043-S1, HL075190, and HL108232 (to Y.-X. Wang).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
V.R.Y., L.M., L.J., Y.-M.Z., and Y.-X.W. conceived and designed research; V.R.Y., T.S., L.M., and L.J. performed experiments; V.R.Y., T.S., L.M., L.J., and Y.-M.Z. analyzed data; V.R.Y., T.S., L.M., L.J., Y.-M.Z., and Y.-X.W. interpreted results of experiments; V.R.Y., T.S., L.M., and L.J. prepared figures; V.R.Y. and T.S. drafted manuscript; V.R.Y., T.S., Y.-M.Z., and Y.-X.W. edited and revised manuscript; Y.-M.Z. and Y.-X.W. approved final version of manuscript.
REFERENCES
- 1.Aaronson PI. Hypoxic pulmonary vasoconstriction is/is not mediated by increased production of reactive oxygen species. J Appl Physiol (1985) 101: 1000–1005, 2006. doi: 10.1152/japplphysiol.00680.2006. [DOI] [PubMed] [Google Scholar]
- 2.Aaronson PI, Robertson TP, Knock GA, Becker S, Lewis TH, Snetkov V, Ward JP. Hypoxic pulmonary vasoconstriction: mechanisms and controversies. J Physiol 570: 53–58, 2006. doi: 10.1113/jphysiol.2005.098855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Aley PK, Murray HJ, Boyle JP, Pearson HA, Peers C. Hypoxia stimulates Ca2+ release from intracellular stores in astrocytes via cyclic ADP ribose-mediated activation of ryanodine receptors. Cell Calcium 39: 95–100, 2006. doi: 10.1016/j.ceca.2005.09.009. [DOI] [PubMed] [Google Scholar]
- 4.Berridge MJ. Inositol trisphosphate and calcium signalling. Nature 361: 315–325, 1993. doi: 10.1038/361315a0. [DOI] [PubMed] [Google Scholar]
- 5.Berridge MJ, Bootman MD, Roderick HL. Calcium signalling: dynamics, homeostasis and remodelling. Nat Rev Mol Cell Biol 4: 517–529, 2003. doi: 10.1038/nrm1155. [DOI] [PubMed] [Google Scholar]
- 6.Bootman MD, Collins TJ, Peppiatt CM, Prothero LS, MacKenzie L, De Smet P, Travers M, Tovey SC, Seo JT, Berridge MJ, Ciccolini F, Lipp P. Calcium signalling—an overview. Semin Cell Dev Biol 12: 3–10, 2001. doi: 10.1006/scdb.2000.0211. [DOI] [PubMed] [Google Scholar]
- 7.Bradford JR, Dean HP. The pulmonary circulation. J Physiol 16: 34–158, 1894. doi: 10.1113/jphysiol.1894.sp000493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Broughton BR, Jernigan NL, Norton CE, Walker BR, Resta TC. Chronic hypoxia augments depolarization-induced Ca2+ sensitization in pulmonary vascular smooth muscle through superoxide-dependent stimulation of RhoA. Am J Physiol Lung Cell Mol Physiol 298: L232–L242, 2010. doi: 10.1152/ajplung.00276.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chandel NS, Maltepe E, Goldwasser E, Mathieu CE, Simon MC, Schumacker PT. Mitochondrial reactive oxygen species trigger hypoxia-induced transcription. Proc Natl Acad Sci USA 95: 11715–11720, 1998. doi: 10.1073/pnas.95.20.11715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.de Frutos S, Duling L, Alò D, Berry T, Jackson-Weaver O, Walker M, Kanagy N, González Bosc L. NFATc3 is required for intermittent hypoxia-induced hypertension. Am J Physiol Heart Circ Physiol 294: H2382–H2390, 2008. doi: 10.1152/ajpheart.00132.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.de Frutos S, Spangler R, Alò D, Bosc LV; de FS . NFATc3 mediates chronic hypoxia-induced pulmonary arterial remodeling with α-actin up-regulation. J Biol Chem 282: 15081–15089, 2007. doi: 10.1074/jbc.M702679200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dingemans KP, Wagenvoort CA. Pulmonary arteries and veins in experimental hypoxia. An ultrastructural study. Am J Pathol 93: 353–368, 1978. [PMC free article] [PubMed] [Google Scholar]
- 13.Dipp M, Nye PC, Evans AM. Hypoxic release of calcium from the sarcoplasmic reticulum of pulmonary artery smooth muscle. Am J Physiol Lung Cell Mol Physiol 281: L318–L325, 2001. doi: 10.1152/ajplung.2001.281.2.L318. [DOI] [PubMed] [Google Scholar]
- 14.Fan C, Su Q, Li Y, Liang L, Angelini DJ, Guggino WB, Johns RA. Hypoxia-induced mitogenic factor/FIZZ1 induces intracellular calcium release through the PLC-IP3 pathway. Am J Physiol Lung Cell Mol Physiol 297: L263–L270, 2009. doi: 10.1152/ajplung.90416.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gelband CH, Gelband H. Ca2+ release from intracellular stores is an initial step in hypoxic pulmonary vasoconstriction of rat pulmonary artery resistance vessels. Circulation 96: 3647–3654, 1997. doi: 10.1161/01.CIR.96.10.3647. [DOI] [PubMed] [Google Scholar]
- 16.González-Pacheco FR, Caramelo C, Castilla MA, Deudero JJ, Arias J, Yagüe S, Jiménez S, Bragado R, Alvarez-Arroyo MV. Mechanism of vascular smooth muscle cells activation by hydrogen peroxide: role of phospholipase Cγ. Nephrol Dial Transplant 17: 392–398, 2002. doi: 10.1093/ndt/17.3.392. [DOI] [PubMed] [Google Scholar]
- 17.Grayson TH, Haddock RE, Murray TP, Wojcikiewicz RJ, Hill CE. Inositol 1,4,5-trisphosphate receptor subtypes are differentially distributed between smooth muscle and endothelial layers of rat arteries. Cell Calcium 36: 447–458, 2004. doi: 10.1016/j.ceca.2004.04.005. [DOI] [PubMed] [Google Scholar]
- 18.Hong JH, Moon SJ, Byun HM, Kim MS, Jo H, Bae YS, Lee SI, Bootman MD, Roderick HL, Shin DM, Seo JT. Critical role of phospholipase Cγ1 in the generation of H2O2-evoked [Ca2+]i oscillations in cultured rat cortical astrocytes. J Biol Chem 281: 13057–13067, 2006. doi: 10.1074/jbc.M601726200. [DOI] [PubMed] [Google Scholar]
- 19.Hopkins N, McLoughlin P. The structural basis of pulmonary hypertension in chronic lung disease: remodelling, rarefaction or angiogenesis? J Anat 201: 335–348, 2002. doi: 10.1046/j.1469-7580.2002.00096.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Icardi L, Mori R, Gesellchen V, Eyckerman S, De Cauwer L, Verhelst J, Vercauteren K, Saelens X, Meuleman P, Leroux-Roels G, De Bosscher K, Boutros M, Tavernier J. The Sin3a repressor complex is a master regulator of STAT transcriptional activity. Proc Natl Acad Sci USA 109: 12,058–12,063, 2012. doi: 10.1073/pnas.1206458109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jernigan NL, Herbert LM, Walker BR, Resta TC. Chronic hypoxia upregulates pulmonary arterial ASIC1: a novel mechanism of enhanced store-operated Ca2+ entry and receptor-dependent vasoconstriction. Am J Physiol Cell Physiol 302: C931–C940, 2012. doi: 10.1152/ajpcell.00332.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jernigan NL, Walker BR, Resta TC. Reactive oxygen species mediate RhoA/Rho kinase-induced Ca2+ sensitization in pulmonary vascular smooth muscle following chronic hypoxia. Am J Physiol Lung Cell Mol Physiol 295: L515–L529, 2008. doi: 10.1152/ajplung.00355.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jones RD, Morice AH. Hydrogen peroxide—an intracellular signal in the pulmonary circulation: involvement in hypoxic pulmonary vasoconstriction. Pharmacol Ther 88: 153–161, 2000. doi: 10.1016/S0163-7258(00)00089-9. [DOI] [PubMed] [Google Scholar]
- 24.Kantores C, McNamara PJ, Teixeira L, Engelberts D, Murthy P, Kavanagh BP, Jankov RP. Therapeutic hypercapnia prevents chronic hypoxia-induced pulmonary hypertension in the newborn rat. Am J Physiol Lung Cell Mol Physiol 291: L912–L922, 2006. doi: 10.1152/ajplung.00480.2005. [DOI] [PubMed] [Google Scholar]
- 25.Korde AS, Yadav VR, Zheng YM, Wang YX. Primary role of mitochondrial Rieske iron-sulfur protein in hypoxic ROS production in pulmonary artery myocytes. Free Radic Biol Med 50: 945–952, 2011. doi: 10.1016/j.freeradbiomed.2011.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li XQ, Zheng YM, Rathore R, Ma J, Takeshima H, Wang YX. Genetic evidence for functional role of ryanodine receptor 1 in pulmonary artery smooth muscle cells. Pflugers Arch 457: 771–783, 2009. doi: 10.1007/s00424-008-0556-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liao B, Zheng YM, Yadav VR, Korde AS, Wang YX. Hypoxia induces intracellular Ca2+ release by causing ROS-mediated dissociation of FKBP12.6 with ryanodine receptor 2 in pulmonary artery myocytes. Antioxid Redox Signal 14: 37–47, 2010. doi: 10.1089/ars.2009.3047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lin MJ, Leung GP, Zhang WM, Yang XR, Yip KP, Tse CM, Sham JS. Chronic hypoxia-induced upregulation of store-operated and receptor-operated Ca2+ channels in pulmonary arterial smooth muscle cells: a novel mechanism of hypoxic pulmonary hypertension. Circ Res 95: 496–505, 2004. doi: 10.1161/01.RES.0000138952.16382.ad. [DOI] [PubMed] [Google Scholar]
- 29.Littler CM, Morris KG Jr, Fagan KA, McMurtry IF, Messing RO, Dempsey EC. Protein kinase C-epsilon-null mice have decreased hypoxic pulmonary vasoconstriction. Am J Physiol Heart Circ Physiol 284: H1321–H1331, 2003. doi: 10.1152/ajpheart.00795.2002. [DOI] [PubMed] [Google Scholar]
- 30.Liu JQ, Sham JS, Shimoda LA, Kuppusamy P, Sylvester JT. Hypoxic constriction and reactive oxygen species in porcine distal pulmonary arteries. Am J Physiol Lung Cell Mol Physiol 285: L322–L333, 2003. doi: 10.1152/ajplung.00337.2002. [DOI] [PubMed] [Google Scholar]
- 31.Liu Z, Zhang J, Li P, Chen SR, Wagenknecht T. Three-dimensional reconstruction of the recombinant type 2 ryanodine receptor and localization of its divergent region 1. J Biol Chem 277: 46712–46719, 2002. doi: 10.1074/jbc.M208124200. [DOI] [PubMed] [Google Scholar]
- 32.Marchant JS, Taylor CW. Cooperative activation of IP3 receptors by sequential binding of IP3 and Ca2+ safeguards against spontaneous activity. Curr Biol 7: 510–518, 1997. doi: 10.1016/S0960-9822(06)00222-3. [DOI] [PubMed] [Google Scholar]
- 33.McMurtry IF, Petrun MD, Reeves JT. Lungs from chronically hypoxic rats have decreased pressor response to acute hypoxia. Am J Physiol Heart Circ Physiol 235: H104–H109, 1978. [DOI] [PubMed] [Google Scholar]
- 34.Moudgil R, Michelakis ED, Archer SL. Hypoxic pulmonary vasoconstriction. J Appl Physiol (1985) 98: 390–403, 2005. doi: 10.1152/japplphysiol.00733.2004. [DOI] [PubMed] [Google Scholar]
- 35.Nagaoka T, Morio Y, Casanova N, Bauer N, Gebb S, McMurtry I, Oka M. Rho/Rho kinase signaling mediates increased basal pulmonary vascular tone in chronically hypoxic rats. Am J Physiol Lung Cell Mol Physiol 287: L665–L672, 2004. doi: 10.1152/ajplung.00050.2003. [DOI] [PubMed] [Google Scholar]
- 36.Norton CE, Broughton BR, Jernigan NL, Walker BR, Resta TC. Enhanced depolarization-induced pulmonary vasoconstriction following chronic hypoxia requires EGFR-dependent activation of NAD(P)H oxidase 2. Antioxid Redox Signal 18: 1777–1788, 2013. doi: 10.1089/ars.2012.4836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Norton CE, Jernigan NL, Kanagy NL, Walker BR, Resta TC. Intermittent hypoxia augments pulmonary vascular smooth muscle reactivity to NO: regulation by reactive oxygen species. J Appl Physiol (1985) 111: 980–988, 2011. doi: 10.1152/japplphysiol.01286.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pak O, Aldashev A, Welsh D, Peacock A. The effects of hypoxia on the cells of the pulmonary vasculature. Eur Respir J 30: 364–372, 2007. doi: 10.1183/09031936.00128706. [DOI] [PubMed] [Google Scholar]
- 39.Patel S, Joseph SK, Thomas AP. Molecular properties of inositol 1,4,5-trisphosphate receptors. Cell Calcium 25: 247–264, 1999. doi: 10.1054/ceca.1999.0021. [DOI] [PubMed] [Google Scholar]
- 40.Platoshyn O, Golovina VA, Bailey CL, Limsuwan A, Krick S, Juhaszova M, Seiden JE, Rubin LJ, Yuan JX. Sustained membrane depolarization and pulmonary artery smooth muscle cell proliferation. Am J Physiol Cell Physiol 279: C1540–C1549, 2000. doi: 10.1152/ajpcell.2000.279.5.C1540. [DOI] [PubMed] [Google Scholar]
- 41.Rathore R, Zheng YM, Li XQ, Wang QS, Liu QH, Ginnan R, Singer HA, Ho YS, Wang YX. Mitochondrial ROS-PKCε signaling axis is uniquely involved in hypoxic increase in [Ca2+]i in pulmonary artery smooth muscle cells. Biochem Biophys Res Commun 351: 784–790, 2006. doi: 10.1016/j.bbrc.2006.10.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rathore R, Zheng YM, Niu CF, Liu QH, Korde A, Ho YS, Wang YX. Hypoxia activates NADPH oxidase to increase [ROS]i and [Ca2+]i through the mitochondrial ROS-PKCε signaling axis in pulmonary artery smooth muscle cells. Free Radic Biol Med 45: 1223–1231, 2008. doi: 10.1016/j.freeradbiomed.2008.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Resta TC, Broughton BR, Jernigan NL. Reactive oxygen species and RhoA signaling in vascular smooth muscle: role in chronic hypoxia-induced pulmonary hypertension. Adv Exp Med Biol 661: 355–373, 2010. doi: 10.1007/978-1-60761-500-2_23. [DOI] [PubMed] [Google Scholar]
- 44.Sommer N, Dietrich A, Schermuly RT, Ghofrani HA, Gudermann T, Schulz R, Seeger W, Grimminger F, Weissmann N. Regulation of hypoxic pulmonary vasoconstriction: basic mechanisms. Eur Respir J 32: 1639–1651, 2008. doi: 10.1183/09031936.00013908. [DOI] [PubMed] [Google Scholar]
- 45.Tasker PN, Michelangeli F, Nixon GF. Expression and distribution of the type 1 and type 3 inositol 1,4, 5-trisphosphate receptor in developing vascular smooth muscle. Circ Res 84: 536–542, 1999. doi: 10.1161/01.RES.84.5.536. [DOI] [PubMed] [Google Scholar]
- 46.Taylor CW. Kinetics of inositol 1,4,5-trisphosphate-stimulated Ca2+ mobilization. Adv Second Messenger Phosphoprotein Res 26: 109–142, 1992. [PubMed] [Google Scholar]
- 47.Taylor CW, Genazzani AA, Morris SA. Expression of inositol trisphosphate receptors. Cell Calcium 26: 237–251, 1999. doi: 10.1054/ceca.1999.0090. [DOI] [PubMed] [Google Scholar]
- 48.Thomsen AR, Hvidtfeldt M, Bräuner-Osborne H. Biased agonism of the calcium-sensing receptor. Cell Calcium 51: 107–116, 2012. doi: 10.1016/j.ceca.2011.11.009. [DOI] [PubMed] [Google Scholar]
- 49.Vermassen E, Fissore RA, Nadif Kasri N, Vanderheyden V, Callewaert G, Missiaen L, Parys JB, De Smedt H. Regulation of the phosphorylation of the inositol 1,4,5-trisphosphate receptor by protein kinase C. Biochem Biophys Res Commun 319: 888–893, 2004. doi: 10.1016/j.bbrc.2004.05.071. [DOI] [PubMed] [Google Scholar]
- 50.Voelkel NF, Mizuno S, Bogaard HJ. The role of hypoxia in pulmonary vascular diseases: a perspective. Am J Physiol Lung Cell Mol Physiol 304: L457–L465, 2013. doi: 10.1152/ajplung.00335.2012. [DOI] [PubMed] [Google Scholar]
- 51.Wahl MI, Jones GA, Nishibe S, Rhee SG, Carpenter G. Growth factor stimulation of phospholipase C-γ1 activity. Comparative properties of control and activated enzymes. J Biol Chem 267: 10447–10456, 1992. [PubMed] [Google Scholar]
- 52.Wang QS, Zheng YM, Dong L, Ho YS, Guo Z, Wang YX. Role of mitochondrial reactive oxygen species in hypoxia-dependent increase in intracellular calcium in pulmonary artery myocytes. Free Radic Biol Med 42: 642–653, 2007. doi: 10.1016/j.freeradbiomed.2006.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wang XT, McCullough KD, Wang XJ, Carpenter G, Holbrook NJ. Oxidative stress-induced phospholipase C-γ1 activation enhances cell survival. J Biol Chem 276: 28364–28371, 2001. doi: 10.1074/jbc.M102693200. [DOI] [PubMed] [Google Scholar]
- 54.Wang YX, Zheng YM. ROS-dependent signaling mechanisms for hypoxic Ca2+ responses in pulmonary artery myocytes. Antioxid Redox Signal 12: 611–623, 2010. doi: 10.1089/ars.2009.2877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Waypa GB, Guzy R, Mungai PT, Mack MM, Marks JD, Roe MW, Schumacker PT. Increases in mitochondrial reactive oxygen species trigger hypoxia-induced calcium responses in pulmonary artery smooth muscle cells. Circ Res 99: 970–978, 2006. doi: 10.1161/01.RES.0000247068.75808.3f. [DOI] [PubMed] [Google Scholar]
- 56.Waypa GB, Schumacker PT. Role for mitochondrial reactive oxygen species in hypoxic pulmonary vasoconstriction. Novartis Found Symp 272: 176–192, 2006. [PubMed] [Google Scholar]
- 57.Weigand L, Sylvester JT, Shimoda LA. Mechanisms of endothelin-1-induced contraction in pulmonary arteries from chronically hypoxic rats. Am J Physiol Lung Cell Mol Physiol 290: L284–L290, 2006. doi: 10.1152/ajplung.00449.2004. [DOI] [PubMed] [Google Scholar]
- 58.Weir EK, Archer SL. The mechanism of acute hypoxic pulmonary vasoconstriction: the tale of two channels. FASEB J 9: 183–189, 1995. doi: 10.1096/fasebj.9.2.7781921. [DOI] [PubMed] [Google Scholar]
- 59.Weir EK, Olschewski A. Role of ion channels in acute and chronic responses of the pulmonary vasculature to hypoxia. Cardiovasc Res 71: 630–641, 2006. doi: 10.1016/j.cardiores.2006.04.014. [DOI] [PubMed] [Google Scholar]
- 60.Welsh DJ, Scott P, Plevin R, Wadsworth R, Peacock AJ. Hypoxia enhances cellular proliferation and inositol 1,4, 5-triphosphate generation in fibroblasts from bovine pulmonary artery but not from mesenteric artery. Am J Respir Crit Care Med 158: 1757–1762, 1998. doi: 10.1164/ajrccm.158.6.9706054. [DOI] [PubMed] [Google Scholar]
- 61.Yadav VR, Song T, Joseph L, Mei L, Zheng YM, Wang YX. Important role of PLC-γ1 in hypoxic increase in intracellular calcium in pulmonary arterial smooth muscle cells. Am J Physiol Lung Cell Mol Physiol 304: L143–L151, 2013. doi: 10.1152/ajplung.00310.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhang WM, Yip KP, Lin MJ, Shimoda LA, Li WH, Sham JS. ET-1 activates Ca2+ sparks in PASMC: local Ca2+ signaling between inositol trisphosphate and ryanodine receptors. Am J Physiol Lung Cell Mol Physiol 285: L680–L690, 2003. doi: 10.1152/ajplung.00067.2003. [DOI] [PubMed] [Google Scholar]
- 63.Zheng YM, Mei QB, Wang QS, Abdullaev I, Lai FA, Xin HB, Kotlikoff MI, Wang YX. Role of FKBP12.6 in hypoxia- and norepinephrine-induced Ca2+ release and contraction in pulmonary artery myocytes. Cell Calcium 35: 345–355, 2004. doi: 10.1016/j.ceca.2003.09.006. [DOI] [PubMed] [Google Scholar]
- 64.Zheng YM, Wang QS, Liu QH, Rathore R, Yadav V, Wang YX. Heterogeneous gene expression and functional activity of ryanodine receptors in resistance and conduit pulmonary as well as mesenteric artery smooth muscle cells. J Vasc Res 45: 469–479, 2008. doi: 10.1159/000127438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zheng YM, Wang QS, Rathore R, Zhang WH, Mazurkiewicz JE, Sorrentino V, Singer HA, Kotlikoff MI, Wang YX. Type-3 ryanodine receptors mediate hypoxia-, but not neurotransmitter-induced calcium release and contraction in pulmonary artery smooth muscle cells. J Gen Physiol 125: 427–440, 2005. doi: 10.1085/jgp.200409232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ziino AJ, Ivanovska J, Belcastro R, Kantores C, Xu EZ, Lau M, McNamara PJ, Tanswell AK, Jankov RP. Effects of rho-kinase inhibition on pulmonary hypertension, lung growth, and structure in neonatal rats chronically exposed to hypoxia. Pediatr Res 67: 177–182, 2010. doi: 10.1203/PDR.0b013e3181c6e5a7. [DOI] [PubMed] [Google Scholar]