

Abstract
Chronic hypoxia (CH) augments basal and endothelin-1 (ET-1)-induced pulmonary vasoconstrictor reactivity through reactive oxygen species (ROS) generation and RhoA/Rho kinase (ROCK)-dependent myofilament Ca2+ sensitization. Because ROCK promotes actin polymerization and the actin cytoskeleton regulates smooth muscle tension, we hypothesized that actin polymerization is required for enhanced basal and ET-1-dependent vasoconstriction after CH. To test this hypothesis, both end points were monitored in pressurized, endothelium-disrupted pulmonary arteries (fourth-fifth order) from control and CH (4 wk at 0.5 atm) rats. The actin polymerization inhibitors cytochalasin and latrunculin attenuated both basal and ET-1-induced vasoconstriction only in CH vessels. To test whether CH directly alters the arterial actin profile, we measured filamentous actin (F-actin)-to-globular actin (G-actin) ratios by fluorescent labeling of F-actin and G-actin in fixed pulmonary arteries and actin sedimentation assays using homogenized pulmonary artery lysates. We observed no difference in actin polymerization between groups under baseline conditions, but ET-1 enhanced actin polymerization in pulmonary arteries from CH rats. This response was blunted by the ROS scavenger tiron, the ROCK inhibitor fasudil, and the mDia (RhoA effector) inhibitor small-molecule inhibitor of formin homology domain 2. Immunoblot analysis revealed an effect of CH to increase both phosphorylated (inactive) and total levels of the actin disassembly factor cofilin but not phosphorylated cofilin-to-total cofilin ratios. We conclude that actin polymerization contributes to increased basal pulmonary arterial constriction and ET-1-induced vasoconstrictor reactivity after CH in a ROS- and ROCK-dependent manner. Our results further suggest that enhanced ET-1-mediated actin polymerization after CH is dependent on mDia but independent of changes in the phosphorylated cofilin-to-total cofilin ratio.
NEW & NOTEWORTHY This research is the first to demonstrate a role for actin polymerization in chronic hypoxia-induced basal pulmonary arterial constriction and enhanced agonist-induced vasoconstrictor activity. These results suggest that a reactive oxygen species-Rho kinase-actin polymerization signaling pathway mediates this response and may provide a mechanistic basis for the vasoconstrictor component of pulmonary hypertension.
Keywords: actin polymerization, chronic hypoxia, pulmonary hypertension, reactive oxygen species, Rho kinase
INTRODUCTION
The adult pulmonary circulation consists of a low-pressure, low-resistance vasculature that normally exhibits little to no basal myogenic tone (21). However, chronic hypoxia (CH), which is associated with prolonged high-altitude exposure and chronic obstructive pulmonary diseases, increases pulmonary vascular resistance and leads to pulmonary hypertension (44). The resulting increase in afterload on the right heart causes right ventricular hypertrophy and can ultimately lead to right heart failure and death. Pulmonary artery narrowing attributable to acute hypoxic pulmonary vasoconstriction is widely thought to significantly contribute to elevated vascular resistance (45). However, additional vasoconstrictor influences, including elevated basal vascular smooth muscle (VSM) tone and enhanced sensitivity to vasoconstrictor stimuli such as endothelin-1 (ET-1), are thought to play an important role in CH-induced pulmonary hypertension (17, 29, 30, 43).
VSM contraction is largely determined by the phosphorylation state of the regulatory protein myosin light chain (MLC), which is regulated by the balance of activities of MLC kinase (MLCK) and MLC phosphatase (MLCP). Like in the systemic circulation, an increase in intracellular Ca2+ concentration ([Ca2+]i) in pulmonary arterial smooth muscle cells (PASMCs) promotes contraction through the activation of MLCK (21). Alternatively, inhibition of MLCP elicits contraction by increasing MLC phosphorylation (10). This increase in MLC phosphorylation and contraction occurring independent of changes in VSM [Ca2+]i is referred to as Ca2+ sensitization. In addition to Ca2+ sensitization, dynamic reorganization of the actin cytoskeleton is crucial for force generation in smooth muscle (18, 49) at actin-myosin cross bridges and force transmission to the plasma membrane, where cortical actin filaments bind to and reinforce integrins and focal adhesion proteins (21). We and others have previously shown that the RhoA/Rho kinase (ROCK) pathway is an important mediator of vasoconstriction in pulmonary hypertension (3, 4, 24, 38), and ROCK inhibitors significantly reduce pulmonary arterial pressure in CH rats (13, 19, 33, 35). ROCK directly phosphorylates and inhibits the regulatory myosin phosphatase-targeting subunit of MLCP (MYPT1), thereby increasing MLC phosphorylation. However, ROCK also increases levels of polymerized actin via phosphorylation of LIM kinase (LIMK) and subsequently cofilin. Cofilin plays an essential role in regulating actin dynamics, and its activities are regulated by the phosphorylation state of its NH2-terminal serine 3 (18). In the dephosphorylated (active) state, cofilin binds actin filaments and severs them. However, serine 3 phosphorylation blocks the ability of cofilin to bind filamentous actin (F-actin), thereby preventing its actin filament disassembly activity (46). Consequently, phosphorylated (inactive) cofilin is usually associated with increased actin phosphorylation, although active cofilin has also been shown to contribute to actin polymerization by increasing the available pool of actin monomers (2, 50). Furthermore, RhoA is known to activate the formin molecule mDia, which directly catalyzes actin nucleation and polymerization (47). Therefore, the primary aim of this study was to test the hypothesis that actin polymerization contributes to augmented basal and agonist-induced pulmonary arterial constriction after CH, occurring via the RhoA/ROCK signaling pathway.
Endogenous reactive oxygen species (ROS) are important regulators of pulmonary VSM phenotype and contractility after CH. Although overall ROS generation has been reported to be reduced in isolated lungs from CH rats (39), chronic ROS inhibition with the superoxide dismutase mimetic tempol inhibits right ventricular hypertrophy and pulmonary arterial remodeling in CH rats (12) and both ROS production and vascular remodeling in CH mice (32). ROS have also been implicated in the vasoconstrictor component of CH-induced pulmonary hypertension (17, 20, 29), with superoxide anion () mediating enhanced RhoA activity and ROCK-mediated vasoconstrictor responsiveness to ET-1 in small pulmonary arteries from CH rats (24). However, whether ROS contribute to CH-induced basal pulmonary arterial tone after CH is unknown and represents an additional focus of the present study.
METHODS
All protocols were approved by the Institutional Animal Care and Use Committee of the University of New Mexico Health Sciences Center and abide by National Institutes of Health guidelines for animal use.
Experimental Groups
Adult male Sprague-Dawley rats (~12 wk old, Harlan Industries) were randomly divided between control and hypoxic groups. Animals within the hypoxic group were maintained for 4 wk in a hypobaric chamber at 0.5 atm barometric pressure (PB) (PB ~380 mmHg and ambient Po2 ~80 mmHg) (41). The chamber was opened 3 times/wk to provide animals with food, water, and clean bedding. Control animals were maintained at ambient PB (PB ~630 mmHg and ambient Po2 ~132 mmHg, Albuquerque, NM) for a similar duration. All animals had ab libitum access to food and water and were housed on a 12:12-h light-dark cycle.
Isolated Small Pulmonary Artery Preparation
Small pulmonary arteries were isolated and cannulated for the simultaneous assessment of vasoreactivity and vessel wall [Ca2+]i as previously described (3). Rats were anesthetized with pentobarbital sodium (200 mg/kg ip), and the left lung was removed and immediately placed in ice-cold physiological saline solution (PSS) containing the following (in mM): 129.80 NaCl, 5.40 KCl, 0.83 MgSO4, 19.00 NaHCO3, 1.80 CaCl2 and 5.50 glucose. A pulmonary artery [100–200-μm internal diameter (ID), fourth-fifth order] of ~1-mm length was dissected free and transferred to a vessel chamber (CH-1, Living Systems) containing ice-cold PSS. The proximal end of the artery was cannulated with a tapered glass pipette, secured in place with a single strand of silk ligature, and gently flushed with PSS to remove any blood from the lumen. The vessel lumen was rubbed with a strand of moose mane to disrupt the endothelium before securing the distal end of the artery onto a second cannula. These microcannulae were created from borosilicate glass tubes [Sutter Instruments, outer diameter (OD) 1.2 mm and ID 0.69 mm] that were tapered to an approximate OD of 100 μm (Sutter Instruments P-30 Vertical Micropipette Puller). The vessel was stretched longitudinally to approximate in situ length and initially pressurized with PSS in the lumen to 12 mmHg with a servo-controlled peristaltic pump (Living Systems). All arteries were studied under no-flow conditions, achieved by closing the distal stopcock. Arteries were required to hold a steady pressure on switching off the servo-control function to verify the absence of leaks; any vessel with apparent leaks was discarded. The vessel chamber was transferred to the stage of an inverted microscope (Nikon Eclipse TS100), and the vessel was continuously superfused with PSS equilibrated with a gas mixture containing 10% O2, 6% CO2, and balance N2 (37°C). A vessel chamber cover was positioned to permit this same gas mixture to flow over the top of the chamber bath. We have previously reported that this gas mixture yields an approximate superfusate pH of 7.40, Po2 of 57 mmHg, and Pco2 of 31 mmHg (22). Bright-field videos of vessels were obtained with an IonOptix camera (CCD100M). Dimensional analysis was performed by IonOptix IonWizard software to measure vessel ID. To determine vessel viability, endothelium-disrupted arteries were preconstricted with UTP (5 μM, Sigma) to ~30% of baseline ID. A lack of a vasodilatory response to acetylcholine (10 μM, Sigma) confirmed endothelial disruption.
Measurement of Vessel Wall [Ca2+]i
Pressurized vessels were loaded abluminally with the cell-permeable Ca2+-sensitive fluorescent indicator fura-2 AM (Invitrogen) for 45 min at room temperature in the dark followed by a 20-min washout period. Fura-2-loaded vessels were alternately excited at 340 and 380 nm at a frequency of 1 Hz with an IonOptix Hyperswitch dual excitation light source (IonOptix), and the respective 510-nm emissions were collected with a photomultiplier tube. Background-subtracted 340-to-380-nm emission ratios were calculated with IonOptix IonWizard software and collected continuously throughout the experiment. Vessel wall [Ca2+]i was expressed as the ratio of background-subtracted 510-nm emission fluorescence intensity attributable to excitation at 340 nm over excitation at 380 nm (F340/F380).
Isolated Vessel Protocols
Role of ROS and actin polymerization in the development of basal tone after CH.
We have previously documented a critical contribution of to enhanced ET-1-mediated vasoconstrictor reactivity after CH (24). However, whether similarly contributes to increased basal pulmonary arterial tone in this setting is unknown. To address this possibility, after initially pressurizing the artery to 12 mmHg as described above, we determined pressure-dependent vasoconstriction by exposing isolated arteries from control and CH rats to a series of 10-mmHg pressure steps, beginning at 5 mmHg and reaching a maximum of 45 mmHg in Ca2+-replete PSS. Each pressure step lasted 5 min. Passive diameter was determined by repeating the pressure steps after 1 h of superfusion with Ca2+-free PSS [containing (in mM) 129.80 NaCl, 5.40 KCl, 0.83 MgSO4, 19.00 NaHCO3, 5.50 glucose, and 3.00 EGTA] and after flushing the lumen with Ca2+-free PSS followed by the reestablishment of no-flow conditions. Basal arterial constriction was calculated as the difference in ID between Ca2+-free and Ca2+-replete conditions expressed as a percentage of ID in Ca2+-free PSS at each pressure. Experiments were conducted in the presence of the scavenger tiron (10 mM, Sigma) (3, 24) or vehicle (PSS) to determine the role of ROS in this response.
To test the hypothesis that actin polymerization is required for CH-induced increases in basal pulmonary arterial tone, we repeated pressure-response curves in arteries from CH rats after treatment with the actin polymerization inhibitors cytochalasin B (CytB; 10 μM, Enzo) (11) and latrunculin B (LatB; 300 nM, Enzo) or vehicle (0.1% DMSO). These inhibitors or vehicle solutions were applied abluminally to the PSS bath in which the vessels were studied. Whereas CytB blocks the addition of actin subunits to existing actin filaments (6), LatB inhibits polymerization by sequestering actin monomers (7).
Contribution of actin polymerization to ET-1-induced vasoconstriction after CH.
To evaluate mechanisms of ET-1-dependent vasoconstriction independent of changes in vessel wall [Ca2+]i, we clamped [Ca2+]i in some arteries by permeabilizing with the Ca2+ ionophore ionomycin (3 μM, Sigma), as previously described (36). All Ca2+-permeabilized vessels were equilibrated at an internal pressure of 12 mmHg with PSS containing a calculated free Ca2+ concentration of 300 nM as previously described (24). This concentration of Ca2+ was chosen to provide optimal vasoreactivity to ET-1 while having minimal effects on basal Ca2+-dependent vasoconstriction based on a previous study (24). Vasoconstrictor responses to increasing concentrations of ET-1 (10−10−10−7 M) were measured in Ca2+-clamped vessels in the presence and absence of CytB or vehicle. Fura-2 ratios were monitored throughout all experiments to confirm Ca2+ clamp.
Fluorescence Microscopy for F-actin and G-actin in Intact Pulmonary Arteries
Because isolated vessel experiments do not provide a direct measure of the actin profile of control and CH vessels, we performed additional experiments to determine whether exposure to CH enhances levels of polymerized actin in pulmonary arteries. Relative actin polymerization levels can be expressed F-actin-to-globular actin (G-actin) ratios, so a fluorescence microscopy approach was used to image and quantify relative F-actin and G-actin levels within pulmonary arteries. Pulmonary arteries (fourth-fifth order) were isolated, cannulated, and subjected to ET-1-dependent vasoconstriction under Ca2+-permeabilized conditions as described above. After the conclusion of the vessel study, the vessel was depressurized by removal from the vessel chamber in the continued presence of ET-1, snap frozen in liquid nitrogen, and stored at −80°C until ready to use for F-actin and G-actin labeling.
On the day of labeling, arteries were fixed in ice-cold 4% paraformaldehyde, rinsed in ice-cold PBS, and permeabilized (0.2% Triton X-100, 30 min at 4°C). Arteries were incubated overnight at 4°C in the presence of Alexa Fluor phalloidin-568 (5 U/ml, ThermoFisher) to stain F-actin filaments and Alexa Fluor DNAse-488 (9 μg/ml, ThermoFisher) to label G-actin followed by incubation with the nuclear stain TO-PRO3 (0.5 μM, 20 min, room temperature, ThermoFisher). Vessels were then washed in ice-cold PBS, mounted on poly-l-lysine slides (ThermoFisher) with Prolong Gold Antifade Mountant (ThermoFisher), and coverslipped. Slides were stored at 4°C in the dark until image analysis was performed.
Arteries were imaged using a Leica laser-scanning microscope (TCS SP5) equipped with a ×63 glycerol immersion objective (numerical aperture: 1.30). Alexa Fluor 488-DNase I was excited using an argon laser, and emissions were processed through an Acousto-Optical Beam Splitter (AOBS; Leica) crystal set to an emission bandwidth of 500–550 nm. Alexa Fluor 568-phalloidin was excited using a DPSS 561 laser, and emissions were processed through an AOBS set to an emission bandwidth of 570–620 nm. TO-PRO3 was excited with a HeNe laser, and emissions were processed through AOBS set to 635–666 nm. Z-stacks were obtained at z-steps of 0.5 μm at constant detector gain and offset. Average fluorescence intensity of both phalloidin-568 and DNAse-488 was background subtracted (arteries not treated with phalloidin-568, DNAse-488, or TO-PRO3). To determine average fluorescence intensity of each label, six nonoverlapping rectangular regions of interest (ROIs) were drawn on each vessel. Within each rectangular ROI, a line ROI was drawn on 10 different cells adjacent to the nucleus to avoid detection of nuclear DNAse-488 or phalloidin-568 fluorescence. Cells designated for analysis were first identified in the TO-PRO3 channel, thus providing an unbiased cell selection method for assessment of phalloidin-568 and DNAse-488 fluorescence intensity. Using the line ROI, we assessed the average fluorescence intensity for each label. At each line ROI, we calculated the ratio of average fluorescence intensity of F-actin over that of G-actin. Data are reported as means ± SE; n represents the number of individual vessels (each vessel collected from only 1 animal) that was used for experimentation. Each individual n is an average of 60 technical replicates of F-actin-to-G-actin ratios calculated for each vessel.
Actin Cellular Fractionation and Western Blots
To complement fluorescence microscopic analysis of F-actin-to-G-actin ratios, a cellular fractionation protocol was used to measure F-actin and G-actin levels in pulmonary arteries from each group. First, pulmonary artery branches were isolated from control and CH rats in HBSS (GIBCO) supplemented with 20 μM CaCl2 and 1% penicillin-streptomycin. Unpressurized vessels were transferred to clean 1.5-ml tubes containing HEPES buffer + vehicle, fasudil (10 μM, Calbiochem), tiron (10 mM, Sigma), small-molecule inhibitor of formin homology 2 domain (SMIFH2; 20 μM, Sigma), or CytB (1 μM, Enzo). Vessels were incubated at 37°C for 15 min before the addition of vehicle or ET-1 (10 nM, Sigma) for another 10 min at 37°C. Vessels then were snap frozen in liquid nitrogen and stored at −80°C before homogenization. Actin cellular fractionation was performed per the protocol in the G-Actin/F-Actin In Vivo Assay Biochem Kit (no. BK037, Cytoskeleton). Briefly, vessels were homogenized with glass homogenizers in 200 μl of F-actin stabilization buffer supplemented with protease inhibitors (PIC02, Cytoskeleton) and phosphatase inhibitor cocktail (P0044, Sigma). After lysates had been cleared, 50 μl of the homogenate were saved for cofilin Western blots. The remaining lysate was centrifuged at 100,000 g (Optima TLX Ultracentrifuge, Beckman Coulter), pelleting F-actin while leaving G-actin in the supernatant. The pellet was resuspended in F-actin depolymerization solution in a volume equivalent to the supernatant. Protein concentrations were determined with a spectrophotometer (Nano Drop 2000, Thermo Scientific). Supernatant fractions (8 μg protein), the equivalent volume of pelleted fractions, and actin standards (AKL99, Cytoskeleton) were separated by SDS-PAGE (no. 456-1044, Bio-Rad), transferred onto methanol-activated PVDF membranes (no. 162-0176, Bio-Rad), and blocked in Tris-buffered saline with 0.1% Tween 20 with 5% nonfat milk (Carnation) at room temperature for 1 h. Blots were probed with a pan-actin polyclonal primary antibody (AAN01, Cytoskeleton) in Tris-buffered saline with 0.1% Tween 20 with 0.1% milk at 4°C overnight and horseradish peroxidase-conjugated goat anti-rabbit secondary antibody (1:3,000, no. 172-1019, lot no. 64026773, Bio-Rad) for 1 h at room temperature the next day. Bands were developed with ECL Western blotting substrate (no. 32209, Thermo Scientific), and densitometry quantification was performed using ImageJ (National Institutes of Health).
Phosphorylated Cofilin/Cofilin Western Blots
Because ROCK is known to stimulate actin polymerization indirectly through LIMK phosphorylation and subsequent cofilin phosphorylation, an immunoblot method was used to measure both phosphorylated (p)-cofilin and total cofilin levels in pulmonary artery lysates. Lysates were collected as described above. Total protein extracts were separated by SDS-PAGE and blotted using anti-cofilin phospho-serine 3 (1:500, no. 3311S, lot no. 6, Cell Signaling) and anti-β-actin (1:14,000, no. ab8227, lot no. GR186254-1, Abcam). p-Cofilin blots were stripped and reprobed with anti-cofilin primary antibody (1:5,000, no. ab42824, lot no. GR295201-1, Abcam) to assess total cofilin levels. All bands were detected with horseradish peroxidase-conjugated goat anti-rabbit secondary antibody and ECL as previously described. Densitometry was performed using ImageJ. Values are expressed as ratios of p-cofilin to total cofilin.
Statistical Analysis
All data are expressed as means ± SE. A t-test or two-way ANOVA was used to make comparisons as appropriate. Data expressed as proportions were arcsine transformed before statistical analysis. A Student-Newman-Keuls post hoc test was used when differences were detected by ANOVA. P < 0.05 was accepted as statistically significant.
RESULTS
ROS Contribute to CH-Induced Basal Vasoconstriction
In agreement with a previous study (4), CH vessels exhibited significantly elevated basal constriction compared with control vessels at intraluminal pressures of 15–45 mmHg (Fig. 1A). This vasoconstrictor response in CH arteries was not associated with an increase in vessel wall [Ca2+]i (Fig. 1B). Neither baseline ID (control: 165 ± 14 μm and CH: 147 ± 14 μm, 12-mmHg intraluminal pressure) nor fura-2 emission ratios (Fig. 1B) differed significantly between groups.
Fig. 1.
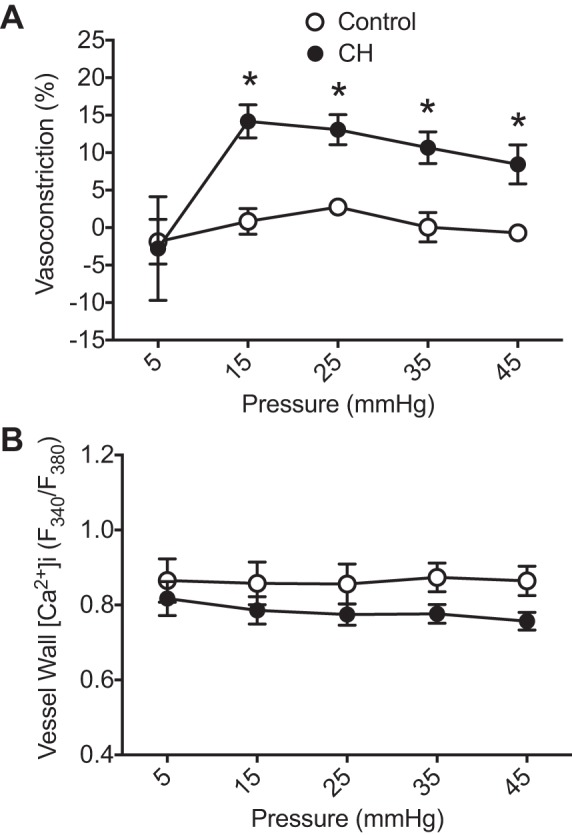
Chronic hypoxia (CH) increases basal pulmonary arterial constriction. A: basal constriction (percentage of passive inner diameter achieved by removal of extracellular Ca2+); B: vessel wall intracellular Ca2+ concentration ([Ca2+]i) [fura 2 340-to-380-nm emission ratios (F340/F380)] as a function of intraluminal pressure in endothelium-disrupted small pulmonary arteries from control and CH rats. n = 6–7/group. *P < 0.05 vs. control.
Pressure-dependent constriction in CH arteries was abolished by the scavenger tiron (3, 36), supporting a contribution of ROS to this response (Fig. 2A). On the basis of a previous study indicating that control arteries do not develop basal tone (4), experiments with tiron were conducted solely in arteries from CH rats. Tiron was without effect on [Ca2+]i, as assessed by fura-2 ratios (data not shown).
Fig. 2.
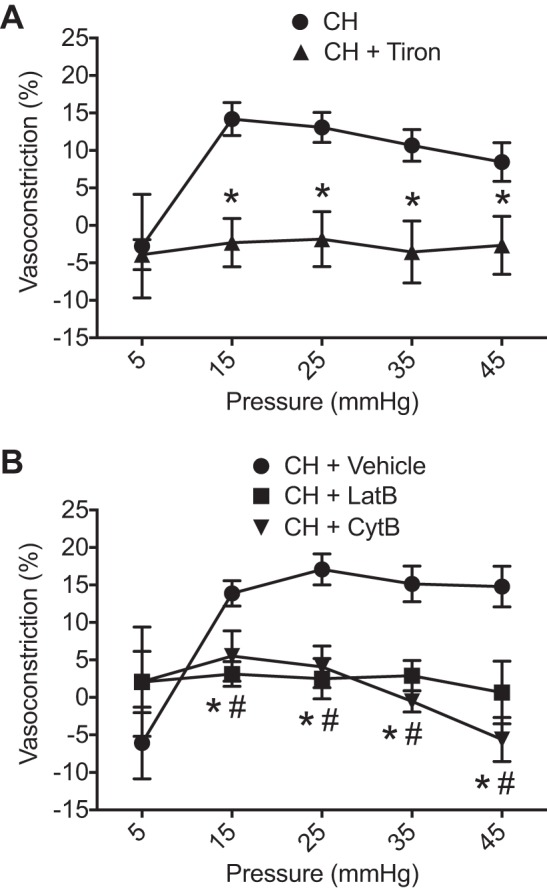
Reactive oxygen species (ROS) and actin polymerization contribute to chronic hypoxia (CH)-induced basal arterial constriction. A: basal constriction (percentage of passive internal diameter achieved by removal of extracellular Ca2+) as a function of intraluminal pressure in CH arteries pretreated with tiron (10 mM). n = 7–8/group. *P < 0.05 vs. CH vehicle. CH vehicle data are from Fig. 1A. B: basal constriction in CH vessels pretreated with actin polymerization inhibitors latrunculin B (LatB; 300 nM) or cytochalasin B (CytB; 10 μM). All experiments were conducted using endothelium-disrupted arteries. n = 5–7/group. *P < 0.05, CH + LatB vs. CH vehicle; #P < 0.05, CH + CytB vs. CH vehicle.
Actin Polymerization Contributes to Basal Pressure-Dependent Vasoconstriction and ET-1-Induced Vasoconstrictor Reactivity After CH
Similar to effects of tiron, the actin polymerization inhibitors LatB and CytB blocked the development of arterial constriction in response to increasing intraluminal pressure (Fig. 2B). Fura-2 ratios were unaltered by administration of actin polymerization inhibitors (data not shown).
To directly evaluate the role of actin polymerization in ET-1-induced vasoconstriction independent of changes in [Ca2+]i, we performed cumulative concentration-response curves to ET-1 in arteries from each group that had been permeabilized to Ca2+ using ionomycin to clamp [Ca2+]i (23, 24). Resting ID did not differ between control (156 ± 18 μm) or CH arteries (153 ± 12 μm). In agreement with our previous study (24), vasoconstrictor sensitivity to ET-1 (10−8–10−7 M) was greater in Ca2+-permeabilized arteries from CH rats compared with control vessels (Fig. 3A). Fura-2 ratios did not increase in response to ET-1 and were similar between groups, confirming the efficacy of ionomycin to clamp [Ca2+]i (Fig. 3B). Consistent with effects of actin polymerization inhibitors to prevent CH-induced basal arterial constriction (Fig. 2B), CytB significantly blunted the vasoconstrictor response to ET-1 in CH vessels (Fig. 4B). In contrast, CytB was without effect on responses to ET-1 control pulmonary arteries (Fig. 4A). Collectively, these data suggest that actin polymerization contributes to both pressure-dependent and ET-1-induced vasoconstriction selectively in arteries from CH rats.
Fig. 3.
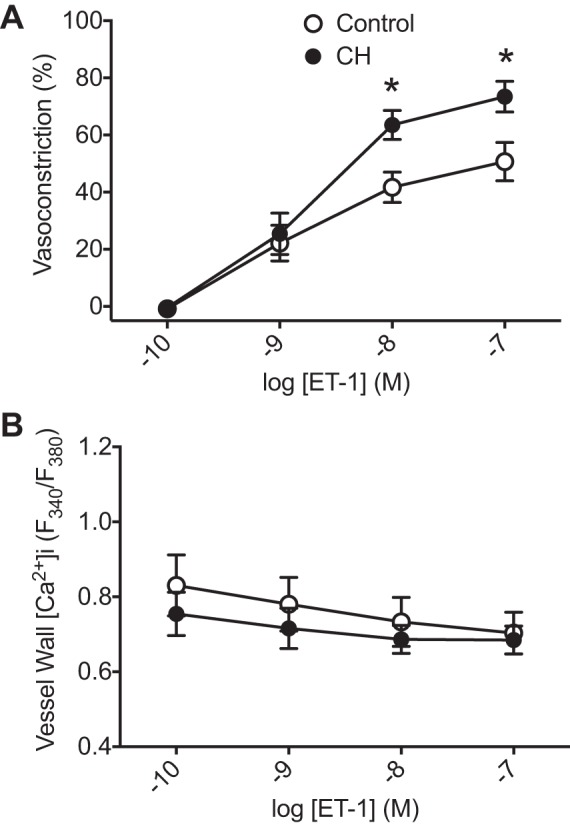
Chronic hypoxia (CH) increases vasoconstrictor sensitivity to endothelin-1 (ET-1). Vasoconstriction (percent baseline internal diameter; A) and fura-2 ratios (B) in endothelium-disrupted, Ca2+-clamped control and CH pulmonary arteries in response to increasing concentrations of ET-1 are shown. n = 5–6/group. *P < 0.05 vs. control.
Fig. 4.
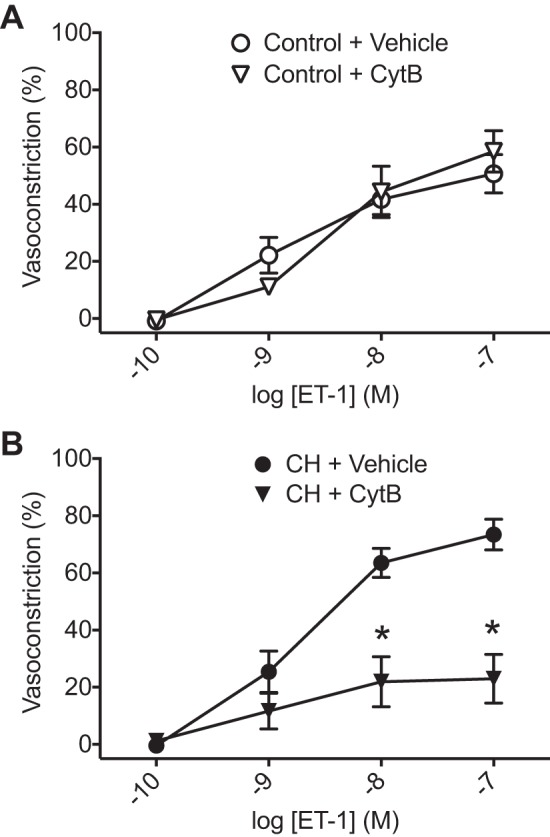
Actin polymerization contributes to enhanced endothelin-1 (ET-1)-induced vasoconstrictor reactivity after chronic hypoxia (CH). Vasoconstrictor responses to ET-1 in Ca2+-permeabilized, endothelium-disrupted pulmonary arteries from control (A) and CH rats (B) in the presence and absence of cytochalasin B (CytB; 10 μM) are shown. Vehicle data are from Fig. 3A. n = 5–6/group. *P < 0.05 vs. CH vehicle.
ROS, ROCK, and mDia Mediate ET-1-Induced Actin Polymerization After CH
Effects of ET-1 to stimulate actin polymerization were compared between small pulmonary arteries from control and CH rats using fluorescent molecular labeling techniques to quantify the ratio of F-actin to G-actin levels (Fig. 5A). Although F-actin-to-G-actin ratios were not different between arteries from control and CH rats under basal conditions, ET-1 produced a significant increase in the F-actin-to-G-actin ratio after CH (Fig. 5B) but not in control arteries.
Fig. 5.

Endothelin-1 (ET-1) stimulates actin polymerization in pulmonary arteries from chronic hypoxia (CH) rats. A: representative fluorescence images of DNaseI 488 (green; G-actin), phalloidin 555 (red; F-actin), and TO-PRO (blue; nuclei) in pulmonary arteries from control and CH rats treated with ET-1 (10 nM) or vehicle. Drawings on the right indicate artery orientation. B: fluorescence intensity was quantified to determine F-actin-to-G-actin ratios. n = 4/group. *P < 0.05 vs. CH vehicle.
In agreement with these findings from fluorescent staining protocols, actin fractionation assays demonstrated no differences in basal F-actin-to-G-actin ratios between control and CH vessels, but treatment with ET-1 significantly enhanced actin polymerization in CH pulmonary arteries compared with both CH vehicle-treated arteries and control ET-1-treated arteries (Fig. 6). Pretreatment with the ROCK-specific inhibitor fasudil, the ROS scavenger tiron, the mDia inhibitor SMIFH2, or CytB prevented this response to ET-1 in CH arteries while having no effect on control vessels (Fig. 6).
Fig. 6.
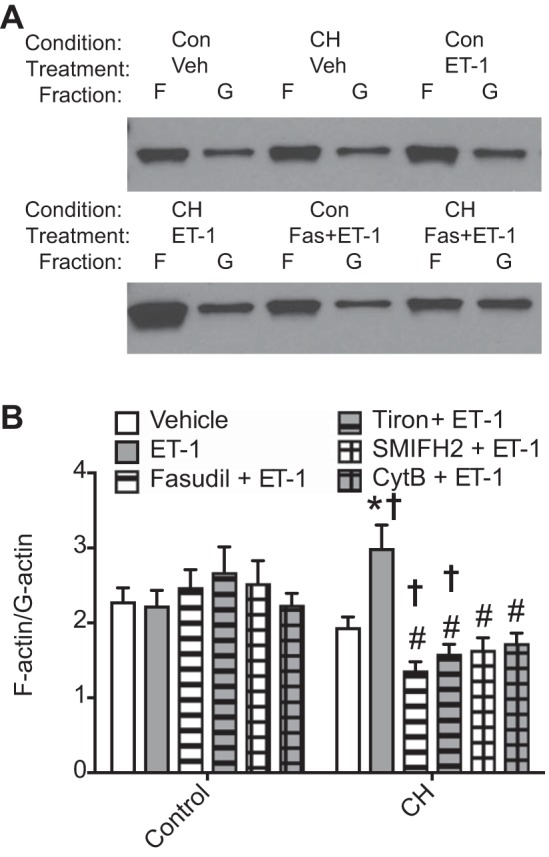
Chronic hypoxia (CH) augments endothelin-1 (ET-1)-stimulated actin polymerization in a Rho kinase-, reactive oxygen species-, and mDia-dependent manner. Intrapulmonary arteries from control (Con) and CH rats were treated with ET-1 (10 nM) or vehicle in the presence and absence of the following inhibitors: fasudil (Fas; 10 μM), tiron (10 mM), small-molecule inhibitor of formin homology domain 2 (SMIFH2; 20 μM), or cytochalasin B (CytB; 1 μM). A: Western blots were performed on fractionated lysates for which actin was probed. Representative blots showing actin in F-actin and G-actin fractions under indicated conditions. B: F-actin and G-actin were quantified by band intensity. n = 7–10/group. *P < 0.05 vs. CH vehicle; †P < 0.05 vs. the respective control; #P < 0.05 vs. CH ET-1.
Cofilin is an actin disassembly factor that is inhibited by LIMK-dependent phosphorylation, contributing to agonist-induced actin polymerization and smooth muscle contraction (1, 18, 46). To determine whether ET-1-induced actin polymerization in CH arteries correlates with cofilin phosphorylation, we measured levels of p-cofilin and total cofilin protein by Western blot analysis in pulmonary artery lysates from each group. No differences in p-cofilin-to-cofilin ratios were detected between control and CH vessels (Fig. 7, A and B) in any treatment group, but overall levels of both p-cofilin and cofilin were higher in CH vessels (Fig. 7, C and D). Neither the ratio of p-cofilin to cofilin nor levels of p-cofilin or total cofilin were altered by ET-1, fasudil, tiron, SMIFH2, or CytB. Taken together, these data indicate that ROS, ROCK, and mDia mediate ET-1-induced actin polymerization after CH, but this is not dependent on greater p-cofilin-to-cofilin ratios.
Fig. 7.

Chronic hypoxia (CH) increases phosphorylated (p-)cofilin and cofilin protein levels but not the p-cofilin-to-cofilin ratio. Western blots were performed on unfractionated lysates of intrapulmonary arteries from control (Con) and CH rats, and p-cofilin and cofilin protein levels were quantified by band intensity and normalized to β-actin protein expression. A: representative blots showing p-cofilin, cofilin, and β-actin levels in control and CH lysates under vehicle (Veh) and endothelin-1 (ET-1)-treated conditions. B: p-cofilin-to-cofilin ratio. SMIFH2, small-molecule inhibitor of formin homology domain 2; CytB, cytochalasin B. C: p-cofilin/β-actin. D: cofilin/β-actin levels. n = 6–9/group. *P < 0.05 CH vs. control.
DISCUSSION
The overall goal of this study was to test the hypothesis that actin polymerization contributes to augmented basal and agonist-induced pulmonary arterial vasoconstriction after CH downstream of the ROS/RhoA/ROCK signaling pathway. Our results indicate that 1) actin polymerization was required for enhanced vasoconstrictor responsiveness in isolated vessels from rats exposed to CH, both at baseline and in response to ET-1; 2) stimulation with ET-1 increased actin polymerization as measured by the F-actin-to-G-actin ratio in pulmonary arteries from CH, but not control, animals; and 3) scavenging of and pharmacological inhibition of ROCK and mDia prevented ET-1-induced actin polymerization in pulmonary arteries from CH animals, independent of alterations in p-cofilin-to-cofilin ratios. Collectively, these findings suggest that actin polymerization contributes to augmented pulmonary vasoreactivity after CH in a ROS- and ROCK-dependent manner (see Fig. 8).
Fig. 8.

Schematic of the proposed signaling pathway for actin polymerization-mediated vasoconstriction after chronic hypoxia (CH). Pathways in gray were hypothesized but not supported by experimental results. CytB, cytochalasin B; ETR, endothelin-1 receptor; ECM, extracellular matrix; ET-1, endothelin-1; LatB, latrunculin B; LIMK, LIM kinase; PASMC, pulmonary arterial smooth muscle cell; ROCK, Rho kinase; SMIFH2, small-molecule inhibitor of formin homology domain 2.
Previous reports have indicated that can activate RhoA in PASMCs (28) and that CH facilitates the coupling of receptor stimulation or depolarizing stimuli to RhoA activation and subsequent Ca2+ sensitization (3, 24). Although an earlier study from our laboratory demonstrated an effect of CH to induce pressure-dependent tone in small pulmonary arteries that is similarly RhoA mediated (4), the contribution of ROS to this response is unknown. Considering that increasing pressure in CH arteries leads to PASMC membrane depolarization independent of Ca2+ influx, we presently set out to address the potential contribution of ROS to basal tone in arteries from pulmonary hypertensive rats. In this study, the scavenger tiron was used to deplete ROS generated in arterioles isolated from control and CH rats. Consistent with previous studies implicating ROS in augmented agonist- and depolarization-induced pulmonary arterial constriction after CH (3, 24), tiron significantly blunted the vasoconstriction observed in CH arteries, confirming that generation is necessary for hypoxia-induced basal vasoconstriction.
ROS have previously been shown to mediate Ca2+ sensitization in pulmonary VSM after CH in a RhoA/ROCK-dependent manner (3, 24, 28). Elevated ROS after CH augment ET-1-induced pulmonary artery vasoconstriction, and this response is attenuated, but not abolished, by pharmacological ROCK inhibition and MLCK inhibition (24). Furthermore, ET-1-induced increases in MYPT1 phosphorylation were significantly reduced by ROCK inhibition (24). These results suggest that pulmonary vasoconstriction after CH is at least partially mediated by ROCK-dependent phosphorylation and inactivation of MYPT1 and subsequent increased activity of MLC and PASMC contraction.
It has also been well documented that the RhoA/ROCK pathway lies upstream of actin polymerization (31), and polymerization of actin filaments is important for contraction and force generation in smooth muscle (46). The role of filamentous actin in actomyosin cross-bridge cycling is well established and widely recognized as the primary mechanism for tension development and shortening in muscle cells, but a more recent study has highlighted a role for actin polymerization and cytoskeletal dynamics in smooth muscle contraction that extend beyond actomyosin interactions and cross-bridge cycling (18). The pharmacological agents CytB and LatB, which inhibit actin polymerization by capping the ends of growing actin filaments and sequestering free globular actin monomers, respectively, have been widely used to assess the role of actin polymerization in smooth muscle cell contraction. These inhibitors significantly inhibit contractility and force generation in airway smooth muscle (34, 48), systemic VSM (5, 40), and uterine and intestinal smooth muscle (37, 42). Fediuk et al. (15, 16) also demonstrated a role for actin polymerization-mediated constriction in pulmonary arteries in response to thromboxane in a porcine model of persistent pulmonary hypertension of the newborn. Much of this evidence suggests that actin polymerization occurring in a submembranous region of the smooth muscle cell enhances contractility by more strongly transmitting tension generated by cross-bridge cycling (14, 18, 49). On the basis of these studies, RhoA/ROCK-induced actin polymerization may provide an alternative parallel pathway to cause contraction independent of changes in [Ca2+]i and MLC activity in pulmonary arteries.
Pharmacological treatment with either CytB or LatB abolished CH-induced basal vasoconstriction, and pretreatment with CytB prevented the augmented pulmonary artery vasoconstrictor response to ET-1 that has previously been observed after CH (24). Taken together, these results suggest that actin polymerization is required for elevated basal pulmonary arterial tone and ET-1 vasoconstrictor sensitivity after CH exposure. Because actin polymerization measurements are complicated by the dynamic nature of the actin cytoskeleton, F-actin-to-G-actin ratios were used as a proxy for relative levels of polymerized actin. Interestingly, both immunofluorescent and cellular fractionation methods determined that pulmonary arteries from CH animals show elevated actin polymerization levels in response to acute ET-1 treatment but not at baseline. This lack of difference in baseline F-actin-to-G-actin ratios between control and CH vessels may be due to the fact that the pulmonary arteries were not pressurized before ET-1 stimulation, and pressure may be a stimulus for actin polymerization. Alternatively, PASMC contraction may exhibit a greater dependency on levels of polymerized actin after CH, analogous to its dependence on ROCK-dependent Ca2+ sensitization. ROCK does not play an appreciable role in either depolarization-stimulated or ET-1-induced constriction in control arteries. However, on the basis of previous studies, CH allows coupling of these stimuli to ROS- and ROCK-dependent constriction (3, 4, 23, 24). In the present study, the acute development of tone in response to intraluminal pressure and the significant increase in actin polymerization in response to acute exposure to ET-1 point to a very rapid, dynamic actin polymerization response in CH vessels. Importantly, this response was abolished by scavenging of with tiron, pharmacological inhibition of ROCK and mDia with fasudil and SMIFH2, respectively, or inhibition of actin polymerization with CytB. These results suggest that the actin polymerization response to ET-1 is dependent on ROS and two RhoA effectors, ROCK and mDia.
mDia directly promotes actin polymerization by nucleating filaments in concert with profilin (47). Alternatively, ROCK activation leads to actin polymerization through an indirect method. ROCK phosphorylates LIMK, which then phosphorylates cofilin (1). Cofilin is known to sever actin filaments, but, when it becomes phosphorylated (inactivated), these activities are inhibited, allowing actin filaments to extend. However, dephosphorylated (active) cofilin also contributes to actin filament assembly by replenishing the globular actin monomers that are necessary for polymerization (2). Zhao et al. (50) previously showed that stimulation of tracheal smooth muscle with acetylcholine causes contraction via dephosphorylation of cofilin. When a constitutively phosphorylated cofilin mutant, cofilin S3E, is overexpressed, acetylcholine is unable to stimulate actin polymerization and contraction because of the lack of available actin monomers. Traditionally, an elevated p-cofilin-to-cofilin ratio indicates signaling through the ROCK-LIMK-cofilin pathway. To test the hypothesis that CH-induced actin polymerization is dependent on this pathway, Western blots were performed to measure levels of p-cofilin and cofilin in pulmonary artery homogenates. Surprisingly, there were no differences in p-cofilin-to-cofilin ratios between control and CH pulmonary arteries, although others have previously reported a less robust relationship between cofilin phosphorylation and actin polymerization in pulmonary arteries than expected (15). In contrast, we observed overall higher levels of both p-cofilin and cofilin in CH vessels compared with control vessels, indicative of a larger pool of cofilin, which may more readily respond to stimuli. Upregulation of cofilin expression has previously been observed by Dai et al. (9) in a monocrotaline model of pulmonary hypertension, where it was proposed that high levels of cofilin correspond to a more dynamic subcortical actin network in motile PASMCs. This high level of cofilin expression may also explain why there was no basal elevation in polymerized actin levels in CH vessels and why the polymerization response to ET-1 was so rapid. Finally, because inhibition of mDia significantly attenuated actin polymerization in CH arteries, and the phosphorylation state of cofilin has no regulatory effect on mDia activity, it appears that the actin polymerization response to ET-1 may be mediated by the combined influences of these distinct signaling pathways.
In conclusion, this study supports a previously undescribed role of actin polymerization to mediate CH-induced basal pulmonary arterial tone and vasoreactivity to ET-1 in a ROS-, ROCK-, and mDia-dependent manner. These results advance our understanding of the signaling mechanisms involved in CH-induced vasoconstriction and associated pulmonary hypertension. Our results suggest that inhibition of ROCK may target both Ca2+ sensitization and actin polymerization pathways, either in series or in parallel. Actin polymerization may represent a component of Ca2+ sensitization because it may provide greater interaction between myosin and actin, such that a greater contraction is achieved for a given level of Ca2+. Furthermore, because mDia lies parallel to and not downstream of ROCK, inhibition of mDia-mediated actin polymerization may provide an additional target for reducing vasoconstrictor responsiveness after CH.
Future studies should address whether this signaling pathway is active in response to other vasoreactive stimuli. This novel finding that actin polymerization plays a particular role in CH-induced pulmonary vasoconstriction leads to some interesting questions left to be answered. Traditionally, actin polymerization in the vasculature has been studied in the context of smooth muscle cell contractility. The actin that interacts with myosin generates tension and cell shortening and is usually referred to as contractile actin (49). Clearly, some basal level of actin polymerization is needed to form actin-myosin cross bridges, and this is a very stable group of filaments. Previous studies have suggested that the actin already bound to tropomyosin at cross bridges is resistant to cofilin and at least partially protected against disassembly by cytochalasin and latrunculin (8, 25). It is unlikely that the CytB treatments performed in this study targeted depolymerization of these already-existing actin filaments for two reasons: 1) F-actin-to-G-actin ratios in CytB-treated pulmonary artery lysates were not different from vehicle-treated vessels (Fig. 6) and 2) CytB-treated control pulmonary arteries constricted in response to ET-1 in a manner similar to vehicle-treated vessels (Fig. 4). However, it is possible that after CH, either ET-1 treatment or increased vascular pressure induces the assembly of additional contractile units by increasing contractile actin polymerization. Such activities would be especially sensitive to the actin polymerization inhibitors CytB and LatB because they prevent assembly of new actin filaments. Alternatively, ET-1 treatment after CH exposure may instead strengthen force transmission of already-existing contractile units to the extracellular matrix by increasing actin polymerization near the cell cortex (see Fig. 8), which has been observed in other studies in VSM (14, 26, 27). It is possible that exposure to CH relies on this dynamic actin pool for elevated arterial tone and sensitivity to agonists. Additional studies examining actin isoform levels and actin filament subcellular localization will be needed to address this question.
GRANTS
This work was supported by National Institutes of Health Grants R01-HL-132883, R01-HL-088192, and T32-HL-007736 (to T. C. Resta) and K12-GM-088021 (to A. Wandinger-Ness).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
L.W.-C., M.S., J.R.S., B.R.B., J.B.S., L.V.G.B., N.L.J., B.R.W., and T.C.R. conceived and designed research; L.W.-C., M.S., and J.R.S. performed experiments; L.W.-C., M.S., J.R.S., and T.C.R. analyzed data; L.W.-C., M.S., J.R.S., B.R.B., J.B.S., L.V.G.B., N.L.J., B.R.W., and T.C.R. interpreted results of experiments; L.W.-C., M.S., and J.R.S. prepared figures; L.W.-C., M.S., and J.R.S. drafted manuscript; L.W.-C., M.S., J.R.S., B.R.B., J.B.S., L.V.G.B., N.L.J., B.R.W., and T.C.R. edited and revised manuscript; L.W.-C., M.S., J.R.S., B.R.B., J.B.S., L.V.G.B., N.L.J., B.R.W., and T.C.R. approved final version of manuscript.
ACKNOWLEDGMENTS
The authors thank Minerva Murphy for technical assistance.
REFERENCES
- 1.Bamburg JR, Bernstein BW. Roles of ADF/cofilin in actin polymerization and beyond. F1000 Biol Rep 2: 62, 2010. doi: 10.3410/B2-62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bamburg JR, McGough A, Ono S. Putting a new twist on actin: ADF/cofilins modulate actin dynamics. Trends Cell Biol 9: 364–370, 1999. doi: 10.1016/S0962-8924(99)01619-0. [DOI] [PubMed] [Google Scholar]
- 3.Broughton BR, Jernigan NL, Norton CE, Walker BR, Resta TC. Chronic hypoxia augments depolarization-induced Ca2+ sensitization in pulmonary vascular smooth muscle through superoxide-dependent stimulation of RhoA. Am J Physiol Lung Cell Mol Physiol 298: L232–L242, 2010. doi: 10.1152/ajplung.00276.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Broughton BR, Walker BR, Resta TC. Chronic hypoxia induces Rho kinase-dependent myogenic tone in small pulmonary arteries. Am J Physiol Lung Cell Mol Physiol 294: L797–L806, 2008. doi: 10.1152/ajplung.00253.2007. [DOI] [PubMed] [Google Scholar]
- 5.Cipolla MJ, Gokina NI, Osol G. Pressure-induced actin polymerization in vascular smooth muscle as a mechanism underlying myogenic behavior. FASEB J 16: 72–76, 2002. doi: 10.1096/cj.01-0104hyp. [DOI] [PubMed] [Google Scholar]
- 6.Cooper JA. Effects of cytochalasin and phalloidin on actin. J Cell Biol 105: 1473–1478, 1987. doi: 10.1083/jcb.105.4.1473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Coué M, Brenner SL, Spector I, Korn ED. Inhibition of actin polymerization by latrunculin A. FEBS Lett 213: 316–318, 1987. doi: 10.1016/0014-5793(87)81513-2. [DOI] [PubMed] [Google Scholar]
- 8.Creed SJ, Bryce N, Naumanen P, Weinberger R, Lappalainen P, Stehn J, Gunning P. Tropomyosin isoforms define distinct microfilament populations with different drug susceptibility. Eur J Cell Biol 87: 709–720, 2008. doi: 10.1016/j.ejcb.2008.03.004. [DOI] [PubMed] [Google Scholar]
- 9.Dai YP, Bongalon S, Tian H, Parks SD, Mutafova-Yambolieva VN, Yamboliev IA. Upregulation of profilin, cofilin-2 and LIMK2 in cultured pulmonary artery smooth muscle cells and in pulmonary arteries of monocrotaline-treated rats. Vascul Pharmacol 44: 275–282, 2006. doi: 10.1016/j.vph.2005.11.008. [DOI] [PubMed] [Google Scholar]
- 10.Dakshinamurti S. Regulation of myosin light chain phosphatase and pulmonary arterial relaxation. Can J Physiol Pharmacol 83: 893–898, 2005. doi: 10.1139/y05-087. [DOI] [PubMed] [Google Scholar]
- 11.de Frutos S, Diaz JM, Nitta CH, Sherpa ML, Bosc LV. Endothelin-1 contributes to increased NFATc3 activation by chronic hypoxia in pulmonary arteries. Am J Physiol Cell Physiol 301: C441–C450, 2011. doi: 10.1152/ajpcell.00029.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Elmedal B, de Dam MY, Mulvany MJ, Simonsen U. The superoxide dismutase mimetic, tempol, blunts right ventricular hypertrophy in chronic hypoxic rats. Br J Pharmacol 141: 105–113, 2004. doi: 10.1038/sj.bjp.0705580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fagan KA, Oka M, Bauer NR, Gebb SA, Ivy DD, Morris KG, McMurtry IF. Attenuation of acute hypoxic pulmonary vasoconstriction and hypoxic pulmonary hypertension in mice by inhibition of Rho-kinase. Am J Physiol Lung Cell Mol Physiol 287: L656–L664, 2004. doi: 10.1152/ajplung.00090.2003. [DOI] [PubMed] [Google Scholar]
- 14.Fediuk J, Dakshinamurti S. A role for actin polymerization in persistent pulmonary hypertension of the newborn. Can J Physiol Pharmacol 93: 185–194, 2015. doi: 10.1139/cjpp-2014-0413. [DOI] [PubMed] [Google Scholar]
- 15.Fediuk J, Gutsol A, Nolette N, Dakshinamurti S. Thromboxane-induced actin polymerization in hypoxic pulmonary artery is independent of Rho. Am J Physiol Lung Cell Mol Physiol 302: L13–L26, 2012. doi: 10.1152/ajplung.00016.2011. [DOI] [PubMed] [Google Scholar]
- 16.Fediuk J, Sikarwar AS, Nolette N, Dakshinamurti S. Thromboxane-induced actin polymerization in hypoxic neonatal pulmonary arterial myocytes involves Cdc42 signaling. Am J Physiol Lung Cell Mol Physiol 307: L877–L887, 2014. doi: 10.1152/ajplung.00036.2014. [DOI] [PubMed] [Google Scholar]
- 17.Fike CD, Slaughter JC, Kaplowitz MR, Zhang Y, Aschner JL. Reactive oxygen species from NADPH oxidase contribute to altered pulmonary vascular responses in piglets with chronic hypoxia-induced pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol 295: L881–L888, 2008. doi: 10.1152/ajplung.00047.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gunst SJ, Zhang W. Actin cytoskeletal dynamics in smooth muscle: a new paradigm for the regulation of smooth muscle contraction. Am J Physiol Cell Physiol 295: C576–C587, 2008. doi: 10.1152/ajpcell.00253.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hyvelin JM, Howell K, Nichol A, Costello CM, Preston RJ, McLoughlin P. Inhibition of Rho-kinase attenuates hypoxia-induced angiogenesis in the pulmonary circulation. Circ Res 97: 185–191, 2005. doi: 10.1161/01.RES.0000174287.17953.83. [DOI] [PubMed] [Google Scholar]
- 20.Jankov RP, Kantores C, Pan J, Belik J. Contribution of xanthine oxidase-derived superoxide to chronic hypoxic pulmonary hypertension in neonatal rats. Am J Physiol Lung Cell Mol Physiol 294: L233–L245, 2008. doi: 10.1152/ajplung.00166.2007. [DOI] [PubMed] [Google Scholar]
- 21.Jernigan NL, Resta TC. Calcium homeostasis and sensitization in pulmonary arterial smooth muscle. Microcirculation 21: 259–271, 2014. doi: 10.1111/micc.12096. [DOI] [PubMed] [Google Scholar]
- 22.Jernigan NL, Resta TC, Walker BR. Contribution of oxygen radicals to altered NO-dependent pulmonary vasodilation in acute and chronic hypoxia. Am J Physiol Lung Cell Mol Physiol 286: L947–L955, 2004. doi: 10.1152/ajplung.00215.2003. [DOI] [PubMed] [Google Scholar]
- 23.Jernigan NL, Walker BR, Resta TC. Chronic hypoxia augments protein kinase G-mediated Ca2+ desensitization in pulmonary vascular smooth muscle through inhibition of RhoA/Rho kinase signaling. Am J Physiol Lung Cell Mol Physiol 287: L1220–L1229, 2004. doi: 10.1152/ajplung.00196.2004. [DOI] [PubMed] [Google Scholar]
- 24.Jernigan NL, Walker BR, Resta TC. Reactive oxygen species mediate RhoA/Rho kinase-induced Ca2+ sensitization in pulmonary vascular smooth muscle following chronic hypoxia. Am J Physiol Lung Cell Mol Physiol 295: L515–L529, 2008. doi: 10.1152/ajplung.00355.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Khaitlina S, Fitz H, Hinssen H. The interaction of gelsolin with tropomyosin modulates actin dynamics. FEBS J 280: 4600–4611, 2013. doi: 10.1111/febs.12431. [DOI] [PubMed] [Google Scholar]
- 26.Kim HR, Graceffa P, Ferron F, Gallant C, Boczkowska M, Dominguez R, Morgan KG. Actin polymerization in differentiated vascular smooth muscle cells requires vasodilator-stimulated phosphoprotein. Am J Physiol Cell Physiol 298: C559–C571, 2010. doi: 10.1152/ajpcell.00431.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kim HR, Leavis PC, Graceffa P, Gallant C, Morgan KG. A new method for direct detection of the sites of actin polymerization in intact cells and its application to differentiated vascular smooth muscle. Am J Physiol Cell Physiol 299: C988–C993, 2010. doi: 10.1152/ajpcell.00210.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Knock GA, Snetkov VA, Shaifta Y, Connolly M, Drndarski S, Noah A, Pourmahram GE, Becker S, Aaronson PI, Ward JP. Superoxide constricts rat pulmonary arteries via Rho-kinase-mediated Ca2+ sensitization. Free Radic Biol Med 46: 633–642, 2009. doi: 10.1016/j.freeradbiomed.2008.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liu JQ, Zelko IN, Erbynn EM, Sham JS, Folz RJ. Hypoxic pulmonary hypertension: role of superoxide and NADPH oxidase (gp91phox). Am J Physiol Lung Cell Mol Physiol 290: L2–L10, 2006. doi: 10.1152/ajplung.00135.2005. [DOI] [PubMed] [Google Scholar]
- 30.Luke T, Maylor J, Undem C, Sylvester JT, Shimoda LA. Kinase-dependent activation of voltage-gated Ca2+ channels by ET-1 in pulmonary arterial myocytes during chronic hypoxia. Am J Physiol Lung Cell Mol Physiol 302: L1128–L1139, 2012. doi: 10.1152/ajplung.00396.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mack CP, Somlyo AV, Hautmann M, Somlyo AP, Owens GK. Smooth muscle differentiation marker gene expression is regulated by RhoA-mediated actin polymerization. J Biol Chem 276: 341–347, 2001. doi: 10.1074/jbc.M005505200. [DOI] [PubMed] [Google Scholar]
- 32.Matsui H, Shimosawa T, Itakura K, Guanqun X, Ando K, Fujita T. Adrenomedullin can protect against pulmonary vascular remodeling induced by hypoxia. Circulation 109: 2246–2251, 2004. doi: 10.1161/01.CIR.0000127950.13380.FD. [DOI] [PubMed] [Google Scholar]
- 33.McNamara PJ, Murthy P, Kantores C, Teixeira L, Engelberts D, van Vliet T, Kavanagh BP, Jankov RP. Acute vasodilator effects of Rho-kinase inhibitors in neonatal rats with pulmonary hypertension unresponsive to nitric oxide. Am J Physiol Lung Cell Mol Physiol 294: L205–L213, 2008. doi: 10.1152/ajplung.00234.2007. [DOI] [PubMed] [Google Scholar]
- 34.Mehta D, Gunst SJ. Actin polymerization stimulated by contractile activation regulates force development in canine tracheal smooth muscle. J Physiol 519: 829–840, 1999. doi: 10.1111/j.1469-7793.1999.0829n.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nagaoka T, Morio Y, Casanova N, Bauer N, Gebb S, McMurtry I, Oka M. Rho/Rho kinase signaling mediates increased basal pulmonary vascular tone in chronically hypoxic rats. Am J Physiol Lung Cell Mol Physiol 287: L665–L672, 2004. doi: 10.1152/ajplung.00050.2003. [DOI] [PubMed] [Google Scholar]
- 36.Norton CE, Broughton BR, Jernigan NL, Walker BR, Resta TC. Enhanced depolarization-induced pulmonary vasoconstriction following chronic hypoxia requires EGFR-dependent activation of NAD(P)H oxidase 2. Antioxid Redox Signal 18: 1777–1788, 2013. doi: 10.1089/ars.2012.4836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Obara K, Yabu H. Effect of cytochalasin B on intestinal smooth muscle cells. Eur J Pharmacol 255: 139–147, 1994. doi: 10.1016/0014-2999(94)90092-2. [DOI] [PubMed] [Google Scholar]
- 38.Oka M, Homma N, Taraseviciene-Stewart L, Morris KG, Kraskauskas D, Burns N, Voelkel NF, McMurtry IF. Rho kinase-mediated vasoconstriction is important in severe occlusive pulmonary arterial hypertension in rats. Circ Res 100: 923–929, 2007. doi: 10.1161/01.RES.0000261658.12024.18. [DOI] [PubMed] [Google Scholar]
- 39.Reeve HL, Michelakis E, Nelson DP, Weir EK, Archer SL. Alterations in a redox oxygen sensing mechanism in chronic hypoxia. J Appl Physiol 90: 2249–2256, 2001. doi: 10.1152/jappl.2001.90.6.2249. [DOI] [PubMed] [Google Scholar]
- 40.Rembold CM, Tejani AD, Ripley ML, Han S. Paxillin phosphorylation, actin polymerization, noise temperature, and the sustained phase of swine carotid artery contraction. Am J Physiol Cell Physiol 293: C993–C1002, 2007. doi: 10.1152/ajpcell.00090.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Resta TC, Chicoine LG, Omdahl JL, Walker BR. Maintained upregulation of pulmonary eNOS gene and protein expression during recovery from chronic hypoxia. Am J Physiol Heart Circ Physiol 276: H699–H708, 1999. [DOI] [PubMed] [Google Scholar]
- 42.Shaw L, Ahmed S, Austin C, Taggart MJ. Inhibitors of actin filament polymerisation attenuate force but not global intracellular calcium in isolated pressurised resistance arteries. J Vasc Res 40: 1–10, 2003. doi: 10.1159/000068940. [DOI] [PubMed] [Google Scholar]
- 43.Shimoda LA, Laurie SS. HIF and pulmonary vascular responses to hypoxia. J Appl Physiol 116: 867–874, 2014. doi: 10.1152/japplphysiol.00643.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Stenmark KR, Fagan KA, Frid MG. Hypoxia-induced pulmonary vascular remodeling: cellular and molecular mechanisms. Circ Res 99: 675–691, 2006. doi: 10.1161/01.RES.0000243584.45145.3f. [DOI] [PubMed] [Google Scholar]
- 45.Sylvester JT, Shimoda LA, Aaronson PI, Ward JP. Hypoxic pulmonary vasoconstriction. Physiol Rev 92: 367–520, 2012. doi: 10.1152/physrev.00041.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tang DD, Anfinogenova Y. Physiologic properties and regulation of the actin cytoskeleton in vascular smooth muscle. J Cardiovasc Pharmacol Ther 13: 130–140, 2008. doi: 10.1177/1074248407313737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Thumkeo D, Watanabe S, Narumiya S. Physiological roles of Rho and Rho effectors in mammals. Eur J Cell Biol 92: 303–315, 2013. doi: 10.1016/j.ejcb.2013.09.002. [DOI] [PubMed] [Google Scholar]
- 48.Tseng S, Kim R, Kim T, Morgan KG, Hai CM. F-actin disruption attenuates agonist-induced [Ca2+], myosin phosphorylation, and force in smooth muscle. Am J Physiol Cell Physiol 272: C1960–C1967, 1997. doi: 10.1152/ajpcell.1997.272.6.C1960. [DOI] [PubMed] [Google Scholar]
- 49.Yamin R, Morgan KG. Deciphering actin cytoskeletal function in the contractile vascular smooth muscle cell. J Physiol 590: 4145–4154, 2012. doi: 10.1113/jphysiol.2012.232306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhao R, Du L, Huang Y, Wu Y, Gunst SJ. Actin depolymerization factor/cofilin activation regulates actin polymerization and tension development in canine tracheal smooth muscle. J Biol Chem 283: 36522–36531, 2008. doi: 10.1074/jbc.M805294200. [DOI] [PMC free article] [PubMed] [Google Scholar]