

Abstract
Ammonia plays a central role in the life and death of all living organisms and has been studied for over 100 yr. Ammonia is necessary for growth and development, but it is toxic in excess, and, as a result, differing methods of ammonia neutralization have evolved. After physiological and pathological stress to the heart, tissue ammonia levels rise. Local ammonia neutralization may be inadequate, and excess ammonia may exert its toxic effects. Phenylbutyrate (PBA), which is Federal Drug Administration approved for the treatment of elevated blood ammonia in urea cycle disorders, provides an accessory pathway for ammonia excretion. Recently, PBA has also been found to prevent specific cardiomyopathies. The central theme presents the hypothesis that stress to the myocardium from a variety of environmental sources causes injury, cell death, necrosis, and ammonia production. Ammonia, if not neutralized, exerts downstream toxic effects. Here, data are presented showing that neutralization with PBA alone and PBA combined with angiotensin-converting enzyme inhibition prevent and reverse pathophysiology associated with specific cardiomyopathies.
NEW & NOTEWORTHY Ammonia produced after myocardial injury is hypothesized to be an upstream stress contributing to the pathophysiology of heart failure, effects that may be attenuated by a documented ammonia-reducing treatment. Reversal of heart failure can be achieved using an angiotensin-converting enzyme inhibitor combined with an ammonia-reducing treatment.
Keywords: ammonia neutralization, cardiac hypertrophy and failure, phenylbutyrate
INTRODUCTION
Current therapy for the treatment of heart failure involves, foremost, the identification and treatment of causative factors. When these are not identified and controlled, the active and passive properties of heart muscle become impaired and heart failure may develop. While there are many treatments that will improve the circulation and symptoms of heart failure, few will reverse or improve myocardial pathology and dysfunction once developed.
Current strategies to improve impaired myocardium include an exploration of cardiac stresses and their effects on downstream pathways and responses that have evolved to attenuate the effects of stress. Recently, phenylbutyrate (PBA) has been found to prevent and reverse specific cardiomyopathies. Mechanisms studied for the therapeutic action of PBA include histone deacetylase (HDAC) inhibition and endoplasmic reticulum (ER) stress attenuation. Since a major action of PBA is to enhance urinary clearance of ammonia, a role for ammonia, as an upstream stress, is hypothesized. If an upstream stress can be identified and treated, then therapy directed at downstream responses might assume a secondary role. Ammonia is both ubiquitous and potentially toxic but has received little attention as a possible stress related to heart disease.
AMMONIA
Nitrogen (N2) is the most abundant gas in the atmosphere and is inert. In biological nitrogen fixation, microorganisms catalyze the conversion to ammonia, the reduced form of nitrogen. In water, ammonia is in equilibrium with unionized NH4. Ammonia is assimilated and used by cells for anabolic amino acid, nucleotide and protein synthesis, growth, and repair.
Biological processes also result in catabolic ammonia production by cells. Thus, there is normal ammonia turnover. NH3 and not total ammonia is responsible for growth and death, and cells are unable to adapt to high ammonia concentrations (19). Because of its central role in metabolism and, conversely, its potential toxicity, ammonia has been studied for over 100 yr, and mechanisms that have evolved to neutralize ammonia have received considerable attention.
In animals, ammonia is primarily produced in the gastrointestinal tract after digestion. The portal system transports ammonia to the liver, where excess ammonia is converted to urea and excreted by the kidney. Blood ammonia is highest in the arterial system, and many tissues extract ammonia (16, 39).
Excess ammonia is neutralized by transamination and deamination, primarily by enzymatic conversion of ammonia and glutamic acid to glutamine. In the liver, glutamine is enzymatically converted back to ammonia and glutamic acid. Excess ammonia enters the urea cycle and is converted to urea, which can be excreted in the urine. With genetic abnormalities of the urea cycle or advanced liver disease with impaired urea cycle activity, blood ammonia rises. Excess ammonia not cleared from the circulation can lead to clinical manifestations of ammonia toxicity, most commonly toxic encephalopathy or hepatic coma.
AMMONIA AND THE HEART
With exercise, blood ammonia production may increase fourfold (10). In experimental myocardial infarction, tissue ammonia rises two- to threefold in both control and infarcted regions of the heart (37). Coronary sinus ammonia has been measured in experimental coronary ischemia, with an increase in ammonia production found (14). Rat hearts perfused with nitrogen 15 isotope over a range of ammonia concentrations demonstrate ammonia neutralization primarily by glutamic acid with glutamine formation. Isoproterenol-induced necrosis led to an elevated tissue ammonia with a decrease in ammonia neutralization (30). Myocardial protection was improved by glutamic acid addition to the isolated perfused rat heart during 30-min ischemic arrest and resulted in less ammonia accumulation and improved functional recovery, although ammonia levels remained high (29). Thus, despite ammonia-neutralizing mechanisms, ammonia levels have been shown to increase in blood and tissue with physiological and pathological stress. Blood concentrations of ammonia are mildly elevated in patients with heart failure, with an overall increase in ammonia extraction by tissues (3). By measuring coronary arteriovenous differences, variable cardiac uptake and release of ammonia into the coronary sinus were found in patients with heart failure (8). Increased myocardial urea release into the coronary sinus has been demonstrated in patients with heart failure, and it has been suggested that the induction of urea cycle activity may be a cardiac adaptation to augment ammonia neutralization (35).
While increased cardiac ammonia production generally does not appear to be found in the coronary sinus of patients with heart failure, it is clear that the myocardium produces ammonia during stress, injury, death and necrosis (11). Even though average coronary arteriovenous differences in ammonia are not found in patients with heart failure, concentrations of ammonia are likely highest in the interstitial space near the site of injury or stress (30, 37), and local ammonia neutralization capacity may be exceeded.
DOWNSTREAM EFFECTS OF AMMONIA
During the development of cardiac failure, myocardial performance is progressively impaired, leading to circulatory decline. Heart failure may result from many stresses; if the stress persists, downstream pathways and compensatory mechanisms are activated.
Effects of ammonia on downstream actions have been studied in the brain to understand toxic encephalopathy. Here, it has been found that ammonia-induced oxidative stress is mediated by N-methyl-d-aspartate (NMDA) receptors and that blockade of these receptors prevents ammonia-induced changes in superoxide dismutase, glutathione peroxidase, and catalase (17).
Ammonia has also been shown to produce oxidative stress, ER stress, and apoptosis (18).
Oxidative stress also activates pathways considered to be important contributors to heart failure (34). Of the many pathways and responses that have evolved in response to stress, several are considered here in relation to the treatment of cardiac hypertrophy and failure.
HDAC Inhibition
HDAC inhibitors increase the acetylation of histones and enhance transcription and protection from oxidative stress (33). Studies have demonstrated that HDAC inhibition attenuates cardiac hypertrophy (25) and may be useful in the treatment of heart disease (2).
ER Stress and the Unfolded Protein Response
An array of genetic and environmental factors, including oxidative stress, can result in ER stress and the unfolded protein response (UPR), which at first ameliorates stress produced by the accumulation of unfolded proteins.
If the UPR capacity is exceeded, apoptotic cell death may occur. It has been suggested that ER stress is an important and promising target for cardiovascular disease including heart failure (24).
Autophagy
Research has been carried out on the role of autophagy with respect to cell survival and death in cardiovascular disease (26). Autophagy is an intracellular degradation system that delivers cytoplasmic constituents to the lysome and plays a variety of physiological and pathological roles.
Ammonia has been shown to stimulate autophagy through the UPR (15).
Thus, enhancing ammonia clearance may reduce ammonia and UPR-induced autophagy.
Neurohormonal Stimulation
A major downstream response to stress is neurohormonal stimulation, which may temporarily aid the normal heart by increasing pressure and flow but is deleterious to the failing heart (31, 36).
Therefore, attenuation of this stress response using angiotensin-converting enzyme (ACE) inhibitors and β-blockers has been shown to be effective in the management of heart failure and are widely used (23). These treatments improve survival and generally reduce further damage; however, structure/function remains impaired with little recovery, reversal generally does not take place (6), and patient mortality remains high (13).
If downstream actions, for example, ER stress and the UPR, result in cell death and associated cell breakdown, additional ammonia and ammonia-induced stress result. If ammonia is an upstream stress, then increasing ammonia clearance may help prevent or reverse downstream consequences.
PHENYLBUTYRATE
The drug PBA is an aromatic fatty acid that is Federal Drug Administration approved for the treatment of urea cycle disorders. More recently, PBA has been studied as a treatment for elevated blood ammonia in advanced liver disease (32). PBA is converted to phenylacetate, which conjugates with glutamine to form phenacetylglutamine and is excreted in the urine.
Thus, PBA provides an accessory pathway for ammonia removal.
Pharmacodynamic studies in mice have demonstrated the PBA to phenylacetate conversion in vivo and that both PBA and phenylacetate appear in tissues including the kidney, liver, heart, muscle, and lungs as well as plasma (22).
PREVENTION AND TREATMENT OF HEART FAILURE WITH PBA
Prevention of Cardiac Pathophysiology and Heart Failure with PBA
Prevention of adriamycin cardiotoxicity was demonstrated in mice by PBA pretreatment. Findings were thought to be due to HDAC inhibition (9) or by the chaperone action of PBA in preventing or reducing ER stress (12). PBA has also been thought to decrease pressure-overload hypertrophy by alleviating ER stress (20, 27). PBA has been reported to attenuate myocardial reperfusion injury in mice through the chaperone action of PBA on the UPR (38). Isoproterenol-induced cardiac fibrosis, myocyte loss, and collagen deposition were prevented by PBA administration (1).
Treatment of Heart Failure With Combined ACE Inhibition and PBA
Studies in the aging spontaneously hypertensive rat (SHR) have characterized a model of chronic hypertension and the progression to heart failure (4, 28). Studies of the mechanisms, prevention, and treatment of heart failure in the SHR are summarized in Ref. 5. ACE inhibition stabilized pathophysiology with improved survival; however, reversal of structural and functional changes associated with heart failure was not seen (6).
To test treatments that might reverse myocardial pathology in the SHR, interventions were studied during the period of ACE-induced stabilization. With combined ACE inhibition and PBA, the manifestations of heart failure were reversed and striking recovery was observed (7). It was found, however, that PBA treatment alone was ineffective with respect to animal survival. This is consistent with data demonstrating lack of efficacy of PBA alone in mice with aortic constriction, severe left ventricular hypertropy, and failure (21) with presumed neurohormonal stimulation. Lack of efficacy suggests that with extensive stress, both upstream and downstream responses to PBA alone are inadequate to neutralize the profound stress associated with neurohormonal stimulation. However, if stress is attenuated by ACE inhibition, the capacity of both upstream and downstream responses may no longer be exceeded and recovery may take place (Fig. 1).
Fig. 1.
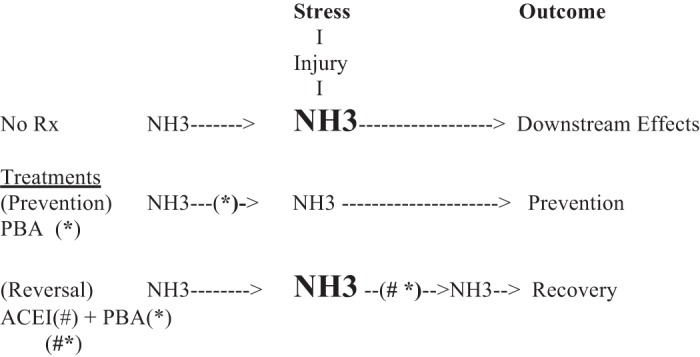
Overview of the hypothesized role of ammonia in the progression to heart failure and its prevention and reversal with treatment. First, this table is best viewed beginning with the concept that stress, from a variety of causes (pressure overload, chemotherapy, myocardial ischemia, or other) induces myocardial injury with ammonia production from the breakdown of cellular material (NH3 → NH3). Second, NH3, if not neutralized, results in downstream stress, including 1) neurohumoral activation, 2) endoplasmic reticulum stress and the unfolded protein response, 3) histone deacetylation and 4) other. Downstream actions lead to compensation or cell death (24). Third, the following treatment effects are shown: 1) treatment before stress [phenylbutyrate (*PBA)] prevents adverse cardiac effects and 2) treatment after stress [angiotensin-converting enzyme inhibition (#ACEI) + *PBA] is associated with reversal of heart failure. ACEI stabilizes the failing heart (6, 13), while ACEI + PBA (#*) allows recovery (7).
SUMMARY
The data are consistent with the hypothesis that cardiac stress induces a shift in the ammonia balance such that ammonia production exceeds neutralization capacity and that ammonia, if not neutralized, represents a toxic stress activating downstream pathways, which lead to compensation or cell death.
A reduction of stress is suggested to shift the balance from apoptosis and cell death to growth and repair. Since PBA is associated with the prevention and reversal of cardiac hypertrophy and failure, PBA treatment deserves further consideration, particularly since this drug is already approved for human use (urea cycle disorders). Combined with the attenuation of stress from angiotensin II, by ACE inhibition, PBA is suggested as a novel addition to the treatment of heart failure.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
O.H.L.B. conceived and designed research; O.H.L.B. performed experiments; O.H.L.B. analyzed data; O.H.L.B. interpreted results of experiments; O.H.L.B. drafted manuscript; O.H.L.B. edited and revised manuscript; O.H.L.B. approved final version of manuscript.
ACKNOWLEDGMENTS
This work is dedicated to Dr. Wesley W. Brooks, who passed away in 2015. Our long collaboration has provided insights into mechanisms underlying heart failure and its treatment.
REFERENCES
- 1.Ayala P, Montenegro J, Vivar R, Letelier A, Urroz PA, Copaja M, Pivet D, Humeres C, Troncoso R, Vicencio JM, Lavandero S, Díaz-Araya G. Attenuation of endoplasmic reticulum stress using the chemical chaperone 4-phenylbutyric acid prevents cardiac fibrosis induced by isoproterenol. Exp Mol Pathol 92: 97–104, 2012. doi: 10.1016/j.yexmp.2011.10.012. [DOI] [PubMed] [Google Scholar]
- 2.Berry JM, Cao DJ, Rothermel BA, Hill JA. Histone deacetylase inhibition in the treatment of heart disease. Expert Opin Drug Saf 7: 53–67, 2008. doi: 10.1517/14740338.7.1.53. [DOI] [PubMed] [Google Scholar]
- 3.Bessman AN, Evans JM. The blood ammonia in congestive heart failure. Am Heart J 50: 715–719, 1955. doi: 10.1016/0002-8703(55)90178-2. [DOI] [PubMed] [Google Scholar]
- 4.Bing OHL, Brooks WW, Robinson KG, Slawsky MT, Hayes JA, Litwin SE, Sen S, Conrad CH. The spontaneously hypertensive rat as a model of the transition from compensated left ventricular hypertrophy to failure. J Mol Cell Cardiol 27: 383–396, 1995. doi: 10.1016/S0022-2828(08)80035-1. [DOI] [PubMed] [Google Scholar]
- 5.Bing OHL, Conrad CH, Boluyt MO, Robinson KG, Brooks WW. Studies of prevention, treatment and mechanisms of heart failure in the aging spontaneously hypertensive rat. Heart Fail Rev 7: 71–88, 2002. doi: 10.1023/A:1013753907135. [DOI] [PubMed] [Google Scholar]
- 6.Brooks WW, Bing OH, Robinson KG, Slawsky MT, Chaletsky DM, Conrad CH. Effect of angiotensin-converting enzyme inhibition on myocardial fibrosis and function in hypertrophied and failing myocardium from the spontaneously hypertensive rat. Circulation 96: 4002–4010, 1997. doi: 10.1161/01.CIR.96.11.4002. [DOI] [PubMed] [Google Scholar]
- 7.Brooks WW, Shen S, Conrad CH, Goldstein RH, Deng LL, Bing OHL. Transcriptional changes associated with recovery from heart failure in the SHR. J Mol Cell Cardiol 49: 390–401, 2010. doi: 10.1016/j.yjmcc.2010.06.002. [DOI] [PubMed] [Google Scholar]
- 8.Chazov E, Smirnov VN, Mazaev AV, Asafov GB, Gukowski DU, Krikov VI. Myocardial ammonia metabolism in patients with heart disease as revealed by coronary sinus catheterization study. Circulation 47: 1327–1334, 1973. doi: 10.1161/01.CIR.47.6.1327. [DOI] [PubMed] [Google Scholar]
- 9.Daosukho C, Chen Y, Noel T, Sompol P, Nithipongvanitch R, Velez JM, Oberley TD, St Clair DK. Phenylbutyrate, a histone deacetylase inhibitor, protects against adriamycin-induced cardiac injury. Free Radic Biol Med 42: 1818–1825, 2007. doi: 10.1016/j.freeradbiomed.2007.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dawson AM. Regulation of blood ammonia. Gut 19: 504–509, 1978. doi: 10.1136/gut.19.6.504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Donaldson AE, Lamont IL. Biochemistry changes that occur after death: potential markers for determining post-mortem interval. PLoS One 8: e82011, 2013. doi: 10.1371/journal.pone.0082011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fu HY, Sanada S, Matsuzaki T, Liao Y, Okuda K, Yamato M, Tsuchida S, Araki R, Asano Y, Asanuma H, Asakura M, French BA, Sakata Y, Kitakaze M, Minamino T. Chemical endoplasmic reticulum chaperone alleviates doxorubicin-induced cardiac dysfunction. Circ Res 118: 798–809, 2016. doi: 10.1161/CIRCRESAHA.115.307604. [DOI] [PubMed] [Google Scholar]
- 13.Giles TD. Renin-angiotensin system modulation for treatment and prevention of cardiovascular diseases: toward an optimal therapeutic strategy. Rev Cardiovasc Med 8, Suppl 2: S14–S21, 2007. [PubMed] [Google Scholar]
- 14.Hacker TA, Renstrom B, Paulson D, Liedtke AJ, stanley WC. Ischemia produces an increase in ammonia output in swine myocardium. Cardioscience 5: 255–260, 1994. [PubMed] [Google Scholar]
- 15.Harder LM, Bunkenborg J, Andersen JS. Inducing autophagy: a comparative phosphoproteomic study of the cellular response to ammonia and rapamycin. Autophagy 10: 339–355, 2014. doi: 10.4161/auto.26863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Huizenga JR, Gips CH, Tangerman A. The contribution of various organs to ammonia formation: a review of factors determining the arterial ammonia concentration. Ann Clin Biochem 33: 23–30, 1996. doi: 10.1177/000456329603300103. [DOI] [PubMed] [Google Scholar]
- 17.Kosenko E, Kaminski Y, Lopata O, Muravyov N, Felipo V. Blocking NMDA receptors prevents the oxidative stress induced by acute ammonia intoxication. Free Radic Biol Med 26: 1369–1374, 1999. doi: 10.1016/S0891-5849(98)00339-6. [DOI] [PubMed] [Google Scholar]
- 18.Liang Z, Liu R, Zhao D, Wang L, Sun M, Wang M, Song L. Ammonia exposure induces oxidative stress, endoplasmic reticulum stress and apoptosis in hepatopancreas of pacific white shrimp (Litopenaeus vannamei). Fish Shellfish Immunol 54: 523–528, 2016. doi: 10.1016/j.fsi.2016.05.009. [DOI] [PubMed] [Google Scholar]
- 19.Lüdemann I, Pörtner R, Märkl H. Effect of NH3 on the cell growth of a hybridoma cell line. Cytotechnology 14: 11–20, 1994. doi: 10.1007/BF00772191. [DOI] [PubMed] [Google Scholar]
- 20.Luo T, Chen B, Wang X. 4-PBA prevents pressure overload-induced myocardial hypertrophy and interstitial fibrosis by attenuating endoplasmic reticulum stress. Chem Biol Interact 242: 99–106, 2015. doi: 10.1016/j.cbi.2015.09.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ma J, Luo T, Zeng Z, Fu H, Asano Y, Liao Y, Minamino T, Kitakaze M. Histone deacetylase inhibitor phenylbutyrate exaggerates heart failure in pressure overload independently of HDAC inhibition. Sci Rep 6: 1–12, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Marahatta A, Bhandary B, Lee MR, Kim DS, Lee YC, Kim SR, Kim HR, Chae HJ. Determination of phenylbutyric acid and its metabolite phenylacetic acid in different tissues of mouse by liquid chromatography with tandem mass spectrometry and its application in drug tissue distribution. J Chromatogr B Analyt Technol Biomed Life Sci 903: 118–125, 2012. doi: 10.1016/j.jchromb.2012.07.004. [DOI] [PubMed] [Google Scholar]
- 23.McMurray JJ, Adamopoulos S, Anker AD, Auricchio A, Böhm M, Dickstein K, Falk V, Filippatos G, Fonseca C, Gomez-Sanchez MA, Jaarsma T, Køber L, Lip GY, Maggioni AP, Parkhomenko A, Pieske BM, Popesc BA, Rønnevik PK, Rutten FH, Schwitter J, Seferovic P, Stepinska J, Trindade PT, Voors AA, Zannad F, Zeiher A; Task Force for the Diagnosis and Treatment of Acute and Chronic Heart Failure 2012 of the European Society of Cardiology, Bax JJ, Baumgartner H, Ceconi C, Dean V, Deaton C, Fagard R, Funck-Brentano C, Hasdai D, Hoes A, Kirchhof P, Knuuti J, Kolh JP, McDonagh T, Moulin C, Popescu BA, Reiner Z, Sechtem U, Sirnes PA, Tendera M, Torbicki A, Vahanian A, Windecker S, McDonagh T, Sechtem U, Bonet LA, Avraamides PT, Ben Lamin HA, Brignole M, Coca A, Cowburn P, Dargie H, Elliott P, Flachskamp FA, Guida GF, Hardman S, Iung B, Merkely B, Mueller C, Nanas JN, Nielsen OW, Ørn S, Parissis JT, Ponikowski P; ESC Committee for Practice Guidelines . ESC guidelines for the diagnosis and treatment of acute and chronic heart failure 2012: the Task Force for the Diagnosis and Treatment of Acute and Chronic Heart Failure 2012 of the European Society of Cardiology. Developed in collaboration with the Heart Failure Association (HFA) of the ESC. Eur Heart J 33: 1787–1847, 2012. doi: 10.1093/eurjhf/hfs105. [DOI] [PubMed] [Google Scholar]
- 24.Minamino T, Komuro I, Kitakaze M. Endoplasmic reticulum stress as a therapeutic target in cardiovascular disease. Circ Res 107: 1071–1082, 2010. doi: 10.1161/CIRCRESAHA.110.227819. [DOI] [PubMed] [Google Scholar]
- 25.Ooi JYY, Tuano NK, Rafehi H, Gao X-M, Ziemann M, Du X-J, El-Osta A. HDAC inhibition attenuates cardiac hypertrophy by acetylation and deacetylation of target genes. Epigenetics 10: 418–430, 2015. doi: 10.1080/15592294.2015.1024406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Orogo AM, Gustafsson AB. Therapeutic targeting of autophagy: potential and concerns in treating cardiovascular disease. Circ Res 116: 489–503, 2015. doi: 10.1161/CIRCRESAHA.116.303791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Park CS, Cha H, Kwon EJ, Sreenivasaiah PK, Kim DH. The chemical chaperone 4-phenylbutyric acid attenuates pressure-overload cardiac hypertrophy by alleviating endoplasmic reticulum stress. Biochem Biophys Res Commun 421: 578–584, 2012. doi: 10.1016/j.bbrc.2012.04.048. [DOI] [PubMed] [Google Scholar]
- 28.Pfeffer MA, Pfeffer JM, Frohlich ED. Pumping ability of the hypertrophying left ventricle of the spontaneously hypertensive rat. Circ Res 38: 423–429, 1976. doi: 10.1161/01.RES.38.5.423. [DOI] [PubMed] [Google Scholar]
- 29.Pisarenko OI, Solomatina ES, Studneva IM, Ivanov VE, Kapelko VI, Smirnov VN. Protective effect of glutamic acid on cardiac function and metabolism during cardioplegia and reperfusion. Basic Res Cardiol 78: 534–543, 1983. doi: 10.1007/BF01906464. [DOI] [PubMed] [Google Scholar]
- 30.Pisarenko OI, Minkovskiĭ EB, Studneva IM. [Neutralization of ammonia in cardiac muscle]. Biull Eksp Biol Med 89: 289–291, 1980. doi: 10.1007/BF00834227. [DOI] [PubMed] [Google Scholar]
- 31.Riegger AJ. Hormones in heart failure−regulation and counterregulation. Eur Heart J 12, Suppl D: 190–192, 1991. [DOI] [PubMed] [Google Scholar]
- 32.Rockey DC, Vierling JM, Mantry P, Ghabril M, Brown RS Jr, Alexeeva O, Zupanets IA, Grinevich V, Baranovsky A, Dudar L, Fadieienko G, Kharchenko N, Klaryts’ka I, Morozov V, Grewal P, McCashland T, Reddy KG, Reddy KR, Syplyviy V, Bass NM, Dickinson K, Norris C, Coakley D, Mokhtarani M, Scharschmidt BF; HALT-HE Study Group . Randomized, double-blind, controlled study of glycerol phenylbutyrate in hepatic encephalopathy. Hepatology 59: 1073–1083, 2014. doi: 10.1002/hep.26611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sartori L, Minucci S. Tackling oxidative stress by a direct route: a new job for HDAC inhibitors? Chem Biol 22: 431–432, 2015. doi: 10.1016/j.chembiol.2015.04.003. [DOI] [PubMed] [Google Scholar]
- 34.Searles CD. The nitric oxide pathway and oxidative stress in heart failure. Congest Heart Fail 8: 142–147, 2002. doi: 10.1111/j.1527-5299.2002.00715.x. [DOI] [PubMed] [Google Scholar]
- 35.Smirnov VN, Asafov GB, Cherpachenko NM, Chemousova GB, Chumachenko MN, Ovchinnikov IA, Merimson VG, Rozynov VG, Chunachenko MN. Ammonia neutralization and urea synthesis in cardiac muscle. Circ Res 35, Suppl 3: 58–65, 1974. [PubMed] [Google Scholar]
- 36.Swedberg K, Eneroth P, Kjekshus J, Wilhelmsen L; CONSENSUS Trial Study Group . Hormones regulating cardiovascular function in patients with severe congestive heart failure and their relation to mortality. Circulation 82: 1730–1736, 1990. doi: 10.1161/01.CIR.82.5.1730. [DOI] [PubMed] [Google Scholar]
- 37.Takahashi A. Myocardial protein metabolism following coronary occlusion. Jpn Circ J 31: 581–600, 1967. doi: 10.1253/jcj.31.581. [DOI] [PubMed] [Google Scholar]
- 38.Takatori O, Usui S, Okajima M, Kaneko S, Ootsuji H, Takashima SI, Kobayashi D, Murai H, Furusho H, Takamura M. Sodium 4-phenylbutyrate attenuates myocardial reperfusion injury by reducing the unfolded protein response. J Cardiovasc Pharmacol Ther 22: 283–292, 2017. doi: 10.1177/1074248416679308. [DOI] [PubMed] [Google Scholar]
- 39.Wright G, Noiret L, Olde Damink SWM, Jalan R. Interorgan ammonia metabolism in liver failure: the basis of current and future therapies. Liver Int 31: 163–175, 2011. doi: 10.1111/j.1478-3231.2010.02302.x. [DOI] [PubMed] [Google Scholar]