

Abstract
Myeloproliferative neoplasms arise from hematopoietic stem cells with somatically altered tyrosine kinase signaling. Classification of myeloproliferative neoplasms is based on hematologic, histopathologic, and molecular characteristics including the presence of the BCR-ABL1 and JAK2 V617F. Although thought to be mutually exclusive, a number of cases with co-occurring BCR-ABL1 and JAK2 V617F have been identified. To characterize the clinicopathologic features of myeloproliferative neoplasms with concomitant BCR-ABL1 and JAK2 V617F, and define the frequency of co-occurrence, we conducted a retrospective multi-institutional study. Cases were identified using a search of electronic databases over a decade at 6 major institutions. Of 1570 patients who were tested for both BCR-ABL1 and JAK2 V617F, 6 were positive for both. An additional 5 patients were identified via clinical records providing a total of 11 cases for detailed evaluation. For each case, clinical variables, hematologic and genetic data, and bone marrow histomorphologic features were analyzed. The sequence of identification of the genetic abnormalities varied: five patients were initially diagnosed with a JAK2 V617F+ myeloproliferative neoplasm, one patient initially had BCR-ABL1+ chronic myeloid leukemia, while both alterations were identified simultaneously in 5 patients. Classification of the BCR-ABL1-negative myeloproliferative neoplasms varied, and in some cases, features only became apparent following tyrosine kinase inhibitor therapy. Seven of the 11 patients showed myelofibrosis, in some cases prior to identification of the second genetic alteration. Our data, reflecting the largest reported study comprehensively detailing clinicopathologic features and response to therapy, show that the co-occurrence of BCR-ABL1 and JAK2 V617F is rare, with an estimated frequency of 0.4%, and most often reflects 2 distinct (“composite”) myeloproliferative neoplasms. Although uncommon, it is important to be aware of this potentially confounding genetic combination, lest these features be misinterpreted to reflect resistance to therapy or disease progression, considerations that could lead to inappropriate management.
INTRODUCTION
Myeloproliferative neoplasms arise from hematopoietic stem cells with somatically acquired tyrosine kinase alterations which activate signaling pathways leading to heightened cellular proliferation. The BCR-ABL1 translocation and JAK2, CALR, and MPL alterations play dominant roles in myeloproliferative neoplasm pathogenesis; accordingly, identification of these specific genetic alterations has been incorporated into WHO diagnostic criteria. The BCR-ABL1 translocation is requisite in the development and diagnosis of chronic myeloid leukemia. On the other hand, the gain of function JAK2 V617F mutation drives disease but has a less directly defined mechanistic relationship across myeloproliferative neoplasm phenotypes. It nevertheless has an essential diagnostic role and is variably identified in polycythemia vera (~95%), primary myelofibrosis (~60%), and essential thrombocythemia (~50%)(1, 2).
Recently, several isolated case reports(3–24) and small case series(25–36) have described patients with co-occurring JAK2 V617F and BCR-ABL1. Because of the inherently small sample size in prior reports, the frequency of co-occurrence, the temporal order of acquisition, and clonal relationship of these alterations remain poorly understood. Furthermore, there is a lack of a systematic analysis of the clinical presentation, hematologic findings, and histopathologic features of patients with these neoplasms, frequently leading to diagnostic and therapeutic uncertainty in this patient population. To address these unknowns, we performed a multi-institutional study of patients evaluated for a suspected myeloproliferative neoplasm at 6 major academic medical centers. Here we present the unique features of 11 cases found to be both JAK2 V617F and BCR-ABL1 positive.
MATERIALS AND METHODS
Patient population
Patients were identified through a combination of systematic search of pathology databases as well as clinical record review. We searched molecular and anatomic pathology databases from three of the six participating institutions for patients with suspected myeloproliferative neoplasm who underwent molecular testing for both JAK2 V617F and BCR-ABL1 over a 10-year period, between July 2005 and June 2015. Additional patients with JAK2+ BCR-ABL1+ myeloproliferative neoplasm were identified via review of clinical records from the other three institutions. Case 7 has previously been reported (27) and the morphologic features of case 8 were highlighted in an image-based publication (37). Institutional Review Board approval was obtained at all participating institutions.
BCR-ABL1 and JAK2 V617F mutation detection
t(9;22) and/or BCR-ABL1 analysis was performed by routine metaphase cytogenetics, reverse transcription polymerase chain reaction and fluorescent in-situ hybridization according to standard operating protocols for clinical diagnosis. JAK2 V617F mutation was detected by an allelic discrimination assay or DNA sequencing on either bone marrow or peripheral blood specimens (Table 1). When archival specimens were accessible, molecular testing was retrospectively performed at earlier time points in an attempt to determine when specific molecular alterations might have been acquired.
TABLE 1.
Clinical and pathologic features, and response to therapy in patients with BCR-ABL1+, JAK2 V617+ myeloproliferative neoplasms
| Initial Neoplasm | Composite (BCR-ABL1+, JAK2 V617F+) MPN | Response to treatment of composite (BCR-ABL1+, JAK2 V617F+) MPN | ||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Pt # | Sex | Age | Initial Dx | BCR-ABL1 (+/−) | JAK2 V617F (+/−) Spec Type | Initial Tx | Time to Dx (Mo) | MPN Hist Features | WBC (×103/μL) | Hgb (g/dL) | Plt (×103/μL) | BCR- ABL1 (+/−) | t(9;22) (% +) | FISH (% +) | JAK2 V617F (+/−) Spec Type | JAK2 Mut allele (%) | F/U time (Mo) | Tx | WBC (×103/μL) | Hgb (g/dL) | Plt (×103/μL) | MMR | JAK2 V617F (+/−) Spec Type | JAK2 Mut allele (%) | Course | Alive/Dead |
| 1 | F | 48 | PMF | – | +* PB | none | 129 | CML | 142 | 8.3 | 557 | + e13a2 | 100 | 94 | + BM | 10–50 | 48 | Nil, Hy, Rux | 5.9 | 9.3 | 141 | Yes | + BM | 78 | MF | A |
| 2 | F | 66 | ET | – | + BM | Hy, Rux | 45 | Post-ET MF; CML | 42.0 | 10.2 | 38 | + e13a2 | 80 | ND | + BM | 24.0 | 23 | Im, Bos, Rux | 12.5 | 10.1 | 39 | No | ND | ND | MF | A |
| 3 | F | 48 | PV | – | + BM | Phleb, Hy, IFN | 58 | Post-PV MF; CML | 26.1 | 13.4 | 66 | + e13a2 | 100 | ND | + BM | 24.4 | 25 | Im, IFN, Rux | 16.1 | 13.9 | 190 | No | ND | ND | MF | A |
| 4 | F | 60 | PV | – | + PB | Hy, Th, Rux | 107 | Post-PV MF; CML | 60.2 | 8.8 | 77 | + e1a2 | 80 | 63 | + PB | >50 | 6 | Im, Hy, Th, Rux | 17.4 | 8.6 | 92 | No | ND | ND | MF | D |
| 5 | M | 76 | PV | – | ND§ | Hy | 73 | CML (AP) | 23.3 | 10 | 32 | + e1a2 | 100 | 82 | + PB | <10 | 4 | Im, Hy | 25.4 | 8.5 | 86 | No | + PB | <10 | CML (AP) | D |
| 6 | F | 49 | CML | + | ND | Im IFN | 109 | MPN, NOS | 6.9 | 12.1 | 858 | + e14a2 | – | ND | + BM | ND | 68 | Im, Das | 18.0 | 10.1 | 114 | No | + BM | 28 | MF | A |
| 7 | M | 70 | NA | NA | NA | NA | NA | CML; MF | 18.5 | 11.7 | 389 | + e14a2 | 60 | ND | + BM | ND | 76 | Im, Nil, Das, Hy Rad | 20.0 | NA | NA | No | + PB | 7.6 | MF | D |
| 8 | M | 68 | NA | NA | NA | NA | NA | CML | 13.1 | 13.6 | 670 | + e14a2 | 95 | ND | + BM | <10 | 43 | Im, Das, An | 7.2 | 11.0 | 554 | Yes | + BM | >25 | Rem | A |
| 9 | M | 49 | NA | NA | NA | NA | NA | CML; MF | 72.9 | 9.7 | 268 | + e14a2 | 100 | ND | + PB | ND | 32 | Im, Das, Allo | 35.9 | 11.8 | 91 | Yes | − BM | <1 | CML (AP) | D |
| 10 | F | 81 | NA | NA | NA | NA | NA | CML; ET | 15.7 | 13.2 | 351 | + e14a2 | 75 | ND | + BM | 18.3 | 109 | Im, Nil, An, Hy | 15.2 | 9.4 | 189 | No | ND | ND | MF CML (BP) | D |
| 11 | F | 73 | NA | NA | NA | NA | NA | CML; PV | 12.8 | 13.3 | 432 | + e1a2 | 10 | 5 | + BM | ND | 17 | Im, Phleb | 14.5 | 12.5 | 409 | No | + PB | ND | Rem | A |
Group 1: JAK2 V617F detected first, BCR-ABL1 second; Group 2: BCR-ABL1 detected first, JAK2 V617F second; Group 3: BCR-ABL1 and JAK2 V617F detected simultaneously
+, positive; −, negative; A, alive; Allo, allogeneic stem cell transplant; An, anagrelide; AP, accelerated phase; BP, blast phase; BM, bone marrow; Bos, bosutinib; CML, chronic myeloid leukemia; D, dead; Das, dasatinib; Dx, diagnosis; ET, essential thrombocythemia; F, female; F/U, follow-up; Hgb, hemoglobin; Hist, histologic; Hy, hydroxyurea; IFN, interferon-α; Im, imatinib mesylate; M, male; Mo, months; MF, myelofibrosis; MMR, major molecular response; MPN, NOS, myeloproliferative neoplasm, not otherwise specified; Nil, nilotinib; NA, not applicable; ND, not determined; PB, peripheral blood; Phleb, therapeutic phlebotomy; Plt, platelet count; PMF, primary myelofibrosis; PV, polycythemia vera; Rad, radiation; Rem, remission; Rux, ruxolitinib; Th, thalidomide; Tx, treatment; WBC, white blood cell count
Testing performed subsequent to initial diagnosis, but before evidence of CML emerged.
Patient diagnosed with PV prior to 2005 based on laboratory, hematologic, and histopathologic criteria.
Clinicopathologic Analysis
For each patient with concurrent JAK2 V617F and BCR-ABL1, demographic (age, gender), clinical, hematologic (peripheral blood counts), genetic (cytogenetic profile, molecular genetic results), and bone marrow histomorphologic features were acquired at diagnosis and follow-up. When histologic material was still available, the authors reviewed the cases from their institution in order to confirm, clarify and expand upon the reported histomorphologic findings. As this was a retrospective study, treatment decisions (therapy received, dosage) were based on the practice of each institution. Treatments included first, second and third generation tyrosine kinase inhibitors, interferon, JAK inhibitors, hydroxyurea, anagrelide, thalidomide, therapeutic phlebotomy, and allogeneic stem cell transplant. Outcome measures (response to therapy and survival) were measured from time of diagnosis of each neoplasm.
RESULTS
Patient Characteristics
Of 1570 patients (from 3 institutions) who had a suspected myeloproliferative neoplasm and were tested concurrently for BCR-ABL1 and JAK2 V617F, 105 (6.7%) tested positive for BCR-ABL1 only, 454 (28.9%) tested positive for JAK2 V617F only, while 6 (0.4%) tested positive for both. Over the same time period, with data only available from 2 institutions, 1695 patients were tested for BCR-ABL1 only, of whom 659 (38.9%) tested positive while in 2437 patients who were tested for JAK2 V617F only, 732 (30.0%) tested positive.
In addition to these patients identified via search of pathology databases, 5 more patients were identified via review of clinical records, for a total of 11 patients (Table 1). Four of the patients were male, 7 were female. The median age at initial diagnosis was 66 years (range 48-81 years). Three patients had a history of prior and/or concurrent non-hematopoietic neoplasm, one of whom received radiation therapy.
Molecular findings at initial presentation
Of the 11 patients with co-occurring JAK2 V617F and BCR-ABL1, 6 had a single molecular alteration identified at initial myeloproliferative neoplasm diagnosis, with the second alteration identified at a later time point. The mean time to detection of the second alteration was 87 months (range 45 - 129 months). In the majority of cases (5 of 6), a JAK2 V617F+ myeloproliferative neoplasm was diagnosed prior to detection of BCR-ABL1. In each case, an initial BCR-ABL1 assay was negative. One patient had documented BCR-ABL1+ chronic myeloid leukemia with the identification of JAK2 V617F occurring at a later date, though this patient had no JAK2 study performed at the initial diagnosis of chronic myeloid leukemia and retrospective analysis could not be performed. Both JAK2 V617F and BCR-ABL1 were identified simultaneously in the remaining 5 patients.
Based on the time course of the identified abnormalities, the cases can thus be categorized into three groups: Group 1 = 5 patients (patients 1-5) with JAK2+ myeloproliferative neoplasm who subsequently acquired BCR-ABL1; Group 2 = 1 patient (patient 6) with BCR-ABL1+ chronic myeloid leukemia with JAK2 V617F detected subsequently; and Group 3 = 5 patients (patients 7-11) who had both BCR-ABL1 and JAK2 V617F identified simultaneously at initial diagnosis. The findings in these three groups are presented in detail below. Laboratory and histopathologic features of representative patients from each group are highlighted in Figures 1–4; laboratory and molecular parameters at specific time points are highlighted in Table 1.
FIGURE 1. Patient 1, Time course of laboratory and histopathologic features in a patient with longstanding JAK2 V617F+ primary myelofibrosis who subsequently acquired BCR-ABL1. Temporary switch to chronic myeloid leukemia phenotype followed by Tyrosine kinase inhibitor induced reversion to primary myelofibrosis.
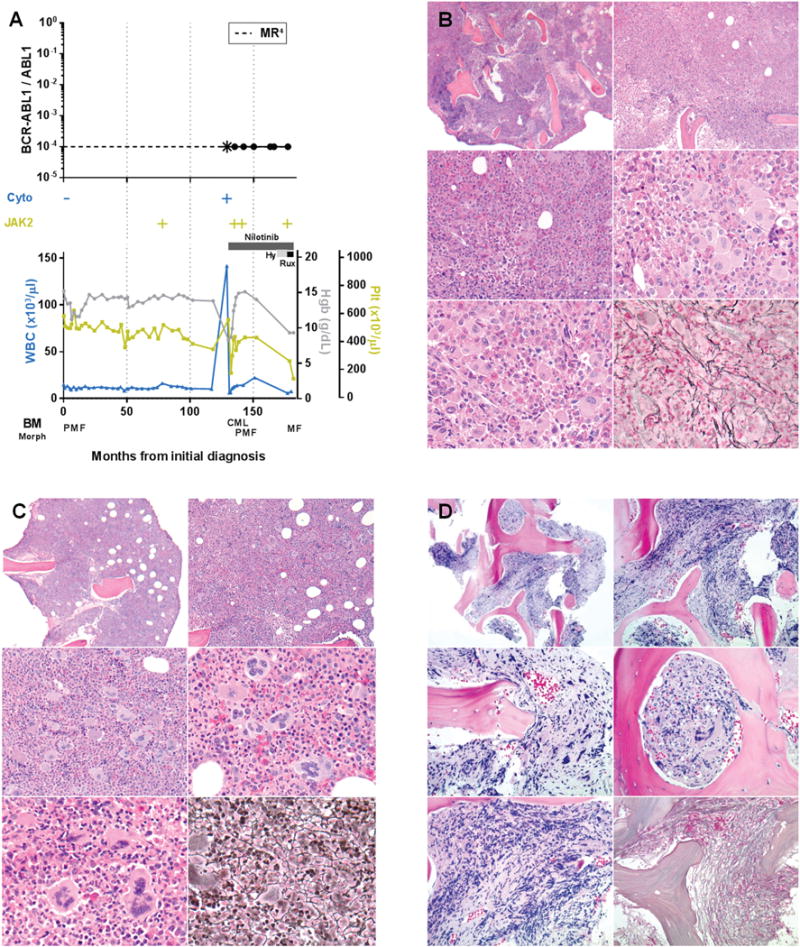
A. Time course of BCR-ABL1 transcript levels (log scale), cytogenetic t(9;22) results (Cyto), JAK2 V617F status, treatment, peripheral blood counts, and bone marrow morphology, Hy = hydroxyurea, Rux = ruxolitinib, *the first quantitative BCR-ABL1 assay performed after 3 months of tyrosine kinase inhibitor therapy revealed that molecular response4 had been attained. B. Bone marrow core biopsy at identification of BCR-ABL1 (month 129) showing predominant features of chronic myeloid leukemia. Hypercellular (top), increased M:E ratio, increased hypolobated megakaryocytes with clustering (middle, bottom left), mildly increased reticulin fibrosis, grade 1/3 (bottom right). C. Bone marrow core biopsy after 6 months of tyrosine kinase inhibitor therapy (month 135) showing predominant features of primary myelofibrosis. Hypercellular (top), mildly increased large megakaryocytes with bulbous nuclei (middle, bottom left), increased reticulin fibrosis, grade 1-2/3 (bottom right). D. Bone marrow core biopsy following tyrosine kinase inhibitor and hydroxyurea therapy (month 177) showing progression to fibrotic phase primary myelofibrosis. Focally hypercellular (top), increased hyperchromatic megakaryocytes (middle, bottom left), markedly increased reticulin fibrosis, grade 3/3 (bottom right).
FIGURE 4. Patient 9, Time course of laboratory and histopathologic features in a patient with simultaneously identified BCR-ABL1 and JAK2 V617F. Mixed histomorphologic features with progression to accelerated phase chronic myeloid leukemia followed by allogeneic transplant induced remission.

A. Time course of BCR-ABL1 transcript levels (log scale), cytogenetic t(9;22) results (Cyto), JAK2 V617F status, treatment, peripheral blood counts, and bone marrow morphology, Allo Tx = allogeneic transplant. B. Bone marrow core biopsy at initial diagnosis (month 0) showing features of chronic myeloid leukemia and myelofibrosis. Hypercellular (top), markedly increased hypolobated megakaryocytes with clustering (middle, bottom left), increased reticulin fibrosis, grade 3/3 (bottom right). C. Bone marrow core biopsy after 15 months of tyrosine kinase inhibitor therapy (month 15) showing accelerated phase chronic myeloid leukemia. Hypercellular (top), increased myeloblasts (5-10%) (middle), atypical hypolobated megakaryocytes (bottom left), decreased reticulin fibrosis, grade 2/3 (bottom right). D. Bone marrow core biopsy following stem cell transplant (month 23). Normocellular (top), trilineage hematopoiesis, no morphologic evidence of JAK2+ BCR-ABL1+ myeloproliferative neoplasm (middle, bottom left), grade 0/3 reticulin fibrosis (bottom right).
Group 1: JAK2 V617F detected first, BCR-ABL1 second (patients 1-5)
Laboratory findings and treatment during JAK2+ myeloproliferative neoplasm disease phase
Of the 5 patients who initially presented with JAK2 V617F+ myeloproliferative neoplasm, one (patient 1) had primary myelofibrosis, one (patient 2) had essential thrombocythemia, and three (patients 3-5) had polycythemia vera. The mean time to identification of BCR-ABL1 was 82 months (range 45 - 129 months). Four patients (patients 2-5) underwent cytoreductive treatment with hydroxyurea. Patients 2 and 4 additionally received ruxolitinib. Interferon-alpha was administered to patient 3 and thalidomide was administered to patient 4. Patient 1 received no therapy. Response to therapy varied, but JAK2 V617F remained detectable in those patients tested.
Bone marrow histology during JAK2+ myeloproliferative neoplasm disease phase
Bone marrow findings prior to acquisition of BCR-ABL1 were compatible with the diagnosed Ph-negative myeloproliferative neoplasm. Per diagnostic pathology reports, those patients with polycythemia vera showed hypercellularity with panmyelosis. Patients 3 and 4 progressed to post-polycythemic myelofibrosis prior to identification of BCR-ABL1. The patient with essential thrombocythemia additionally showed progression to post-essential thrombocythemia myelofibrosis. Of note, all three patients with myelofibrosis had concurrent negative BCR-ABL1 testing at the time myelofibrosis was noted. Patient 1 was initially diagnosed with primary myelofibrosis over ten years prior to acquisition of BCR-ABL1. In no case were there any overt histomorphologic harbingers that might have been predictive of the subsequent development of chronic myeloid leukemia.
Laboratory hematology findings at time of identification of BCR-ABL1
In all patients, complete blood counts revealed an increase in the WBC count at the time of BCR-ABL1 acquisition (see patient 1, Figure 1) from a mean baseline of 16.7 ×103/μL (range 6.3 - 31.6 ×103/μL) to a mean WBC of 58.7 ×103/μL (range 23.3 – 142 ×103/μL). Patients 1, 2, and 4 showed circulating myeloid precursors with basophilia. Patient 3 showed only peripheral neutrophilia without left-shift and patient 5 showed granulocytes in all phases of maturation with increased numbers of circulating myeloblasts, which prompted a bone marrow study that showed left-shifted granulocytic hyperplasia and 10-15% blasts. The blasts displayed a myeloid immunophenotype. Patients 2-5 showed progressive thrombocytopenia, (mean 53 ×103/μL; range 32 - 77 ×103/μL, decreased from baseline mean 230 ×103/μL; range 88 - 349 ×103/μL), though three of these patients had known prior progression to myelofibrosis. Among these same four patients, hemoglobin levels dropped from a mean of 12.3 g/dL (range 8.4 - 15.2 g/dL) to 10.6 g/dL (range 8.3 – 13.4 g/dL). Patient 1 showed new onset thrombocytosis (557 ×103/μL, increased from 349 ×103/μL) and anemia (Hgb: 8.3 g/dL, decreased from 13.8 g/dL).
Genetic findings at time of identification of BCR-ABL1
Patients 2-5 were undergoing treatment for the JAK2+ myeloproliferative neoplasm at the time of BCR-ABL1 identification, with a range of mutant JAK2 V617F allele levels detected. Patient 4 had a JAK2 V617F mutant allele frequency of >50%, patients 1-3 had mutant allele frequencies between 10-50%, while patient 5 had a mutant allele burden of <10%. All 5 patients displayed high levels of t(9;22) and/or BCR-ABL1 positive cells at this phase of their disease with bone marrow cytogenetic studies showing t(9;22) in a mean of 86% metaphases (range 80% to 100%), while FISH showed a mean of 76% positive cells (range 63 - 94%) in the 3 patients tested. All patients had BCR-ABL1 transcripts detected by qualitative RT-PCR; patients 4 and 5 showed the e1a2 transcript only (Table 1).
Bone marrow histology at time of identification of BCR-ABL1
Patients 2-4 had shown increased reticulin fibrosis (two myelofibrosis grade 2/3, one myelofibrosis grade 3/3) prior to detection of BCR-ABL1. Upon acquisition of BCR-ABL1, bone marrow histology in all 5 cases showed morphologic features that were generally consistent with, and in some cases essentially indistinguishable from, those seen in chronic myeloid leukemia (Figure 1), though in cases 2-4 there was persistent fibrosis. All marrows showed hypercellularity, and four of five (patients 1, 2, 4, and 5) displayed granulocytic hyperplasia while patient 3 showed panmyelosis. Megakaryocytic atypia was variably prominent with the most common morphology being small, hypolobated forms ("dwarf" megakaryocytes), though large atypical megakaryocytes were additionally present in the marrow of patient 4. Patient 5 showed 10-15% myeloblasts and left-shifted granulocytic hyperplasia, findings which were interpreted to represent chronic myeloid leukemia in accelerated phase. Other than the myelofibrosis and the single case with large atypical megakaryocytes, there were no morphologic findings that could be attributable to the presence mutant JAK2. While morphologic features of chronic myeloid leukemia most often predominated, final interpretation generally acknowledged the dual contribution of both genetic lesions.
Laboratory and molecular response to treatment of JAK2+ BCR-ABL1+ myeloproliferative neoplasm
All patients started tyrosine kinase inhibitor therapy upon identification of BCR-ABL1, and in some cases, therapy directed towards the Ph-negative neoplasm was terminated. Response of the BCR-ABL1-harboring clones to tyrosine kinase inhibitor therapy varied. In patient 1, confirmed molecular response4 (quantitative molecular response showing a 4 log decrease in BCR-ABL1 transcripts) was achieved and maintained (Figure 1) while in the remaining four cases, suboptimal molecular responses were noted. In two cases (patients 4 and 5), including the patient who presented with accelerated phase chronic myeloid leukemia, tyrosine kinase inhibitor therapy failed to achieve a single log reduction in BCR-ABL1 transcripts in the short follow-up period (tested at 3 and 4 months, respectively) prior to death. Patients 2 and 3 showed molecular evidence of tyrosine kinase inhibitor failure with only a single log reduction at latest follow-up (tested at 18 and 25 months, respectively).
Assessment of the response of the JAK2+ harboring cells is complicated by less frequent testing and semi-quantitative nature of the assay. Patients 1 and 5 had positive JAK2 studies following tyrosine kinase inhibitor treatment; the other three patients were not tested. Patient 1, who showed BCR-ABL1 molecular response4, experienced an apparent increase in mutant JAK2 allele burden (initially interpreted to be monoallelic on a semi-quantitative assay, subsequently found to have 78% mutated JAK2 alleles by next generation sequencing) (Figure 1). Patient 5, with accelerated phase chronic myeloid leukemia, had a JAK2 V617F allele burden of <10% at identification of BCR-ABL1 as well as four months later, a finding which likely reflects the relative dominance of the BCR-ABL1 driven disease during that period of time.
All patients showed mild to moderate peripheral blood count abnormalities at latest follow-up variably involving all three myeloid lineages. Patient 1 showed an initial normalization of peripheral counts followed by progressive anemia and thrombocytopenia (Figure 1). Patients 4 and 5 never established complete normalization of blood counts in the limited follow-up period prior to death (at 4 and 6 months following identification of BCR-ABL1, respectively). Leukocytosis, thrombocytopenia, and anemia were the most common group-wide findings.
Bone marrow morphologic response to treatment of JAK2+ BCR-ABL1+ myeloproliferative neoplasm
Post-therapy bone marrow evaluation was only available for patient 1. In this patient, who had a longstanding history of primary myelofibrosis, an initial morphologic response to tyrosine kinase inhibitor therapy was evident after 6 months of therapy, with a decrease in the granulocytic hyperplasia and left-shift, reverting the apparent phenotype from that of chronic myeloid leukemia back to primary myelofibrosis. While molecular studies showed complete abrogation of BCR-ABL1 transcripts, a subsequent bone marrow biopsy after 4 years of tyrosine kinase inhibitor therapy showed progressive myelofibrosis (Figure 1). Of the remaining four patients, patients 4 and 5 did not have post-therapy bone marrow biopsies due to disease progression and death, and patients 2 and 3 had incomplete follow-up.
Outcome
At latest documented follow-up, patients 1-3 in this group are alive. Patient 1 has complications of fibrotic stage myelofibrosis (progressive pancytopenia, splenomegaly and profound constitutional symptoms) warranting JAK inhibitor therapy (Figure 1). Patients 2 and 3 were clinically stable with limited follow-up. Two patients have died: Patient 5 succumbed 4 months after diagnosis of chronic myeloid leukemia in accelerated phase (77 months after initial diagnosis of polycythemia vera) and patient 4 died 6 months after acquisition of BCR-ABL1 (113 months following initial diagnosis of polycythemia vera) due to complications of progressive myelofibrosis.
Group 2: BCR-ABL1 detected first, JAK2 V617F second (patient 6)
Laboratory findings and treatment during chronic myeloid leukemia disease phase
One of our eleven patients (patient 6) initially presented with BCR-ABL1+ chronic myeloid leukemia in chronic phase prior to the subsequent detection of JAK2 V617F, 109 months following initial diagnosis. Cytogenetics showed a 45-46,XX,del(3)(?),t(9;22)(q34;q11.2),-10 at diagnosis. After tyrosine kinase inhibitor therapy was initiated, molecular response3 was initially achieved, but persistently positive, with low level BCR-ABL1 transcripts detected up until the time that JAK2 V617F was identified (range 0.02% - 1.08% IS). While this patient maintained essentially normal WBC counts, rising platelet counts (from 242 ×103/μL to 638 ×103/μL) were noted during the two years preceding detection of JAK2 V617F.
Bone marrow histology during chronic myeloid leukemia disease phase Prior to JAK2
V617F identification, this patient's bone marrow biopsies showed histologic features consistent with chronic myeloid leukemia, including granulocytic hyperplasia and left shifted myeloid series. There were no overt morphologic features of Ph-negative myeloproliferative neoplasm.
Laboratory hematology findings at time of identification of JAK2
V617F A CBC was notable for marked thrombocytosis (Plt 858 ×103/μL) without leukocytosis or anemia (WBC 6.9 ×103/μL, Hgb 12.1 g/dL). Ovalocytes and dacrocytes were noted on peripheral blood smear.
Genetic findings at time of identification of JAK2 V617F
Molecular studies showed a moderate response to tyrosine kinase inhibitor therapy at the time of JAK2 V617F identification: BCR-ABL1 transcripts showed a 2.4 log reduction from 100% IS. Cytogenetic analysis was negative for t(9;22), but interestingly showed a new karyotypic anomaly: 46,XX,add(17)(p12)[17]/46,XX[3]. The first JAK2 study performed on this patient, 109 weeks following their diagnosis of chronic myeloid leukemia, was qualitative only. This patient had never been tested for a JAK2 mutation during her earlier chronic myeloid leukemia disease phase.
Bone marrow histology at time of identification of JAK2 V617F
Bone marrow biopsy performed at the time of JAK2 V617F identification showed a hypercellular marrow with increased numbers of morphologically atypical megakaryocytes that included hyperlobated, hypolobated and hyperchromatic forms. The marrow showed grade 2/3 reticulin fibrosis. A diagnosis of myeloproliferative neoplasm, not otherwise specified was rendered here, acknowledging the challenges in arriving at a definitive classification in this setting.
Laboratory and molecular response to treatment of JAK2+ BCR-ABL1+ myeloproliferative neoplasm
Over the subsequent years, the patient was treated with first and second generation tyrosine kinase inhibitors but never received a JAK inhibitor. During the first six years of follow-up, white blood cell counts remained within normal limits. Platelet counts were initially elevated (858 ×103/μL), but trended downward (to 139 ×103/μL). BCR-ABL1 transcript levels remained persistently low positive (range 0.004% to1.74% IS). Bone marrow cytogenetic studies showed persistence of the cytogenetic abnormality on the short arm of chromosome 17, though t(9:22) remained negative. The only quantitative JAK2 study revealed a mutant allele burden of 27.6% four years after initial JAK2 V617F identification.
Bone marrow morphologic response to treatment of JAK2+ BCR-ABL1+ myeloproliferative neoplasm
Follow-up bone marrow studies performed over the subsequent three years showed persistent hypercellularity, with a normal to mildly elevated M:E ratio. Megakaryocytes were increased and clustered. Reticulin fibrosis remained increased (grade 2/3). Blasts were not increased at the last bone marrow evaluation.
Outcome
The patient remains alive on therapy, although comorbidities, including development of medulloblastoma, have necessitated admission to a skilled nursing facility. At most recent follow-up, this patient shows leukocytosis (18.0 ×103/μL), anemia (10.1 g/dL), and thrombocytopenia (114 ×103/μL).
Group 3: BCR-ABL1 and JAK2 V617F detected simultaneously (patients 7-11)
Laboratory hematology findings at diagnosis of JAK2+ BCR-ABL1+ myeloproliferative neoplasm
These patients presented with variable peripheral blood abnormalities. Patients 7, 8, 10, and 11 had moderately elevated WBC counts (mean 15.1 ×103/μL, range 12.8 - 18.5 ×103/μL) (Figures 2 and 3), while patient 9 presented with marked leukocytosis (72.9 ×103/μL) (Figure 4). Patient 9 was anemic (9.7 g/dL) while the other four had mostly normal hemoglobin levels (mean 13.0 g/dL, range 11.7 - 13.6 g/dL). Patient 8 showed thrombocytosis (670 ×103/μL) while the other four had platelet counts less than 450 ×103/μL (mean 360 ×103/μL, range 268 - 432 ×103/μL).
FIGURE 2. Patient 7, Time course of laboratory and histopathologic features in a patient with simultaneously identified BCR-ABL1 and JAK2 V617F. Mixed morphologic features at both initial diagnosis and follow-up with progression to marrow failure.
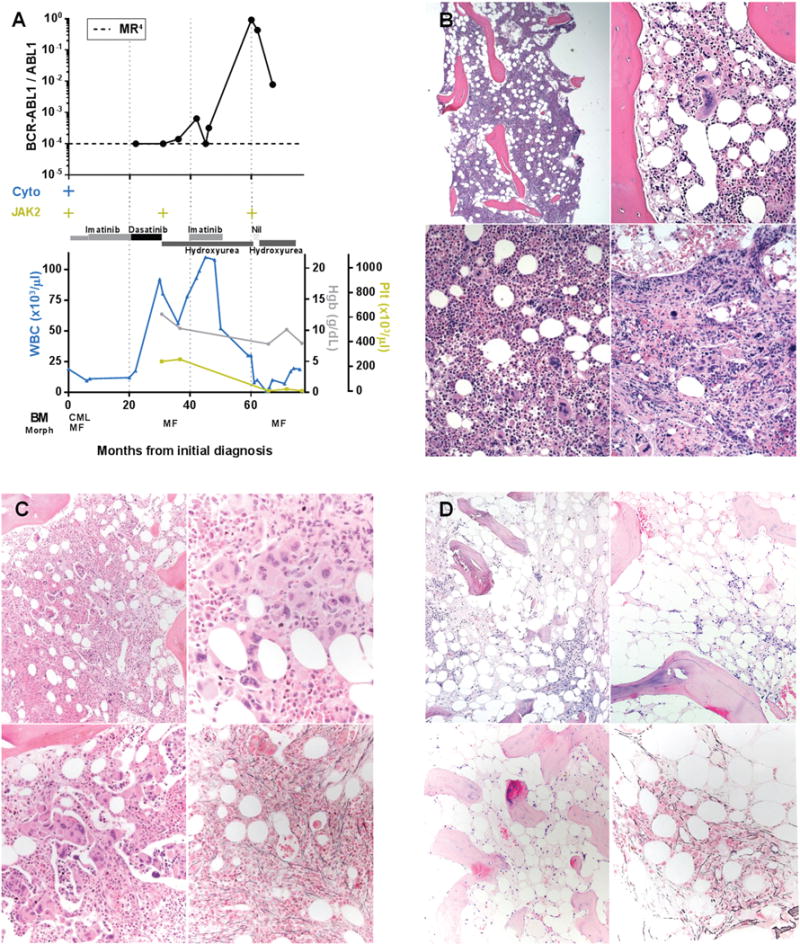
A. Time course of BCR-ABL1 transcript levels (log scale), cytogenetic t(9;22) results (Cyto), JAK2 V617F status, treatment, peripheral blood counts, and bone marrow morphology, Nil = nilotinib. B. Bone marrow core biopsy at initial diagnosis (month 0) showing features of chronic myeloid leukemia and myelofibrosis. Hypercellular (top left), mixed population of small hypolobated megakaryocytes and larger forms with atypical morphology (bulbous "cloud-like" nuclei, hyperlobation) with some clustering (top right, lower). C. Bone marrow core biopsy following tyrosine kinase inhibitor therapy showing predominant features of myelofibrosis (month 31). Hypercellular (top left), marked granulocytic hyperplasia, increased atypical megakaryocytes with variably bulbous "cloud-like" morphology (top right), many intrasinusoidal megakaryocytes (bottom left), increased reticulin fibrosis, grade 2/3 (bottom right). D. Bone marrow core biopsy following tyrosine kinase inhibitor and hydroxyurea therapy (month 65). Hypocellular (~10%) (top), osteosclerosis and residual reticulin fibrosis (bottom).
FIGURE 3. Patient 8, Time course of laboratory and histopathologic features in a patient with simultaneously identified BCR-ABL1 and JAK2 V617F. Tyrosine kinase inhibitor therapy for BCR-ABL1 unmasks an essential thrombocythemia phenotype37.
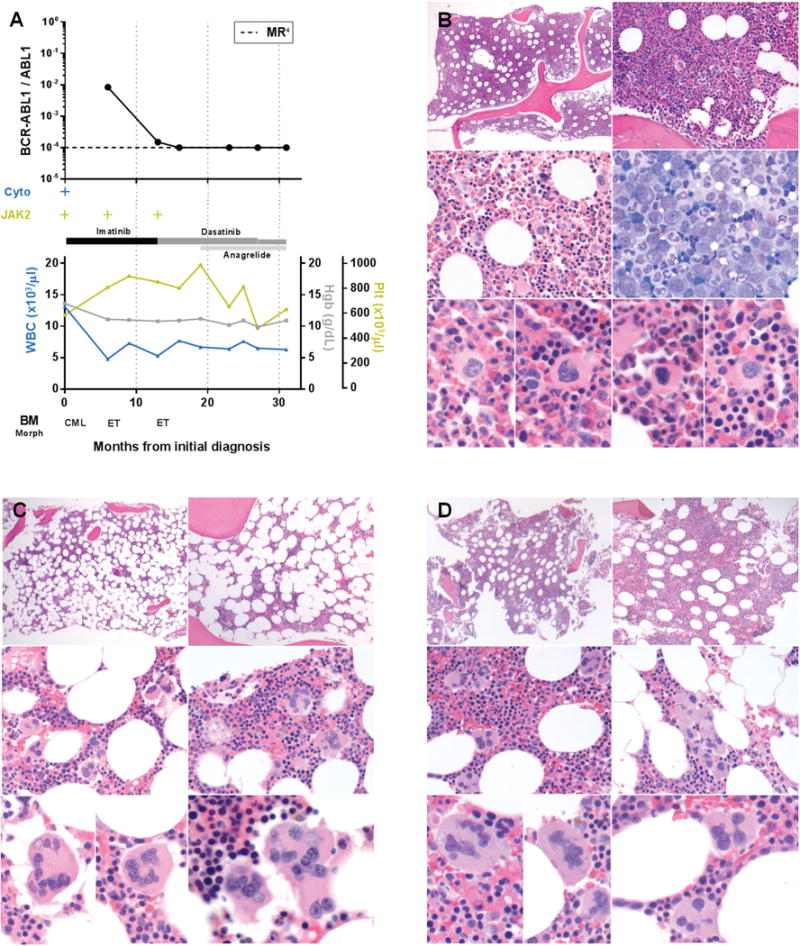
A. Time course of BCR-ABL1 transcript levels (log scale), cytogenetic t(9;22) results (Cyto), JAK2 V617F status, treatment, peripheral blood counts, and bone marrow morphology. B. Bone marrow core biopsy at initial diagnosis (month 0) showing predominant features of chronic myeloid leukemia. Hypercellular (top), elevated M:E ratio (middle), small hypolobated megakaryocytes (bottom). C and D. Bone marrow core biopsy after tyrosine kinase inhibitor therapy (month 6 and month 13, respectively) showing predominant features of essential thrombocythemia. Normocellular (C) to mildly hypercellular (D), hyperlobated “staghorn” megakaryocytes with clustering (middle and bottom).
Genetic findings at diagnosis of JAK2+ BCR-ABL1+ myeloproliferative neoplasm
Both BCR-ABL1 and JAK2 alterations were identified at initial diagnosis. Patients 7, 8, 9, and 10 displayed high levels of t(9;22) and/or BCR-ABL1 positive cells, with metaphase analysis showing a mean of 83% positive cells (range 60 -100%). In patient 11, cytogenetic analysis revealed 46,XX,t(9;22)(q34;q11.2)[2]/46,XX[19] and only 5% BCR-ABL1 positivity by FISH; RT-PCR in this patient showed the e1a2 BCR-ABL1 transcript only. Measurements of JAK2 levels were available for patients 8 and 10, and revealed a mutant allele fraction of <20%.
Bone marrow histology at diagnosis of JAK2+ BCR-ABL1+ myeloproliferative neoplasm
Bone marrow findings in this group were heterogeneous with relatively few consistent group-wide findings. All marrows were hypercellular and M:E ratio was mildly increased in each case (mean 3:8 to 1, range 3.4 - 5 to 1). Megakaryocytes showed atypical morphology and localization, though the atypical features varied by case. Patient 7 showed some small hypolobated megakaryocytes as well as some with bulbous or cloud-like nuclei with moderate loose clustering and grade 2/3 reticulin fibrosis (Figure 2). In patients 8 and 11, megakaryocytes showed predominantly prototypic chronic myeloid leukemia morphology (Figure 3). Conversely, patient 9 showed marked megakaryocytic hyperplasia (accounting for ~35% of total cellularity) with large clusters of hypolobated and hyperchromatic forms (Figure 4). Patient 9 showed 3/3 reticulin fibrosis with osteosclerosis, patients 7, 10, and 11 showed grade 2/3 fibrosis, and patient 8 showed grade 0/3. Overall, patient 8 showed features most consistent with chronic phase chronic myeloid leukemia (Figure 3), while the other four had nonspecific myeloproliferative features (Figures 2 and 4).
Laboratory and molecular response to treatment of JAK2+ BCR-ABL1+ myeloproliferative neoplasm
All patients started tyrosine kinase inhibitor therapy upon diagnosis; no additional therapy for the Ph-negative myeloproliferative neoplasm was started at that time. Patient 7 initially showed > 3 log reduction in BCR-ABL1 transcripts during tyrosine kinase inhibitor therapy; however, BCR-ABL1 transcripts increased when tyrosine kinase inhibitor therapy was temporarily withdrawn (Figure 2). Patient 8 achieved and maintained molecular response4 (Figure 3). Patient 9 failed both first and second generation tyrosine kinase inhibitors before advancing to an allogeneic stem cell transplant. Following transplant, BCR-ABL1 transcripts became undetectable (Figure 4). Patient 10 showed molecular evidence of disease progression (with BCR-ABL1 now showing a <1 log reduction) following acquisition of the imatinib-resistant p.E255K and p.T315I ABL1 mutations. Patient 11, who had only low level BCR-ABL1 disease burden at diagnosis (5% BCR-ABL1 positivity by FISH) showed relatively stable low level BCR-ABL1 transcripts equivalent to a 2 log “reduction” from the standardized baseline throughout treatment.
Patients 7, 8, and 10 subsequently received therapy more specifically targeting the Ph-negative myeloproliferative neoplasm and follow-up JAK2 studies were performed for patients 7, 8, 9, and 11. Patients 7, 8 and 11 remained positive for JAK2 V617F, consistent with the present dearth of treatments that effectively target JAK2 V617F clones. Mutated JAK2 allele percentage remained relatively low (7-8%) over the course of treatment for patient 7 (Figure 2). Following tyrosine kinase inhibitor therapy but prior to initiation of anagrelide, semi-quantitative studies for patient 8 showed an apparent increase in the percentage of mutant JAK2 alleles (from <10% to >25%), with a corresponding 2.4-log reduction in BCR-ABL1 transcripts; no JAK2 studies were performed after anagrelide therapy (Figure 3). Surprisingly, follow-up on patient 9, who did not receive treatment specific for Ph-negative myeloproliferative neoplasm, demonstrated a disappearance of the JAK2 V617F clone. However, this study was performed at disease progression to accelerated phase chronic myeloid leukemia with a tyrosine kinase inhibitor- resistant BCR-ABL1 clone, suggesting preferential expansion of an independent clone (Figure 4).
Despite therapeutic intervention, patient 7 developed progressive anemia and thrombocytopenia with moderate leukocytosis prior to death (Figure 2). Following tyrosine kinase inhibitor therapy, patient 8 achieved and maintained normalized white counts (13.1 to 4.8 ×103/μL) but showed increased thrombocytosis (670 to 809 ×103/μL). Platelet levels remained elevated until anagrelide therapy was initiated (Figure 3). Patient 9 showed a WBC response following tyrosine kinase inhibitor therapy (72.9 to 3.2 ×103/μL) which was sustained for over two years prior to the development of leukocytosis (35.9 ×103/μL) and soon after died (Figure 4). Patient 10 showed a transient improvement of hematologic parameters at last evaluation (WBC 15.2 ×103/μL, Hgb 9.4 g/dL, Plt 189 ×103/μL) before his discharge from the hospital and subsequent death from disease complications. Patient 11 maintained low-level leukocytosis throughout the follow-up period of 17 months (range 12.8 to 16.3 ×103/μL) with an essentially normal hemoglobin (range 12.5 - 13.6 g/dL) and high normal platelet count (range 409 - 440 ×103/μL).
Bone marrow morphologic response to treatment of JAK2+ BCR-ABL1+ myeloproliferative neoplasm
All patients had post-therapy bone marrow evaluation with a wide array of morphologic responses. After many modifications in therapy over 5 years, a bone marrow biopsy performed on patient 7 was hypocellular (~10%) with residual myelofibrosis and osteosclerosis (Figure 2). Patient 8, whose initial bone marrow revealed morphologic features consistent with chronic myeloid leukemia, showed normalization of M:E ratio and a predominance of hyperlobated “staghorn” megakaryocytes with the disappearance of the “dwarf” megakaryocytes after 6 and 13 months of tyrosine kinase inhibitor therapy. Therapy had essentially unmasked an essential thrombocythemia phenotype (Figure 3)(37). Patient 9, whose initial marrow revealed features of both chronic myeloid leukemia and myelofibrosis and grade 3/3 myelofibrosis, showed progression to an apparent accelerated phase tyrosine kinase inhibitor-resistant chronic myeloid leukemia with persistent (but mildly decreased) myelofibrosis after 15 months of therapy (Figure 4). Following an allogeneic stem cell transplant, the marrow showed no residual evidence of a myeloproliferative neoplasm (Figure 4). Patient 10, whose follow-up study consisted of a bone marrow aspirate smear only, showed progression to blast phase chronic myeloid leukemia (51% myeloblasts). Patient 11 showed no significant changes in bone marrow morphology after 4 months of tyrosine kinase inhibitor therapy.
Outcome
At most recent follow-up, patients 8 and 11 are alive with clinically stable disease. Patient 7 died 76 months after initial diagnosis secondary to complications of pneumonia, patient 10 died from complications of disease following progression to blast phase chronic myeloid leukemia and patient 9 died 32 months following diagnosis of his myeloproliferative neoplasm due to complications of bone marrow transplant.
DISCUSSION
We identified 11 patients with co-occurring BCR-ABL1 and JAK2 V617F mutations and provide a comprehensive analysis of their clinical, hematologic, pathological and genetic features. This study reflects the most detailed report to date of such cases. While clinical courses varied, in the absence of tyrosine kinase inhibitor therapy, patients generally demonstrated clinicopathologic features of chronic myeloid leukemia, suggesting that BCR-ABL1 has comparatively dominant transforming potential in this setting. Many patients showed progression to myelofibrosis, raising the possibility the two mutant tyrosine kinases may accelerate disease progression. Our study highlights the importance of the recognition of this scenario to prevent inappropriate diagnosis and management.
Frequency of occurrence
Our review of 1570 patients with suspected myeloproliferative neoplasm who underwent concurrent testing for both BCR-ABL1 and JAK2 V617F identified 6 (0.4%) who tested positive for both. An additional 5 cases were identified from clinical records. Two previous studies have provided estimates for the frequency of BCR-ABL1+ JAK2+ myeloproliferative neoplasms, though their selected cohorts differ from our own. One reported a frequency of 0.2%, a value that reflects a BCR-ABL1+ JAK2+ cohort (23 patients) amongst all patients (10,875) who were diagnosed with a myeloproliferative neoplasm (9134 JAK2 V617F+, 1487 BCR-ABL1+, and 254 MPL+)(38). Conversely, another group reported a frequency of 2.5% (8/314), a value generated by testing 314 patients with BCR-ABL+ chronic myeloid leukemia for JAK2 V617F from three centers in Italy(33). The disparity in these three reported frequencies likely reflects ascertainment bias as a consequence of the differences in the study design as well as variance in testing practices between institutions and countries. Our study population is the broadest: all patients with a clinical suspicion of a myeloproliferative neoplasm who were concurrently tested for BCR-ABL1 and JAK2 V617F, either at presentation or during follow up. Using this population, we establish a frequency of JAK2+ BCR-ABL1+ myeloproliferative neoplasm which is most clinically relevant during the initial phase of workup, given that it includes both patients who ultimately test positive as well as those who ultimately test negative for both BCR-ABL1 and JAK2 V617F. The other study populations are narrower – they evaluated only patients with prior myeloproliferative neoplasm diagnoses. Those selected populations (any patient with a mutation-positive myeloproliferative neoplasm or any patient with prior diagnosis of BCR-ABL1+ chronic myeloid leukemia) more specifically assess the likelihood of finding an additional genetic alteration amongst those patients who already have one documented. Finally, it is clear that clinical testing practices affect these calculated frequencies which likely vary widely between countries, institutions, and even clinicians. The factors which may prompt a clinician to test for both BCR-ABL1 and JAK2 V617F at initial workup, rather than one or the other, may be influenced by clinical parameters, amongst others factors. Likewise, the clinical and/or laboratory findings which prompt additional testing in a patient with a prior myeloproliferative neoplasm diagnosis are largely dependent on clinician initiative.
The striking difference in the percentage of patients who tested positive for BCR-ABL1 amongst those who were tested for BCR-ABL1 only (38.9%) and those that were tested for both BCR-ABL1 and JAK2 V617F (6.7%) in our study may be due to a number of interrelated factors. Since the hematologic features of chronic myeloid leukemia are more readily recognized by the ordering physician, a single BCR-ABL1 assay but not a JAK2 assay is likely to be requested in this setting. By contrast, patients with a suspected JAK2 V617F-positive, BCR-ABL1-negative myeloproliferative neoplasms often require a negative BCR-ABL1 assay in order to fulfill WHO diagnostic criteria.
Order of mutation identification
Six of the eleven patients in our study were noted to apparently acquire the second mutation sequentially. Of these, five were positive for JAK2 V617F and negative for BCR-ABL1 initially, with the subsequent acquisition of BCR-ABL1, while the other was positive for BCR-ABL1 initially, with JAK2 V617F only detected subsequently, noting that this patient was not tested for a JAK2 V617F initially. These differences are not altogether surprising since a patient with a suspicion of a myeloproliferative neoplasm other than chronic myeloid leukemia is more likely to be tested for both (to seek the presence of a JAK2 V617F and absence of BCR-ABL1), while one with a suspicion for chronic myeloid leukemia is more likely to be tested for BCR-ABL1 only at the time of presentation.
Clonal composition
Numerous prior reports have questioned the clonal composition of myeloproliferative neoplasms harboring both BCR-ABL1 and JAK2 V617F. Some studies have postulated that the two aberrations exist in the same clone or emerging subclones(4, 7, 9, 16, 35, 36) while others have argued for the presence of two independently coexisting neoplasms (i.e. "composite tumor")(3, 10, 18, 21, 34). It is becoming increasingly clear from prior studies that each of these proposed theories is possible, i.e. BCR-ABL1 and JAK2 V617F can exist in completely independent clones or the same clone, but with the majority of cases likely reflecting the presence of unrelated, independent clones.
We believe that our clinical data support this emerging consensus of the presence of a composite myeloproliferative neoplasm. Patient 8 (Figure 3), who presented with simultaneous detection of BCR-ABL1 and JAK2 V617F, showed initial clinical and morphologic features of chronic myeloid leukemia prior to tyrosine kinase inhibitor therapy. After tyrosine kinase inhibitor therapy effectively decreased BCR-ABL1 transcripts and normalized WBC count, JAK2 allele burden and platelet counts increased and subsequent bone marrow histomorphology showed features of essential thrombocythemia. Similarly, patient 1 (Figure 1), who had a longstanding history of clinically and hematologically stable primary myelofibrosis, experienced a sudden unexplained leukocytosis prompting additional workup which identified BCR-ABL1. At the time of BCR-ABL1 identification, bone marrow histology showed features of chronic myeloid leukemia. FISH and cytogenetic studies pointed towards substantial BCR-ABL1 disease burden. Subsequent tyrosine kinase inhibitor therapy eliminated all hematologic and molecular genetic evidence of BCR-ABL1 driven disease within 3 months, and bone marrow morphology reverted to that of primary myelofibrosis. In both of these patients, the response to tyrosine kinase inhibitor therapy argues for the presence of two distinct clones.
Relative dominance of mutation
Given the coexistence of two driver mutations in this patient population, it is important to understand the relative contribution of BCR-ABL1 and mutant JAK2 in producing the clinicopatholgic phenotype. Many have postulated that the BCR-ABL1 tyrosine kinase is a stronger driver of cellular proliferation and may more effectively influence phenotype(13, 16, 26, 33). JAK2 V617F, it follows, would play a smaller role in shaping the hematologic and morphologic phenotype. Within our cohort, a few key findings allow us to deduce the relative role of these two drivers. Amongst our patients with initially diagnosed JAK2+ myeloproliferative neoplasm, most of whom were under treatment, we noted marked left-shifted leukocytosis at the time of emergence of BCR-ABL1. Concurrently, bone marrow histology often showed morphologic features of chronic myeloid leukemia. These morphologic and hematologic findings were substantiated by the cytogenetic observation that t(9;22) was present in nearly all cells. Simultaneously, JAK2 V617F was generally present in only a minor subset of cells, as exemplified by patient 5. These findings suggest that BCR-ABL1 has dominant transforming potential in the setting of diminished mutationally activated JAK2. We also noted, in a subset of patients, that tyrosine kinase inhibitor therapy unmasked a Ph-negative myeloproliferative neoplasm phenotype. This observation suggests that essential clearance of the dominant BCR-ABL1 clone permits the weaker JAK2 driver to exert its effect. Prior reports have noted similar clinical courses(3, 27, 30, 33).
Histologic features suggesting the presence of concurrent BCR-ABL1 and JAK2 V617F
We sought to identify histologic features that might hint at the presence of multiple underlying genetic aberrations. Given the heterogeneous and often overlapping histologic features of myeloproliferative neoplasms, this presented a unique challenge. While there are no specific features that will decisively point to coexistent mutations, a few findings should raise the index of suspicion. Cases which exhibit mixed bone marrow cytologic features (e.g. both "dwarf" and "cloud-like" megakaryocytes) (Figure 2) or which show unexpected changes bone marrow histomorphology (e.g. marked granulocytic hyperplasia and hypolobated megakaryocytes in a patient with known primary myelofibrosis) (Figure 1) should prompt consideration of the possible coexistence of BCR-ABL1 and JAK2 V617F.
Laboratory features suggesting the presence of concurrent BCR-ABL1 and JAK2 V617F
Similarly, we sought to identify molecular and laboratory features which suggest the presence of coexistent mutations. Longitudinal molecular studies play an important role in monitoring disease progression and response to treatment in patients with myeloproliferative neoplasms, particularly those with chronic myeloid leukemia and we noted a few specific scenarios where molecular markers of disease burden and hematologic parameters were incongruent. In the one patient with a history of chronic myeloid leukemia who subsequently acquired JAK2 V617F, tyrosine kinase inhibitor therapy had acheived a >3 log reduction in BCR-ABL1 transcripts. Despite this apparent therapeutic success, platelet counts unexpectedly rose, a finding which ultimately led to the appropriate identification of JAK2 V617F. JAK2 mutant allele burden is less often used as a marker for disease status in Ph-negative myeloproliferative neoplasms and was not tested with sufficient frequency to reliably identify unexpected disease progression. However, in both patient 1 and patient 8, while treatment with tyrosine kinase inhibitor effectively diminished BCR-ABL1 transcripts, a concurrent apparent rise in JAK2 V617F allele burden was noted.
Is the location of the BCR breakpoint relevant?
Although our numbers are low, we noted an unexpectedly high fraction of patients (3 of 11) with an e1 breakpoint in BCR, raising the question of the role of the p190 oncoprotein in this subset of patients. In vitro studies suggest the p190 protein has a five-fold increase in tyrosine kinase activity over p210 which may explain its increased association with acute rather than chronic leukemia(39, 40) though no difference in outcome has been identified in the setting of B-lymphoblastic leukemias with p190 vs p210(41). Additional studies have reported a monocytosis in a subset of chronic myeloid leukemia patients with e1a2 translocation(41, 42). Amongst our three patients with p190, two died within 6 months of BCR-ABL1 identification. One patient developed accelerated phase chronic myeloid leukemia, the other showed progressive myelofibrosis. The third patient remains clinically well at last follow-up. None of these three patients showed sustained monocytosis.
Disease progression
It is worth noting that 7 of our 11 patients progressed to myelofibrosis, raising the possibility that these patients are more prone to progression. This finding also supports a concept that these cases exist within the more genetically complex end of the spectrum seen in myeloproliferative neoplasms. While polycythemia vera, essential thrombocythemia and to a lesser degree chronic myeloid leukemia have a propensity to develop fibrosis, it seems reasonable that two mutant tyrosine kinases might act in concert to accelerate fibrosis. In those cases where both lesions had been identified, patients often received a tyrosine kinase inhibitor only. While tyrosine kinase inhibitors are very effective for chronic myeloid leukemia, no similarly active tyrosine kinase inhibitor selectively targets JAK2 V617F. Thus, while one component of the patient's disease may be adequately treated, the second genetic alteration may not be. In our cohort, patient 1 (Figure 1) progressed to myelofibrosis despite a > 4.5 log reduction in BCR-ABL1 driven disease, indicating the dominant role that the mutant JAK2 played in development of the myelofibrosis. These findings are supported by similar reports in a subset of BCR-ABL1+ JAK2+ patients(16, 17).
In summary, this is, to our knowledge the largest reported study comprehensively detailing clinicopathologic features evident when BCR-ABL1 and JAK2 V617F co-occur. In some cases, features of an undiagnosed concurrent myeloproliferative neoplasm only manifest following treatment of the dominant myeloproliferative neoplasm. Although uncommon, it is important to be aware of this potentially confounding genetic combination, since these hematologic features may be misinterpreted to reflect resistance to therapy or disease progression, considerations that could lead to inappropriate management.
Acknowledgments
The authors would like to thank Cordelia Sever MD, and Fan Yang MD PhD, for contributing bone marrow histology images, and Mohammad Vasef, MD, for performing additional molecular testing, for patient 7.
Footnotes
DISCLOSURE/DUALITY OF INTEREST
The authors have no disclosures or dualities of interest to declare
References
- 1.Swerdlow SH, Campo E, Harris NL, et al. WHO classification of tumours of haematopoietic and lymphoid tissues. Lyon: International Agency for Research on Cancer; 2008. pp. 29–60. ((World Health Organization classification of tumours), Chapter 2, Myeloproliferative Neoplasms). [Google Scholar]
- 2.Arber DA, Orazi A, Hasserjian R, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. 2016;127:2391–405. doi: 10.1182/blood-2016-03-643544. [DOI] [PubMed] [Google Scholar]
- 3.Bee PC, Gan GG, Nadarajan VS, et al. A man with concomitant polycythaemia vera and chronic myeloid leukemia: the dynamics of the two disorders. Int J Hematol. 2010;91:136–9. doi: 10.1007/s12185-009-0471-6. [DOI] [PubMed] [Google Scholar]
- 4.Bocchia M, Vannucchi AM, Gozzetti A, et al. Insights into JAK2-V617F mutation in CML. Lancet Oncol. 2007;8:864–6. doi: 10.1016/S1470-2045(07)70295-4. [DOI] [PubMed] [Google Scholar]
- 5.Bornhauser M, Mohr B, Oelschlaegel U, et al. Concurrent JAK2(V617F) mutation and BCR-ABL translocation within committed myeloid progenitors in myelofibrosis. Leukemia. 2007;21:1824–6. doi: 10.1038/sj.leu.2404730. [DOI] [PubMed] [Google Scholar]
- 6.Cambier N, Renneville A, Cazaentre T, et al. JAK2V617F-positive polycythemia vera and Philadelphia chromosome-positive chronic myeloid leukemia: one patient with two distinct myeloproliferative disorders. Leukemia. 2008;22:1454–5. doi: 10.1038/sj.leu.2405088. [DOI] [PubMed] [Google Scholar]
- 7.Campiotti L, Appio L, Solbiati F, et al. JAK2-V617F mutation and Philadelphia positive chronic myeloid leukemia. Leuk Res. 2009;33:e212–3. doi: 10.1016/j.leukres.2009.06.011. [DOI] [PubMed] [Google Scholar]
- 8.Caocci G, Atzeni S, Orru N, et al. Response to imatinib in a patient with chronic myeloid leukemia simultaneously expressing p190(BCR-ABL) oncoprotein and JAK2V617F mutation. Leuk Res. 2010;34:e27–9. doi: 10.1016/j.leukres.2009.08.009. [DOI] [PubMed] [Google Scholar]
- 9.Conchon MR, Costa JL, Novaes MM, et al. Simultaneous detection of JAK2 V617F mutation and Bcr-Abl translocation in a patient with chronic myelogenous leukemia. Int J Hematol. 2008;88:243–5. doi: 10.1007/s12185-008-0131-2. [DOI] [PubMed] [Google Scholar]
- 10.Gattenlohner S, Volker HU, Etschmann B, et al. BCR-ABL positive chronic myeloid leukemia with concurrent JAK2(V617F) positive myelodysplastic syndrome/myeloproliferative neoplasm (RARS-T) Am J Hematol. 2009;84:306–7. doi: 10.1002/ajh.21296. [DOI] [PubMed] [Google Scholar]
- 11.Grisouard J, Ojeda-Uribe M, Looser R, et al. Complex subclone structure that responds differentially to therapy in a patient with essential thrombocythemia and chronic myeloid leukemia. Blood. 2013;122:3694–6. doi: 10.1182/blood-2013-07-516385. [DOI] [PubMed] [Google Scholar]
- 12.Hussein K, Bock O, Seegers A, et al. Myelofibrosis evolving during imatinib treatment of a chronic myeloproliferative disease with coexisting BCR-ABL translocation and JAK2V617F mutation. Blood. 2007;109:4106–7. doi: 10.1182/blood-2006-12-061135. [DOI] [PubMed] [Google Scholar]
- 13.Inami M, Inokuchi K, Okabe M, et al. Polycythemia associated with the JAK2V617F mutation emerged during treatment of chronic myelogenous leukemia. Leukemia. 2007;21:1103–4. doi: 10.1038/sj.leu.2404591. [DOI] [PubMed] [Google Scholar]
- 14.Inokuchi K, Yamaguchi H, Tamai H, et al. Disappearance of Both the BCR/ABL1 Fusion Gene and the JAK2V617F Mutation with Dasatinib Therapy in a Patient with Imatinib-Resistant Chronic Myelogenous Leukemia. J Clin Exp Hematop. 2012;52:145–7. doi: 10.3960/jslrt.52.145. [DOI] [PubMed] [Google Scholar]
- 15.Jallades L, Hayette S, Tigaud I, et al. Emergence of therapy-unrelated CML on a background of BCR-ABL-negative JAK2V617F-positive chronic idiopathic myelofibrosis. Leuk Res. 2008;32:1608–10. doi: 10.1016/j.leukres.2008.03.004. [DOI] [PubMed] [Google Scholar]
- 16.Krämer A, Reiter A, Kruth J, et al. JAK2-V617F mutation in a patient with Philadelphia-chromosome-positive chronic myeloid leukaemia. Lancet Oncol. 2007;8:658–60. doi: 10.1016/S1470-2045(07)70206-1. [DOI] [PubMed] [Google Scholar]
- 17.Pardini S, Fozza C, Contini S, et al. A case of coexistence between JAK2V617F and BCR/ABL. Eur J Haematol. 2008;81:75–6. doi: 10.1111/j.1600-0609.2008.01063.x. [DOI] [PubMed] [Google Scholar]
- 18.Pastore F, Schneider S, Christ O, et al. Impressive thrombocytosis evolving in a patient with a BCR-ABL positive CML in major molecular response during dasatinib treatment unmasks an additional JAK2V617F. Exp Hematol Oncol. 2013;2:24. doi: 10.1186/2162-3619-2-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pingali SRK, Mathiason MA, Lovrich SD, et al. Emergence of chronic myelogenous leukemia from a background of myeloproliferative disorder: JAK2V617F as a potential risk factor for BCR-ABL translocation. Clin Lymphoma Myeloma. 2009;9:E25–9. doi: 10.3816/CLM.2009.n.080. [DOI] [PubMed] [Google Scholar]
- 20.Qin YW, Yang YN, Li S, et al. Coexistence of JAK2V617F Mutation and BCR-ABL Translocation in a Pregnant Woman with Essential Thrombocythemia. Indian J Hematol Blood Transfus. 2014;30:331–4. doi: 10.1007/s12288-014-0385-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tefferi A, Levitt R, Lasho TL, et al. Postimatinib therapy emergence of a new JAK2V617F clone and subsequent development of overt polycythemia vera in a patient with chronic myelogenous leukaemia. Eur J Haematol. 2010;85:86–7. doi: 10.1111/j.1600-0609.2010.01458.x. [DOI] [PubMed] [Google Scholar]
- 22.Toogeh G, Ferdowsi S, Naadali F, et al. Concomitant presence of JAK2 V617F mutation and BCR-ABL translocation in a pregnant woman with polycythemia vera. Med Oncol. 2011;28:1555–8. doi: 10.1007/s12032-010-9570-8. [DOI] [PubMed] [Google Scholar]
- 23.Xu W, Chen B, Tong X. Chronic myeloid leukemia patient with co-occurrence of BCR-ABL junction and JAK2 V617F mutation. Int J Hematol. 2014;99:87–90. doi: 10.1007/s12185-013-1480-z. [DOI] [PubMed] [Google Scholar]
- 24.Zhou A, Knoche EM, Engle EK, et al. Concomitant JAK2 V617F-positive polycythemia vera and BCR-ABL-positive chronic myelogenous leukemia treated with ruxolitinib and dasatinib. Blood Cancer J. 2015;5:e351. doi: 10.1038/bcj.2015.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cappetta M, Perez V, Zubillaga MN, et al. Concomitant detection of BCR-ABL translocation and JAK2 V617F mutation in five patients with myeloproliferative neoplasm at diagnosis. Int J Lab Hematol. 2013;35:e4–5. doi: 10.1111/ijlh.12010. [DOI] [PubMed] [Google Scholar]
- 26.Hussein K, Bock O, Theophile K, et al. Chronic myeloproliferative diseases with concurrent BCR ABL junction and JAK2V617F mutation. Leukemia. 2008;22:1059–62. doi: 10.1038/sj.leu.2404993. [DOI] [PubMed] [Google Scholar]
- 27.Hummel JM, Kletecka MCF, Sanks JK, et al. Concomitant BCR-ABL1 translocation and JAK2(V617F) mutation in three patients with myeloproliferative neoplasms. Diagn Mol Pathol. 2012;21:176–83. doi: 10.1097/PDM.0b013e318246975e. [DOI] [PubMed] [Google Scholar]
- 28.Kim YK, Shin MG, Kim HR, et al. Simultaneous occurrence of the JAK2V617F mutation and BCR-ABL gene rearrangement in patients with chronic myeloproliferative disorders. Leuk Res. 2008;32:993–5. doi: 10.1016/j.leukres.2007.10.018. [DOI] [PubMed] [Google Scholar]
- 29.Laibe S, Tadrist Z, Arnoulet C, et al. A myeloproliferative disorder may hide another one. Leuk Res. 2009;33:1133–6. doi: 10.1016/j.leukres.2009.01.034. [DOI] [PubMed] [Google Scholar]
- 30.Lee YJ, Moon JH, Shin HC, et al. Two CML patients who subsequently developed features of essential thrombocythemia with JAK2-V617F mutation while in complete cytogenetic remission after treatment with imatinib mesylate. Int J Hematol. 2013;97:804–7. doi: 10.1007/s12185-013-1326-8. [DOI] [PubMed] [Google Scholar]
- 31.Mirza I, Frantz C, Clarke G, et al. Transformation of Polycythemia Vera to Chronic Myelogenous Leukemia. Arch Pathol Lab Med. 2007;131:1719–24. doi: 10.5858/2007-131-1719-TOPVTC. [DOI] [PubMed] [Google Scholar]
- 32.Park SH, Chi HS, Cho YU, et al. Two cases of myeloproliferative neoplasm with a concurrent JAK2 (V617F) mutation and BCR/ABL translocation without chronic myelogenous leukemia phenotype acquisition during hydroxyurea treatment. Ann Lab Med. 2013;33:229–32. doi: 10.3343/alm.2013.33.3.229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pieri L, Spolverini A, Scappini B, et al. Concomitant occurrence of BCR-ABL and JAK2V617F mutation. Blood. 2011;118:3445–6. doi: 10.1182/blood-2011-07-365007. [DOI] [PubMed] [Google Scholar]
- 34.Veronese L, Tchirkov A, Richard-Pebrel C, et al. A thrombocytosis occurring in Philadelphia positive CML in molecular response to imatinib can reveal an underlying JAK2(V617F) myeloproliferative neoplasm. Leuk Res. 2010;34:e94–6. doi: 10.1016/j.leukres.2009.09.025. [DOI] [PubMed] [Google Scholar]
- 35.Wang X, Tripodi J, Kremyanskaya M, et al. BCR-ABL1 is a secondary event after JAK2V617F in patients with polycythemia vera who develop chronic myeloid leukemia. Blood. 2013;121:1238–9. doi: 10.1182/blood-2012-11-467787. [DOI] [PubMed] [Google Scholar]
- 36.Xiao X, Zhang Y, Zhang GS, et al. Coexistence of JAK2V617F mutation and BCR-ABL1 transcript in two Chinese patients with chronic myelogenous leukemia. Acta Haematol. 2011;127:47–9. doi: 10.1159/000331564. [DOI] [PubMed] [Google Scholar]
- 37.Soderquist C, Bagg A. Coexistent BCR-ABL1 and JAK2 V617F: converting CML dwarves to ET staghorns with imatinib therapy. Blood. 2014;124:2463. doi: 10.1182/blood-2014-06-585141. [DOI] [PubMed] [Google Scholar]
- 38.Martin-Cabrera P, Haferlach C, Kern W, et al. BCR-ABL1-positive and JAK2 V617F-positive clones in 23 patients with both aberrations reveal biologic and clinical importance. Br J Haematol. 2016 doi: 10.1111/bjh.13932. [DOI] [PubMed] [Google Scholar]
- 39.Lugo TG, Pendergast AM, Muller AJ, et al. Tyrosine kinase activity and transformation potency of bcr-abl oncogene products. Science. 1990;247:1079–82. doi: 10.1126/science.2408149. [DOI] [PubMed] [Google Scholar]
- 40.Clarkson B, Strife A, Wisniewski D, et al. Chronic myelogenous leukemia as a paradigm of early cancer and possible curative strategies. Leukemia. 2003;17:1211–62. doi: 10.1038/sj.leu.2402912. [DOI] [PubMed] [Google Scholar]
- 41.Ohsaka A, Shiina S, Kobayashi M, et al. Philadelphia chromosome-positive chronic myeloid leukemia expressing p190(BCR-ABL) Intern Med. 2002;41:1183–7. doi: 10.2169/internalmedicine.41.1183. [DOI] [PubMed] [Google Scholar]
- 42.Melo JV, Myint H, Galton DA, et al. P190BCR-ABL chronic myeloid leukaemia: the missing link with chronic myelomonocytic leukaemia? Leukemia. 1994;8:208–11. [PubMed] [Google Scholar]