

Abstract
Macrophages have emerged as promising therapeutic targets in cancer. Within tumor tissue, macrophages foster tumor development, invasion, and metastasis. As the phenotype of macrophages is inherently pliable and dependent on cues received from the surrounding microenvironment, macrophages co-evolve with malignant and other non-malignant cells during cancer progression. In doing so, they establish a microenvironment that is therapeutically resistant and thwarts the productivity of T cell immunosuveillance. Strategies designed to deplete, inhibit, or redirect macrophages with anti-tumor activity are being explored to reverse the pro-tumor properties of macrophages that are commonly observed in cancer. In this review, we discuss our current understanding of the mechanisms that regulate macrophage recruitment to tumors, their impact on the tumor microenvironment, and their promise as therapeutic targets for improving the efficacy of cytotoxic- and immune-based therapies.
Keywords: Macrophages, cancer, tumor, resistance, immunotherapy, chemotherapy
1. Introduction
Macrophages are differentiated cells arising from the mononuclear myeloid lineage and can be found within many tissues where they mediate important biological functions including host defense against pathogens, removal of cellular debris, wound repair, tissue remodeling, and general maintenance of tissue homeostasis (Gordon and Martinez, 2010). Tissue-resident macrophages largely arise from the yolk-sac and seed tissues (e.g. liver, brain, skin, heart, kidneys, and pancreas) during the earliest stages of embryonic development. These macrophages persist into adulthood (Davies et al., 2013; Epelman et al., 2014). In cancer, embryonic-derived tissue macrophages have been found to support tumor development and progression (Zhu et al., 2017). However, tissue macrophages can also arise from tissue-infiltrating peripheral blood monocytes which develop in the bone marrow and are attracted to tissues by chemokines (Figure 1). Under normal homeostatic conditions, inflammatory monocytes arising from the peripheral blood traverse healthy tissue, but may then recirculate back into the peripheral blood (Jakubzick et al., 2014). However, in an inflamed setting, monocytes infiltrate tissues and differentiate into macrophages. In cancer, tumor-infiltrating monocytes contribute to therapeutic resistance and immune suppression (Kalbasi et al., 2016; Mitchem et al., 2013; Nywening et al., 2016).
Figure 1. Origins of tissue resident macrophages.
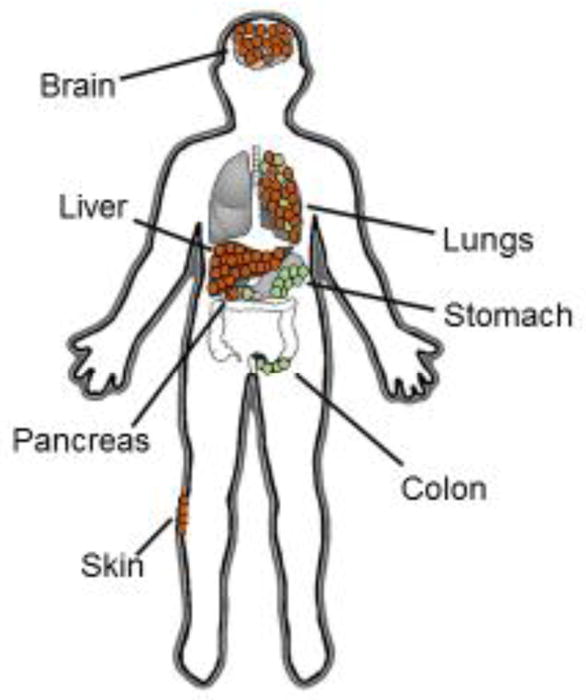
In the steady state, tissue macrophages are either (i) yolk sac- or fetal liver-derived (shown in brown) or (ii) monocyte-derived from the bone marrow (shown in green). Macrophages residing in the brain, liver, lungs, and red pulp of the spleen are predominately derived from the yolk sac with Langerhans cells of the skin derived from fetal liver. In contrast, resident macrophages in the gastrointestinal tract are mainly monocyte-derived. In the setting of inflammation, monocyte-derived macrophages infiltrate tissues and co-exist with tissue resident macrophages.
Within tissues, the phenotype and function (i.e. polarization status) of macrophages is directed by signals received from their surrounding microenvironment (Epelman et al., 2014). For example, macrophage phagocytosis of cellular material within tissues can imprint macrophages with a unique transcriptional signature and in doing so, contribute to the heterogeneity of tissue macrophages (A-Gonzalez et al., 2017). This inherent plasticity in macrophage biology has major implications and can be a critical determinant of the natural evolution of a disease, such as cancer.
Monocyte frequency in the peripheral blood has been suggested as a prognostic factor in patients with cancer (Liu et al., 2017). Monocytes are precursors for tumor-associated macrophages and their increased presence within tumors is most often associated with a poor prognosis (Qian and Pollard, 2010). Consistent with this notion, in patients with pancreatic cancer, low peripheral blood monocyte levels correlate with improved overall survival (Sanford et al., 2013). Similarly, resected cancers displaying a heavy infiltrate of macrophages show a greater likelihood for disease recurrence (Balermpas et al., 2014; Mahmoud et al., 2012; Shabo et al., 2009; Shabo et al., 2008; Wei et al., 2014). Macrophage infiltration in solid tumors is also associated with lower overall survival rates (DeNardo et al., 2011). The prognostic significance defined by the presence of macrophages in tumors is due to the capacity of macrophages to support many of the hallmarks of cancer (Hanahan and Weinberg, 2011) and mediate protective and pathogenic functions that can contribute to the development of an immunosuppressive microenvironment that surrounds malignant cells (Murray and Wynn, 2011). This microenvironment can thwart the development and productivity of anti-tumor immunity and inhibit the efficacy of cytotoxic therapies. Thus, macrophages are pivotal determinants of cancer biology.
During the earliest stages of tumor development, macrophages may, in some settings, acquire a tumor-suppressing phenotype that is associated with the production of pro-inflammatory cytokines such as interleukin (IL)-12, which can support anti-tumor cytotoxic T lymphocyte (CTL) recruitment and activation (Zaynagetdinov et al., 2011). Tumor-suppressive macrophages can produce inducible nitric oxide synthase (iNOS) and demonstrate tumoricidal activity (Eue et al., 1998). In lung cancer, the phenotype of tumor-infiltrating macrophages has been suggested as a prognostic indicator of overall survival (Yuan et al., 2015). Thus, tumor biology is most significantly impacted by the phenotype of tumor-infiltrating macrophages, rather than the mere presence of macrophages.
Given the nearly universal involvement of macrophages in cancer development and progression, macrophages have emerged as a promising therapeutic target for cancer. In this review, we discuss our current and emerging understanding of factors that regulate macrophage biology in tumors and strategies to manipulate macrophages for therapeutic benefit.
2. Macrophage plasticity
Within tissues, the specific phenotype (i.e. polarization status) of activated macrophages is determined by signals received from the surrounding microenvironment. The extracellular milieu that encompasses cells can produce a broad range of cellular signals and as such, this milieu is critical for instructing macrophages with vastly distinct functional properties. Phenotypical profiles of macrophages are manifested by differential expression of cell surface markers, production of cytokines and enzymes, and biological functions (e.g. metabolism, phagocytosis, and antigen presentation). However, given the expansive array of potential macrophage phenotypes, classification and characterization of macrophage subtypes has been challenging (Murray et al., 2014). One common classification strategy relies on in vitro findings determined using distinct cytokine stimuli to activate macrophages. Using this cytokine-differentiation approach, macrophages have been broadly classified as either M1 (i.e. classically activated), or M2 (i.e. alternatively activated) macrophages (Mantovani et al., 2002; Martinez and Gordon, 2014).
Traditionally, M1 macrophages are polarized by a combination of the inflammatory cytokine interferon-γ (IFN-γ) and the component of the outer membrane of gram negative bacteria, lipopolysaccharide (LPS). Alternatively, stimulation via granulocyte-macrophage colony stimulating factor (GM-CSF) can direct macrophages with an M1 phenotype (Joshi et al., 2014; Martinez and Gordon, 2014; Mills et al., 2000). M1 macrophages are pro-inflammatory and characterized by glycolytic metabolism (Galvan-Pena and O’Neill, 2014), the expression of CD86, MHC class II, and iNOS, as well as the production of specific cytokines, including IL-1, IL-6, IL-12, and TNF-α, as well as the production of reactive oxygen and nitric oxygen species. M1 macrophages are crucial for effective control of bacterial infections, but can also mediate anti-tumor activity (Qian and Pollard, 2010).
Conversely, M2 macrophages are traditionally polarized by anti-inflammatory cytokines, such as IL-4 and IL-13, or alternatively, by colony stimulating factor 1 (CSF1) (Joshi et al., 2014; Martinez and Gordon, 2014; Mills et al., 2000). These macrophages are characterized by increased fatty acid oxidation (Galvan-Pena and O’Neill, 2014), express high levels of macrophage scavenger receptor 1 (CD204), the mannose receptor (CD206), arginase 1, and low levels of MHC class II. M2 macrophages produce high levels of the cytokine IL-10, as well as vascular endothelial growth factor (VEGF) and matrix metalloproteinase 9 (MMP9). In addition to their involvement in anti-inflammatory processes, M2 macrophages have been associated with pro-tumorigenic functions, including angiogenesis and promoting tumor growth, survival and metastasis (Qian and Pollard, 2010), and have been shown to be important in wound healing responses (Epelman et al., 2014). M2 macrophages have also been further classified into subsets: M2a, M2b, M2c, and M2d (Murray et al., 2014). This further classification illustrates the complexity of phenotypes that can be manifested by macrophages and has led to the terminology of ‘M1-like’ and ‘M2-like’ to describe this range of biological phenotypes that can be observed in vivo. In general, tumor-associated macrophages have been described as displaying an M2-like phenotype, based on high expression of the mannose receptor and production of IL-10 and VEGF, with a relatively low production of cytokines attributed to M1-like macrophages, such as IL-1, IL-6, IL-12, and TNF-α.
The use of the M1/M2 classification for macrophages provides an easy, albeit oversimplified, strategy for defining macrophage biology. However, a major shortcoming of this strategy is the inability to account for variations in macrophage phenotype based on spatial localization within tissues. For example, TIE2+ macrophages, which reside adjacent to endothelial cells, show specific functional properties. Specifically, they are involved in regulation of angiogenesis in cancer (Chen et al., 2016a; Mazzieri et al., 2011). Thus, while the M1/M2 classification is a mainstay for many investigators as they seek to describe the biology of macrophages in cancer, it is critical to be mindful of particular factors that can influence macrophage phenotype and function including macrophage origin, spatial localization within tissues, and pathological state of the tissue. Each of these elements must be considered to accurately classify macrophages in cancer.
In this review, we classify macrophages broadly into tumor-promoting or tumor-suppressing macrophages. These extreme subsets are based on the capacity of macrophages to mediate a spectrum of activities that define tissue inflammation, tumorigenesis, wound healing, angiogenesis, and fibrosis (Davies et al., 2013; Epelman et al., 2014; Long and Beatty, 2013). Here, we also discuss ways to shift the polarity of tumor-infiltrating macrophages from tumor-promoting to tumor-suppressing.
2.1. Mechanisms that recruit macrophages to tumors
Monocytes are recruited to solid tumors via various mechanisms, including chemokine-driven chemotaxis and extracellular matrix components acting as chemoattractants. Because the majority of monocytes infiltrating tumors are Ly6Chi inflammatory monocytes that express high levels of CCR (C-C chemokine receptor) 2, trafficking and recruitment of monocytes to tumor tissue is significantly controlled by CCL (C-C chemokine ligand) 2 (Epelman et al., 2014; Franklin et al., 2014; Mitchem et al., 2013; Qian et al., 2011). CCL2 production is commonly upregulated in solid tumors (Lim et al., 2016) and has a significant role in fostering monocyte recruitment to tumors. In addition, CCL2 can enhance monocyte mobilization from the bone marrow to the peripheral blood (Sanford et al., 2013). However, other chemokines can also interact with CCR2 and in doing so, promote monocyte infiltration into tissues (Lim et al., 2016). For example, CCL7 can bind CCR2 and support the recruitment of CCR2 expressing monocytes (Shi and Pamer, 2011). In addition, other chemokines such as CCL5 that are produced by solid tumors can act as chemoattractants for monocytes (Balkwill, 2004; Murdoch et al., 2004). Blockade of CCR5 has been found to induce tumor regressions in both mice and humans with colorectal carcinoma that is dependent on a shift in the polarity of tumor-infiltrating macrophages from tumor promoting to tumor suppressing (Halama et al., 2016). Thus, chemokines are important regulators of monocyte recruitment to tumors.
In addition to chemokines, extracellular matrix components present within the stroma that surrounds malignant cells can act to recruit and retain monocytes as macrophages within tissues. For example, type I collagen is significantly overproduced in many solid malignancies and can act as a chemoattractant for monocytes. Consistent with this, monocytes and macrophages are commonly lured to type I collagen rich regions (Parks et al., 2004). Moreover, the extent of macrophage migration in tissues occurs in a type I collagen concentration dependent manner (Postlethwaite and Kang, 1976). Notably, this collagen-dependent recruitment does not require intact collagen fibers, as collagen fragments can also attract monocytes and macrophages (Mundy et al., 1981). The ability of collagen fragments to recruit monocytes and macrophages has particular relevance in cancer as collagen within the tumor microenvironment is in a constant state of production and breakdown.
Like type I collagen, hyaluronic acid (HA) has also been shown to contribute to monocyte mobilization and trafficking into tissues (Kobayashi et al., 2010). HA is deposited in the stroma of many cancers and is involved in enhancing the stiffness of the matrix. Disrupting HA production by conditional gene targeting of the HA synthase 2 gene in stromal fibroblasts has been found to impair the recruitment of macrophages to tumors (Kobayashi et al., 2010). Thus, the extracellular matrix that surrounds malignant cells is critical to the recruitment of monocytes and their retention in cancer tissues.
2.2. Soluble factors produced by malignant and non-malignant cells regulate macrophage biology
Macrophages are a key component of cancer-associated inflammation which is a major contributor to many of the hallmarks of cancer (Hanahan and Weinberg, 2011). During the earliest stages of tumor development, macrophages may initially display a tumor-suppressing phenotype due to the presence of pro-inflammatory cytokines present within an inflamed tissue. These early tumor-suppressing macrophages can act to promote anti-tumor immune responses that may seek to help eliminate tumor cells (Zaynagetdinov et al., 2011). However, this early inflammatory response can also stimulate the expression of adhesion molecules, such as ICAM-1, that can subsequently, serve as a chemoattractant for further recruitment of macrophages (Liou et al., 2015). With tumor progression, macrophages within tumors almost invariably assume a tumor-promoting phenotype (Qian and Pollard, 2010).
The interaction between macrophages and tumor cells has commonly been studied under in vitro conditions using conditioned media produced during tumor cell culture. In this setting, macrophages upregulate expression of markers associated with an M2-like phenotype, including IL-10, VEGF, and MMP9 (Sica and Mantovani, 2012). This process occurs independent of direct cell-to-cell interaction and thus, illustrates the importance of tumor-derived factors in regulating macrophage biology.
Within the tumor microenvironment, factors such as hypoxia may also influence the phenotype and function of macrophages. For instance, hypoxia-induced factors, including Semaphorin 3a, are known to attract macrophages to and retain them within tumor regions that demonstrate low oxygen tension (Casazza et al., 2013). To this end, hypoxia is well-recognized to stimulate a transcriptional response pathway that is mediated in large part by hypoxia-inducible factor (HIF) proteins, HIF1 and HIF2. HIF1 induces the release of the chemoattractant SDF1α, also known as CXCL (C-X-C ligand) 12, and in doing so, supports the recruitment of CXCR (C-X-C receptor) 4+ macrophage populations and the subsequent release of soluble VEGF through increased macrophage-derived MMP-9 activity (Du et al., 2008). This hypoxia-regulated response promotes macrophage entrapment within hypoxic regions of tumors and supports angiogenesis and immunosuppression (Casazza et al., 2013; Du et al., 2008). However, hypoxia can also influence the phenotypic fate of macrophages by promoting anaerobic glycolysis within tissues and the subsequent release of lactic acid. Colegio et al. showed that lactic acid can serve as a metabolic substrate for tumor-associated macrophages and as a result, stimulate a HIF1α-dependent and IL-4-independent increase in macrophage expression of tumor-promoting factors including arginase 1 and VEGF (Colegio et al., 2014). HIF1α has also been shown to support macrophage glycolysis and ATP generation important for chronic inflammatory states (Cramer et al., 2003). In addition, the shift in the metabolic state of macrophages induced in the setting of hypoxia may be critical to defining their response to pro-infilammatory stimuli (Mills et al., 2016). Together, these findings highlight the complexity of molecules within the tumor microenvironment that can contribute to macrophage behavior.
Cytokines produced by non-malignant cells present within the tumor microenvironment are also important determinants of macrophage phenotype. In this regard, IL-4 and IL-13, leukemia inhibitory factor, IL-6, and lactic acid have each demonstrated roles in regulating macrophage polarization in vivo (Colegio et al., 2014; Duluc et al., 2007; Hagemann et al., 2006; Roca et al., 2009; Stein et al., 1992). Similarly, transforming growth factor-beta (TGF-β) can impact macrophage function within tumors by inhibiting tumor-infiltrating macrophages from acquiring an M1-like phenotype (Haak-Frendscho et al., 1990). Thus, macrophage biology is intricately controlled within tumors by both malignant and non-malignant cells.
3. Macrophage biology within the tumor microenvironment
During cancer development, malignant cells often become encased in dense fibrosis, referred to as desmoplasia. This fibrotic reaction is observed in many types of solid tumors and is driven by an overproduction and accumulation of excess extracellular matrix proteins, including fibrillar collagen type I and type III proteins and fibronectin (Cox and Erler, 2014; Wynn, 2008). Tumor fibrosis is dependent on fibroblasts which are the major producers of extracellular matrix proteins (Wynn and Ramalingam, 2012). Like macrophages, fibroblasts can be induced with either a pro- or anti-fibrotic phenotype. Fibroblast biology is also pliable and is dependent on factors present within their surrounding microenvironment (Cox and Erler, 2014; Wynn and Ramalingam, 2012). As such, macrophages and fibroblasts may co-evolve within tumors with the resulting tumor fibrosis contributing to tumor growth, survival, and metastasis.
3.1. Tumor fibrosis regulates macrophage phenotype
The recruitment, differentiation status and activation of macrophages within tissues is strongly influenced by extracellular matrix proteins (Egeblad et al., 2010; Kobayashi et al., 2010; Mundy et al., 1981; Postlethwaite and Kang, 1976). In particular, the rigidity of the extracellular matrix can alter the polarization state and subsequently, the function of macrophages (McWhorter et al., 2015). For example, increased extracellular matrix rigidity can facilitate the polarization of macrophages toward an M2-like phenotype as seen by an increased expression of CD204 and arginase 1 (McWhorter et al., 2013; Stahl et al., 2013). Similarly, culturing macrophages in vitro on a type I collagen matrix has been found to decrease the tumoricidal activity of macrophages (Kaplan, 1983). Glycosaminoglycans (e.g. hyaluronic acid), which are key extracellular matrix components of tumor fibrosis, can also influence macrophage phenotype (Rayahin et al., 2015) by inducing an M2-like polarization of tumor-associated macrophages (Kuang et al., 2007). Thus, the matrix that surrounds malignant cells can shape the phenotype of tumor-infiltrating macrophages.
Aside from the physical aspect that fibrosis contributes to the tumor microenvironment, it can also promote hypoxia which in turn, can paralyze the migration of macrophages within tumor tissues as discussed above and skew their differentiation toward a tumor-promoting phenotype (Henze and Mazzone, 2016). Under hypoxic conditions, tumor cells may respond by releasing exosomes containing microRNAs, which can induce macrophages with tumor-promoting properties (Chen et al., 2017). Activated cancer-associated fibroblasts have also been shown to contribute to the differentiation of monocytes and the polarity of tumor-associated macrophages by releasing soluble factors, such as IL-6. Here, IL-6 produced by fibroblasts has been shown to stimulate the differentiation of monocytes into macrophages (Chomarat et al., 2000). Moreover, when cultured in fibroblast conditioned media, monocytes have been found to differentiate into macrophages with tumor promoting properties (Mathew et al., 2016). This potential cross-talk between fibroblasts and macrophages provides further support for their co-evolution within tumors as malignant cells seek to orchestrate a microenvironment that is conducive to tumor development and progression.
3.2. Macrophage regulate fibroblast phenotype and activation
While fibroblasts can influence macrophage phenotype, macrophages can also impact the biology of fibroblasts. For example, during earliest stages of tumor development, IL-1β produced by macrophages has been found to activate cancer associated fibroblasts, which in turn can stimulate recruitment and polarization of a new wave of tumor infiltrating macrophages (Erez et al., 2010). This reciprocal interaction between macrophages and fibroblasts is appreciated in the context of wound repair and tissue fibrosis where macrophages produce TGF-β and platelet-derived growth factor which together activate fibroblasts and promote their proliferation (Pakyari et al., 2013). Similarly, in the setting of cancer, activated fibroblasts are potent contributors to fibrosis and can be activated by tissue macrophages via TGF-β (Pickup et al., 2013). This cross-talk between macrophages and fibroblasts has also recently been shown to be a mechanism for altering the liver microenvironment with increased susceptibility to cancer cell metastasis (Costa-Silva et al., 2015). Thus, reciprocal interactions between fibroblasts and macrophages are fundamental to tumor development and cancer cell spread to distant organs.
3.3. Macrophage orchestrate extracellular matrix production and remodeling
The accumulation of fibrosis in tumor tissue is regulated by the balance of extracellular matrix protein production and degradation. Macrophages are major orchestrators of this balance. For example, Afik and colleagues showed in a mouse model of colorectal cancer that tumor-associated macrophages can contribute directly to extracellular matrix production as well as the cross-linking of collagen fibers (Afik et al., 2016). Macrophages are also major producers of matrix metalloproteinases (MMPs) as well as tissue inhibitor of matrix metalloproteinases (TIMPs). These proteins act to support and inhibit, respectively, the breakdown of extracellular matrix. For instance, MMP9 is a gelatinase produced by macrophages that can selectively degrade type IV collagen associated with the basement membrane and in doing so, support tumor invasion and subsequent metastasis (Huang et al., 2002). The ability of macrophages to both support the deposition and breakdown of fibrosis is a remarkable property dependent on their phenotype. This has been clearly demonstrated in models of liver fibrosis where macrophages are required for both deposition of fibrosis in the setting of chemical injury as well as subsequent resolution of fibrosis upon removal of the chemical insult (Duffield et al., 2005). In this model of chemical-induced liver fibrosis, collagen degradation and tissue repair was shown to be dependent on macrophage production of MMP13 (Fallowfield et al., 2007). Similarly, in a model of pancreatic cancer, it has been found that macrophages can stimulate the production of MMP13 in tumor tissue, and in doing so, shift tumors from chemotherapy resistant to chemotherapy sensitive (Long et al., 2016). In the steady-state, this cross-talk between macrophages and fibrosis is dependent, at least in part, on elevated activity of focal adhesion kinase (FAK) which has been shown to be an important determinant of the fibrotic and immunosuppressive microenvironment observed in pancreatic cancer (Jiang et al., 2016). Disruption of FAK activity alters the expression of myeloid chemoattractants within tumors and with sustained inhibition is associated with a decrease in myeloid recruitment and fibrosis (Jiang et al., 2016). Thus, macrophages hold a strong governance over tissue matrix composition which can influence the natural history of cancer progression and therapeutic outcomes.
4. Macrophages as targets for cancer therapy
Macrophages are major contributors of therapeutic resistance in solid tumors. For example, macrophages can support malignant cell survival in the setting of cytotoxic stress by regulating angiogenesis and by inducing an immunosuppressive gene signature in tumors (Chen et al., 2016a; Kaneda et al., 2016; Mazzieri et al., 2011). In addition, macrophages can contribute to the physical architecture (i.e. collagen and HA) of the tumor microenvironment which can limit effective delivery of cytotoxic drugs (Olive et al., 2009; Provenzano et al., 2012). For these reasons, macrophages have emerged as a promising therapeutic target in cancer.
Strategies to target macrophages in cancer have focused on approaches that are designed to (i) deplete, (ii) inhibit or (iii) redirect macrophages (Liu et al., 2017) (Figure 2). Macrophages can be depleted from the tumor microenvironment by blocking their recruitment using inhibitors of chemokine signaling pathways (e.g. CCR2), by blocking growth factors (e.g. CSF1/CSF-1R) important for macrophage survival, or by directly targeting macrophage subsets for cell death (e.g. trabectedin) (Germano et al., 2013; Ries et al., 2014; Sanford et al., 2013). The functions of tumor-infiltrating macrophages can also be inhibited by targeting signaling pathways (e.g. JAK/STAT), cytokines secreted by macrophages (e.g. IL-6) and immunosuppressive molecules produced by macrophages (e.g. IDO, arginase I) (Chang et al., 2001; Lesina et al., 2011; Long et al., 2017; Munn and Mellor, 2007). Finally, the inherent pliability of tumor-associated macrophages offers an opportunity to redirect macrophages with anti-tumor properties. For example, macrophages can be harnessed for anti-tumor activity by disengaging molecules that inhibit macrophage biology (e.g. CD47, HDAC IIa) or by directly activating macrophages with anti-tumor properties (e.g. IFN-γ, CD40, TLR agonists) (Beatty et al., 2017; Chao et al., 2012b; Guerriero et al., 2017). Here, we discuss each of these strategies for targeting macrophages for cancer therapy.
Figure 2. Targeting macrophage biology in cancer.
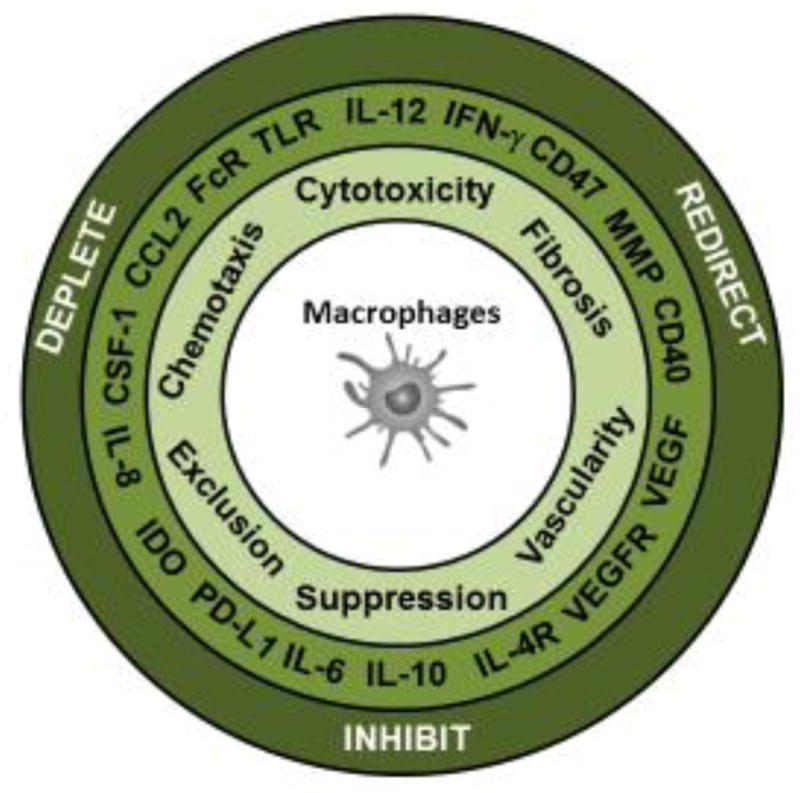
Macrophages can mediate an array of biological activities in cancer (e.g. chemotaxis, direct cytotoxicity against tumor cells, regulation of fibrosis and vascularity as well as suppression and exclusion of T cells. Multiple molecules are associated with each of these activities and serve as targets for therapeutic intervention involving strategies that inhibit, deplete or redirect macrophages in cancer.
4.1. Depleting macrophages from tumors
4.1.1 Targeting the CCL2/CCR2 pathway
Peripheral blood monocytes are actively recruited to tumors by chemokines produced within the tumor microenvironment. Thus, strategies to disrupt chemokine signaling are being studied as an approach to restrict myeloid infiltration into tumors and their subsequent pro-tumor functions. CCR2 is expressed at elevated levels on a subset of monocytes which infiltrate tumors in response to the chemokine CCL2. In melanoma, overexpression of CCL2 by tumor cells is associated with an enhanced presence of tumor-associated macrophages and rapid tumor progression (Gazzaniga et al., 2007). In CCR2 deficient mice, macrophage recruitment to orthotopically implanted pancreatic cancer is also markedly reduced and is associated with slowed tumor growth (Sanford et al., 2013).
The importance of CCL2 in regulating macrophage recruitment to tumors has been studied using neutralizing antibodies to CCL2. For example, neutralization of CCL2 with a monoclonal antibody (mAb) decreases overall tumor burden in models of pancreas, prostate, breast and lung cancer (Loberg et al., 2007; Mitchem et al., 2013; Qian et al., 2011; Zhao et al., 2013). Similarly, neutralizing CCL2 mAbs have been shown to decease macrophage accumulation in a model of glioma and in doing so, improve overall survival (Zhu et al., 2011).
CCL2/CCR2 blockade can also improve the efficacy of cytotoxic therapies. For example, CCL2 neutralization enhances the efficacy of radiotherapy in a model of pancreas cancer by blocking the infiltration of pro-tumor macrophages which are recruited in response to radiation treatment to support tumor cell survival (Kalbasi et al., 2016). CCR2 inhibitors have also been shown in some settings to stimulate T cell infiltration raising the possibility that this therapeutic target may combine with inhibitors of immune checkpoint molecules (e.g. PD-1 and CTLA-4) that act to enhance the activity of endogenous anti-tumor T cell immunity (Mitchem et al., 2013). In this study, blockade of CCR2 signaling with a small molecule inhibitor was found to enhance the efficacy of chemotherapy in mice. Similarly, an early phase clinical study has demonstrated promise for combining CCR2 inhibitors with chemotherapy for treatment of patients with locally advanced and borderline resectable pancreatic carcinoma (Nywening et al., 2016). Thus, derailing monocyte recruitment to tumors by inhibiting chemokine signaling via CCR2 has emerged as an important therapeutic approach in cancer.
4.1.1 Targeting CSF-1/CSF-1R
The survival of macrophages within tumors is dependent on growth factors such as colony-stimulating factor 1 (CSF-1). Strategies to disrupt CSF-1 signaling via the CSF-1 receptor (CSF-1R) produces macrophage depletion in some tumors. In a model of pancreatic cancer, blockade of CSF1R signaling depleted macrophages within tumors, and when combined with chemotherapy, significantly slowed tumor growth (Mitchem et al., 2013). Genetic deletion of CSF-1 using CSF-1−/− mice is also associated with decreased accumulation of macrophages within tumors and reduced tumor outgrowth (Pyonteck et al., 2012).
The importance of CSF-1 for macrophage survival in tumors has been seen across several malignancies. However, in some settings, inhibition of CSF-1 signaling is associated with a shift in macrophage phenotype rather than a decrease in macrophage survival. For example, in a model of glioma, CSF-1R inhibition using a small molecule inhibitor was found to redirect tumor-infiltrating macrophages with anti-tumor activity (Pyonteck et al., 2013). In this case, macrophage survival in the absence of CSF-1R signaling was supported by additional signals, including IFN-γ and GM-CSF, present within the tumor microenvironment. In a model of liver cancer, CSF1R blockade was also observed to shift the phenotype of macrophages from tumor-promoting to tumor-suppressing, resulting in decreased tumor growth (Ao et al., 2017).
Depletion of tumor-infiltrating macrophages using CSF-1R blockade has been found to induce T cell dependent anti-tumor activity (DeNardo et al., 2011; Zhu et al., 2014). Based on this finding, several groups have recently demonstrated the potential to combine CSF-1R blockade to deplete macrophages with immunotherapy, including dendritic cell vaccination and antagonistic antibodies targeting PD-1 and CTLA-4 (Dammeijer et al., 2017; Holmgaard et al., 2016; Mao et al., 2016). Based on this work, ongoing clinical studies are evaluating the use of both antibodies and small molecule inhibitors to target the CSF1/CSF1R pathway as a strategy for altering macrophage biology in tumors and improving the activity of both cytotoxic chemotherapy and immunotherapy.
4.2. Inhibiting pro-tumor activity by tumor macrophages
Tumor-infiltrating macrophages commonly show activation through multiple signaling pathways including signal transducer and activator of transcription-3 (STAT3) which is associated with pro-carcinogenic inflammation (Corcoran et al., 2011; Lesina et al., 2011; Yu et al., 2009). Elevated phosphorylated STAT3 levels in tumors is correlated with decreased overall survival in patients with advanced solid malignancies (Nagathihalli et al., 2015). While multiple stimuli can activate STAT3 signaling including VEGF, LIF, IL-17, IL-11 and IL-6 among others, IL-6 is a potent and major regulator of STAT3 activation. However, blockade of IL-6 signaling using IL-6 receptor blocking antibodies or genetic deletion of IL-6 only partially inhibits STAT3 activation in tumors with little impact on STAT3 activation in myeloid cells (Long et al., 2017). This finding supports the notion that multiple signaling pathways may converge to induce STAT3 activation in macrophages in vivo.
The skewing of macrophages toward an M2-like phenotype via IL-4 and IL-13 signaling is directed through the STAT6 pathway. Like STAT3, STAT6 expression is upregulated in macrophages in vivo and has been shown to enhance tumor growth in a mouse model of pancreas cancer (Yan et al., 2016). In breast cancer, STAT6 activation inhibits T cell-dependent anti-tumor immunity (Sasaki et al., 2008). Tumor-derived soluble factors can also stimulate STAT6 activation in macrophages leading to an increase in CD206 expression associated with an M2-like phenotype (Chen et al., 2016b). In addition, STAT3 and STAT6 signaling pathways have been shown to synergize to upregulate cathepsin secretion by macrophages which can support tumor cell invasion and metastasis (Yan et al., 2016). Thus, strategies to selectively inhibit STAT activation in macrophages may yield therapeutic benefit.
Inhibition of STAT signaling has been shown to improve the efficacy of cytotoxic chemotherapy (Long et al., 2017; Nagathihalli et al., 2015) and to inhibit the progression of premalignant lesions to advanced carcinoma (Corcoran et al., 2011; Lesina et al., 2011). As a result, ongoing clinical trials are evaluating the use of selective JAK1 inhibitors as well as non-selective JAK1/2 inhibitors as strategies to reverse STAT signaling in tumors (Liu et al., 2017). More selective strategies to reverse STAT activation mediated by distinct molecules, such as VEGF and IL-6, are also being evaluated. For example, IL-6 receptor blockade has emerged as a therapeutic approach to inhibit macrophage activation associated with cytokine release syndrome seen in the setting of adoptive cell therapy with chimeric antigen receptor-modified T cells (Maude et al., 2014). Blockade of IL-6 receptor can also enhance the activity of chemotherapy in a spontaneous model of pancreatic cancer [Long et al MCT 2017]. Thus, the STAT pathways and the stimuli that mediate their activation are important targets for modulating macrophage and tumor biology.
4.3. Redirecting tumor-infiltrating macrophages with anti-tumor activity
The inherent plasticity of macrophage phenotype suggests the possibility that tumor-infiltrating macrophages may be redirected with anti-tumor activity. Strategies to shift the phenotype of macrophages in cancer have addressed approaches to (i) deliver activation signals (e.g. CD40, IFN-γ, TLR agonists) and (ii) relieve inhibitory signals (e.g. CSF1R, CD47, HDAC IIa) that control the phenotypical fate of macrophages (Beatty et al., 2017; Chao et al., 2012b; Guerriero et al., 2017; Pyonteck et al., 2013).
Under in vitro conditions, macrophages can be induced with anti-tumor activity upon exposure to IFN-γ and LPS. In this setting, macrophages mediate anti-tumor activity by releasing tumoricidal soluble factors, including reactive oxygen and nitrogen species as well as cytokines, such as TNF-α (Long and Beatty, 2013). Macrophages stimulated with IFN-γ and LPS can also directly mediate tumor cell lysis as well as phagocytose tumor cells (Feng et al., 2015).
IFN-γ is well-recognized as a key activator of macrophage anti-tumor activity. However, toxicity associated with systemic delivery of IFN-γ in patients has precluded its development as a therapeutic approach (Zaidi and Merlino, 2011). Nonetheless, strategies capable of stimulating the production of IFN-γ in vivo can also redirect tumor-infiltrating macrophages with anti-tumor activity. For example, treatment with an agonistic CD40 monoclonal antibody stimulates a cytokine release syndrome marked by increased levels of IFN-γ. In this setting, tumor infiltrating monocytes are redirected by systemic IFN-γ to mediate anti-tumor activity and enhance the sensitivity of tumors to cytotoxic chemotherapy (Beatty et al., 2011; Long et al., 2016).
Toll-like receptor (TLR) agonists have also been studied for their capacity to activate macrophages with tumor suppressive activity. Activation of macrophages with TLR agonists can stimulate the release of soluble factors capable of inducing tumor cell death (Buhtoiarov et al., 2006). In addition, TLR agonists have been found to stimulate macrophages to phagocytose tumor cells (Feng et al., 2015). One example of a TLR agonist that has been studied for its ability to activate macrophages with anti-tumor activity is CpG (Krieg, 2008). CpG is a short synthetic single-stranded DNA molecule containing unmethylated cytosine (C) and guanine (G) deoxynucleotides that activates macrophages via TLR9. Repeated administration of CpG in vivo produces a macrophage activation syndrome (Behrens et al., 2011). As monotherapy, CpG has shown activity in some cancer models. However, in combination with additional macrophage-directed therapies, CpG has produced potent anti-tumor responses. For example, in combination with anti-IL-10 receptor blockade, CpG treatment has been shown to induce a rapid macrophage-dependent tumor debulking within one day of treatment in multiple models of cancer (Guiducci et al., 2005). Similarly, in combination with anti-CD40, CpG has demonstrated macrophage-dependent anti-tumor activity with macrophages mediating direct killing of tumor cells in a model of melanoma (Buhtoiarov et al., 2006). The capacity of CpG to activate macrophages with tumor-suppressive properties has led to its translation for cancer therapy. When delivered intratumorally or via intradermal/subcutaneous routes of delivery, TLR9 agonists have been generally well-tolerated with the most common adverse events associated with local injection-site reactions and a systemic flu-like reaction that occurs within 24 hours and generally resolves within 48 hours of treatment (Krieg, 2007). Treatment with a TLR9 agonist in patients with metastatic solid tumors has produced stable disease as the best overall response indicating that additional therapeutic maneuvers will be required to realize the potential immunomodulatory activity of TLR9 agonists (Weihrauch et al., 2015). Nonetheless, TLR9 and other TLR agonists represent a promising category of macrophage-targeted therapies for redirecting tumor-infiltrating myeloid cells with tumor suppressive activity.
As macrophage phenotype is regulated by a balance of activating and inhibitory signals, strategies that block inhibitory cues (e.g. HDAC IIa and CD47) have also been studied for their capacity to redirect macrophages with anti-tumor activity. For example, inhibition of class IIa histone deacytelases (HDAC IIa) can induce macrophages with phagocytic and immunostimulatory properties capable of reducing tumor burden and suppressing metastasis in a spontaneous model of breast carcinoma (Guerriero et al., 2017). By altering macrophage phenotype, HDAC IIa inhibition was also found to enhance the efficacy of cytotoxic chemotherapy and immune checkpoint blockade, suggesting a role for macrophage phenotype in defining therapeutic efficacy in cancer.
Upon entry into the tumor microenvironment, myeloid cells encounter a variety of factors that can influence their ultimate fate. Some of these factors (e.g. CSF1), as discussed above, may shape the activity of macrophages. However, other signals act to directly inhibit the activity of macrophages. For example, CD47 is a membrane bound protein that interacts with Sirp-alpha expressed on macrophages to inhibit phagocytosis. CD47 expression is increased on many solid tumors (Willingham et al., 2012) and has been studied as an immune checkpoint molecule capable of regulating macrophage anti-tumor activity (Chao et al., 2012a). In xenograft studies, blockade of CD47, using CD47 blocking antibodies or engineered high affinity Sirp-alpha monomers, has shown capacity to stimulate macrophages with robust anti-tumor activity capable of inhibiting tumor growth (Gholamin et al., 2017; Weiskopf et al., 2013). CD47 blockade is associated with enhanced macrophage phagocytosis of tumor cells and has been suggested to increase tumor immunogenicity through improved cross-presentation of tumor antigens to tumor-specific T cells (Liu et al., 2015). Multiple clinical trials are ongoing to study the safety and preliminary efficacy of this strategy for cancer.
4.4. Translating macrophage-directed therapies in cancer
As major orchestrators of the tumor microenvironment, macrophages shape the sensitivity of cancer to cytotoxic- and immune-based therapies (Figure 3). For example, the fibrotic reaction that surrounds malignant cells can limit the effectiveness of drug delivery. Macrophages, though, can be induced to modulate this fibrotic reaction. Specifically, systemic activation of the CD40 pathway stimulates macrophages to facilitate fibrosis degradation in an MMP-dependent manner. In doing so, macrophages condition the tumor microenvironment with enhanced sensitivity to cytotoxic chemotherapy (Beatty et al., 2011; Beatty et al., 2013; Long et al., 2016). Similarly, HDAC IIa inhibition modulates macrophage phenotype leading to normalization of tumor vasculature and enhanced sensitivity to chemotherapy (Guerriero et al., 2017). CCR2 inhibition by blocking macrophage recruitment in response to cytotoxic stress has also been shown to enhance the efficacy of radiation therapy and chemotherapy (Kalbasi et al., 2016; Mitchem et al., 2013). Thus, macrophages are promising targets for reversing therapeutic resistance and improving the tumor debulking potential of cytotoxic therapies.
Figure 3. Macrophages regulate therapeutic resistance in cancer.

Macrophage phenotype is associated with cancer sensitivity to therapeutic intervention. Tumor promoting macrophages orchestrate a microenvironment that is supportive of tumor resistance. However, the inherent pliability of macrophage phenotype presents an opportunity to shift the polarity of macrophages from tumor-promoting to tumor-suppressing by targeting molecules (e.g. CD40, TLRs, IFN-γ, CD47, HDAC IIa, IL-4, IL-13, and IL-10) involved in regulating macrophage phenotype. Tumor suppressing macrophages can prime malignant cells for increased sensitivity to cytotoxic and immune based therapies as well as modulate the stromal microenvironment for enhanced therapeutic efficacy.
Macrophages are also being studied for their capacity to regulate the efficacy of T cell immunotherapy. For example, tumor-associated macrophages can inhibit productive T cell immunosurveillance in cancer by producing multiple immunoregulatory enzymes, such as arginase I and indoleamine 2,3 dioxygenase which deplete key amino acids (arginine and tryptophan, respectively) required for effector T cell activity (Chang et al., 2001; Munn and Mellor, 2007). Macrophages can also regulate T cell entry into tumors and the efficacy of vaccine therapies (Beatty et al., 2015). In addition, macrophages can directly suppress effector T cells via the expression of immune checkpoint molecules (e.g. PDL1) (Kuang et al., 2009). Thus, strategies that deplete, inhibit or redirect macrophages are being combined with inhibitors of immune checkpoint molecules (e.g. PD-1/PD-L1 and CTLA-4 blocking antibodies) in an effort to harness the treatment response durability that can be achieved with T cell immunotherapy (Beatty and Gladney, 2015; Liu et al., 2017).
5. Conclusions
Macrophages are key components of the innate immune system and are necessary for normal tissue function. Their capacity to acquire tumor-promoting or tumor-suppressing activity is important for shaping cancer cell biology. During tumor development and progression, macrophages almost invariably assume tumor-promoting activity that supports malignant cell survival. Preclinical modeling of macrophages in cancer has produced an in-depth understanding of the inherent plasticity of macrophage phenotype, mechanisms of macrophage-dependent pro-tumor activity, and the discreet signals that regulate macrophage recruitment to tissues and their subsequent survival. This knowledge has revealed novel opportunities for intervening on macrophage biology to improve the efficacy of cancer therapies, including cytotoxic chemotherapy and immunotherapy. Nonetheless, many knowledge gaps still exist particularly in better identifying macrophage subsets in vivo that have therapeutic importance as well as defining how a tumor and its surrounding microenvironment adapts in the setting of macrophage-directed therapies. Investigation into this biology will be a critical next step for successfully translating macrophages as targets for cancer therapy.
Highlights.
Macrophage infiltration in solid tumors is a poor prognostic feature.
Macrophages are mediators of therapeutic resistance in cancer.
Macrophage biology is inherently pliable.
Macrophages are targets for enhancing cytotoxic and immune-based therapies.
Acknowledgments
This work was supported by the National Institutes of Health grant [R01 CA197916].
Abbreviations
- CCL
C-C chemokine ligand
- CCR
C-C chemokine receptor
- CSF
colony stimulating factor
- CTL
cytotoxic T lymphocyte
- CTLA-4
cytotoxic T lymphocyte antigen 4
- CXCL
C-X-C motif chemokine ligand
- CXCR
C-X-C motif chemokine receptor
- FAK
focal adhesion kinase
- GM-CSF
granulocyte-macrophage colony stimulating factor
- HDAC
histone deacytelase
- HIF
Hypoxia inducible factor
- IDO
indolemine 2,3 dioxygenase
- IFN
interferon
- IL
interleukin
- iNOS
inducible nitric oxide synthase
- JAK
janus kinase
- LIF
leukemia inhibitory factor
- LPS
lipopolysaccharide
- mAb
monoclonal antibody
- MHC
major histocompatibility complex
- MMP
matrix metalloproteinase
- PD-1
programmed cell death-1
- STAT
signal transducer and activator of transcription
- TGF
transforming growth factor
- TIMP
tissue inhibitor of matrix metalloproteinase
- TLR
toll-like receptor
- TNF
tumor necrosis factor
- VEGF
vascular endothelial growth factor
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- A-Gonzalez N, Quintana JA, Garcia-Silva S, Mazariegos M, Gonzalez de la Aleja A, Nicolas-Avila JA, Walter W, Adrover JM, Crainiciuc G, Kuchroo VK, Rothlin CV, Peinado H, Castrillo A, Ricote M, Hidalgo A. Phagocytosis imprints heterogeneity in tissue-resident macrophages. J Exp Med. 2017;214:1281–1296. doi: 10.1084/jem.20161375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Afik R, Zigmond E, Vugman M, Klepfish M, Shimshoni E, Pasmanik-Chor M, Shenoy A, Bassat E, Halpern Z, Geiger T, Sagi I, Varol C. Tumor macrophages are pivotal constructors of tumor collagenous matrix. J Exp Med. 2016;213:2315–2331. doi: 10.1084/jem.20151193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ao JY, Zhu XD, Chai ZT, Cai H, Zhang YY, Zhang KZ, Kong LQ, Zhang N, Ye BG, Ma DN, Sun HC. Colony-Stimulating Factor 1 Receptor Blockade Inhibits Tumor Growth by Altering the Polarization of Tumor-Associated Macrophages in Hepatocellular Carcinoma. Mol Cancer Ther. 2017;16:1544–1554. doi: 10.1158/1535-7163.MCT-16-0866. [DOI] [PubMed] [Google Scholar]
- Balermpas P, Rodel F, Liberz R, Oppermann J, Wagenblast J, Ghanaati S, Harter PN, Mittelbronn M, Weiss C, Rodel C, Fokas E. Head and neck cancer relapse after chemoradiotherapy correlates with CD163+ macrophages in primary tumour and CD11b+ myeloid cells in recurrences. Br J Cancer. 2014;111:1509–18. doi: 10.1038/bjc.2014.446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balkwill F. Cancer and the chemokine network. Nat Rev Cancer. 2004;4:540–50. doi: 10.1038/nrc1388. [DOI] [PubMed] [Google Scholar]
- Beatty GL, Chiorean EG, Fishman MP, Saboury B, Teitelbaum UR, Sun W, Huhn RD, Song W, Li D, Sharp LL, Torigian DA, O’Dwyer PJ, Vonderheide RH. CD40 agonists alter tumor stroma and show efficacy against pancreatic carcinoma in mice and humans. Science. 2011;331:1612–6. doi: 10.1126/science.1198443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beatty GL, Gladney WL. Immune escape mechanisms as a guide for cancer immunotherapy. Clin Cancer Res. 2015;21:687–92. doi: 10.1158/1078-0432.CCR-14-1860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beatty GL, Li Y, Long KB. Cancer immunotherapy: activating innate and adaptive immunity through CD40 agonists. Expert Rev Anticancer Ther. 2017;17:175–186. doi: 10.1080/14737140.2017.1270208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beatty GL, Torigian DA, Chiorean EG, Saboury B, Brothers A, Alavi A, Troxel AB, Sun W, Teitelbaum UR, Vonderheide RH, O’Dwyer P. A phase I study of an agonist CD40 monoclonal antibody (CP-870,893) in combination with gemcitabine in patients with advanced pancreatic ductal adenocarcinoma. Clin Cancer Res. 2013;19:6286–95. doi: 10.1158/1078-0432.CCR-13-1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beatty GL, Winograd R, Evans RA, Long KB, Luque SL, Lee JW, Clendenin C, Gladney WL, Knoblock DM, Guirnalda PD, Vonderheide RH. Exclusion of T Cells From Pancreatic Carcinomas in Mice Is Regulated by Ly6C(low) F4/80(+) Extratumoral Macrophages. Gastroenterology. 2015;149:201–10. doi: 10.1053/j.gastro.2015.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behrens EM, Canna SW, Slade K, Rao S, Kreiger PA, Paessler M, Kambayashi T, Koretzky GA. Repeated TLR9 stimulation results in macrophage activation syndrome-like disease in mice. J Clin Invest. 2011;121:2264–77. doi: 10.1172/JCI43157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buhtoiarov IN, Lum HD, Berke G, Sondel PM, Rakhmilevich AL. Synergistic activation of macrophages via CD40 and TLR9 results in T cell independent antitumor effects. J Immunol. 2006;176:309–18. doi: 10.4049/jimmunol.176.1.309. [DOI] [PubMed] [Google Scholar]
- Casazza A, Laoui D, Wenes M, Rizzolio S, Bassani N, Mambretti M, Deschoemaeker S, Van Ginderachter JA, Tamagnone L, Mazzone M. Impeding macrophage entry into hypoxic tumor areas by Sema3A/Nrp1 signaling blockade inhibits angiogenesis and restores antitumor immunity. Cancer Cell. 2013;24:695–709. doi: 10.1016/j.ccr.2013.11.007. [DOI] [PubMed] [Google Scholar]
- Chang CI, Liao JC, Kuo L. Macrophage arginase promotes tumor cell growth and suppresses nitric oxide-mediated tumor cytotoxicity. Cancer Res. 2001;61:1100–6. [PubMed] [Google Scholar]
- Chao MP, Majeti R, Weissman IL. Programmed cell removal: a new obstacle in the road to developing cancer. Nat Rev Cancer. 2012a;12:58–67. doi: 10.1038/nrc3171. [DOI] [PubMed] [Google Scholar]
- Chao MP, Weissman IL, Majeti R. The CD47-SIRPalpha pathway in cancer immune evasion and potential therapeutic implications. Curr Opin Immunol. 2012b;24:225–32. doi: 10.1016/j.coi.2012.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Li J, Wang F, Dai C, Wu F, Liu X, Li T, Glauben R, Zhang Y, Nie G, He Y, Qin Z. Tie2 Expression on Macrophages Is Required for Blood Vessel Reconstruction and Tumor Relapse after Chemotherapy. Cancer Res. 2016a;76:6828–6838. doi: 10.1158/0008-5472.CAN-16-1114. [DOI] [PubMed] [Google Scholar]
- Chen W, Xu Y, Zhong J, Wang H, Weng M, Cheng Q, Wu Q, Sun Z, Jiang H, Zhu M, Ren Y, Xu P, Chen J, Miao C. MFHAS1 promotes colorectal cancer progress by regulating polarization of tumor-associated macrophages via STAT6 signaling pathway. Oncotarget. 2016b;7:78726–78735. doi: 10.18632/oncotarget.12807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Ying X, Wang X, Wu X, Zhu Q, Wang X. Exosomes derived from hypoxic epithelial ovarian cancer deliver microRNA-940 to induce macrophage M2 polarization. Oncol Rep. 2017;38:522–528. doi: 10.3892/or.2017.5697. [DOI] [PubMed] [Google Scholar]
- Chomarat P, Banchereau J, Davoust J, Palucka AK. IL-6 switches the differentiation of monocytes from dendritic cells to macrophages. Nat Immunol. 2000;1:510–4. doi: 10.1038/82763. [DOI] [PubMed] [Google Scholar]
- Colegio OR, Chu NQ, Szabo AL, Chu T, Rhebergen AM, Jairam V, Cyrus N, Brokowski CE, Eisenbarth SC, Phillips GM, Cline GW, Phillips AJ, Medzhitov R. Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature. 2014;513:559–63. doi: 10.1038/nature13490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corcoran RB, Contino G, Deshpande V, Tzatsos A, Conrad C, Benes CH, Levy DE, Settleman J, Engelman JA, Bardeesy N. STAT3 plays a critical role in KRAS-induced pancreatic tumorigenesis. Cancer Res. 2011;71:5020–9. doi: 10.1158/0008-5472.CAN-11-0908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa-Silva B, Aiello NM, Ocean AJ, Singh S, Zhang H, Thakur BK, Becker A, Hoshino A, Mark MT, Molina H, Xiang J, Zhang T, Theilen TM, Garcia-Santos G, Williams C, Ararso Y, Huang Y, Rodrigues G, Shen TL, Labori KJ, Lothe IM, Kure EH, Hernandez J, Doussot A, Ebbesen SH, Grandgenett PM, Hollingsworth MA, Jain M, Mallya K, Batra SK, Jarnagin WR, Schwartz RE, Matei I, Peinado H, Stanger BZ, Bromberg J, Lyden D. Pancreatic cancer exosomes initiate pre-metastatic niche formation in the liver. Nat Cell Biol. 2015;17:816–26. doi: 10.1038/ncb3169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox TR, Erler JT. Molecular pathways: connecting fibrosis and solid tumor metastasis. Clin Cancer Res. 2014;20:3637–43. doi: 10.1158/1078-0432.CCR-13-1059. [DOI] [PubMed] [Google Scholar]
- Cramer T, Yamanishi Y, Clausen BE, Forster I, Pawlinski R, Mackman N, Haase VH, Jaenisch R, Corr M, Nizet V, Firestein GS, Gerber HP, Ferrara N, Johnson RS. HIF-1alpha is essential for myeloid cell-mediated inflammation. Cell. 2003;112:645–57. doi: 10.1016/s0092-8674(03)00154-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dammeijer F, Lievense LA, Kaijen-Lambers ME, van Nimwegen M, Bezemer K, Hegmans JP, van Hall T, Hendriks RW, Aerts JG. Depletion of Tumor-Associated Macrophages with a CSF-1R Kinase Inhibitor Enhances Antitumor Immunity and Survival Induced by DC Immunotherapy. Cancer Immunol Res. 2017;5:535–546. doi: 10.1158/2326-6066.CIR-16-0309. [DOI] [PubMed] [Google Scholar]
- Davies LC, Jenkins SJ, Allen JE, Taylor PR. Tissue-resident macrophages. Nat Immunol. 2013;14:986–95. doi: 10.1038/ni.2705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeNardo DG, Brennan DJ, Rexhepaj E, Ruffell B, Shiao SL, Madden SF, Gallagher WM, Wadhwani N, Keil SD, Junaid SA, Rugo HS, Hwang ES, Jirstrom K, West BL, Coussens LM. Leukocyte complexity predicts breast cancer survival and functionally regulates response to chemotherapy. Cancer Discov. 2011;1:54–67. doi: 10.1158/2159-8274.CD-10-0028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du R, Lu KV, Petritsch C, Liu P, Ganss R, Passegue E, Song H, Vandenberg S, Johnson RS, Werb Z, Bergers G. HIF1alpha induces the recruitment of bone marrow-derived vascular modulatory cells to regulate tumor angiogenesis and invasion. Cancer Cell. 2008;13:206–20. doi: 10.1016/j.ccr.2008.01.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duffield JS, Forbes SJ, Constandinou CM, Clay S, Partolina M, Vuthoori S, Wu S, Lang R, Iredale JP. Selective depletion of macrophages reveals distinct, opposing roles during liver injury and repair. J Clin Invest. 2005;115:56–65. doi: 10.1172/JCI22675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duluc D, Delneste Y, Tan F, Moles MP, Grimaud L, Lenoir J, Preisser L, Anegon I, Catala L, Ifrah N, Descamps P, Gamelin E, Gascan H, Hebbar M, Jeannin P. Tumor-associated leukemia inhibitory factor and IL-6 skew monocyte differentiation into tumor-associated macrophage-like cells. Blood. 2007;110:4319–30. doi: 10.1182/blood-2007-02-072587. [DOI] [PubMed] [Google Scholar]
- Egeblad M, Rasch MG, Weaver VM. Dynamic interplay between the collagen scaffold and tumor evolution. Curr Opin Cell Biol. 2010;22:697–706. doi: 10.1016/j.ceb.2010.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Epelman S, Lavine KJ, Randolph GJ. Origin and functions of tissue macrophages. Immunity. 2014;41:21–35. doi: 10.1016/j.immuni.2014.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erez N, Truitt M, Olson P, Arron ST, Hanahan D. Cancer-Associated Fibroblasts Are Activated in Incipient Neoplasia to Orchestrate Tumor-Promoting Inflammation in an NF-kappaB-Dependent Manner. Cancer Cell. 2010;17:135–47. doi: 10.1016/j.ccr.2009.12.041. [DOI] [PubMed] [Google Scholar]
- Eue I, Kumar R, Dong Z, Killion JJ, Fidler IJ. Induction of nitric oxide production and tumoricidal properties in murine macrophages by a new synthetic lipopeptide JBT3002 encapsulated in liposomes. J Immunother. 1998;21:340–51. doi: 10.1097/00002371-199809000-00002. [DOI] [PubMed] [Google Scholar]
- Fallowfield JA, Mizuno M, Kendall TJ, Constandinou CM, Benyon RC, Duffield JS, Iredale JP. Scar-associated macrophages are a major source of hepatic matrix metalloproteinase-13 and facilitate the resolution of murine hepatic fibrosis. J Immunol. 2007;178:5288–95. doi: 10.4049/jimmunol.178.8.5288. [DOI] [PubMed] [Google Scholar]
- Feng M, Chen JY, Weissman-Tsukamoto R, Volkmer JP, Ho PY, McKenna KM, Cheshier S, Zhang M, Guo N, Gip P, Mitra SS, Weissman IL. Macrophages eat cancer cells using their own calreticulin as a guide: roles of TLR and Btk. Proc Natl Acad Sci U S A. 2015;112:2145–50. doi: 10.1073/pnas.1424907112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franklin RA, Liao W, Sarkar A, Kim MV, Bivona MR, Liu K, Pamer EG, Li MO. The cellular and molecular origin of tumor-associated macrophages. Science. 2014;344:921–5. doi: 10.1126/science.1252510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galvan-Pena S, O’Neill LA. Metabolic reprograming in macrophage polarization. Front Immunol. 2014;5:420. doi: 10.3389/fimmu.2014.00420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gazzaniga S, Bravo AI, Guglielmotti A, van Rooijen N, Maschi F, Vecchi A, Mantovani A, Mordoh J, Wainstok R. Targeting tumor-associated macrophages and inhibition of MCP-1 reduce angiogenesis and tumor growth in a human melanoma xenograft. J Invest Dermatol. 2007;127:2031–41. doi: 10.1038/sj.jid.5700827. [DOI] [PubMed] [Google Scholar]
- Germano G, Frapolli R, Belgiovine C, Anselmo A, Pesce S, Liguori M, Erba E, Uboldi S, Zucchetti M, Pasqualini F, Nebuloni M, van Rooijen N, Mortarini R, Beltrame L, Marchini S, Fuso Nerini I, Sanfilippo R, Casali PG, Pilotti S, Galmarini CM, Anichini A, Mantovani A, D’Incalci M, Allavena P. Role of macrophage targeting in the antitumor activity of trabectedin. Cancer Cell. 2013;23:249–62. doi: 10.1016/j.ccr.2013.01.008. [DOI] [PubMed] [Google Scholar]
- Gholamin S, Mitra SS, Feroze AH, Liu J, Kahn SA, Zhang M, Esparza R, Richard C, Ramaswamy V, Remke M, Volkmer AK, Willingham S, Ponnuswami A, McCarty A, Lovelace P, Storm TA, Schubert S, Hutter G, Narayanan C, Chu P, Raabe EH, Harsh Gt, Taylor MD, Monje M, Cho YJ, Majeti R, Volkmer JP, Fisher PG, Grant G, Steinberg GK, Vogel H, Edwards M, Weissman IL, Cheshier SH. Disrupting the CD47-SIRPalpha anti-phagocytic axis by a humanized anti-CD47 antibody is an efficacious treatment for malignant pediatric brain tumors. Sci Transl Med. 2017:9. doi: 10.1126/scitranslmed.aaf2968. [DOI] [PubMed] [Google Scholar]
- Gordon S, Martinez FO. Alternative activation of macrophages: mechanism and functions. Immunity. 2010;32:593–604. doi: 10.1016/j.immuni.2010.05.007. [DOI] [PubMed] [Google Scholar]
- Guerriero JL, Sotayo A, Ponichtera HE, Castrillon JA, Pourzia AL, Schad S, Johnson SF, Carrasco RD, Lazo S, Bronson RT, Davis SP, Lobera M, Nolan MA, Letai A. Class IIa HDAC inhibition reduces breast tumours and metastases through anti-tumour macrophages. Nature. 2017;543:428–432. doi: 10.1038/nature21409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guiducci C, Vicari AP, Sangaletti S, Trinchieri G, Colombo MP. Redirecting in vivo elicited tumor infiltrating macrophages and dendritic cells towards tumor rejection. Cancer Res. 2005;65:3437–46. doi: 10.1158/0008-5472.CAN-04-4262. [DOI] [PubMed] [Google Scholar]
- Haak-Frendscho M, Wynn TA, Czuprynski CJ, Paulnock D. Transforming growth factor-beta 1 inhibits activation of macrophage cell line RAW 264.7 for cell killing. Clin Exp Immunol. 1990;82:404–10. doi: 10.1111/j.1365-2249.1990.tb05461.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagemann T, Wilson J, Burke F, Kulbe H, Li NF, Pluddemann A, Charles K, Gordon S, Balkwill FR. Ovarian cancer cells polarize macrophages toward a tumor-associated phenotype. J Immunol. 2006;176:5023–32. doi: 10.4049/jimmunol.176.8.5023. [DOI] [PubMed] [Google Scholar]
- Halama N, Zoernig I, Berthel A, Kahlert C, Klupp F, Suarez-Carmona M, Suetterlin T, Brand K, Krauss J, Lasitschka F, Lerchl T, Luckner-Minden C, Ulrich A, Koch M, Weitz J, Schneider M, Buechler MW, Zitvogel L, Herrmann T, Benner A, Kunz C, Luecke S, Springfeld C, Grabe N, Falk CS, Jaeger D. Tumoral Immune Cell Exploitation in Colorectal Cancer Metastases Can Be Targeted Effectively by Anti-CCR5 Therapy in Cancer Patients. Cancer Cell. 2016;29:587–601. doi: 10.1016/j.ccell.2016.03.005. [DOI] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–74. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- Henze AT, Mazzone M. The impact of hypoxia on tumor-associated macrophages. J Clin Invest. 2016;126:3672–3679. doi: 10.1172/JCI84427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmgaard RB, Brachfeld A, Gasmi B, Jones DR, Mattar M, Doman T, Murphy M, Schaer D, Wolchok JD, Merghoub T. Timing of CSF-1/CSF-1R signaling blockade is critical to improving responses to CTLA-4 based immunotherapy. Oncoimmunology. 2016;5:e1151595. doi: 10.1080/2162402X.2016.1151595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang S, Van Arsdall M, Tedjarati S, McCarty M, Wu W, Langley R, Fidler IJ. Contributions of stromal metalloproteinase-9 to angiogenesis and growth of human ovarian carcinoma in mice. J Natl Cancer Inst. 2002;94:1134–42. doi: 10.1093/jnci/94.15.1134. [DOI] [PubMed] [Google Scholar]
- Jakubzick C, Gautier EL, Gibbings SL, Sojka DK, Schlitzer A, Johnson TE, Ivanov S, Duan Q, Bala S, Condon T, van Rooijen N, Grainger JR, Belkaid Y, Ma’ayan A, Riches DW, Yokoyama WM, Ginhoux F, Henson PM, Randolph GJ. Minimal differentiation of classical monocytes as they survey steady-state tissues and transport antigen to lymph nodes. Immunity. 2014;39:599–610. doi: 10.1016/j.immuni.2013.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang H, Hegde S, Knolhoff BL, Zhu Y, Herndon JM, Meyer MA, Nywening TM, Hawkins WG, Shapiro IM, Weaver DT, Pachter JA, Wang-Gillam A, DeNardo DG. Targeting focal adhesion kinase renders pancreatic cancers responsive to checkpoint immunotherapy. Nat Med. 2016;22:851–60. doi: 10.1038/nm.4123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi S, Singh AR, Zulcic M, Bao L, Messer K, Ideker T, Dutkowski J, Durden DL. Rac2 controls tumor growth, metastasis and M1-M2 macrophage differentiation in vivo. PLoS One. 2014;9:e95893. doi: 10.1371/journal.pone.0095893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalbasi A, Komar CA, Tooker GM, Liu M, Lee JW, Gladney WL, Ben-Josef E, Beatty GL. Tumor-derived CCL2 mediates resistance to radiotherapy in pancreatic ductal adenocarcinoma. Clin Cancer Res. 2016 doi: 10.1158/1078-0432.CCR-16-0870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaneda MM, Messer KS, Ralainirina N, Li H, Leem CJ, Gorjestani S, Woo G, Nguyen AV, Figueiredo CC, Foubert P, Schmid MC, Pink M, Winkler DG, Rausch M, Palombella VJ, Kutok J, McGovern K, Frazer KA, Wu X, Karin M, Sasik R, Cohen EE, Varner JA. PI3Kgamma is a molecular switch that controls immune suppression. Nature. 2016;539:437–442. doi: 10.1038/nature19834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan G. In vitro differentiation of human monocytes. Monocytes cultured on glass are cytotoxic to tumor cells but monocytes cultured on collagen are not. J Exp Med. 1983;157:2061–72. doi: 10.1084/jem.157.6.2061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi N, Miyoshi S, Mikami T, Koyama H, Kitazawa M, Takeoka M, Sano K, Amano J, Isogai Z, Niida S, Oguri K, Okayama M, McDonald JA, Kimata K, Taniguchi S, Itano N. Hyaluronan deficiency in tumor stroma impairs macrophage trafficking and tumor neovascularization. Cancer Res. 2010;70:7073–83. doi: 10.1158/0008-5472.CAN-09-4687. [DOI] [PubMed] [Google Scholar]
- Krieg AM. Development of TLR9 agonists for cancer therapy. J Clin Invest. 2007;117:1184–94. doi: 10.1172/JCI31414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krieg AM. Toll-like receptor 9 (TLR9) agonists in the treatment of cancer. Oncogene. 2008;27:161–7. doi: 10.1038/sj.onc.1210911. [DOI] [PubMed] [Google Scholar]
- Kuang DM, Wu Y, Chen N, Cheng J, Zhuang SM, Zheng L. Tumor-derived hyaluronan induces formation of immunosuppressive macrophages through transient early activation of monocytes. Blood. 2007;110:587–95. doi: 10.1182/blood-2007-01-068031. [DOI] [PubMed] [Google Scholar]
- Kuang DM, Zhao Q, Peng C, Xu J, Zhang JP, Wu C, Zheng L. Activated monocytes in peritumoral stroma of hepatocellular carcinoma foster immune privilege and disease progression through PD-L1. J Exp Med. 2009;206:1327–37. doi: 10.1084/jem.20082173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesina M, Kurkowski MU, Ludes K, Rose-John S, Treiber M, Kloppel G, Yoshimura A, Reindl W, Sipos B, Akira S, Schmid RM, Algul H. Stat3/Socs3 activation by IL-6 transsignaling promotes progression of pancreatic intraepithelial neoplasia and development of pancreatic cancer. Cancer Cell. 2011;19:456–69. doi: 10.1016/j.ccr.2011.03.009. [DOI] [PubMed] [Google Scholar]
- Lim SY, Yuzhalin AE, Gordon-Weeks AN, Muschel RJ. Targeting the CCL2-CCR2 signaling axis in cancer metastasis. Oncotarget. 2016;7:28697–710. doi: 10.18632/oncotarget.7376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liou GY, Doppler H, Necela B, Edenfield B, Zhang L, Dawson DW, Storz P. Mutant KRAS-induced expression of ICAM-1 in pancreatic acinar cells causes attraction of macrophages to expedite the formation of precancerous lesions. Cancer Discov. 2015;5:52–63. doi: 10.1158/2159-8290.CD-14-0474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu M, Kalbasi A, Beatty GL. Functio Laesa: Cancer Inflammation and Therapeutic Resistance. J Oncol Pract. 2017;13:173–180. doi: 10.1200/JOP.2016.020347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Pu Y, Cron K, Deng L, Kline J, Frazier WA, Xu H, Peng H, Fu YX, Xu MM. CD47 blockade triggers T cell-mediated destruction of immunogenic tumors. Nat Med. 2015;21:1209–15. doi: 10.1038/nm.3931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loberg RD, Ying C, Craig M, Day LL, Sargent E, Neeley C, Wojno K, Snyder LA, Yan L, Pienta KJ. Targeting CCL2 with systemic delivery of neutralizing antibodies induces prostate cancer tumor regression in vivo. Cancer Res. 2007;67:9417–24. doi: 10.1158/0008-5472.CAN-07-1286. [DOI] [PubMed] [Google Scholar]
- Long KB, Beatty GL. Harnessing the antitumor potential of macrophages for cancer immunotherapy. Oncoimmunology. 2013;2:e26860. doi: 10.4161/onci.26860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long KB, Gladney WL, Tooker GM, Graham K, Fraietta JA, Beatty GL. IFNgamma and CCL2 Cooperate to Redirect Tumor-Infiltrating Monocytes to Degrade Fibrosis and Enhance Chemotherapy Efficacy in Pancreatic Carcinoma. Cancer Discov. 2016;6:400–13. doi: 10.1158/2159-8290.CD-15-1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long KB, Tooker G, Tooker E, Luque SL, Lee JW, Pan X, Beatty GL. IL-6 receptor blockade enhances chemotherapy efficacy in pancreatic ductal adenocarcinoma. Mol Cancer Ther. 2017 doi: 10.1158/1535-7163.MCT-16-0899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahmoud SM, Lee AH, Paish EC, Macmillan RD, Ellis IO, Green AR. Tumour-infiltrating macrophages and clinical outcome in breast cancer. J Clin Pathol. 2012;65:159–63. doi: 10.1136/jclinpath-2011-200355. [DOI] [PubMed] [Google Scholar]
- Mantovani A, Sozzani S, Locati M, Allavena P, Sica A. Macrophage polarization: tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol. 2002;23:549–55. doi: 10.1016/s1471-4906(02)02302-5. [DOI] [PubMed] [Google Scholar]
- Mao Y, Eissler N, Blanc KL, Johnsen JI, Kogner P, Kiessling R. Targeting Suppressive Myeloid Cells Potentiates Checkpoint Inhibitors to Control Spontaneous Neuroblastoma. Clin Cancer Res. 2016;22:3849–59. doi: 10.1158/1078-0432.CCR-15-1912. [DOI] [PubMed] [Google Scholar]
- Martinez FO, Gordon S. The M1 and M2 paradigm of macrophage activation: time for reassessment. F1000Prime Rep. 2014;6:13. doi: 10.12703/P6-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathew E, Brannon AL, Del Vecchio A, Garcia PE, Penny MK, Kane KT, Vinta A, Buckanovich RJ, di Magliano MP. Mesenchymal Stem Cells Promote Pancreatic Tumor Growth by Inducing Alternative Polarization of Macrophages. Neoplasia. 2016;18:142–51. doi: 10.1016/j.neo.2016.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maude SL, Barrett D, Teachey DT, Grupp SA. Managing cytokine release syndrome associated with novel T cell-engaging therapies. Cancer J. 2014;20:119–22. doi: 10.1097/PPO.0000000000000035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazzieri R, Pucci F, Moi D, Zonari E, Ranghetti A, Berti A, Politi LS, Gentner B, Brown JL, Naldini L, De Palma M. Targeting the ANG2/TIE2 axis inhibits tumor growth and metastasis by impairing angiogenesis and disabling rebounds of proangiogenic myeloid cells. Cancer Cell. 2011;19:512–26. doi: 10.1016/j.ccr.2011.02.005. [DOI] [PubMed] [Google Scholar]
- McWhorter FY, Davis CT, Liu WF. Physical and mechanical regulation of macrophage phenotype and function. Cell Mol Life Sci. 2015;72:1303–16. doi: 10.1007/s00018-014-1796-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McWhorter FY, Wang T, Nguyen P, Chung T, Liu WF. Modulation of macrophage phenotype by cell shape. Proc Natl Acad Sci U S A. 2013;110:17253–8. doi: 10.1073/pnas.1308887110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mills CD, Kincaid K, Alt JM, Heilman MJ, Hill AM. M-1/M-2 macrophages and the Th1/Th2 paradigm. J Immunol. 2000;164:6166–73. doi: 10.4049/jimmunol.164.12.6166. [DOI] [PubMed] [Google Scholar]
- Mills EL, Kelly B, Logan A, Costa ASH, Varma M, Bryant CE, Tourlomousis P, Dabritz JHM, Gottlieb E, Latorre I, Corr SC, McManus G, Ryan D, Jacobs HT, Szibor M, Xavier RJ, Braun T, Frezza C, Murphy MP, O’Neill LA. Succinate Dehydrogenase Supports Metabolic Repurposing of Mitochondria to Drive Inflammatory Macrophages. Cell. 2016;167:457–470. e13. doi: 10.1016/j.cell.2016.08.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchem JB, Brennan DJ, Knolhoff BL, Belt BA, Zhu Y, Sanford DE, Belaygorod L, Carpenter D, Collins L, Piwnica-Worms D, Hewitt S, Udupi GM, Gallagher WM, Wegner C, West BL, Wang-Gillam A, Goedegebuure P, Linehan DC, DeNardo DG. Targeting tumor-infiltrating macrophages decreases tumor-initiating cells, relieves immunosuppression, and improves chemotherapeutic responses. Cancer Res. 2013;73:1128–41. doi: 10.1158/0008-5472.CAN-12-2731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mundy GR, DeMartino S, Rowe DW. Collagen and collagen-derived fragments are chemotactic for tumor cells. J Clin Invest. 1981;68:1102–5. doi: 10.1172/JCI110334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munn DH, Mellor AL. Indoleamine 2,3-dioxygenase and tumor-induced tolerance. J Clin Invest. 2007;117:1147–54. doi: 10.1172/JCI31178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murdoch C, Giannoudis A, Lewis CE. Mechanisms regulating the recruitment of macrophages into hypoxic areas of tumors and other ischemic tissues. Blood. 2004;104:2224–34. doi: 10.1182/blood-2004-03-1109. [DOI] [PubMed] [Google Scholar]
- Murray PJ, Allen JE, Biswas SK, Fisher EA, Gilroy DW, Goerdt S, Gordon S, Hamilton JA, Ivashkiv LB, Lawrence T, Locati M, Mantovani A, Martinez FO, Mege JL, Mosser DM, Natoli G, Saeij JP, Schultze JL, Shirey KA, Sica A, Suttles J, Udalova I, van Ginderachter JA, Vogel SN, Wynn TA. Macrophage activation and polarization: nomenclature and experimental guidelines. Immunity. 2014;41:14–20. doi: 10.1016/j.immuni.2014.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray PJ, Wynn TA. Protective and pathogenic functions of macrophage subsets. Nat Rev Immunol. 2011;11:723–37. doi: 10.1038/nri3073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagathihalli NS, Castellanos JA, Shi C, Beesetty Y, Reyzer ML, Caprioli R, Chen X, Walsh AJ, Skala MC, Moses HL, Merchant NB. Signal Transducer and Activator of Transcription 3, Mediated Remodeling of the Tumor Microenvironment Results in Enhanced Tumor Drug Delivery in a Mouse Model of Pancreatic Cancer. Gastroenterology. 2015;149:1932–1943. e9. doi: 10.1053/j.gastro.2015.07.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nywening TM, Wang-Gillam A, Sanford DE, Belt BA, Panni RZ, Cusworth BM, Toriola AT, Nieman RK, Worley LA, Yano M, Fowler KJ, Lockhart AC, Suresh R, Tan BR, Lim KH, Fields RC, Strasberg SM, Hawkins WG, DeNardo DG, Goedegebuure SP, Linehan DC. Targeting tumour-associated macrophages with CCR2 inhibition in combination with FOLFIRINOX in patients with borderline resectable and locally advanced pancreatic cancer: a single-centre, open-label, dose-finding, non-randomised, phase 1b trial. Lancet Oncol. 2016;17:651–662. doi: 10.1016/S1470-2045(16)00078-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olive KP, Jacobetz MA, Davidson CJ, Gopinathan A, McIntyre D, Honess D, Madhu B, Goldgraben MA, Caldwell ME, Allard D, Frese KK, Denicola G, Feig C, Combs C, Winter SP, Ireland-Zecchini H, Reichelt S, Howat WJ, Chang A, Dhara M, Wang L, Ruckert F, Grutzmann R, Pilarsky C, Izeradjene K, Hingorani SR, Huang P, Davies SE, Plunkett W, Egorin M, Hruban RH, Whitebread N, McGovern K, Adams J, Iacobuzio-Donahue C, Griffiths J, Tuveson DA. Inhibition of Hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science. 2009;324:1457–61. doi: 10.1126/science.1171362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pakyari M, Farrokhi A, Maharlooei MK, Ghahary A. Critical Role of Transforming Growth Factor Beta in Different Phases of Wound Healing. Adv Wound Care (New Rochelle) 2013;2:215–224. doi: 10.1089/wound.2012.0406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parks WC, Wilson CL, Lopez-Boado YS. Matrix metalloproteinases as modulators of inflammation and innate immunity. Nat Rev Immunol. 2004;4:617–29. doi: 10.1038/nri1418. [DOI] [PubMed] [Google Scholar]
- Pickup M, Novitskiy S, Moses HL. The roles of TGFbeta in the tumour microenvironment. Nat Rev Cancer. 2013;13:788–99. doi: 10.1038/nrc3603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Postlethwaite AE, Kang AH. Collagen-and collagen peptide-induced chemotaxis of human blood monocytes. J Exp Med. 1976;143:1299–307. doi: 10.1084/jem.143.6.1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Provenzano PP, Cuevas C, Chang AE, Goel VK, Von Hoff DD, Hingorani SR. Enzymatic targeting of the stroma ablates physical barriers to treatment of pancreatic ductal adenocarcinoma. Cancer Cell. 2012;21:418–29. doi: 10.1016/j.ccr.2012.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pyonteck SM, Akkari L, Schuhmacher AJ, Bowman RL, Sevenich L, Quail DF, Olson OC, Quick ML, Huse JT, Teijeiro V, Setty M, Leslie CS, Oei Y, Pedraza A, Zhang J, Brennan CW, Sutton JC, Holland EC, Daniel D, Joyce JA. CSF-1R inhibition alters macrophage polarization and blocks glioma progression. Nat Med. 2013;19:1264–72. doi: 10.1038/nm.3337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pyonteck SM, Gadea BB, Wang HW, Gocheva V, Hunter KE, Tang LH, Joyce JA. Deficiency of the macrophage growth factor CSF-1 disrupts pancreatic neuroendocrine tumor development. Oncogene. 2012;31:1459–67. doi: 10.1038/onc.2011.337. [DOI] [PubMed] [Google Scholar]
- Qian BZ, Li J, Zhang H, Kitamura T, Zhang J, Campion LR, Kaiser EA, Snyder LA, Pollard JW. CCL2 recruits inflammatory monocytes to facilitate breast-tumour metastasis. Nature. 2011;475:222–5. doi: 10.1038/nature10138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian BZ, Pollard JW. Macrophage diversity enhances tumor progression and metastasis. Cell. 2010;141:39–51. doi: 10.1016/j.cell.2010.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rayahin JE, Buhrman JS, Zhang Y, Koh TJ, Gemeinhart RA. High and low molecular weight hyaluronic acid differentially influence macrophage activation. ACS Biomater Sci Eng. 2015;1:481–493. doi: 10.1021/acsbiomaterials.5b00181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ries CH, Cannarile MA, Hoves S, Benz J, Wartha K, Runza V, Rey-Giraud F, Pradel LP, Feuerhake F, Klaman I, Jones T, Jucknischke U, Scheiblich S, Kaluza K, Gorr IH, Walz A, Abiraj K, Cassier PA, Sica A, Gomez-Roca C, de Visser KE, Italiano A, Le Tourneau C, Delord JP, Levitsky H, Blay JY, Ruttinger D. Targeting tumor-associated macrophages with anti-CSF-1R antibody reveals a strategy for cancer therapy. Cancer Cell. 2014;25:846–59. doi: 10.1016/j.ccr.2014.05.016. [DOI] [PubMed] [Google Scholar]
- Roca H, Varsos ZS, Sud S, Craig MJ, Ying C, Pienta KJ. CCL2 and interleukin-6 promote survival of human CD11b+ peripheral blood mononuclear cells and induce M2-type macrophage polarization. J Biol Chem. 2009;284:34342–54. doi: 10.1074/jbc.M109.042671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanford DE, Belt BA, Panni RZ, Mayer A, Deshpande AD, Carpenter D, Mitchem JB, Plambeck-Suess SM, Worley LA, Goetz BD, Wang-Gillam A, Eberlein TJ, Denardo DG, Goedegebuure SP, Linehan DC. Inflammatory Monocyte Mobilization Decreases Patient Survival in Pancreatic Cancer: A Role for Targeting the CCL2/CCR2 Axis. Clin Cancer Res. 2013;19:3404–15. doi: 10.1158/1078-0432.CCR-13-0525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki K, Zhao X, Pardee AD, Ueda R, Fujita M, Sehra S, Kaplan MH, Kane LP, Okada H, Storkus WJ. Stat6 signaling suppresses VLA-4 expression by CD8+ T cells and limits their ability to infiltrate tumor lesions in vivo. J Immunol. 2008;181:104–8. doi: 10.4049/jimmunol.181.1.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shabo I, Olsson H, Sun XF, Svanvik J. Expression of the macrophage antigen CD163 in rectal cancer cells is associated with early local recurrence and reduced survival time. Int J Cancer. 2009;125:1826–31. doi: 10.1002/ijc.24506. [DOI] [PubMed] [Google Scholar]
- Shabo I, Stal O, Olsson H, Dore S, Svanvik J. Breast cancer expression of CD163, a macrophage scavenger receptor, is related to early distant recurrence and reduced patient survival. Int J Cancer. 2008;123:780–6. doi: 10.1002/ijc.23527. [DOI] [PubMed] [Google Scholar]
- Shi C, Pamer EG. Monocyte recruitment during infection and inflammation. Nat Rev Immunol. 2011;11:762–74. doi: 10.1038/nri3070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sica A, Mantovani A. Macrophage plasticity and polarization: in vivo veritas. J Clin Invest. 2012;122:787–95. doi: 10.1172/JCI59643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stahl M, Schupp J, Jager B, Schmid M, Zissel G, Muller-Quernheim J, Prasse A. Lung collagens perpetuate pulmonary fibrosis via CD204 and M2 macrophage activation. PLoS One. 2013;8:e81382. doi: 10.1371/journal.pone.0081382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein M, Keshav S, Harris N, Gordon S. Interleukin 4 potently enhances murine macrophage mannose receptor activity: a marker of alternative immunologic macrophage activation. J Exp Med. 1992;176:287–92. doi: 10.1084/jem.176.1.287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei IH, Harmon CM, Arcerito M, Cheng DF, Minter RM, Simeone DM. Tumor-associated macrophages are a useful biomarker to predict recurrence after surgical resection of nonfunctional pancreatic neuroendocrine tumors. Ann Surg. 2014;260:1088–94. doi: 10.1097/SLA.0000000000000262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weihrauch MR, Richly H, von Bergwelt-Baildon MS, Becker HJ, Schmidt M, Hacker UT, Shimabukuro-Vornhagen A, Holtick U, Nokay B, Schroff M, Wittig B, Scheulen ME. Phase I clinical study of the toll-like receptor 9 agonist MGN1703 in patients with metastatic solid tumours. Eur J Cancer. 2015;51:146–56. doi: 10.1016/j.ejca.2014.11.002. [DOI] [PubMed] [Google Scholar]
- Weiskopf K, Ring AM, Ho CC, Volkmer JP, Levin AM, Volkmer AK, Ozkan E, Fernhoff NB, van de Rijn M, Weissman IL, Garcia KC. Engineered SIRPalpha variants as immunotherapeutic adjuvants to anticancer antibodies. Science. 2013;341:88–91. doi: 10.1126/science.1238856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willingham SB, Volkmer JP, Gentles AJ, Sahoo D, Dalerba P, Mitra SS, Wang J, Contreras-Trujillo H, Martin R, Cohen JD, Lovelace P, Scheeren FA, Chao MP, Weiskopf K, Tang C, Volkmer AK, Naik TJ, Storm TA, Mosley AR, Edris B, Schmid SM, Sun CK, Chua MS, Murillo O, Rajendran P, Cha AC, Chin RK, Kim D, Adorno M, Raveh T, Tseng D, Jaiswal S, Enger PO, Steinberg GK, Li G, So SK, Majeti R, Harsh GR, van de Rijn M, Teng NN, Sunwoo JB, Alizadeh AA, Clarke MF, Weissman IL. The CD47-signal regulatory protein alpha (SIRPa) interaction is a therapeutic target for human solid tumors. Proc Natl Acad Sci U S A. 2012;109:6662–7. doi: 10.1073/pnas.1121623109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wynn TA. Cellular and molecular mechanisms of fibrosis. J Pathol. 2008;214:199–210. doi: 10.1002/path.2277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wynn TA, Ramalingam TR. Mechanisms of fibrosis: therapeutic translation for fibrotic disease. Nat Med. 2012;18:1028–40. doi: 10.1038/nm.2807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan D, Wang HW, Bowman RL, Joyce JA. STAT3 and STAT6 Signaling Pathways Synergize to Promote Cathepsin Secretion from Macrophages via IRE1alpha Activation. Cell Rep. 2016;16:2914–27. doi: 10.1016/j.celrep.2016.08.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu H, Pardoll D, Jove R. STATs in cancer inflammation and immunity: a leading role for STAT3. Nat Rev Cancer. 2009;9:798–809. doi: 10.1038/nrc2734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan A, Hsiao YJ, Chen HY, Chen HW, Ho CC, Chen YY, Liu YC, Hong TH, Yu SL, Chen JJ, Yang PC. Opposite Effects of M1 and M2 Macrophage Subtypes on Lung Cancer Progression. Sci Rep. 2015;5:14273. doi: 10.1038/srep14273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaidi MR, Merlino G. The two faces of interferon-gamma in cancer. Clin Cancer Res. 2011;17:6118–24. doi: 10.1158/1078-0432.CCR-11-0482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaynagetdinov R, Sherrill TP, Polosukhin VV, Han W, Ausborn JA, McLoed AG, McMahon FB, Gleaves LA, Degryse AL, Stathopoulos GT, Yull FE, Blackwell TS. A critical role for macrophages in promotion of urethane-induced lung carcinogenesis. J Immunol. 2011;187:5703–11. doi: 10.4049/jimmunol.1100558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao L, Lim SY, Gordon-Weeks AN, Tapmeier TT, Im JH, Cao Y, Beech J, Allen D, Smart S, Muschel RJ. Recruitment of a myeloid cell subset (CD11b/Gr1 mid) via CCL2/CCR2 promotes the development of colorectal cancer liver metastasis. Hepatology. 2013;57:829–39. doi: 10.1002/hep.26094. [DOI] [PubMed] [Google Scholar]
- Zhu X, Fujita M, Snyder LA, Okada H. Systemic delivery of neutralizing antibody targeting CCL2 for glioma therapy. J Neurooncol. 2011;104:83–92. doi: 10.1007/s11060-010-0473-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y, Herndon JM, Sojka DK, Kim KW, Knolhoff BL, Zuo C, Cullinan DR, Luo J, Bearden AR, Lavine KJ, Yokoyama WM, Hawkins WG, Fields RC, Randolph GJ, DeNardo DG. Tissue-Resident Macrophages in Pancreatic Ductal Adenocarcinoma Originate from Embryonic Hematopoiesis and Promote Tumor Progression. Immunity. 2017;47:597. doi: 10.1016/j.immuni.2017.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y, Knolhoff BL, Meyer MA, Nywening TM, West BL, Luo J, Wang-Gillam A, Goedegebuure SP, Linehan DC, DeNardo DG. CSF1/CSF1R blockade reprograms tumor-infiltrating macrophages and improves response to T-cell checkpoint immunotherapy in pancreatic cancer models. Cancer Res. 2014;74:5057–69. doi: 10.1158/0008-5472.CAN-13-3723. [DOI] [PMC free article] [PubMed] [Google Scholar]