

Abstract
The clinical success of chimeric antigen receptor (CAR) T cell immunotherapy in treating multiple blood cancers has created a need for efficient methods of ex vivo gene delivery to primary human T cells for cell engineering. Here, we synthesize and evaluate a panel of cationic polymers for gene delivery to both cultured and primary human T cells. We show that a subset of comb- and sunflower-shaped pHEMA-g-pDMAEMA polymers can mediate transfection with efficiencies up to 50% in the Jurkat human T cell line with minimal concomitant toxicity (>90% viability). We then optimize primary human T cell transfection conditions including activation time, cell density, DNA dose, culture media, and cytokine treatment. We demonstrate transfection of both CD4+ and CD8+ primary human T cells with messenger RNA and plasmid DNA at efficiencies up to 25 and 18%, respectively, with similarly high viability.
Keywords: Nonviral gene delivery, Cationic polymers, T lymphocytes
Graphical abstract
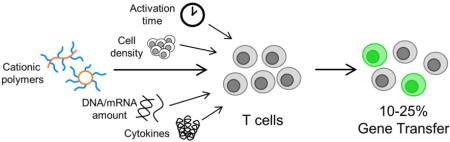
1. Introduction
T lymphocytes are key cells of the adaptive immune system that facilitate recognition and clearance of disease. In cancer, T cells primed with cancer antigens can often effectively eliminate cancer cells [1]. However, tumors can hinder T cell activity by creating a poorly immunogenic environment and expressing anti-inflammatory cytokines and exhaustion signals that push T cells towards a regulatory or exhausted phenotype [2]. Over the last decade, scientists and clinicians have developed new immunotherapies that can overcome these barriers to allow T cells to better recognize and clear tumors [3,4]. One therapy that has shown remarkable promise, especially in hematologic cancers, is a cellular therapy using autologous, genetically-modified T cells that express a chimeric antigen receptor (CAR) [5]. These CARs consist of an extracellular domain that recognizes a surface epitope on cancer cells, and an intracellular domain that contains co-stimulatory signals that trigger T cell expansion, inflammatory cytokine production, and cytotoxic action against cancer cells [6].
To generate a CAR T cell product, primary human T cells must be genetically modified to express the CAR protein. Most clinical trials for CAR T cells use gammaretroviral and lentiviral methods for gene delivery [7]. These viruses are efficient at gene delivery, have low intrinsic immunogenicity, and integrate into the host genome, resulting in permanent CAR expression [8]. However, viral vectors have important limitations: DNA cargo size, potential for insertional mutagenesis, cost and challenge of production, lot-to-lot variability, and increased regulatory requirements make them a costly choice for use in CAR T cell manufacturing [9]. Further, construction and production of viral vectors is time-intensive and often a limiting factor in screening CAR constructs at the research phase. In addition, lentiviral vectors are limited to a cargo size of ~10 kb, which could limit the future ability to introduce additional functionalities into CAR T cells including kill-switches, triggered cytokine production, and multiplexed gene delivery [10]. Non-viral gene delivery methods like electroporation and chemical transfection reagents are an attractive alternative due to their reduced cost, their easy adaptation to various cargo, and their improved safety profile compared to viral vectors but historically have low gene transfer efficiencies in T cells [11,12].
Cationic lipid and polymer-based reagents have been developed extensively as gene delivery vehicles [13]. Unlike viruses, these materials are not limited to a size or type of genetic cargo they can carry. This makes them attractive delivery vehicles, because they can deliver large plasmids (pDNA), as well as messenger RNA (mRNA) and small interfering RNA (siRNA). Polymer-based reagents are especially of interest due to their controllable chemical diversity and shelf stability. Polymers for gene delivery are typically made of cationic monomers that contain primary, secondary, and/or tertiary amine groups that can complex with negatively charged nucleic acid to form condensed polymer-nucleic acid nanoparticle complexes, called “polyplexes” [14]. Polyethylenimine (PEI) and poly(2-dimethylaminoethyl methacrylate) (pDMAEMA) are two classes of cationic polymers that have been studied extensively [15,16]. Branched PEI contains primary, secondary, and tertiary amines at an approximate 1:2:1 ratio whereas pDMAEMA only contains tertiary amines. Polyplexes are formulated with an excess positive charge to promote electrostatic binding to the negative cell membrane [17]. Highly cationic polyplexes are taken up into cells by interaction with surface proteoglycans, followed by internalization via endocytosis [18].
Very few polymers have been developed as gene delivery systems specifically for primary human T cells. Low molecular weight PEI (5 kDa) has been conjugated to transferrin to increase uptake of siRNA in activated primary T cells, which resulted in 50% gene silencing [19,20]. The Freitag group polymerized 20 “arms” of pDMAEMA off a silsesquioxane core and used this star polymer to deliver pDNA or siRNA, obtaining approximately 50% knockdown from siRNA delivery, and 10-50% transfection efficiency with 40-100% viability using pDNA [21]. This group also recently reported 13% transfection efficiency of primary T cells using this material with cell viability of 80% [22]. Most recently, the Stephan group reported a nanoparticle formulation with a poly(β-amino ester) core for DNA condensation and an antibody-conjugated polyglutamic acid shell for receptor-mediated uptake that was successfully used for in vitro and in vivo gene delivery to primary murine T cells, achieving ~3% transfection efficiency in vitro and ~1.5% in vivo [23]. This same nanoparticle system was able to achieve up to ~80% in vitro transfection efficiency of mRNA in primary human T cells [24].
The advances in controlled radical polymerization techniques over the last decade have significantly increased the design space for gene delivery polymers [25–27]. Chemists can specifically tune molecular weight, create complex architectures, and build in environmentally-responsive components [28,29]. In our laboratory, we found that altering the architecture of a pDMAEMA polymer from linear-branched (comb) to cyclic-branched (sunflower) decreased the toxicity and increased the efficiency of gene delivery to multiple cancer cell lines [30]. Including an endosomal lytic peptide in a pH sensitive block of a statistical co-polymer improved endosomal escape of polyplexes and increased subsequent gene expression in many cell lines [31].
The aim of this study was to empirically evaluate a panel of polymer architectures developed recently by our group for efficacy as an ex vivo gene delivery agent to primary human T cells. Polymers identified in this study could hold potential for future applications in adoptive T cell therapy manufacturing. We were surprised to find that the trends in polymer gene transfer efficiency in our previous adherent cell line studies did not hold for either cultured (Jurkat) T cells or primary T cells. In this current work, we identified a specific architecture of comb pDMAEMA polymers that, combined with optimized transfection protocols, showed the highest gene transfer efficiency to cultured and primary human T cells.
2. Materials and methods
2.1. Synthesis of pHEMA15-g-pDMAEMA
pHEMA15-g-pDMAEMA with different degrees of polymerization (DP) of DMAEMA was synthesized by three steps listed in Supplementary Information. Polymers were characterized by gel permeation chromatography (GPC) and nuclear magnetic resonance (1H NMR) (Supplemental Figure 1).
2.2. Polymer preparation
Branched polyethylenimine (Mw ~ 25,000) was purchased from Sigma and diluted to 65 μg/mL in sterile molecular grade H2O for transfection studies. Linear pDMAEMA290, comb and sunflower polymers with a core of pHEMA25, and virus-inspired polymer for endosomal release (VIPER) were synthesized as reported previously by controlled living radical polymerization [30,31]. Polymers were diluted in sterile molecular grade H2O to desired amine concentration for transfection studies.
2.3. Antibodies and plasmids
PE/Cy-7 anti-human CD4 (clone: RPA-T4), APC anti-human CD8 (clone: RPA-T8), and Zombie Violet fixable live-dead stain were purchased from Biolegend and titrated prior to use.
Plasmids were prepared using standard molecular biology techniques. XL10 Gold ultracompetent cells (Stratagene) were transformed with pmaxGFP plasmid (Lonza). A single colony was grown up in an overnight culture, lysed and purified using the NucleoBond Xtra Maxi Endotoxin Free kit (Macherey-Nagel). Purity and concentration were quantified by Nanodrop and a diagnostic gel. Enhanced green fluorescent protein (eGFP) mRNA was purchased from TriLink and stored in aqueous stock solutions at −80°C until use.
2.4. Cell culture conditions
Jurkat cells (human T lymphocyte line) were a kind gift from Dr. Michael Jensen (Seattle Children’s Research Institute). Jurkat cells were cultured in RPMI-1640 supplemented with 10% fetal bovine serum (v/v). Cells were used in transfection studies 18-24 hours after passaging.
Cryopreserved vials of healthy donor primary human T lymphocytes, isolated by magnetic activated cell sorting, were generously provided by Juno Therapeutics. Thawed cells were washed once in basal X-VIVO 15 medium (Lonza) before being cultured in X-VIVO 15 supplemented with 2% KnockOut serum replacement (ThermoFisher) and either recombinant human IL-2 (200 IU/mL) or recombinant human IL-21 (10 ng/mL) (Miltenyi) at a density of 1.5×106/mL. Cells were rested for 2-16 hours before activation with CD3/CD28 Human T Activator beads (DynaBeads, Gibco) at the recommended 1:1 bead:cell ratio. Cells were activated for 0-72 hours prior to transfection. All cells were maintained in a 37°C and 5% CO2 humidified incubator.
2.5. Polyplex formulation
Polyplexes were formed immediately before transfection experiments. Plasmid DNA or mRNA (1.75 and 0.1 mg/mL, respectively) was diluted in a sterile filtered 150 mM NaCl solution (Sigma) after which cationic polymers were added at the desired amine-to-phosphate (N/P) ratio to a final volume of 45 μL. This mixture was vortexed for 10 seconds and incubated at room temperature for 20-30 minutes before transfection.
2.6. Zeta potential and hydrodynamic diameters of polyplexes
Polyplexes were formulated at N/P 3, 5 & 7 with CP-25-16 polymer and 2 μg pmaxGFP plasmid DNA in molecular grade H2O. Polyplexes were diluted to a final volume of 800 μL in 10 mM NaCl and loaded into a DTS1070 Zetasizer cell (Malvern Instruments) for dynamic light scattering and zeta potential measurements using a Zetasizer Nano ZS (Malvern Instruments).
2.7. Transfections
The same transfection protocol was used for Jurkat cells and primary human T cells. Cells were washed once in phosphate buffered saline (PBS) via centrifugation at 300×g for 5 minutes and resuspended in transfection medium 30 minutes prior to transfection. OptiMEM (Gibco), Jurkat culture medium, and T cell culturing medium were all used as transfection media in various experiments. Cells were plated at various concentrations in 250 μL transfection medium in a 24-well TC treated plate and stored in a 37°C and 5% CO2 humidified incubator until transfection. Jurkat cells were plated at 1×106/mL (250 K cells per well), and T cells were plated at 2×106/mL (500 K cells per well), unless otherwise noted in optimization experiments.
Unless specified otherwise, 1.5 μg of nucleic acid was used per well in Jurkat transfections, and 2 μg in primary T cells transfections. Polyplexes or a control 150 mM NaCl solution were added to designated wells dropwise. Plates were returned immediately to the 37°C and 5% CO2 humidified incubator. Cells were cultured for 48 hours prior to flow cytometry analysis.
2.8. Flow cytometry
Cells were transferred to a U-bottom 96-well plate by successive centrifugation and aspiration steps. Cells were washed once with PBS via centrifugation at 300×g for 5 minutes before being resuspended in 100 μL of a 1:500 dilution of Zombie Violet dye in PBS. Cells were incubated in the live/dead stain for 10-15 minutes at room temperature in the dark and subsequently washed twice with PBS with 1% BSA (PBSA from diluted 10% BSA stock, Miltenyi). If antibody staining was used in the experiment, cells were incubated in 100 μL of the diluted antibody cocktail for 20 minutes at room temperature, and washed twice with 1% PBSA. Cells were resuspended in 200 μL of 1% PBSA, and immediately analyzed on a MacsQuant Analyzer flow cytometer (Miltenyi) where at least 10,000 events in the “Live Cells” gate were collected.
Data analysis was performed using FlowJo software (FlowJo, LLC), with serial gating (Supplemental Figure 3). Transfection efficiency was measured as the percentage of live cells expressing GFP fluorescence.
2.9. Statistical analyses
Results are given as mean value ± standard deviation (SD). Multiple t tests, or one-way ANOVA with either Tukey’s or Dunnett’s multiple comparisons posthoc analysis were performed in GraphPad Prism software (Graph Pad Software).
3. Results and Discussion
3.1. Polymer panel screening in Jurkat cell line
Suspension cell lines and primary white blood cells are notoriously challenging to transfect, with no commercially available chemical reagent able to achieve high gene delivery efficiency. A panel of polymers were screened for transfection efficiency and cytotoxicity in the Jurkat human T cell line. This polymer panel included commercially available branched polyethylenimine (bPEI), as well as four distinct polymer architectures synthesized in our lab (Scheme 1). VIPER (virus-inspired polymer for endosomal release) is a diblock copolymer that contains a hydrophilic cationic block for nucleic acid loading and colloidal stability, and a pH sensitive membrane lytic block for endosomal release. This polymer is the best-performing polymer our lab has developed to date, with 80% transfection of HeLa cells with low cytotoxicity (>90% viability) [31]. Sunflower and comb polymers with a core size of 25 (pHEMA25-g-pDMAEMAn) also showed high efficiency in HeLas (50 and 40%, respectively) with low cytotoxicity (>75% viability), whereas the linear pDMAEMA polymer showed gene transfer efficiencies similar to bPEI (10%) in HeLas [30]. One polymer from each architecture was screened for transfection efficiency to Jurkat cells in both serum-containing and serum-free media using a range of polymer to DNA ratios (Supplemental Figure 4). The optimal N/P ratios for serum-containing and serum-free medium conditions were determined to be N/P 15 and N/P 5, respectively, such that the viability of cells stayed over 80% while maximizing transfection efficiency (Figure 1). The higher N/P ratio required in serum-containing media is likely due to the interaction of free polymer with serum proteins; free polymer has been reported to assist in intracellular trafficking [32–34].
Scheme 1.
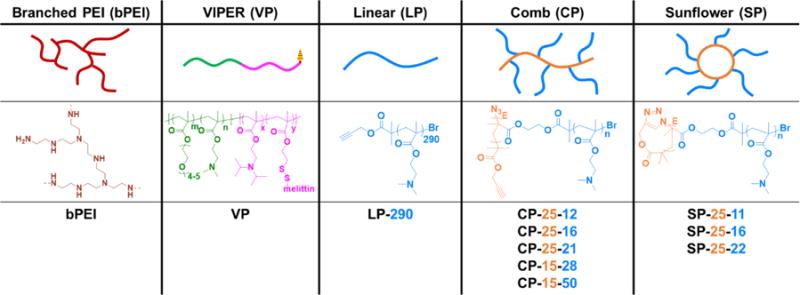
Schematic and chemical structures of polymers evaluated in gene delivery studies. Abbreviated polymer names denote degrees of polymerization (DP) for each block.
Figure 1.
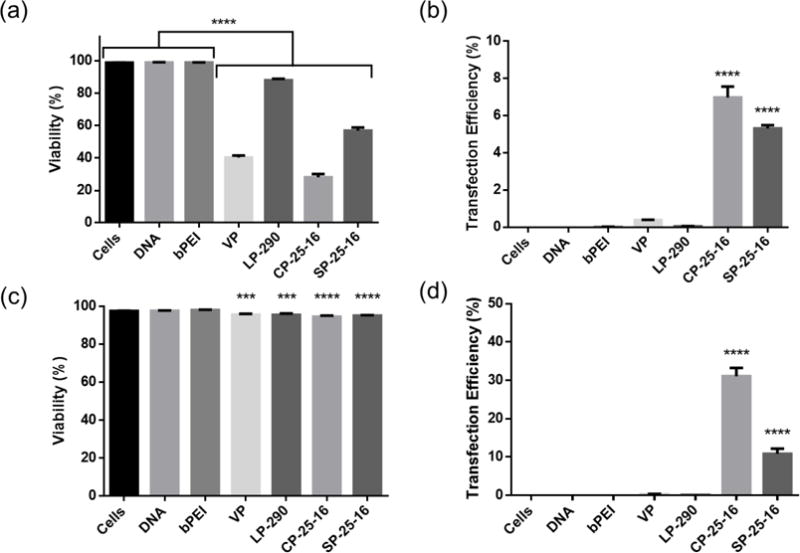
Polymer transfections delivering pmaxGFP plasmid in serum and serum-free medium to Jurkat T cell line. Toxicity (a) and transfection efficiency (b) of cationic polymers in Jurkat human T cell line in serum containing medium at N/P 15. Toxicity (c) and transfection efficiency (d) in serum-free medium at N/P 5. Transfection efficiencies are expressed as percentage of GFP-positive cells. Data are shown as mean ± SD (n=3; 1-way ANOVA with Tukey’s multiple comparisons, *p<0.05, *** p<0.001, ****p<0.0001).
Surprisingly, the trends in polymer performance in adherent cell lines was not recapitulated in Jurkat cells. Comb and sunflower polymers were the only architectures that showed appreciable transfection efficiency in serum-containing or serum-free conditions. We speculate that these differences could be due to differences in uptake and intracellular trafficking mechanisms in suspension cells compared to adherent cells, resulting in different polymer characteristics required for successful gene delivery. A 5-fold increase in transfection efficiency in serum-free media compared to serum-containing media was observed for both the comb and sunflower polymers. These results are expected, as negatively charged serum proteins can nonspecifically bind and disrupt polyplexes prior to endocytosis [35]. In addition, the cell viability was improved for transfections performed in serum-free medium, most likely due to the lower polymer concentration (N/P ratio) required for polyplex delivery. We chose to further evaluate the comb and sunflower polymers in serum-free transfection conditions for all additional Jurkat transfection studies, as the future utility of these polymers will be in well-defined ex vivo transfection protocols.
3.2. Optimization of pDMAEMA polymer architecture for transfection of Jurkat cells
Due to the promising T cell transfection efficiencies mediated by comb and sunflower polymers, we expanded this panel by synthesizing polymers with two core sizes and varying branch lengths per core size (Table 1). The impact of molecular weight and polymer geometry on gene transfer was then explored. Polymers with the same core size of DP 25 had similar transfection efficiencies and cytotoxicities in Jurkat cells (Figure 2 a & b). There were statistically significant differences between core geometries (linear vs. circular) at the two lower branch lengths, with comb polymers outperforming sunflower. Branch length was only significantly different within the same core geometry for the smallest molecular weight sunflower polymer (SP-25-11).
Table 1.
Characterization of synthesized polymers.
| Compositiona | Code | Mn [kDa]a | Mn [kDa]b | PDIb |
|---|---|---|---|---|
| l-pDMAEMA290 | LP-290 | 46 | 53.5 | 1.09 |
| l-pHEMA25-g-(pDMAEMA12)25 | CP-25-12 | 54 | 192 | 1.48 |
| l-pHEMA25-g-(pDMAEMA16)25 | CP-25-16 | 70 | 263 | 1.67 |
| l-pHEMA25-g-(pDMAEMA21)25 | CP-25-21 | 89 | 371 | 1.75 |
| l-pHEMA15-g-(pDMAEMA28)15 | CP-15-28 | 70 | 209 | 1.43 |
| l-pHEMA15-g-(pDMAEMA50)15 | CP-15-50 | 122 | 481 | 1.62 |
| c-pHEMA25-g-(pDMAEMA11)25 | SP-25-11 | 50 | 176 | 1.44 |
| c-pHEMA25-g-(pDMAEMA16)25 | SP-25-16 | 70 | 287 | 1.81 |
| c-pHEMA25-g-(pDMAEMA22)25 | SP-25-22 | 93 | 413 | 1.88 |
| p(OEGMA11-DMAEMA56)-b-p(DIPAMA33-(PDSEMA-g-melittinb)1) | VP-67-34 | 23 | 25 | 1.03 |
Determined by 1HNMR;
Obtained by GPC.
Figure 2.
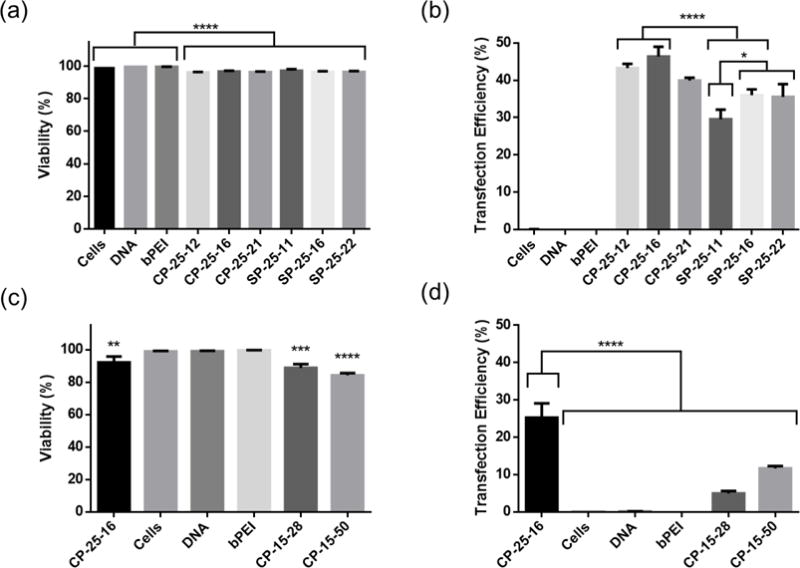
Impacts of polymer architecture on transfection of pmaxGFP plasmid to Jurkat T cell line. Toxicity and transfection efficiency of cationic polymers with varying core shape and branch length (a & b) or core size and branch length (c & d). Transfection efficiencies are expressed as percentage of GFP-positive cells. Data are shown as mean ± SD (n=3; 1-way ANOVA with (a, c & d) Dunnett’s or (b) Tukey’s multiple comparisons, * p<0.05, ** p<0.01, *** p<0.001, ****p<0.0001).
We observed a larger significant difference in transfection efficiencies of comb polymers with varying core sizes (DP 15 vs. 25), with the DP 15 core polymers exhibiting significantly reduced transfection efficiency and greater cytotoxicity (Figure 2 c & d, Supplemental Figure 5). While we did not investigate the mechanism underlying this difference in performance between the two core sizes, we hypothesize that this difference may be due to increased branching on the DP25 polymers. This hypothesis would be consistent with the results from the Müller group, where they have previously shown that star PDMAEMA with a higher number of arms were less toxic and resulted in higher transfection efficiency than polymers with fewer arms at equivalent molecular weights [36]. With these results, we chose to use comb polymers with a core size of DP 25 to optimize transfection conditions for primary human T cells, as they consistently showed the highest transfection efficiencies and viabilities across transfection studies in the Jurkat cell line.
Prior to moving into primary T cell studies, a more refined range of N/P ratios (3, 5, & 7) were evaluated around the highest performing N/P in serum-free transfection conditions. The size and zeta potential of polyplexes formed at these N/P ratios were similar (Supplemental Figure 2). N/P ratios of 5 and 7 resulted in similar viabilities and transfection efficiencies in Jurkat cells and were used for primary T cell studies (Supplementary Figure 6).
3.3. Impact of activation time on primary T cell transfection
Unlike viruses, cationic polymers do not have an active mechanism for transporting DNA into the nucleus of cells. Therefore, transfection efficiency is highly dependent on cell cycling and is most efficient during the G2/M phase of mitosis [37]. Unsurprisingly, transfections to quiescent, unactivated primary human T cells resulted in negligible gene expression (data not shown). To determine the optimal timing for transfections, primary T cells were transfected at various timepoints from 0-72 hours post-activation (Figure 3). In this study, cells were seeded at 250 K cells/well in OptiMEM and transfected with 1 μg of pDNA at N/P ratio of 5 with CP-25-16. The complete T cell medium was supplemented with 200 IU/mL IL-2. Untransfected cells went through the same manipulation steps as transfected cells, but were not treated with polyplexes.
Figure 3.
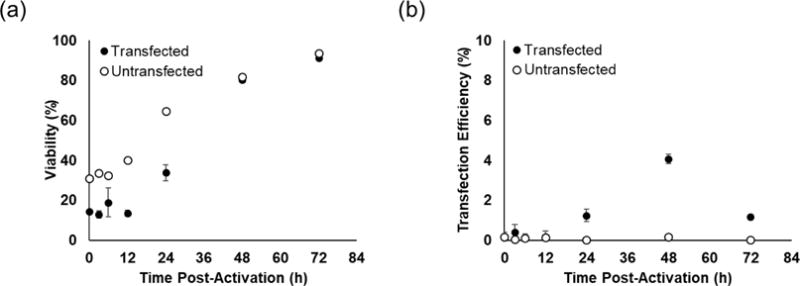
Impact of activation time prior to transfection on primary T cell viability (a) and transfection efficiency (b). Cells were transfected with 1 μg of pmaxGFP pDNA at N/P ratio of 5 using CP-25-16. Transfection efficiencies are expressed as percentage of GFP-positive cells. Data are shown as mean ± SD (n=3).
Significant cell death (>50%) was observed in transfected and control groups manipulated prior to 24-hours. These results are consistent with common in vitro T cell culture practices of leaving T cells untouched for the first 2-3 days after bead-based activation, as cells are more prone to activation-induced apoptosis [38]. Maximum transfection efficiency was reached when performed 48-hours after activation, however, transfection efficiencies were 10-fold lower than those observed in Jurkat cells. We therefore sought to improve transfection efficiencies by optimizing other parameters of the transfection protocol.
3.4. Optimization of transfection conditions for primary T cells
We performed two design of experiment (DOE)-style screening experiments to identify which variables impacted primary T cell transfection and viability. In the first experiment, we screened cell density (125 K, 250 K, or 500 K cells/well), total mass of DNA delivered (1 or 2 μg), and transfection medium (OptiMEM or complete T cell medium with IL-2) (Figure 4 a & b). Higher cell densities and lower DNA doses both improved viability of cells. Transfection efficiency with CP-25-16 was slightly higher when OptiMEM was used as the transfection medium, and was highest for the highest cell density (500 K cells/well) and highest DNA dose (2 μg).
Figure 4.
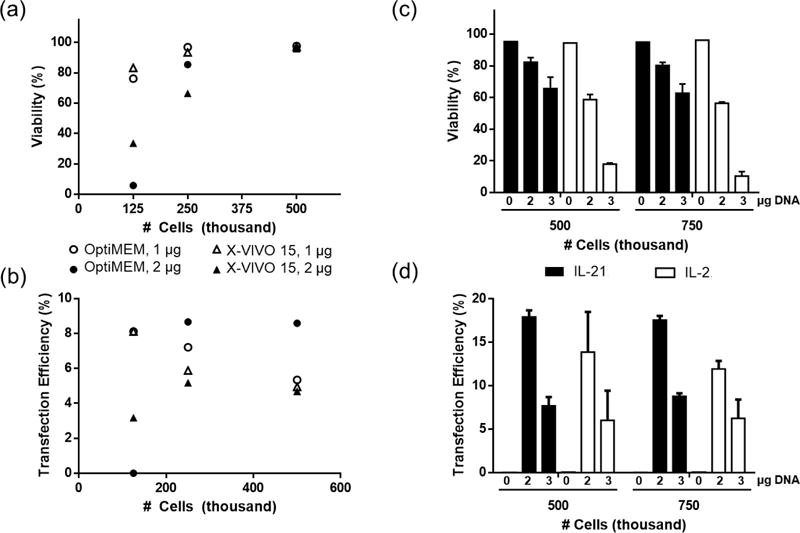
Design of Experiments (DOE) optimization of primary T cell transfection conditions. (a & b) Screening effects of transfection medium, cell density, and DNA dose on viability and transfection efficiency (n=1). (c & d) Screening effects of cytokine supplement in culture medium, cell density, and DNA dose (n=3). Cells were transfected with pmaxGFP pDNA at N/P ratio of 7 using CP-25-16. Transfection efficiencies are expressed as percentage of GFP-positive cells. Data are shown as mean ± SD.
We hypothesized that higher DNA doses could be tolerated at higher cell densities, and perhaps result in even greater transfection efficiencies. In the second optimization experiment we screened cell density (500 K or 750 K cells/well), DNA dose (2 or 3 μg), and cytokine supplement (IL-2 or IL-21). We chose to include cytokine supplement to T cell culture medium as a variable because cytokines can significantly impact the phenotype and differentiation programs of cultured T cells. While IL-2 is the most common cytokine used for outgrowth of T cells, it has been shown that T cells cultured in IL-21 supplemented media can better mediate tumor regression after adoptive transfer [39].
Cells cultured in IL-21 showed an overall higher cell viability and comparable or higher transfection efficiency by CP-25-16 for all cell densities and DNA doses tested (Figure 4 c & d). Increasing the DNA dose to 3 μg significantly reduced the viability of cells cultured in IL-2 and reduced the viability of IL-21 cultured cells. Increasing the DNA dose to 3 μg did not have the desired effect of increasing transfection efficiency. Instead, cells treated with this higher dose showed lower transfection efficiencies for all conditions tested.
The maximum transfection efficiency achieved for delivery of plasmid DNA to primary T cells was 18%. Optimal conditions were 500-750K cells per well in a 24-well-plate format in complete T cell medium supplemented with IL-21, and transfecting cells in OptiMEM with a total dose of 2 μg of DNA 48-hours after activation. These transfection parameters were used to probe the initial utility of these comb polymers for applications relevant to adoptive T cell therapy.
3.5. Delivery of various nucleic acid cargoes to primary T cells
Many future applications of cationic polymers as gene transfer agents for T cell-based therapies will require the use of an integrating technology like transposon/transposase for stable gene expression or CRISPR/Cas9 systems for gene editing [40,41]. The Sleeping Beauty transposon/transposase system has already been used with electroporation to generate stably expressing CAR T cells [11,42,43]. These systems require the parallel delivery of two nucleic acid cargoes: a plasmid or mRNA to express the editing enzyme (Cas9 or transposase) and a plasmid with the gene for insertion (transposon or DNA template). The delivery platform with the best safety profile delivers the editing enzyme gene via mRNA and the gene for insertion by plasmid DNA [44]. The transient expression of the enzyme nearly eliminates the possibility of repeat template excision/re-integration genotoxicities, and prevents the enzyme from integrating itself into the host genome.
To show the utility of cationic polymers for this application, we successfully delivered mRNA and pDNA to primary T cells using the optimized transfection protocol (Figure 5 a & b). We observed a higher level of gene expression from cells transfected with mRNA compared to plasmid DNA, suggesting that nuclear transport is a contributing barrier to higher gene expression with this reagent.
Figure 5.
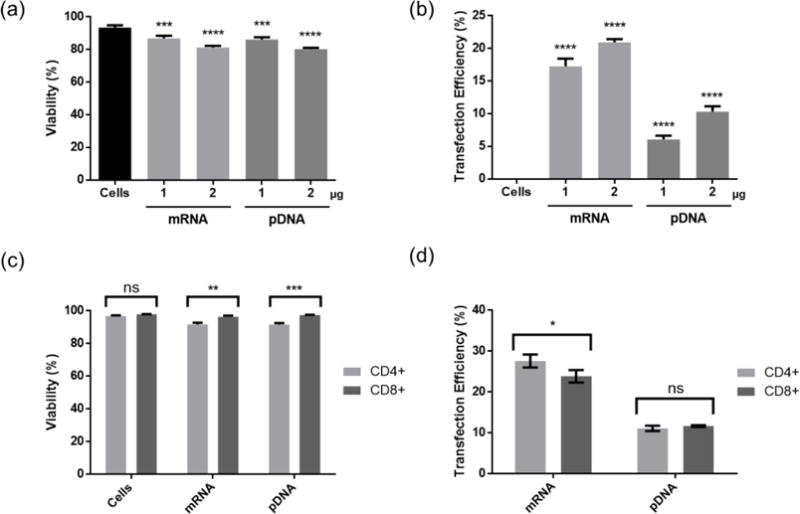
Viability (a) and transfection efficiency (b) of eGFP expressing pDNA and mRNA to a mixture of CD4+ and CD8+ T cells at optimized conditions. Viability (c) and transfection efficiency (d) in CD4+ and CD8+ T cells. Cells were transfected with pDNA at N/P ratio of 7 using CP-25-16. Transfection efficiencies are expressed as percentage of GFP-positive cells. Data are shown as mean ± SD (n =3; (a & b) 1-way ANOVA with Tukey’s multiple comparisons, (c & d) multiple t tests, *p<0.05, * *p<0.01, ***p<0.001, ****p<0.0001).
3.6. Delivery to both CD4+ and CD8+ primary T cells
We wanted to ensure that these comb polymers could transfect both CD4+ “helper” and CD8+ “cytotoxic” T cells, as both subtypes are used in CAR T cell therapy, and initial polymer screening was performed on the CD4+ Jurkat cell line. We stained transfected T cells with fuorescently tagged antibodies for CD4 and CD8 and observed an equivalent proportion of transfected cells in each subset (Figure 5 c & d). There was no difference in transfection efficiency of pDNA and only a small statistical difference in transfection efficiency of mRNA between CD4+ and CD8+ T cells, with a slightly higher level of gene delivery to CD4+ T cells.
These results support the continued development of comb polymers for ex vivo non-viral genetic reprogramming of primary human T cells for the application of CAR T cell manufacturing. Additional polymer and polyplex engineering would be required to create a gene delivery platform for in vivo delivery to prevent non-specific interactions and toxicities caused by the positive charges on the surface of the polyplex. However, the flexible cargo loading of this polymer system makes it attractive for ex vivo gene delivery applications.
4. Conclusion
One of the major opportunities for cost reduction and safety improvement in CAR T cell manufacturing is moving from viral to non-viral methods of genetically reprogramming patient T cells. These studies begin to evaluate shelf-stable, easy to manufacture, untargeted cationic polymers as alternative non-viral gene delivery vehicles for CAR T cell manufacturing. We identified a class of pHEMA-g-pDMAEMA comb and sunflower polymers that can transfect the Jurkat T cell line at high efficiencies of 25-50% in serum-free medium. This could be a very useful tool for the screening experiments performed in the development of new chimeric antigen receptors. Extensive and iterative protein engineering is required for developing new chimeric antigen receptors [45,46]. Having a quick and efficient method to screen expression and binding affinity of various CARs in a T cell line, instead of creating a new viral vector for each one, could significantly reduce the time it takes to develop and perform early validation studies on new CAR constructs. Jurkat cells, like all suspension cell lines, are notoriously challenging to transfect. Though untested, these comb and sunflower polymers may also be able to transfect other suspension cell lines with high efficiency.
Our data demonstrate that these polymers can also transfect primary human T cells with pDNA and mRNA at modest efficiencies up to 18 and 25%, respectively, while maintaining high cell viability (>75%). These efficiencies were reached through DOE-style optimization experiments that yielded a 4-fold increase over initial transfection values. These results support the continued development of comb polymers for non-viral genetic reprogramming of primary human T cells for the application of CAR T cell manufacturing. Future studies will seek to understand the additional barriers to non-viral gene delivery in primary T cells and identify alternative protocol changes that will further increase transfection efficiency to a level approaching those achieved by viral gene delivery methods.
Supplementary Material
Acknowledgments
We thank Dr. Joshua Gustafson (Seattle Children’s Research Institute) for helpful discussions on primary T cell culture. We thank David Peeler, Albert Yen, and the lab of Dr. Shaoyi Jiang for assistance in zeta potential and DLS measurements. This work was supported by the National Institutes of Health [1R01CA177272, 2R01NS064404]. B.R.O. was supported by a National Science Foundation Graduate Research Fellowship [DGE-1256082].
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interest
A patent application has been fled on the sunflower and comb polymers by the University of Washington.
Appendix A. Supplementary Information
References
- 1.Strausberg RL. Tumor microenvironments, the immune system and cancer survival. Genome Biol. 2005;6:211. doi: 10.1186/gb-2005-6-3-211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gajewski T, Meng Y, Harlin H. Immune suppression in the tumor microenvironment. J Immunother. 2006;29:233–240. doi: 10.1097/01.cji.0000199193.29048.56. [DOI] [PubMed] [Google Scholar]
- 3.Banchereau J, Palucka AK. Dendritic cells as therapeutic vaccines against cancer. Nat Rev Immunol. 2005;5:296–306. doi: 10.1038/nri1592. [DOI] [PubMed] [Google Scholar]
- 4.Kalos M, Levine BL, Porter DL, Katz S, Grupp SA, Bagg A, June CH. T cells with chimeric antigen receptors have potent antitumor effects and can establish memory in patients with advanced leukemia. Sci Transl Med. 2011;3:95ra73. doi: 10.1126/scitranslmed.3002842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hay KA, Turtle CJ. Chimeric Antigen Receptor (CAR) T Cells: Lessons Learned from Targeting of CD19 in B-Cell Malignancies. Drugs. 2017;77:237–245. doi: 10.1007/s40265-017-0690-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Barrett DM, Singh N, Porter DL, Grupp SA, June CH. Chimeric antigen receptor therapy for cancer. Annu Rev Med. 2014;65:333–47. doi: 10.1146/annurev-med-060512-150254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Holzinger A, Barden M, Abken H. The growing world of CAR T cell trials: a systematic review. Cancer Immunol Immunother. 2016;65:1–18. doi: 10.1007/s00262-016-1895-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gill S, June CH. Going viral: Chimeric antigen receptor T-cell therapy for hematological malignancies. Immunol Rev. 2015;263:68–89. doi: 10.1111/imr.12243. [DOI] [PubMed] [Google Scholar]
- 9.Wang X, Rivière I. Clinical manufacturing of CAR T cells: foundation of a promising therapy. Mol Ther Oncolytics. 2016;3:16015. doi: 10.1038/mto.2016.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jaspers JE, Brentjens RJ. Development of CAR T cells designed to improve antitumor efficacy and safety. Pharmacol Ther. 2017;178:83–91. doi: 10.1016/j.pharmthera.2017.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Singh H, Figliola MJ, Dawson MJ, Olivares S, Zhang L, Yang G, Maiti S, Manuri P, Senyukov V, Jena B, Kebriaei P, Champlin RE, Huls H, Cooper LJN. Manufacture of Clinical-Grade CD19-Specific T Cells Stably Expressing Chimeric Antigen Receptor Using Sleeping Beauty System and Artificial Antigen Presenting Cells. PLoS One. 2013;8:1–11. doi: 10.1371/journal.pone.0064138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhao Y, Zheng Z, Cohen CJ, Gattinoni L, Palmer DC, Restifo NP, Rosenberg SA, Morgan RA. High-efficiency transfection of primary human and mouse T lymphocytes using RNA electroporation. Mol Ther. 2006;13:151–159. doi: 10.1016/j.ymthe.2005.07.688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yin H, Kanasty RL, Eltoukhy AA, Vegas AJ, Dorkin JR, Anderson DG. Non-viral vectors for gene-based therapy. Nat Rev Genet. 2014;15:541–555. doi: 10.1038/nrg3763. [DOI] [PubMed] [Google Scholar]
- 14.Pack DW, Hoffman AS, Pun S, Stayton PS. Design and development of polymers for gene delivery. Nat Rev Drug Discov. 2005;4:581–93. doi: 10.1038/nrd1775. [DOI] [PubMed] [Google Scholar]
- 15.Boussif O, Lezoualc’h F, Zanta MA, Mergny MD, Scherman D, Demeneix B, Behr JP. A versatile vector for gene and oligonucleotide transfer into cells in culture and in vivo: polyethylenimine. Proc Natl Acad Sci. 1995;92:7297–7301. doi: 10.1073/pnas.92.16.7297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jiang X, Lok MC, Hennink WE. Degradable-Brushed pHEMA – pDMAEMA Synthesized via ATRP and Click Chemistry for Gene Delivery. Bioconjug Chem. 2007:2077–2084. doi: 10.1021/bc0701186. [DOI] [PubMed] [Google Scholar]
- 17.Khalil IA, Kogure K, Akita H, Harashima H. Uptake pathways and subsequent intracellular trafficking in nonviral gene delivery. Pharmacol Rev. 2006;58:32–45. doi: 10.1124/pr.58.1.8.32. [DOI] [PubMed] [Google Scholar]
- 18.Mislick KA, Baldeschwieler JD. Evidence for the role of proteoglycans in cationmediated gene transfer. Proc Natl Acad Sci U S A. 1996;93:12349–12354. doi: 10.1073/pnas.93.22.12349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Xie Y, Kim NH, Nadithe V, Schalk D, Thakur A, Kılıç A, Lum LG, Bassett DJP, Merkel OM. Targeted delivery of siRNA to activated T cells via transferrinpolyethylenimine (Tf-PEI) as a potential therapy of asthma. J Control Release. 2016:120–129. doi: 10.1016/j.jconrel.2016.03.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Xie Y, Killinger B, Moszczynska A, Merkel O. Targeted Delivery of siRNA to Transferrin Receptor Overexpressing Tumor Cells via Peptide Modified Polyethylenimine. Molecules. 2016;21:1334. doi: 10.3390/molecules21101334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schallon A, Synatschke CV, Jérôme V, Müller AHE, Freitag R. Nanoparticulate nonviral agent for the effective delivery of pDNA and siRNA to differentiated cells and primary human T lymphocytes. Biomacromolecules. 2012;13:3463–3474. doi: 10.1021/bm3012055. [DOI] [PubMed] [Google Scholar]
- 22.Raup A, Stahlschmidt U, Jérôme V, Synatschke CV, Müller AHE, Freitag R. Influence of polyplex formation on the performance of star-shaped polycationic transfection agents for mammalian cells. Polymers (Basel) 2016;8 doi: 10.3390/polym8060224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Smith TT, Stephan SB, Moffett HF, McKnight LE, Ji W, Reiman D, Bonagofski E, Wohlfahrt ME, Pillai SPS, Stephan MT. In situ programming of leukaemia-specific T cells using synthetic DNA nanocarriers. Nat Nanotechnol. 2017 doi: 10.1038/nnano.2017.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Moffett HF, Coon ME, Radtke S, Stephan SB, McKnight L, Lambert A, Stoddard BL, Kiem HP, Stephan MT. Hit-and-run programming of therapeutic cytoreagents using mRNA nanocarriers. Nat Commun. 2017;8:389. doi: 10.1038/s41467-017-00505-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ahmed M, Narain R. Progress of RAFT based polymers in gene delivery. Prog Polym Sci. 2013;38:767–790. doi: 10.1016/j.progpolymsci.2012.09.008. [DOI] [Google Scholar]
- 26.Agarwal S, Zhang Y, Maji S, Greiner A. PDMAEMA based gene delivery materials. Mater Today. 2012;15:388–393. doi: 10.1016/S1369-7021(12)70165-7. [DOI] [Google Scholar]
- 27.Chu DSH, Schellinger JG, Shi J, Convertine AJ, Stayton PS, Pun SH. Application of living free radical polymerization for nucleic acid delivery. Acc Chem Res. 2012;45:1089–1099. doi: 10.1021/ar200242z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.York AW, Kirkland SE, McCormick CL. Advances in the synthesis of amphiphilic block copolymers via RAFT polymerization: Stimuli-responsive drug and gene delivery. Adv Drug Deliv Rev. 2008;60:1018–1036. doi: 10.1016/j.addr.2008.02.006. [DOI] [PubMed] [Google Scholar]
- 29.Qiu LY, Bae YH. Polymer architecture and drug delivery. Pharm Res. 2006;23:1–30. doi: 10.1007/s11095-005-9046-2. [DOI] [PubMed] [Google Scholar]
- 30.Cheng Y, Wei H, Tan JKY, Peeler DJ, Maris DO, Sellers DL, Horner PJ, Pun SH. Nano-Sized Sunflower Polycations As Effective Gene Transfer Vehicles. Small. 2016;12:2750–2758. doi: 10.1002/smll.201502930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cheng Y, Yumul RC, Pun SH. Virus-Inspired Polymer for Efficient In Vitro and In Vivo Gene Delivery. Angew Chemie - Int Ed. 2016;55:12013–12017. doi: 10.1002/anie.201605958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Boeckle S, von Gersdorff K, van der Piepen S, Culmsee C, Wagner E, Ogris M. Purification of polyethylenimine polyplexes highlights the role of free polycations in gene transfer. J Gene Med. 2004;6:1102–1111. doi: 10.1002/jgm.598. [DOI] [PubMed] [Google Scholar]
- 33.Saul JM, Wang CHK, Ng CP, Pun SH. Multilayer nanocomplexes of polymer and DNA exhibit enhanced gene delivery. Adv Mater. 2008;20:19–25. doi: 10.1002/adma.200700633. [DOI] [Google Scholar]
- 34.Cai J, Yue Y, Wang Y, Jin Z, Jin F, Wu C. Quantitative study of effects of free cationic chains on gene transfection in different intracellular stages. J Control Release. 2016;238:71–79. doi: 10.1016/j.jconrel.2016.07.031. [DOI] [PubMed] [Google Scholar]
- 35.Buyens K, Lucas B, Raemdonck K, Braeckmans K, Vercammen J, Hendrix J, Engelborghs Y, De Smedt SC, Sanders NN. A fast and sensitive method for measuring the integrity of siRNA-carrier complexes in full human serum. J Control Release. 2008;126:67–76. doi: 10.1016/j.jconrel.2007.10.024. [DOI] [PubMed] [Google Scholar]
- 36.Synatschke CV, Schallon A, Jérôme V, Freitag R, Müller AHE. Influence of polymer architecture and molecular weight of poly(2-(dimethylamino)ethyl methacrylate) polycations on transfection efficiency and cell viability in gene delivery. Biomacromolecules. 2011;12:4247–4255. doi: 10.1021/bm201111d. [DOI] [PubMed] [Google Scholar]
- 37.Tseng WC, Haselton FR, Giorgio TD. Mitosis enhances transgene expression of plasmid delivered by cationic liposomes. Biochim Biophys Acta - Gene Struct Expr. 1999;1445:53–64. doi: 10.1016/S0167-4781(99)00039-1. [DOI] [PubMed] [Google Scholar]
- 38.Kalamasz D, Long SA, Taniguchi R, Buckner JH, Berenson RJ, Bonyhadi M. Optimization of human T-cell expansion ex vivo using magnetic beads conjugated with anti-CD3 and Anti-CD28 antibodies. J Immunother. 2004;27:405–418. doi: 10.1097/00002371-200409000-00010. [DOI] [PubMed] [Google Scholar]
- 39.Hinrichs CS, Spolski R, Paulos CM, Gattinoni L, Kerstann KW, Palmer DC, Klebanoff CA, Rosenberg SA, Leonard WJ, Restifo NP. IL-2 and IL-21 confer opposing differentiation programs to CD8+ T cells for adoptive immunotherapy. Blood. 2008;111:5326–5333. doi: 10.1182/blood-2007-09-113050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Aronovich EL, McIvor RS, Hackett PB. The Sleeping Beauty transposon system: A non-viral vector for gene therapy. Hum Mol Genet. 2011;20:14–20. doi: 10.1093/hmg/ddr140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Eyquem J, Mansilla-Soto J, Giavridis T, van der Stegen SJC, Hamieh M, Cunanan KM, Odak A, Gönen M, Sadelain M. Targeting a CAR to the TRAC locus with CRISPR/Cas9 enhances tumour rejection. Nature. 2017;543:113–117. doi: 10.1038/nature21405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Singh H, Huls H, Cooper LJN. A new approach to gene therapy using Sleeping Beauty to genetically modify clinical-grade T cells to target CD19. Immunol Rev. 2014;257:181–190. doi: 10.1016/j.immuni.2010.12.017.Two-stage. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Monjezi R, Miskey C, Gogishvili T, Schleef M, Schmeer M, Einsele H, Ivics Z, Hudecek M. Enhanced CAR T-cell engineering using non-viral Sleeping Beauty transposition from minicircle vectors. Leukemia. 2016;31:1–9. doi: 10.1038/leu.2016.180. [DOI] [PubMed] [Google Scholar]
- 44.Wilber A, Frandsen JL, Geurts JL, Largaespada DA, Hackett PB, McIvor RS. RNA as a source of transposase for Sleeping Beauty-mediated gene insertion and expression in somatic cells and tissues. Mol Ther. 2006;13:625–630. doi: 10.1016/j.ymthe.2005.10.014. [DOI] [PubMed] [Google Scholar]
- 45.Hudecek M, Lupo-Stanghellini MT, Kosasih PL, Sommermeyer D, Jensen MC, Rader C, Riddell SR. Receptor affinity and extracellular domain modifications affect tumor recognition by ROR1-specifc chimeric antigen receptor T cells. Clin Cancer Res. 2013;19:3153–3164. doi: 10.1158/1078-0432.CCR-13-0330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hudecek M, Sommermeyer D, Kosasih PL, Silva-Benedict A, Liu L, Rader C, Jensen MC, Riddell SR. The Nonsignaling Extracellular Spacer Domain of Chimeric Antigen Receptors Is Decisive for In Vivo Antitumor Activity. Cancer Immunol Res. 2015;3:125–135. doi: 10.1158/2326-6066.CIR-14-0127. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.