

Abstract
Acyl-coenzyme A (CoA) ligases catalyze the activation of carboxylic acids via a two-step reaction of adenylation followed by thioesterification. Here, we report the discovery of a non-adenylating acyl-CoA ligase PtmA2 and the functional separation of an acyl-CoA ligase reaction. Both PtmA1 and PtmA2, two acyl-CoA ligases from the biosynthetic pathway of platensimycin and platencin, are necessary for the two steps of CoA activation. Gene inactivation of ptmA1 and ptmA2 resulted in the accumulation of free acid and adenylate intermediates, respectively. Enzymatic and structural characterization of PtmA2 confirmed its ability to only catalyze thioesterification. Structural characterization of PtmA2 revealed it binds both free acid and adenylate substrates and undergoes the established mechanism of domain alternation. Finally, site-directed mutagenesis restored both the adenylation and complete CoA activation reactions. This study challenges the currently accepted paradigm of adenylating enzymes and inspires future investigations on functionally separated acyl-CoA ligases and their ramifications in biology.
INTRODUCTION
Carboxylic acid activation is ubiquitous in nature and is found throughout the biosyntheses of primary and secondary metabolites. Fatty acids are activated with coenzyme A (CoA) as substrates for β-oxidation1; amino acids are activated as acyl-phosphates or acyl-adenylates in amino acid2 and amino-acyl-tRNA biosynthesis3; short carboxylic acids and amino acids are tethered to an acyl carrier protein (ACP) or peptidyl carrier protein for incorporation into polyketide and nonribosomal peptide natural products, respectively4,5. Adenylation is one of the most common first steps in carboxylic acid activation, and unsurprisingly, adenylate-forming enzymes have undergone numerous classifications6,7. The ANL (acyl-CoA synthetases, NRPS adenylation domains, and luciferase enzymes) superfamily of adenylating enzymes6 can also be grouped together with aminoacyl-tRNA synthetases and NRPS-independent siderophore enzymes into a larger adenylate-forming superfamily7. Recently, the discovery of BioW, a pimeloyl-CoA synthetase with a new structural fold, further expanded the diversity of the superfamily of adenylating enzymes8,9.
In all known cases of carboxylic acid activation by CoA, a single enzyme catalyzes two inseparable “half” reactions: adenylation and thioesterification (Fig. 1a)6,7. Due to the inherent reactivity of the acyl-adenylate intermediate, it appears to be logical and necessary that thioesterification occurs directly after adenylation within the same enzyme active site. Acyl-CoA synthetases, and the ANL family in general, achieve this bifunctionality through an elegant domain alternation strategy in which the C-terminal domain rotates ~140° to present two different faces to the active site (Fig. 1b)6. To facilitate this major conformational change, a hinge residue, typically Asp or Lys, in the A8 core motif (core sequences for ANL members were previously defined10) undergoes main chain torsion angle rotations6,11. The interactions of the N-terminal domain with each face of the dynamic C-terminal domain cause important changes to the active site resulting in half-reaction specificity.
Figure 1. Acyl-CoA ligases catalyze adenylation and thioesterification.
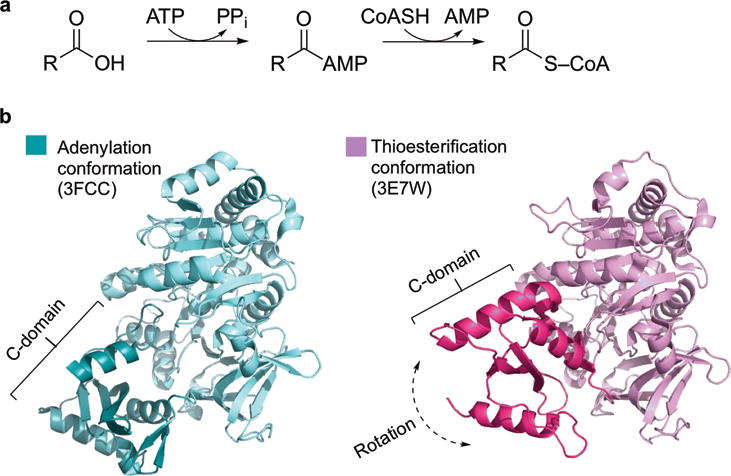
a, Acyl-CoA ligases convert carboxylic acids into CoA thioesters with two half-reactions, adenylation and thioesterification. b, Acyl-CoA ligases catalyze two half-reactions using a domain alternation mechanism. The C-terminal domain rotates to present two different faces to the active site, as exemplified by DltA in the adenylation conformation (3FCC27, teal) and thioesterification conformation (3E7W37, magenta). C-domains are shown in darker colors.
Platensimycin (PTM; 1) and platencin (PTN; 2), potent and selective inhibitors of bacterial and mammalian fatty acid synthases, have emerged as promising drug leads for both antibacterial and antidiabetic therapies12,13. Structurally, PTM and PTN are composed of two distinct scaffolds, a 3-amino-2,4-dihydroxybenzoic acid (ADHBA) and a diterpene-derived ketolide moiety, connected by a flexible propionamide chain (Fig. 2a). The propionamide linkers in PTM and PTN are the result of cleavage of the C-4–C-5 bonds (A-ring cleavage) in the ent-kauranol14 and ent-atiserene scaffolds15, respectively12. After A-ring cleavage, β-oxidation of the six-carbon linker, presumably in the form of an acyl-CoA and resulting in thiolytic loss of a propyl moiety, yields the characteristic ketolide scaffolds of PTM and PTN (Supplementary Fig. 1)16.
Figure 2. Structures of PTM, PTN, and congeners and metabolite profiles of selected S. platensis strains upon LC-MS analysis.
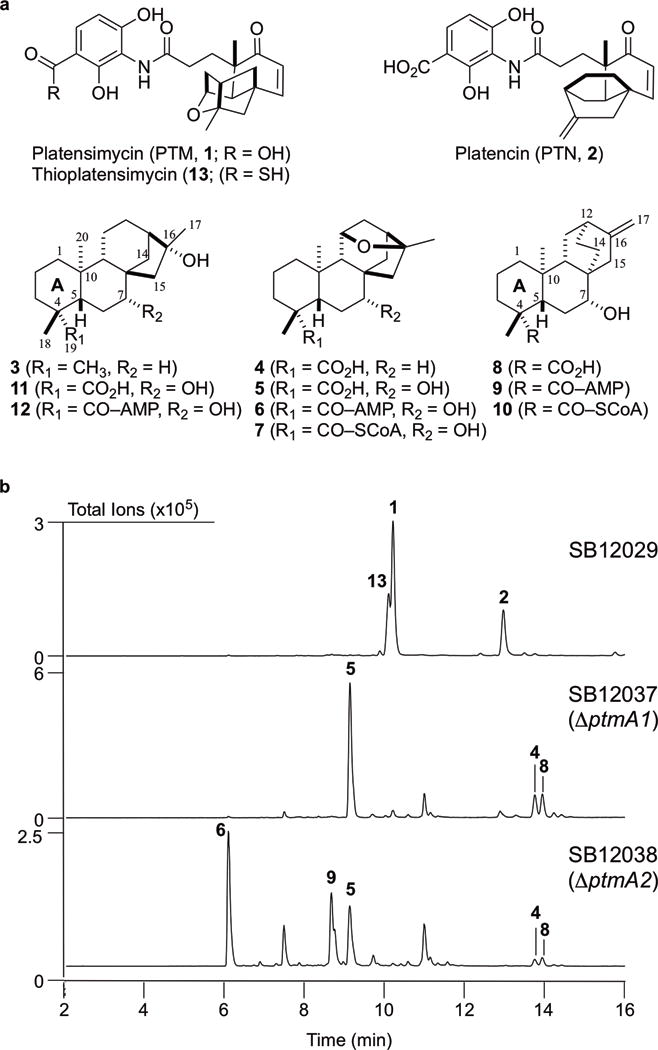
a, Structures of PTM (1), PTN (2), the intermediates isolated in this study (3–10), and previously isolated PTM congeners used for cocrystallography (11 and 12). Intermediates 3–6, 8, and 9 were isolated from the ΔptmA2 mutant S. platensis SB12038; 7 and 10 were isolated from PtmA2 enzymatic reactions; 11 and 12 were previously isolated from the ΔptmO5 mutant S. platensis SB1203614; thioPTM (13) was previously isolated from the dual PTM–PTN overproducer S. platensis SB1202923. b, Crude extracts were analyzed by total ion current (TIC) chromatograms. (16R)-ent-Kauran-16-ol (3) could not be detected by LC-MS. Metabolite profiles are representative of both small and large scale fermentations, each of which were independently performed n>3.
There is substantial evidence suggesting that the C-19 methyl groups of ent-kauranol and ent-atiserene are activated early in the biosynthesis of PTM and PTN (Supplementary Fig. 1). The isolation of (16R)-ent-kaurane-16,19-diol and (11S, 16R)-11,16-epoxy-ent-kauran-19-oic acid (4) from the ΔptmO4 mutant S. platensis SB12030 (ref. 16) and the ent-atiseren-19-oic acid derivatives platencin SL3 and SL4 from the heterologous PTN-producing Streptomyces lividans SB12600 (ref. 17) indicated that oxidation at C-19 occurs prior to A-ring cleavage in PTM and PTN biosynthesis (early oxidation at C-19 mirrors that of gibberellin biosynthesis18). In fact, platencin SL4 possessed an N-acetylcysteamine (S-NAC) moiety at C-19, mimicking CoA activation of the C-19 carboxylic acid17. Finally, two diterpenoid acyl-adenylates, platensimycin ML11 (12) and platencin ML3 (9), were isolated from the ΔptmO5 mutant S. platensis SB12036, supporting CoA activation at C-1914.
Here, we report the discovery of a non-adenylate-forming acyl-CoA ligase, PtmA2, and the natural functional separation of an acyl-CoA synthetase reaction. Two distinct acyl-CoA synthetases, PtmA1 and PtmA2, were determined to be necessary for C-19 activation in PTM and PTN biosynthesis. X-ray crystal structures of PtmA2 revealed that it resembles canonical ANL enzymes and retains its ability to alternate its C-terminal domain, but is missing key catalytic residues for the adenylation step. Rational mutagenesis proved effective in restoring the lost adenylation activity, albeit very inefficiently. The unprecedented functional separation of PtmA2 challenges the well-established model of adenylation enzymes and carboxylic acid activation and provides an opportunity to investigate functionally separated acyl-CoA ligases and their relevance in biology.
RESULTS
Three acyl-CoA ligases in the ptm and ptn gene clusters
We cloned and sequenced the ptm and ptn gene clusters from the PTM–PTN dual producer Streptomyces platensis MA7327 and the PTN producer Streptomyces platensis MA7339, respectively15. There are three distinct acyl-CoA synthetase-encoding genes in both the ptm and ptn gene clusters (Supplementary Fig. 2a). Given the hypothesis of CoA activation for β-oxidation processing (Supplementary Fig. 1), only one acyl-CoA synthetase appeared to be necessary, suggesting that the other two proteins are either unrelated to PTM and PTN biosynthesis, inactive, or involved in a cryptic CoA activation step. BLAST analysis19 of the three acyl-CoA synthetases, PtmA1, PtmA2, and PtmA3, revealed sequence similarities to the phenylacetate-CoA ligase (PaaK), bile acid-CoA ligase (BACL), and fatty acyl-CoA ligases (FadD) subfamilies, respectively. Phylogenetic analysis of PtmA1, PtmA2, and PtmA3 with selected members of the ANL superfamily confirmed these bioinformatics annotations. PtmA1 is the most divergent, showing alignment coverage of only 14% and 55% with PtmA2 and PtmA3, respectively. In comparison, PtmA2 and PtmA3 share 25% protein sequence identity and 38% similarity over 94% coverage (Supplementary Fig. 2b).
Inactivation of ptmA1 and ptmA2 in SB12029
We individually replaced the ptmA1 and ptmA2 genes in the dual PTM–PTN overproducer S. platensis SB1202916 with an apramycin resistance cassette20–22. Both mutants were confirmed by Southern analysis (Supplementary Tables 1-3, Supplementary Fig. 3). The ΔptmA1 and ΔptmA2 recombinant strains S. platensis SB12037 and SB12038, respectively, were then fermented under conditions known for PTM and PTN production, using SB12029 as a positive control (Fig. 2b). Both SB12037 and SB12038 lost production of both PTM and PTN, as well as the thioacid analogue thioplatensimycin23 (13) (Fig. 2b). Several new metabolites were detected from the crude extracts of SB12037 and SB12038, including three metabolites (4, 5, and 8) that were produced by both mutants (Fig. 2b).
A 2.4-L fermentation of SB12038 was performed to isolate the PTM- and PTN-related congeners. Six metabolites were isolated including four PTM congeners (3–6) and two PTN congeners (8 and 9, Fig. 2a), two of which are new (5 and 6) and four are known (3, 4, 8, and 9)14,24. All compounds were identified or determined based on a combination of high-resolution ESIMS (HRESIMS) and 1D (1H and 13C) and 2D NMR (Supplementary Note). The known compounds (16R)–ent-kauran-16-ol (3)14, (11S, 16R)-11, 16-epoxy-ent-kauran-19-oic acid (4)16, (7R)-7-hydroxy-ent-atiseren-19-oic acid (8)24, and the acyl-adenylate PTN ML3 (9)14 were consistent with literature reports. PTM ML 15 (5), a major metabolite in both SB12037 and SB12038, was determined to be the 7R-hydroxy congener of 4. PTM ML16 (6), found only in SB12038, was identified as the C-19 acyl-adenylate of 5 (Supplementary Note).
The identification of the diterpene free acids 4, 5, and 8 from the ΔptmA1 mutant SB12037 implicated PtmA1 as an acyl-CoA synthetase that activates the C-19 carboxylic acid on both the ent-kaurane and ent-atiserene scaffolds of PTM and PTN. The isolation of both the diterpene free acids 4, 5, and 8, and the acyl-adenylates 6 and 9 from the ΔptmA2 mutant SB12038 suggested that only thioesterification activity was disrupted when ptmA2 was inactivated. Our data supports that PtmA1 catalyzes only the first half reaction (i.e., adenylation of free acids) and PtmA2 catalyzes only the second half reaction (i.e., thioesterification of acyl-adenylates). Together, the discrete reactions of PtmA1 and PtmA2 complete the carboxylic acid activation of the diterpenoid intermediates in PTM and PTN biosynthesis (Fig. 3a).
Figure 3. Functional separation of CoA activation in PTM and PTN biosynthesis.
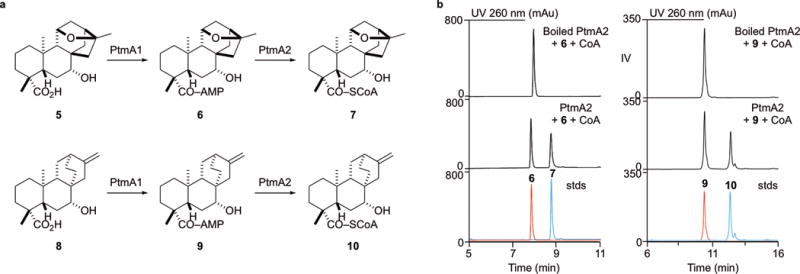
a, PtmA1 and PtmA2 independently catalyze adenylation and thioesterification, respectively, at the C-19 carboxylic acid of PTM and PTN intermediates. b, HPLC chromatograms of thioesterification reactions catalyzed by the non-adenylate-forming acyl-CoA ligase PtmA2. Shown are boiled and enzymatic reactions with PtmA2 and overlaid standards of 6, 7, 9, and 10 (stds). Chromatograms are representative examples of n>3 independent experiments.
PtmA2 is a non-adenylating acyl-CoA ligase
The one common feature among all acyl-CoA ligases, and all members of the superfamily of adenylating enzymes, is their ability to activate a carboxylic acid with ATP to form an acyl-adenylate intermediate6,7. Our in vivo results suggested that PtmA2 requires an acyl-adenylate substrate, instead of a free acid, for activation via CoA. To test this hypothesis, we cloned ptmA2 from S. platensis CB00739 (ref. 25) and heterologously produced PtmA2 in E. coli (Supplementary Fig. 4). When PtmA2 was incubated with free acid 5 in the presence of ATP and Mg2+ or ATP, Mg2+, and CoA, no acyl-adenylate or acyl-CoA products were formed (Supplementary Fig. 5). Incubation of PtmA2 with acyl-adenylate 6 and CoA revealed one new enzymatic product (7, Fig. 3b), which had an [M + H]+ ion at 1084.326, consistent with that of the acyl-CoA analogue of 5. PtmA2 was also capable of transforming the ent-atiserene-derived acyl-adenylate 9 into acyl-CoA 10 (Fig. 3b). Acyl-adenylates 6 and 9 do not convert into 7 or 10, respectively, in the presence of boiled PtmA2 (Fig. 3). Both 7 and 10 were enzymatically prepared, isolated, and characterized by HRESIMS and 1D and 2D NMR (Supplementary Note), confirming their identities. These results, in correlation with the in vivo results comparing the phenotypes of SB12037 and SB12038 with SB12029 as a control, demonstrate that PtmA2 is a non-adenylating acyl-CoA ligase. In spite of extensive effort, all attempts to produce recombinant PtmA1, however, yielded insoluble proteins, forfeiting complementary experiments to demonstrate PtmA1 as an adenylating enzyme in vitro.
PtmA2 equally activates PTM and PTN intermediates
After the sequenced ptm and ptn gene clusters revealed that the ptm biosynthetic gene cluster encodes the genes necessary for production of both PTM and PTN15, a unified biosynthetic pathway was proposed (Supplementary Fig. 1)12. A group of enzymes process the two distinct diterpene-derived scaffolds into the characteristic ketolides and couple them to ADHBA, affording PTM and PTN. Thus, each enzyme must be able to accommodate at least two different substrates. Our isolation of both the ent-kaurane (6) and ent-atiserene (9) acyl-adenylates offered us a unique opportunity to use PtmA2 as a model enzyme to test the unified biosynthesis proposal.
The steady-state kinetics of PtmA2 were evaluated under optimized conditions (Supplementary Fig. 6). The rates (kcat) and Michaelis constants (Km) of the acyl-adenylates and CoA were determined using a nonlinear fit of initial velocities versus substrate concentrations (Table 1, Supplementary Fig. 7). The values of kcat and Km were determined to be 22 s−1 and 1.5 μM for 6, and 21 s−1 and 1.0 μM for 9, respectively. Under saturating conditions of acyl-adenylates, the values of kcat and Km for CoA were 20 s−1 and 6.2 μM (using 6) and 19 s−1 and 2.5 μM (using 9), respectively. The kinetic values for both 6 and 9 suggest that PtmA2 is equally adept at activating two distinct scaffolds and supports a unified biosynthetic pathway for PTM and PTN biosynthesis (Supplementary Fig. 1). PtmA2 was also able to accept nonnative adenylate substrates as evidenced by its activation of the “open ether” adenylate 12 (Supplementary Fig. 8).
Table 1.
Enzyme kinetics of native PtmA2 and designed mutantsa
| Protein | Substrate | Km (μM) | Vmax (μmol s−1 × 10−2) | kcat (s−1) | kcat/Km (s−1 M−1) |
|---|---|---|---|---|---|
| Native | 6 | 1.5 ± 0.2 | 21.5 ± 0.4 | 22 | 1.5 × 107 |
| CoAb | 6.2 ± 0.6 | 9.7 ± 0.3 | 20 | 3.2 × 106 | |
| 9 | 1.0 ± 0.1 | 20.6 ± 0.3 | 21 | 2.1 × 107 | |
| CoAc | 2.5 ± 0.4 | 9.3 ± 0.3 | 19 | 7.6 × 106 | |
|
| |||||
| E416A | 6 | 0.42 ± 0.11 | 10.7 ± 0.3 | 0.21 | 5.0 × 105 |
| CoAb | 7.8 ± 1.2 | 8.9 ± 0.4 | 0.18 | 2.3 × 104 | |
|
| |||||
| A497K | 6 | 1.6 ± 0.2 | 8.9 ± 0.4 | 26 | 1.6 × 107 |
| CoAb | 6.8 ± 1.0 | 7.9 ± 0.4 | 32 | 4.7 × 106 | |
|
| |||||
| P-loop | 6 | 0.58 ± 0.12 | 5.1 ± 0.1 | 10 | 1.7 × 107 |
| CoAb | 70 ± 8 | 14.8 ± 0.4 | 7.4 | 1.1 × 105 | |
|
| |||||
| A497K/P-loop | 6 | 0.71± 0.11 | 14.3 ± 0.3 | 14 | 2.0 × 107 |
| CoAb | 69 ± 11 | 9.9 ± 0.4 | 10 | 1.4 × 105 | |
Values are the means of three independent assays and reported with standard deviations.
Kinetics determined with saturating concentration (100 μM) of 6.
Kinetics determined with saturating concentration (100 μM) of 9.
Structure of PtmA2 reveals cause for loss of function
Given the inability of PtmA2 to catalyze adenylation, we set out to establish the biochemical origin for this lack of activity. We hypothesized that PtmA2 is (i) unable to bind the diterpenoid free acid and/or ATP, (ii) missing catalytic residues responsible for adenylation, or (iii) locked in the thioester-forming conformation. To address these questions, we determined the crystal structures of unliganded PtmA2 (2.23 Å resolution, PDB entry 5E7Q), and in complex, separately, with free acid 11 (2.42 Å, 5UPT) and adenylates 6 (2.05 Å, 5UPS) and 12 (1.92 Å, 5UPQ). Data collection and refinement statistics are listed in Supplementary Table 4; omit maps of the active sites are shown in Supplementary Figure 9.
PtmA2 forms a two-domain architecture similar to other acyl-CoA synthetases6, consisting of a large (407 residues) N-terminal domain and a small (115 residues) C-terminal domain connected by the A8 motif (Fig. 4a and Supplementary Fig. 10). The N-terminal domain of PtmA2 forms an αβαβα structure with two β-sheets (six- or seven-stranded) flanking two α-helices and ends with a four-stranded antiparallel distorted β-sheet (Fig. 4a). The C-terminal domain consists of a central three-stranded distorted β-sheet surrounded by three small α-helices (Fig. 4a). All four structures were supported as dimeric enzymes with dimers in the asymmetric units or based on PISA analysis26. PtmA2 was determined to be a dimer in solution (Supplementary Fig. 11).
Figure 4. Overall structure and active sites of PtmA2 in both the adenylation and thioesterification conformations.
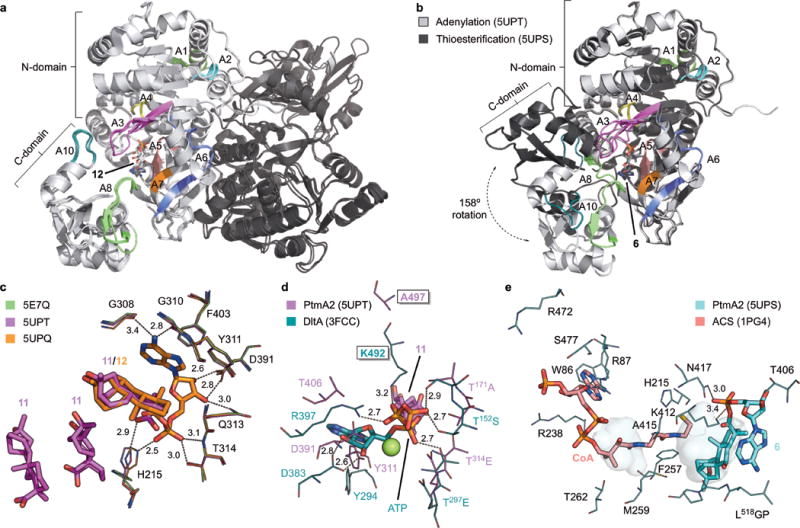
a, Superposition of the three structures of PtmA2 in the adenylation conformation (5E7Q, 5UPQ, and 5UPT). The dimer (monomers shown in light and dark gray) interface is along the length of both N-terminal domains. The core motifs are colored and labeled. Adenylate 12 is bound in the active site cavity. b, Monomers of the adenylation (5UPT, light gray) and thioesterification (5UPS, dark gray) conformations. The C-terminal domains diverge at the A8 hinge residue resulting in a 158° rotation. The core motifs are colored and labeled for the thioesterification conformation. Adenylate 6 is bound in the active site cavity. c, The adenylation active site in the apo form (5E7Q, green), in complex with three molecules of 11 (5UPT, magenta), and in complex with 12 (5UPQ, orange). d, The putative ATP binding site in PtmA2 (5UPT, magenta) overlaid with ATP, Mg2+ (green sphere), and residues from DltA (3FCC, teal)28. Ala497 (Lys492 in DltA) was proposed to be the residue that prevents adenylation from occurring. Free acid 11 is behind ATP. e, The putative CoA binding site in PtmA2 (5UPS, cyan) overlaid with CoA and interacting residues in ACS (1PG4, salmon)38. Without CoA bound, the pantetheine tunnel (shown as pockets) in PtmA2 is blocked by Phe257, Met259, and Ala415. All ligands are shown as sticks. Residues discussed in the text are depicted as lines. Dashes indicate residue–ligand interactions with distances in Å listed.
PtmA2 was found to be in the adenylation conformation in the unliganded structure, as well as when complexed with 11 or 12 (Fig. 4a). Only minor differences were observed between each of the structures in the adenylation conformation (root-mean-square deviation (rmsd) values of <0.423 Å between each structure). Conversely, PtmA2 was in the thioesterification conformation when in complex with 6 (Fig. 4b). While the N-terminal domain remains unchanged between the two conformations (rmsd value of 0.262 Å for the N-terminal domains of unliganded and in complex with 6), the C-terminal domain rotates ~158° (Fig. 4b, Supplementary Fig. 12). Thus, PtmA2 retains the canonical ability of acyl-CoA ligases to undergo domain alternation to provide two different faces to the active site.
In the adenylation conformation, the A3 (phosphate-binding loop or P-loop), A4, A5, A7, A8, and A10 motifs of PtmA2 form the walls of the active site cavity (Fig. 4a). Of the three structures in the adenylation conformation, there is only one difference in the active site between unliganded and liganded states. The imidazole ring of His215 (A4 motif) flips ~75° to face the C-19 carboxylic acid of 11 (2.9 Å) and 12 (3.8 Å) and the phosphate of 12 (2.5 Å). The “flipped out” His215 in unliganded PtmA2 mirrors that of the A4 aromatic residue in other acyl-CoA ligases when in the thioesterification conformation6.
Adenylate 12 sits in the cavity with its 20-carbon aliphatic cage surrounded by hydrophobic residues (Fig. 4c). The NH-ε2 of His215 (A4 motif) and both the side chain OH and main chain NH of Thr314 (A5) form hydrogen bonds with oxygens in the phosphate moiety. The side chains of Asp391 (A7) and Gln313 (A5) hydrogen bond with the 2´- and 3´-hydroxyl groups of ribose. Phe403 (A8) π-stacks with the adenine ring while the main chain carbonyls of Gly308 and Gly310 (A5) hydrogen bond with its 6-amino group. As opposed to members of the ANL superfamily27, PtmA2 does not possess a conserved loop that forms a planar backbone that stacks against the N-terminal face of the adenine ring. The folded orientation of adenylates 6 and 12 suggests that the hydrophobic diterpenoid moiety intramolecularly stacks with the adenine ring (Fig. 4c). Three molecules of “open ether” free acid 11 bind in the active site (Fig. 4c). One molecule of 11 superposes with the diterpenoid moiety of 12; the other two molecules of 11 are each positioned ~4 Å away from one another, lying in the pocket opposite from where the adenine ring of 12 is bound. The superposable molecule of 11 can easily be imagined as the likely binding orientation for adenylation, suggesting that an inability to bind free acid does not prevent PtmA2 from catalyzing adenylation.
We did not obtain diffraction quality crystals of PtmA2 in the presence of ATP and are therefore unable to determine if and how ATP binds in the adenylation conformation. Comparison of PtmA2 with the d-alanyl carrier protein ligase DltA from Bacillus cereus in complex with ATP (3FCC, 4.2 Å rmsd)28 revealed that PtmA2 retains most of the residues that bind ATP and the divalent cation (Fig. 4d). In PtmA2, the T171A motif in the P-loop, Y311GxTE motif in A5, Asp391 in A7, and Thr406 in A8 can easily be envisaged as making interactions with ATP and Mg2+. The universally conserved Lys492 in DltA, which typically binds the ribose moiety but is also near the β-phosphate6, is replaced with Ala497 in PtmA2 (Fig. 4d).
In the thioesterification conformation, the A10 and A8 motifs are rotated away from and into the active site, respectively, to form a new face opposite of the N-terminal domain (Fig. 4b). Adenylate 6 assumes the same position as adenylate 12 in the adenylation conformation (Fig. 4e). Accordingly, the hydrophobic pocket, phosphate-binding Thr314, ribose-binding Gln313 and Asp391, and adenine-binding Phe403, Gly308, and Gly310 are essentially unchanged between the two conformations. The major differences surrounding the adenylate substrate are that His215 is “flipped out” and the presence of residues from the A8 motif (Fig. 4e). The carboxylic acid of Glu416 forms a hydrogen bond with the imidazole of His215 to facilitate removing His215 from the pantetheine tunnel. The side chains of Thr406, Met410, Lys412, and Asn417 are positioned near the phosphate and ribose moieties, with Lys412 and Asn417 hydrogen-bonding the phosphate oxygens. A C-terminal loop of L518GP also abuts the adenine ring and diterpenoid moiety.
The CoA-binding pocket of acyl-CoA ligases typically consists of a solvent-accessible nucleotide binding site and a pantetheine tunnel allowing the free thiol access to the adenylate substrate6. This pocket is formed at the interface of the N- and C-terminal domains with residues from the rotated A8 motif making contact with the pantetheine arm. Across the superfamily, residues for CoA binding are not as conserved as those that bind ATP or adenylate, but trends are present: The pantetheine tunnel is moderately hydrophobic, aromatic residues stack with the adenine ring, and positively charged side chains bind the phosphate groups. In PtmA2, the pantetheine tunnel is blocked by Phe257, Met259, and Ala415 (Fig. 4e). On the surface of the protein, where the nucleotide likely binds, lies Trp86 and several charged residues, which likely bind the adenine ring and CoA phosphates, respectively. His215 is positioned near the putative site of the CoA thiol group and may contribute a positive dipole to reduce the pKa of the thiol6,29, suggesting its importance in thioesterification.
Mutagenesis restores the adenylation activity of PtmA2
Structural analysis supported that PtmA2 interconverts between the adenylation and thioesterification conformations (Fig. 4b), binds both diterpenoid free acids and acyl-adenylates, and has an ATP- and Mg2+-binding pocket that retains most of the canonical interactions found in the ANL superfamily. The most conspicuous difference between the ANL superfamily and the non-adenylating PtmA2 based on structure (Fig. 4d) and primary sequence alignment (Fig. 5a) is the lack of a Lys in the A10 motif (Ala497 in PtmA2). This universally conserved Lys is essential for adenylation and was proposed to track the negative charge through the first half reaction6,30. Other differences highlighted by sequence alignment is that the typically Ser/Thr-rich P-loop and A8 hinge region are quite different in PtmA2 (Fig. 5a), although the latter does not seem to negatively affect changes in conformation.
Figure 5. Reconstitution of the lost adenylation activity of PtmA2 via site-directed mutagenesis.
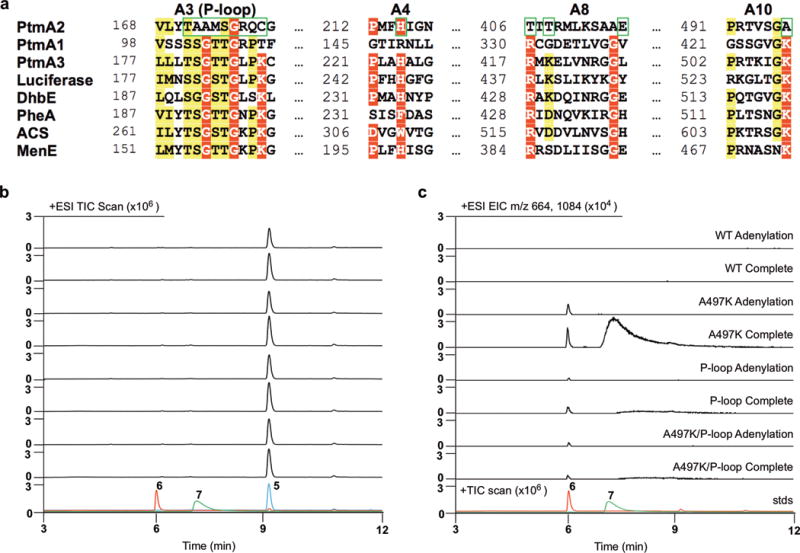
a, Sequence alignment of PtmA2 and selected acyl-CoA synthetases. A3, A4, A8, and A10 motifs are shown with mutated residues highlighted with green boxes. See Supplementary Figure 10 for full sequence alignment. b,c, Total ion chromatograms (TICs; b) and extracted ion chromatograms (EICs; c) of PtmA2 mutant enzyme reactions. Shown are adenylation and complete (both adenylation and thioesterification) reactions with native and mutant PtmA2, and overlaid standards of 5, 6, and 7 (stds). See Supplementary Figures 14, 15, and 17 for reactions of other PtmA2 mutants, using free acid 8, and negative controls using boiled PtmA2. Chromatograms are representative examples of n>3 independent experiments.
We constructed a series of site-directed mutants of PtmA2 to investigate the thioesterification half reaction and attempt to restore the adenylation half reaction. His215 and Glu416, which are implicated in the thioesterification reaction6, were both mutated to Ala. All mutants except H215A were able to catalyze the thioesterification of adenylate 6 (Supplementary Fig. 13). The lack of detectable product by H215A supports its catalytic role in thioesterification. The E416A mutant showed a 3-fold decrease in the Km of 6 despite not directly contacting 6, and an ~100-fold decrease in kcat/Km, supporting its noncatalytic role in orienting His215 (Table 1)6.
We hypothesized that an A497K mutant, a P-loop exchange, a A497K/P-loop double mutant, or a T406R/T408K (A8 hinge region) mutant may restore the lost adenylation activity of PtmA2. The P-loop of PtmA2 (T171AAMSGRQC) was replaced with a consensus P-loop sequence from PtmA1 and other members of the ANL superfamily, resulting in a new P-loop of T171SGTSGRPK. Thioesterification was not substantially impacted by any of these mutations; only the mutants with the exchanged P-loop showed 10-fold increases in the Km of CoA (Table 1). It is unclear why changes to the P-loop, which is not in spatial proximity to the CoA binding site, causes this change in Km. All mutants were tested for adenylation activity alone or both partial reactions with diterpenoid free acid 5 (Fig. 5b, c; Supplementary Fig. 14). As expected, native PtmA2, H215A, and E416A did not form any detectable amount of adenylate 6 or CoA thioester 7; the T406R/T408K mutant also did not restore adenylation activity (Supplementary Fig. 14). Importantly, A497K, the P-loop mutant, and the A497K/P-loop double mutant all showed formation of both adenylate 6 and CoA thioester 7 (Fig. 5b,c); free acid 8 was also converted into adenylate 9 or CoA thioester 10 (Supplementary Fig. 15). Adenylation and thioesterification is most evident in the A497K mutant. As the rate of thioesterification did not change in the A497K mutant (Table 1), the minute amount of 6 and 7 generated from 5 is presumably due inefficient adenylation. Since both the A497K or the P-loop mutants alone restore adenylation activity, but the A497K/P-loop double mutant does not show a cumulative effect, it is clear that the evolution of PtmA2 into a non-adenylating acyl-CoA ligase altered more than just these two sites.
DISCUSSION
PtmA2 represents a new class of non-adenylating acyl-CoA ligases. At first glance, PtmA2 appeared to be a member of the canonical ANL superfamily of adenylating enzymes based on bioinformatics, sequence, and structure (Supplementary Fig. 2b). However, the accumulation of adenylates after in vivo gene inactivation of ptmA2 in S. platensis SB12029 hinted that PtmA2 may not catalyze the first half-reaction. Detailed examination of the sequence and structure of PtmA2 revealed only minor differences in the active site of PtmA2; the most obvious difference was the absence of a universally conserved Lys that is only involved in the adenylation half-reaction. Native PtmA2 was confirmed to only catalyze thioesterification and is the first reported non-adenylating acyl-CoA ligase. Mutation of Ala497 to Lys restored the ability of PtmA2 to catalyze both the adenylation reaction and the complete conversion of free carboxylic acid to acyl-CoA, albeit very inefficiently. The discovery and functional and structural characterization of PtmA2 challenge the established paradigm that acyl-CoA ligases are all capable of performing the adenylation half-reaction.
A BLAST search19 revealed that PtmA2-like non-adenylating acyl-CoA ligases are widespread in nature. Excluding the known and putative PTM–PTN dual-producing strains, the top 20 hits all possess core motifs nearly identical to those in PtmA2 (Supplementary Fig. 16, overall sequence identities ≥67%). Notably, all of these PtmA2-like proteins have either Ala or Gly in the conserved Lys position, implying their inability to catalyze adenylation. It is unclear if these enzymes are involved in primary or secondary metabolism, but a separate enzyme that catalyzes the initial adenylation reaction is required. Indeed, many of the strains with the PtmA2 homologues contain PtmA1-like counterparts in genetic proximity. This study inspires future investigations of functionally separated acyl-CoA ligases, as well as other multifunctional enzymes, and their relevance in biology.
Functional inactivation of acyl-CoA ligases in nature is not unprecedented. The adenylation activity of acetyl-CoA synthetases from both prokaryotes and eukaryotes is modulated via the posttranslational acetylation of the catalytic Lys in the A10 motif31,32. Reversible acetylation allows an organism to control when adenylation occurs, while not impacting thioesterification. The fatty acyl-AMP ligases (FAALs), a subfamily of ANL enzymes, lost their ability to catalyze thioesterification with CoA; instead, they transfer the activated adenylate directly to the ACP of polyketide synthases33. Although these FAALs retained the CoA binding pocket, a unique insertion motif prohibits proper domain alternation and therefore CoA binding34–36. Rational mutagenesis of FAALs resulted in the conversion of FAALs to canonical fatty acid CoA ligases, and vice versa34,36.
The evolution of PtmA2 into a CoA-forming-specific enzyme is fundamentally different. Acetylation is reversible regulation and enzymes such as FAALs evolved to utilize fatty acyl-adenylates in other biosynthetic pathways. PtmA2 still requires its substrate to be activated via adenylation, but has apparently lost its own ability to adenylate diterpenoid free acids. Thus, PtmA2 requires the presence of an adenylation-only ligase. Together, PtmA1 and PtmA2 constitute the functional equivalent of a canonical acyl-CoA ligase. The idea that nature evolved two distinct enzymes to catalyze half-reactions when one enzyme is capable of performing both is counterintuitive. Two enzymes, instead of one, may create an additional layer of regulation, provide improved substrate selectivity, or enhance catalytic efficiency. It is logical to assume that the evolutionary ancestor of PtmA2 possessed the ability to catalyze both half-reactions, considering that a single point mutation, K497A, would render adenylation ineffective. It is also clear based on phylogenetic analysis that PtmA1 (PaaK-like ligase) and PtmA2 (BACL-like ligase) are not the result of recent gene duplication (i.e., occurring after the ptm and ptn gene clusters formed). Thus, PtmA1 and PtmA2 provide a model system to study the evolution of two discrete enzymes that, together, constitute a functional acyl-CoA synthetase.
ONLINE METHODS
General experimental procedures
All 1H, 13C, and 2D NMR (1H-1H COSY, 1H-13C HSQC, 1H-13C HMBC, 1H-1H ROESY) experiments were run on a Bruker Avance III Ultrashield 700 at 700 MHz for 1H and 175 MHz for 13C nuclei. Preparative HPLC was performed on an Agilent 1260 Prep Infinity LC with an MWD detector equipped with an Agilent Eclipse XDB-C18 column (250 mm × 21.2 mm, 7 μm). LC-MS was performed on an Agilent 1260 Infinity LC coupled to a 6230 TOF (HRESI) equipped with an Agilent Poroshell 120 EC-C18 column (50 mm × 4.6 mm, 2.7 μm) with a constant temperature of 40 °C. Analytical HPLC was performed on an Agilent 1260 Infinity LC with a DAD detector equipped with an Agilent Poroshell 120 EC-C18 column (50 mm × 4.6 mm, 2.7 μm) with a constant temperature of 35 °C. Optical rotations were obtained using an AUTOPOL IV automatic polarimeter (Rudolph Research Analytical). UV was measured with a NanoDrop 2000C spectrophotometer (Thermo Scientific). IR spectra were attained using a Spectrum One FT-IR spectrophotometer (PerkinElmer).
Bacterial strains, plasmids, and chemicals
Strains, plasmids, and PCR primers used in this study are listed in Supplementary Tables 1–3, respectively. PCR primers were obtained from Sigma-Aldrich. Enzymes for cloning were purchased from NEB and used following the protocols provided by the manufacturer. DNA gel extraction and plasmid preparation kits were purchased from Omega Bio-Tek. DNA sequencing was conducted by Eton Bioscience. The REDIRECT Technology kit for PCR-targeting gene replacement was provided by the John Innes Center (Norwich, U.K.)20. Escherichia coli ET12567/pUZ800222 was used as the host for intergeneric conjugation with S. platensis SB1202916. Other chemicals, biochemical, and media components were purchased from standard commercial sources.
Culture conditions
E. coli strains harboring plasmids or cosmids were grown in lysogeny broth (LB) with appropriate antibiotic selection21. Streptomyces strains were grown on solid ISP4 medium at 28 °C or cultured in liquid tryptic soy broth (TSB) at 28 °C and 250 rpm, with appropriate antibiotic selection, if needed21. E. coli–Streptomyces conjugations were plated onto ISP4 medium supplemented with 10 mM MgCl2. Fermentation of S. platensis recombinant strains were conducted as described previously14,16. Briefly, fresh spores of Streptomyces strains were inoculated into TSB seed medium and cultured for 2 d. For metabolite production, strains were cultured for 7 d in liquid medium for PTM production, which contains 70 g L−1 soluble starch, 15 g L−1 soybean flour, 5 g L−1 MOPS, 5 g L−1 CaCO3, 15 g L−1 MnCl2·4H2O, and 30 mg L−1 (NH4)6Mo7O24·4H2O, pH 7.339. The fermentation medium was inoculated with 4% (v/v) seed culture and 3% (w/v) Amberlite XAD-16 resin (Sigma-Aldrich).
Inactivation of ptmA1 and ptmA2 in SB12029 affording the ΔptmA1 and ΔptmA2 mutants SB12037 and SB12038, respectively
Gene inactivations of ptmA1 and ptmA2 in S. platensis SB12029 were carried out as previously described14,16,25. Briefly, ptmA1 and ptmA2 were replaced, separately, with the aac(3)IV + oriT cassette from pIJ773 using λRED-mediated PCR-targeting gene replacement in E. coli BW25113/pIJ79020 harboring pBS12064, a cosmid containing a portion of the ptm gene cluster. The genotypes of the resultant ΔptmA1 and ΔptmA2 mutant cosmids, pBS12065 and pBS12066, respectively, were confirmed by PCR, transformed into E. coli ET12567/pUZ800222, and introduced into S. platensis SB12029 by intergeneric conjugation. Double crossover homologous recombination between SB12029 and pBS12065 or pBS12066 resulted in the isolation of apramycin-resistant and kanamycin-sensitive mutants SB12037 (ΔptmA1) and SB12038 (ΔptmA2), respectively. The genotypes of SB12037 and SB12038 were confirmed by Southern analyses.
Identification of metabolites from SB12037 (ΔptmA1) and SB12038 (ΔptmA2)
For small-scale fermentations of SB12037 and SB12038, extraction of natural products from resin followed previously reported procedures14,16. Briefly, after harvesting the resin from the fermentation broth, the resin was washed with water and extracted with CH3OH three times. The CH3OH extract was used directly for LC-MS analysis. LC-MS chromatography was conducted using a flow rate of 0.4 mL min−1 and an 18 min solvent gradient from 5–100% CH3CN in H2O containing 0.1% formic acid.
Isolation and structural elucidation of PTM and PTN biosynthetic intermediates/congeners (3–6, 8, and 9) from SB12038
For a large-scale fermentation (2.4 L) of SB12038, six 2.0 L baffled flasks, each containing 400 mL of production medium with 4% (v/v) seed culture and 20 g of Amberlite XAD-16 resin, were incubated for 7 d. The harvested and washed resin was exhaustively extracted with acetone. Acetone was removed in vacuo and the resulting red gum (6.15 g) was subjected to vacuum flash chromatography (VFC) over silica gel (65 × 50 mm, 66.0 g), yielding nine fractions (nf1–nf9) by sequential elutions with 250 mL each of hexanes (nf1, 8.1 mg dry weight), hexanes-EtOAc (7:3, v/v; nf2, 643.3 mg), hexanes-EtOAc (1:1, v/v; nf3, 281.1 mg), EtOAc (nf4, 489.6 mg), EtOAc-MeOH (8:2, v/v; nf5, 654.4 mg), EtOAc-MeOH (7:3, v/v; nf6, 180.0 mg), EtOAc-MeOH (6:4, v/v; nf7, 172.8 mg), EtOAc-MeOH (5:5, v/v; nf8, 547.7 mg), and MeOH (nf9, 673.4 mg). Fraction nf2 was further purified using VFC over a silica gel column (43 × 50 mm, 28.0 g) and eluted with hexanes-EtOAc (9:1, v/v) to yield compounds 3 (109.7 mg) and 4 (110.0 mg). Compound 5 easily precipitated from a methanolic solution of nf5 and was purified by washing the centrifuged precipitate with methanol three times, yielding pure 5 (89.2 mg). Compounds 6 and 9 were purified from nf8 by preparative HPLC using a 20 min elution system of 5–50% CH3CN in H2O containing 0.1% formic acid at a flow rate of 20 mL min−1 with UV detection at 260 nm. Compound 6 (27.6 mg) and 9 (12.5 mg) eluted at Rt 10.7 min and 16.4 min, respectively. A portion (231.0 mg) of nf4 was separated over a silica RediSep Rf flash chromatography column (4 g, Teledyne Isco) using hexanes-acetone (8:2, v/v) as eluent to yield compound 8 (5.4 mg).
Compounds 3, 4, 8, and 9 were determined to be the known diterpenoids (16R)-ent-kauran-16-ol, (11S, 16S)-11, 16-epoxy-ent-kauran-19-oic acid, (7R)-7-hydroxy-ent-atiseren-19-oic acid, and platencin ML3, respectively, based on 1H and 13C NMR and HRESIMS analysis (Supplementary Note).
Gene cloning
For enzyme assays, ptmA2 from S. platensis CB00739 (NCBI accession, ptm gene cluster KJ189771; PtmA2 protein AIW55550) was amplified by PCR from genomic DNA with Q5 DNA polymerase (NEB) following the protocol produced by the manufacturer using the 739A2_F and 739A2_R primers (Supplementary Table 3). The PCR product was purified, treated with T4 polymerase, and cloned into pBS3080 according to ligation-independent procedures to afford pBS12067. For site-directed mutagenesis of ptmA2, the ptmA2 gene from pBS12067 was amplified in two steps by primer extension using the 739A2_F and 739A2_R primers with internal primers containing the desired mutation(s) (Supplementary Table 3). The mutant ptmA2 genes were then cloned into pBS3080 as described above yielding pBS12068–pBS12073.
For protein crystallography, ptmA2 was amplified with KOD Hot Start DNA polymerase (Novagen) in amplification buffer supplemented with betaine to a final concentration of 2.5 M using the 739xtalA2_F and 739xtalA2_R primers (Supplementary Table 3). The PCR product was treated and cloned into pMCSG68 as described above to afford pBS12074.
Gene expression and protein production and purification
For enzyme activity assays, pBS12067 was transformed into E. coli BL21(DE3) (Life Technologies) and grown in 1 L of lysogeny broth (LB) at 37 °C with shaking at 250 rpm until an OD600 of 0.6 was reached. The culture was cooled to 4 °C, gene expression was induced with the addition of 0.25 mM isopropyl β-d-1-thiogalactopyranoside (IPTG), and the cells were grown overnight at 18 °C with shaking. After harvesting the cells by centrifugation at 4000 g for 15 min at 4 °C, the pellet was resuspended in lysis buffer (100 mM Tris, pH 8.0, containing 300 mM NaCl, 15 mM imidazole, and 10% glycerol), lysed by sonication, and centrifuged at 15,000 g for 20 min at 4 °C. The supernatant containing PtmA2 was purified by nickel-affinity chromatography using an ÄKTA FPLC system (GE Healthcare Biosciences) equipped with a HisTrap column. The resultant protein with an N-terminal His6-tag was desalted using a HiPrep desalting column (GE Healthcare Biosciences) in 50 mM Tris buffer, pH 7.8, containing 100 mM NaCl, 50 mM KCl, and 5% glycerol and concentrated using an Amicon Ultra-15 concentrator (Millipore). Protein concentrations were determined from the absorbance at 280 nm using a molar absorptivity constant (ε280 = 97,860 M−1·cm−1). Each of the PtmA2 mutants was produced and purified as described for native PtmA2.
For protein crystallization, pBS12074 was transformed into E. coli BL21(DE3)-Gold (Stratagene) and grown in 1 L of enriched M9 medium40 at 37 °C with shaking at 200 rpm until an OD600 of 1.0 was reached. Methionine biosynthetic inhibitory amino acids (25 mg·L−1 each of l-valine, l-isoleucine, l-leucine, l-lysine, l-threonine, l-phenylalanine) and 90 mg·L−1 of l-selenomethionine (SeMet, Orion Enterprises) were added to the culture, which was then cooled to 4 °C for 60 min. Gene expression was induced with 0.5 mM IPTG and the cells were grown overnight at 18 °C with shaking. After harvesting the cells by centrifugation at 4500 rpm for 25 min at 4 °C, the pellet was resuspended in lysis buffer (50 mM HEPES, pH 8.0, containing 500 mM NaCl, 20 mM imidazole, 10 mM β-mercaptoethanol, and 5% glycerol), lysed, and purified using Ni-NTA Immobilized Metal Affinity Chromatography (IMAC 1) and the ÄKTAxpress system (GE Healthcare Biosciences). The N-terminal His6-tag was then cleaved from purified PtmA2 using recombinant His6-tagged TEV protease. After an additional IMAC step (IMAC 2) to remove the protease, affinity tag, and uncut PtmA2, the resultant cut PtmA2 was concentrated using an Amicon Ultra-15 concentrator (Millipore) in 20 mM HEPES, pH 8.0, containing 250 mM NaCl, and 2 mM DTT.
Enzymatic activity of PtmA2
Preliminary incubations were performed in 50 mM Tris, pH 7.8, containing 50 mM MgCl2, 1 mM diterpenoid free acid or adenylate substrates, 1 mM ATP (for free acids only), 0.5 mM CoA, and 1 μM PtmA2 in a total volume of 50 μL. After incubation at 25 °C for 3 h, 50 μL of methanol was added to quench the reaction. The reaction mixture was then centrifuged at 10,000 g for 10 min at 4 °C and the supernatant was injected and analyzed by LC-MS and analytical HPLC with UV detection at 260 nm. LC-MS was conducted using a flow rate of 0.4 mL min−1 and an 18 min solvent gradient from 5–100% CH3CN in H2O containing 0.1% formic acid. HPLC was conducted using a flow rate of 1 mL min−1 and a 6 min solvent gradient from 5–40% CH3CN in 0.2 M ammonium acetate at 35 °C. The reaction conditions for PtmA2 were optimized by monitoring CoA ester production using the HPLC method. Buffers (citrate, phosphate, Tris, borate), pH (5.0–10.0), and EDTA (at 10 mM) were tested for their effects on the activity of PtmA2.
Enzymatic synthesis of CoA esters (7 and 10)
To a 4-mL reaction mixture of 50 mM Tris buffer, pH 7.8, containing 5 μM of PtmA2 and 3.8 mM of CoA (11.7 mg), adenylates 6 (6.6 mg, 2.5 mM) or 9 (4.3 mg, 1.67 mM) were added and incubated overnight at 25 °C. The reaction mixtures were then purified by preparative HPLC using a 20 min elution system of 5–50% CH3CN in H2O containing 0.1% formic acid at a flow rate of 20 mL min−1 with UV detection at 260 nm. Platensimycin ML17 (7, 7.6 mg, 71%), the CoA ester of 5, and platencin ML4 (10, 2.3 mg, 32%), the CoA ester of 8, eluted at Rt = 12.2 and 16.9 min, respectively.
Kinetic studies of PtmA2
Although reaction condition optimization suggested pH 9.0 was optimal, poor stability of PtmA2 required all kinetic assays to be performed in Tris buffer, pH 7.8. Concentrations of native PtmA2 or mutants, diterpenoid adenylates, and CoA varied for each reaction depending on enzyme rate and substrate Km. Each 50 μL reaction was incubated at 25 °C for 10 min and quenched with 50 μL of CH3OH. After centrifugation, the reaction mixtures with 7 were analyzed by HPLC as described above and the integrated area under curve (AUC) at 260 nm was calculated. The reaction mixtures with 10 were analyzed by HPLC using a 6 min solvent gradient from 20–55% CH3CN in 0.2 M ammonium acetate. A standard curve of CoA product was used to convert AUC into the amount of product formed. Each kinetic assay was performed in triplicate.
Analytical size-exclusion chromatography
The molecular weight (MW) and monomeric state of PtmA2 in solution was determined by size-exclusion chromatography using a HiLoad 16/600 (16 × 600 mm) column (GE Healthcare) connected to a GE Healthcare ÄKTA pure HPLC system. The column was pre-equilibrated with two column volumes of 50 mM Tris, pH 8.0, containing 300 mM NaCl, and calibrated with ribonuclease A (13.7 kDa), ovalbumin (44 kDa), conalbumin (75 kDa), aldolase (158 kDa), and ferritin (440 kDa). The chromatography was carried out at 4 °C at a flow rate of 1 mL·min−1. Data analysis was performed using the Unicorn 7.0.2 software (GE Healthcare).
Protein crystallization
SeMet-labeled PtmA2 was screened for crystallization conditions using a Mosquito liquid dispenser (TTP Labtech) and a sitting drop vapor diffusion technique in 96-well CrystalQuick plates (Greiner Bio-one). The protein was screened against the MCSG 1–4 screens (Microlytic) at 16 °C. For each condition, 0.4 μL of protein [36 mg·mL−1 for unliganded, in complex with platensimycin ML9 (11)14, and in complex with platensimycin ML11 (12)14; 25 mg·mL−1 in complex with 6] and 0.4 μL of crystallization formulation were mixed and then equilibrated against 140 μL of the reservoir solution. Crystals of unliganded PtmA2 appeared under a number of conditions; the best crystals were harvested from 0.1 M MES buffer, pH 6.5 containing 1.6 M magnesium sulfate appeared after two months and were used for structure solution at a resolution of 2.23 Å. Crystals of PtmA2 in complex with 12 (3 mM) were harvested from 0.1 M bis-tris propane buffer, pH 7.0 containing 1.5 M lithium chloride and used for structure solution at a resolution of 2.42 Å. Crystals of PtmA2 in complex with 6 (2.2 mM) were harvested from 0.2 M sodium formate, pH 7.2 containing 20% PEG 3350 and used for structure solution at a resolution of 2.05 Å. Crystals of PtmA2 in complex with 11 (3 mM) were harvested from 0.1 M bis-tris propane buffer, pH 7.0 containing 1.5 M lithium sulfate and 0.01 M hexamine cobalt (III) chloride and used for structure solution at a resolution of 1.92 Å. Crystals selected for data collection were soaked in the crystallization buffer supplemented with either 20% of ethylene glycol (for 6) or 25% glycerol (for unliganded, 11, and 12), and flash-frozen in liquid nitrogen.
Data collection, structure determination, and refinement
Single-wavelength X-ray diffraction data were collected at the peak wavelength of 0.9792 Å at 100 K temperature at the 19BM and 19ID beamline of the Structural Biology Center at the Advanced Photon Source at Argonne National Laboratory using the program SBCcollect. The intensities were integrated and scaled with the HKL-3000 suite41. The structure of unliganded PtmA2 was determined by single-wavelength anomalous dispersion (SAD) phasing using the AutoSol/AutoBuild phasing pipeline42 from the PHENIX suite43. The structures of liganded PtmA2 were determined by molecular replacement using the HKL-3000 suite41 incorporating MOLREP44 from the CCP4 suite45 and using the unliganded structure as the starting model. Several rounds of manual adjustments of structure models and refinements using Coot and Refmac from the CCP4 suite45 were done. The stereochemistry of the structures were validated using the PHENIX suite43, incorporating MolProbity tools46. The Ramachandran favored/allowed regions (%) for the backbone dihedral angles in the final models of 5E7Q, 5UPQ, 5UPS, and 5UPT are 97.9/100, 95.0/99.9, 97.8/100, and 97.9/100, respectively. A summary of data collection and refinement statistics is given in Supplementary Table 4. Figures were prepared using PyMOL (Schrödinger, LLC).
Phylogenetic Analysis
Protein sequences of acyl-CoA synthetases and homologues were obtained from the NCBI database: PtmA1 (Streptomyces platensis CB00739; AIW55546), PtmA2 (S. platensis CB00739; AIW55550), PtmA3 (S. platensis CB00739; AIW55578), PtmA1 homologue (Streptomyces cellulosae; WP_030660917), PtmA1 homologue (Streptomyces ciscaucasicus; WP_062039468), PtmA1 homologue (Streptomyces mirabilis; WP_037709869), PtmA2 homologue (Streptomyces canus; WP_020120457), PtmA2 homologue (Streptomyces ciscaucasicus; WP_062039429), PtmA2 homologue (Streptomyces sp. Root369; WP_057612788), PtmA3 homologue (Streptomyces viridochromogenes DSM40736; EFL35407), acetyl-CoA synthetase (ACS; Salmonella enterica; Q8ZKF6), bile acid-CoA ligase (BACL; Clostridium scindens; P19409), DhbE (Bacillus subtilis; AAN15214), fatty acyl-AMP ligase 28 (FAAL28; Mycobacterium tuberculosis H37Rv; P9WQ59), FadD10 (M. tuberculosis H37Rv; AIR12820), firefly luciferase (Photinus pyralis; BAF48390), MenE (B. subtilis; P23971), PaaK1 (Burkholderia cenocepacia J2315; CAR50715), PheA (Brevibacillus brevis; AAA58718, residues 1–556). The protein sequences were aligned using Clustal Omega47 and the phylogenetic tree was generated by MEGA 5.2 using the JTT method and the maximum likelihood algorithm48.
Data Availability
Protein Data Bank: Atomic coordinates and structure factor amplitudes for PtmA2 crystal structures were deposited with accession codes 5E7Q, 5UPQ, 5UPS, and 5UPT. All other data generated or analyzed in this study are available within the article and Supplementary Information files. A Life Sciences Reporting Summary for this paper is available.
Supplementary Material
Acknowledgments
This work is supported in part by the National Institute of General Medical Sciences Protein Structure Initiative Grants GM094585 (A. Joachimiak) and GM098248 (G. Phillips), and National Institutes of Health Grants GM109456 (G. Phillips) and GM114353 (B. Shen). The use of Structural Biology Center beamlines at the Advanced Photon Source was supported by U.S. Department of Energy, Office of Biological and Environmental Research grant DE-AC02-06CH11357 (A. Joachimiak). N. Wang is supported in part by the Institute of Applied Ecology, Chinese Academy of Sciences, and a scholarship from the Chinese Scholarship Council (201504910034). J. Rudolf is supported in part by an Arnold O. Beckman Postdoctoral Fellowship. C.-Y. Chang is supported in part by the Fellowship of Academia Sinica–The Scripps Research Institute Postdoctoral Talent Development Program. This is manuscript #29600 from The Scripps Research Institute.
Footnotes
Author contributions. B.S., G.N.P., and A.J. conceived the project; N.W., J.D.R., L.-B.D., G.N.P. and B.S. designed the experiments; N.W. and J.D.R. performed bioinformatics, molecular cloning, protein production and purification, biochemical analysis, and enzyme reactions; J.D.R. constructed the genetic knockouts in Streptomyces; N.W., J.D.R., and L.-B.D. conducted Streptomyces fermentation; N.W. and L.-B.D. performed natural product isolation and structure determination; C.H.-S. and M.E. performed crystallography; J.O. conducted protein structure determination; N.W., J.D.R., L.-B.D., J.O., C.H.-S., M.E., C.-Y.C., G.B., and B.S. analyzed the results; N.W. and J.D.R. wrote the first draft with inputs from all co-authors; J.D.R. and B.S. revised and finalized the manuscript.
Competing financial interests. The authors declare no competing financial interest.
References
- 1.Houten SM, Wanders RJA. A general introduction to the biochemistry of mitochondrial fatty acid β-oxidation. J Inherited Metab Dis. 2010;33:469–477. doi: 10.1007/s10545-010-9061-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Umbarger HE. Amino acid biosynthesis and its regulation. Annu Rev Biochem. 1978;47:533–606. doi: 10.1146/annurev.bi.47.070178.002533. [DOI] [PubMed] [Google Scholar]
- 3.Ibba M, Soll D. Aminoacyl-tRNA synthesis. Annu Rev Biochem. 2000;69:617–650. doi: 10.1146/annurev.biochem.69.1.617. [DOI] [PubMed] [Google Scholar]
- 4.Weissman KJ. The structural biology of biosynthetic megaenzymes. Nat Chem Biol. 2014;11:660–670. doi: 10.1038/nchembio.1883. [DOI] [PubMed] [Google Scholar]
- 5.Hur GH, Vickery CR, Burkart MD. Explorations of catalytic domains in non-ribosomal peptide synthetase enzymology. Nat Prod Rep. 2012;29:1074–1098. doi: 10.1039/c2np20025b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gulick AM. Conformational dynamics in the acyl-CoA synthetases, adenylation domains of non-ribosomal peptide synthetases, and firefly luciferase. ACS Chem Biol. 2009;4:811–827. doi: 10.1021/cb900156h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schmelz S, Naismith JH. Adenylate-forming enzymes. Curr Opin Struct Biol. 2009;19:666–671. doi: 10.1016/j.sbi.2009.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Estrada P, et al. The pimeloyl-CoA synthetase BioW defines a new fold for adenylate-forming enzymes. Nat Chem Biol. 2017;13:668–674. doi: 10.1038/nchembio.2359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang M, et al. Using the pimeloyl-CoA synthetase adenylation fold to synthesize fatty acid thioesters. Nat Chem Biol. 2017;13:660–667. doi: 10.1038/nchembio.2361. [DOI] [PubMed] [Google Scholar]
- 10.Marahiel MA, Stachelhaus T, Mootz HD. Modular peptide synthetases involved in nonribosomal peptide synthesis. Chem Rev. 1997;97:2651–2674. doi: 10.1021/cr960029e. [DOI] [PubMed] [Google Scholar]
- 11.Wu R, Reger AS, Lu X, Gulick AM, Dunaway-Mariano D. The mechanism of domain alternation in the acyl-adenylate forming ligase superfamily member 4-chlorobenzoate: coenzyme A ligase. Biochemistry. 2009;48:4115–4125. doi: 10.1021/bi9002327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rudolf JD, Dong LB, Shen B. Platensimycin and platencin: Inspirations for chemistry, biology, enzymology, and medicine. Biochem Pharmacol. 2017;133:139–151. doi: 10.1016/j.bcp.2016.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dong LB, et al. In vivo instability of platensimycin and platencin: Synthesis and biological evaluation of urea- and carbamate-platensimycin. Bioorg Med Chem. 2017;25:1990–1996. doi: 10.1016/j.bmc.2017.02.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rudolf JD, Dong LB, Manoogian K, Shen B. Biosynthetic origin of the ether ring in platensimycin. J Am Chem Soc. 2016;138:16711–16721. doi: 10.1021/jacs.6b09818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Smanski MJ, et al. Dedicated ent-kaurene and ent-atiserene synthases for platensimycin and platencin biosynthesis. Proc Natl Acad Sci U S A. 2011;108:13498–13503. doi: 10.1073/pnas.1106919108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rudolf JD, Dong LB, Huang T, Shen B. A genetically amenable platensimycin- and platencin-overproducer as a platform for biosynthetic explorations: a showcase of PtmO4, a long-chain acyl-CoA dehydrogenase. Mol BioSyst. 2015;11:2717–2726. doi: 10.1039/c5mb00303b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Smanski MJ, et al. Expression of the platencin biosynthetic gene cluster in heterologous hosts yielding new platencin congeners. J Nat Prod. 2012;75:2158–2167. doi: 10.1021/np3005985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hedden P, Sponsel V. A century of gibberellin research. J Plant Growth Regul. 2015;34:740–760. doi: 10.1007/s00344-015-9546-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 20.Gust B, Challis GL, Fowler K, Kieser T, Chater KF. PCR-targeted Streptomyces gene replacement identifies a protein domain needed for biosynthesis of the sesquiterpene soil odor geosmin. Proc Natl Acad Sci U S A. 2003;100:1541–1546. doi: 10.1073/pnas.0337542100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kieser T, Bibb MJ, Buttner MJ, Chater KF, Hopwood DA. Practical Streptomyces Genetics. 2000;613 [Google Scholar]
- 22.MacNeil DJ, et al. Analysis of Streptomyces avermitilis genes required for avermectin biosynthesis utilizing a novel integration vector. Gene. 1992;111:61–68. doi: 10.1016/0378-1119(92)90603-m. [DOI] [PubMed] [Google Scholar]
- 23.Dong LB, Rudolf JD, Shen B. Antibacterial sulfur-containing platensimycin and platencin congeners from Streptomyces platensis SB12029. Bioorg Med Chem. 2016;24:6348–6353. doi: 10.1016/j.bmc.2016.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Herz W, Kulanthaivel P, Watanabe K. ent-Kauranes and other constituents of three Helianthus species. Phytochemistry. 1983;22:2021–2025. [Google Scholar]
- 25.Hindra, et al. Strain prioritization for natural product discovery by a high-throughput real-time PCR method. J Nat Prod. 2014;77:2296–2303. doi: 10.1021/np5006168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Krissinel E, Henrick K. Inference of macromolecular assemblies from crystalline state. J Mol Biol. 2007;372:774–797. doi: 10.1016/j.jmb.2007.05.022. [DOI] [PubMed] [Google Scholar]
- 27.Gulick AM, Lu X, Dunaway-Mariano D. Crystal structure of 4-chlorobenzoate:CoA ligase/synthetase in the unliganded and aryl substrate-bound states. Biochemistry. 2004;43:8670–8679. doi: 10.1021/bi049384m. [DOI] [PubMed] [Google Scholar]
- 28.Osman KT, Du L, He Y, Luo Y. Crystal structure of Bacillus cereus D-alanyl carrier protein ligase (DltA) in complex with ATP. J Mol Biol. 2009;388:345–355. doi: 10.1016/j.jmb.2009.03.040. [DOI] [PubMed] [Google Scholar]
- 29.Wu R, et al. Mechanism of 4-chlorobenzoate:coenzyme A ligase catalysis. Biochemistry. 2008;47:8026–8039. doi: 10.1021/bi800698m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Horswill AR, Escalante-Semerena JC. Characterization of the propionyl-CoA synthetase (PrpE) enzyme of Salmonella enterica: residue Lys592 is required for propionyl-AMP synthesis. Biochemistry. 2002;41:2379–2387. doi: 10.1021/bi015647q. [DOI] [PubMed] [Google Scholar]
- 31.Starai VJ, Celic I, Cole RN, Boeke JD, Escalante-Semerena JC. Sir2-dependent activation of acetyl-CoA synthetase by deacetylation of active lysine. Science. 2002;298:2390–2392. doi: 10.1126/science.1077650. [DOI] [PubMed] [Google Scholar]
- 32.Schwer B, Bunkenborg J, Verdin RO, Andersen JS, Verdin E. Reversible lysine acetylation controls the activity of the mitochondrial enzyme acetyl-CoA synthetase 2. Proc Natl Acad Sci U S A. 2006;103:10224–10229. doi: 10.1073/pnas.0603968103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Trivedi OA, et al. Enzymic activation and transfer of fatty acids as acyl-adenylates in mycobacteria. Nature. 2004;428:441–445. doi: 10.1038/nature02384. [DOI] [PubMed] [Google Scholar]
- 34.Arora P, et al. Mechanistic and functional insights into fatty acid activation in Mycobacterium tuberculosis. Nat Chem Biol. 2009;5:166–173. doi: 10.1038/nchembio.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang Z, et al. Structural and functional studies of fatty acyl adenylate ligases from E coli and L. pneumophila. J Mol Biol. 2011;406:313–324. doi: 10.1016/j.jmb.2010.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Goyal A, Verma P, Anandhakrishnan M, Gokhale RS, Sankaranarayanan R. Molecular basis of the functional divergence of fatty acyl-AMP ligase biosynthetic enzymes of Mycobacterium tuberculosis. J Mol Biol. 2012;416:221–238. doi: 10.1016/j.jmb.2011.12.031. [DOI] [PubMed] [Google Scholar]
- 37.Yonus H, et al. Crystal Structure of DltA: implications for the reaction mechanism of non-ribosomal peptide synthetase adenylation domains. J Biol Chem. 2008;283:32484–32491. doi: 10.1074/jbc.M800557200. [DOI] [PubMed] [Google Scholar]
- 38.Gulick AM, Starai VJ, Horswill AR, Homick KM, Escalante-Semerena JC. The 1.75 Å crystal structure of acetyl-CoA synthetase bound to adenosine-5′-propylphosphate and coenzyme A. Biochemistry. 2003;42:2866–2873. doi: 10.1021/bi0271603. [DOI] [PubMed] [Google Scholar]
- 39.Shi J, et al. Titer improvement and pilot-scale production of platensimycin from Streptomyces platensis SB12026. J Ind Microbiol Biotechnol. 2016;43:1027–1035. doi: 10.1007/s10295-016-1769-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Stols L, Millard CS, Dementieva I, Donnelly MI. Production of selenomethionine-labeled proteins in two-liter plastic bottles for structure determination. J Struct Funct Genomics. 2006;5:95–102. doi: 10.1023/B:JSFG.0000029196.87615.6e. [DOI] [PubMed] [Google Scholar]
- 41.Minor W, Cymborowski M, Otwinowski Z, Chruszcz M. HKL-3000: the integration of data reduction and structure solution - from diffraction images to an initial model in minutes. Acta Crystallogr, Sect D Biol Crystallogr. 2006;62:859–866. doi: 10.1107/S0907444906019949. [DOI] [PubMed] [Google Scholar]
- 42.Terwilliger TC, et al. Iterative model building, structure refinement and density modification with the PHENIX AutoBuild wizard. Acta Crystallogr, Sect D Biol Crystallogr. 2008;64:61–69. doi: 10.1107/S090744490705024X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Adams PD, et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr, Sect D Biol Crystallogr. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Vagin A, Teplyakov A. Molecular replacement with MOLREP. Acta Crystallogr, Sect D Biol Crystallogr. 2010;66:22–25. doi: 10.1107/S0907444909042589. [DOI] [PubMed] [Google Scholar]
- 45.Winn MD, et al. Overview of the CCP4 suite and current developments. Acta Crystallogr, Sect D Biol Crystallogr. 2011;67:235–242. doi: 10.1107/S0907444910045749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chen VB, et al. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr, Sect D Biol Crystallogr. 2010;66:12–21. doi: 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sievers F, et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol Syst Biol. 2011;7:539. doi: 10.1038/msb.2011.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tamura K, et al. MEGA5: Molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol. 2011;28:2731–2739. doi: 10.1093/molbev/msr121. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Protein Data Bank: Atomic coordinates and structure factor amplitudes for PtmA2 crystal structures were deposited with accession codes 5E7Q, 5UPQ, 5UPS, and 5UPT. All other data generated or analyzed in this study are available within the article and Supplementary Information files. A Life Sciences Reporting Summary for this paper is available.