

Abstract
The catalytic activity of transglutaminase 2 (TG2), a ubiquitously expressed mammalian enzyme, is regulated by multiple post-translational mechanisms. Because elevated activity of TG2 in the extracellular matrix is associated with organ-specific diseases such as celiac disease and renal fibrosis, there is growing therapeutic interest in inhibitors of this enzyme. Cyst-amine, a symmetric disulfide compound, is one of the earliest reported TG2 inhibitors. Despite its widespread use as a tool compound to block TG2 activity in vitro and in vivo, its mechanism of action has remained unclear. Here, we demonstrate that cystamine irreversibly inhibits human TG2 (kinh/Ki = 1.2 mM−1 min−1) via a mechanism fundamentally distinct from those proposed previously. Through mass spectrometric disulfide mapping and site-directed mutagenesis, we show that cystamine promotes the formation of a physiologically relevant disulfide bond between Cys370 and Cys371 that allosterically abrogates the catalytic activity of human TG2. This discovery led us to evaluate clinically useful thiol → disulfide oxidants for TG2 inhibitory activity. It is demonstrated that disulfiram, a relatively safe oral thiuram disulfide, is a fairly potent TG2 inhibitor (kinh/Ki = 8.3 mM−1 min−1) and may therefore provide a practical tool for clinically validating this emerging therapeutic target in intestinal disorders such as celiac disease.
Human transglutaminase 2 (TG2) is a multifunctional member of the mammalian transglutaminase family that is ubiquitously expressed in almost all cell types. Found in both the intra- and extracellular environments of most tissues, TG2 deamidates or transamidates its substrates. Mechanistically, both of these reactions proceed by formation of an acyl–enzyme thioester between Cys277 in the TG2 active site and a Gln residue on the substrate protein or peptide. This is followed by attack of the thioester intermediate by a primary amine, resulting in the formation of an isopeptide bond, or by water, resulting in Gln → Glu conversion. Although its precise physiological functions remain unclear, TG2 is believed to be involved in processes such as wound healing, extracellular matrix assembly, and cell adhesion.1–3
Upregulation of TG2 activity is implicated in a variety of seemingly unrelated inflammatory and fibrotic pathologies such as celiac disease and renal fibrosis.4,5 Because of the suspected role of TG2 in these common maladies, there is increasing interest in development of TG2 inhibitors.6 Although several classes of active site-directed7,8 as well as allosteric9 inhibitors have been discovered and optimized for increased potency and isozyme specificity, arguably the most common TG2 inhibitor used by biologists to date is cystamine (Chart 1). (As of early 2018, there have been more than 200 publications citing the use of this compound as a TG2 inhibitor.) Recently, it has been suggested that cystamine is a competitive amine substrate of TG2-catalyzed transamidation.10,11 However, several decades ago, Lorand and Conrad proposed that cystamine oxidatively inactivated TG2 via thiol–disulfide exchange with the nucleophilic Cys residue in its active site (Cys277 in human TG2).12 Thus, notwithstanding uncertainty regarding its mechanism of action, cystamine-mediated inhibition of TG2 has yielded many important insights into the biology of this enigmatic mammalian protein.13
Chart 1.
Cystamine, a Widely Used TG2 Inhibitor
Through a series of structural, mechanistic, cellular, and animal studies over the past decade, we have elucidated a novel allosteric regulatory mechanism in TG2 that involves a redox triad comprised of Cys230, Cys370, and Cys371, but not the active site Cys277. In this triad, Cys230 acts as an intraenzyme catalyst that promotes formation of a Cys370–Cys371 disulfide bond, which completely abolishes enzymatic activity, thereby maintaining extracellular TG2 in a catalytically dormant state under normal conditions.14 The redox protein cofactor thioredoxin is a specific reductant of this allosteric disulfide bond,15,16 whereas another protein cofactor, ERp57, selectively promotes disulfide bond oxidation.17 In the course of our efforts leading to identification of ERp57 as an inactivator of TG2, we observed that cystine was a more potent oxidant of the human enzyme than glutathione.17 This finding motivated us to re-examine the mechanism by which cystamine inhibits TG2.
As a preliminary step toward ascertaining the mode of action of cystamine, we incubated TG2, recombinantly expressed and purified from Escherichia coli,18 with a 10-fold molar excess of cystamine and withdrew aliquots after 2.5 and 15 min. At these times, cystamine was quickly removed from the protein via centrifugal size exclusion (molecular weight cutoff of 7 kDa), and the concentration of each TG2 aliquot was normalized by spectrophotometry at 280 nm. Then, residual TG2 activity was measured in an established coupled enzyme assay that measures the rate at which TG2 deamidates the synthetic peptidic substrate Cbz-Gln-Gly.19,20 Figure 1A shows a time-dependent decrease in TG2 activity that persisted after removal of the inhibitor, strongly suggesting that cystamine irreversibly inhibits TG2.
Figure 1.

Irreversible inactivation of human TG2 by cystamine. (A) Residual enzyme activity after incubation of TG2 with cystamine (100 μM) for the indicated times, followed by removal of cystamine by size exclusion. (B) Comparative time-dependent inactivation of TG2 by cystamine, cystine, and GSSG. Values are the mean of three technical replicates, and error bars show the standard deviation. Solid lines are fits to a single-exponential decay equation for irreversible enzyme inhibition, as described in the text. (C) Determination of the pseudo-first-order rate constant (k′) as a function of cystamine concentration. Error bars represent the standard error of the fit of the experimental data to the exponential decay equation (eq 1) obtained for the given cystamine concentration (panel B and Figure S1). Data in all panels were collected at a TG2 concentration of 10 μM in 50 mM Tris and 1 mM EDTA at pH 7.6 and 22 °C.
Next, we sought to test whether the rate of cystamine-mediated TG2 inhibition was consistent with a kinetic model in which cystamine oxidizes TG2 via thiol–disulfide exchange. As previously shown for cystine and GSSG, the kinetics of oxidative inactivation of TG2 are sufficiently described by a standard irreversible inhibition model, in which an oxidative inhibitor Iox reacts with enzyme E to generate inhibited enzyme E* according to Scheme 1.17
Scheme 1.
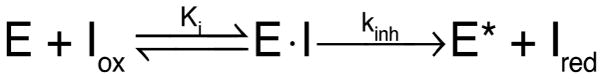
Kinetic Mechanism for Irreversible Enzyme Inhibition by an Oxidative Inhibitor
The integrated rate law that describes the formation of E* as a function of time t is exponential decay eq 1:
| (1) |
where pseudo-first-order rate constant k′ can be converted to a bimolecular rate constant (kinh/Ki) by dividing by initial inhibitor concentration [I0]. Alternatively, separate estimates of kinh and Ki can be made by determining k′ as a function of [I0] and fitting to eq 2:
| (2) |
We incubated TG2 with cystamine and withdrew aliquots after 2.5, 5, 10, 30, 90, and 120 min. As controls, cystine and GSSG were compared. Aliquots of inhibitor-treated TG2 were diluted 10-fold into assay buffer, and the deamidation activity of TG2, relative to enzyme maintained in the absence of oxidant, was measured at each time point (Figure 1B). All inhibition curves gave good fits to eq 1 (R2 > 0.9), and the bimolecular rate constants obtained for cystine and GSSG (Table 1) are in agreement with literature values.15,17 Cystamine was a markedly better oxidant than either cystine or GSSG (Figure 1B and Table 1). Additional time-dependent inhibition experiments were performed in an attempt to separately estimate Ki and kinh (Figure S1); however, we were unable to reach saturating inhibitor concentrations (Figure 1C).
Table 1.
Rate Constants for TG2 Oxidation by Cystamine, Cystine, and GSSGa
| oxidant | kinh/Ki (mM−1 min−1) | Ki (mM) |
|---|---|---|
| cystineb | 0.29 | NDd |
| GSSGb | 0.0033 | NDd |
| cystamineb | 1.2 | >0.6 |
| cystaminec | 1.7 | >0.1 |
Measured in 50 mM Tris and 1 mM EDTA at pH 7.6 and 22 °C.
Determined in a deamidation activity assay.
Determined in a transamidation activity assay.
Not determined.
Thus, the composite parameter (kinh/Ki) was estimated from the slope of the plot of k′ versus [I0] (Table 1). A similar rate constant for cystamine-mediated oxidative inhibition of TG2 was obtained in an established transamidation assay in which TG2 is immobilized on fibronectin,14,17 which more realistically reflects the environment of TG2 in the extracellular matrix of cells (Table 1 and Figure S2).
To establish the precise mechanism of cystamine-mediated TG2 inhibition, we assayed the redox status of the Cys residues of TG2 using a protocol described previously.17 In brief, reduced TG2 was incubated in the presence or absence of a 10-fold molar excess of cystamine for 30 min. Then, reduced Cys residues were labeled with iodoacetamide (IAM). The protein was separated by nonreducing sodium dodecyl sulfate–polyacrylamide gel electrophoresis, and excised gel bands were reduced with dithiothreitol and alkylated with iodoacetic acid (IAA), followed by in-gel trypsinization and analysis by nanoliquid chromatography coupled to Orbitrap mass spectrometry. Then, tryptic peptides of TG2 were identified (Table S1).17 It was anticipated that peptides bearing Cys residues that were oxidized by cystamine would display a decrease in their level of IAM labeling and an increase in their level of IAA labeling relative to those of controls.
As shown in Figure 2A, we observed peptides corresponding to 10 of the 20 Cys residues of TG2 at sufficient precursor ion intensities to permit a semiquantitative estimate of their relative abundances before and after cystamine treatment. The tryptic peptide harboring Cys370 and Cys371 was most oxidized by cystamine (Figure 2A). The disappearance of IAM-labeled Cys370 and Cys371 was accompanied by a relative increase in the level of the corresponding IAA-labeled peptide (Figure 2B). Additionally, there was a decrease in the level of IAM-labeled Cys230 after cystamine treatment, whereas the level of the IAA-labeled form increased (Figure 2B). These results provide strong support that cystamine promotes formation of the Cys370–Cys371 disulfide via a Cys230–Cys370 intermediate. Notably, spectral evidence for a peptide containing IAM-labeled Cys277 was present, but its precursor ion was not present at sufficient abundance to allow semiquantitative analysis; on the other hand, IAA-labeled Cys277 was not observed. Moreover, no evidence for a mixed Cys277–cystamine disulfide was obtained, suggesting that the active site was not targeted by cystamine.
Figure 2.

Cystamine targets the Cys230–Cys370–Cys371 allosteric redox triad of human TG2. (A) Fold decrease in the precursor intensities of iodoacetamide (IAM)-labeled (reduced) Cys residues of TG2 following incubation with cystamine, estimated via mass spectrometry. (B) Extracted ion chromatograms of IAM- and iodoacetic acid (IAA)-labeled tryptic peptides containing Cys370, Cys371, and Cys230 (Table S1). An increase in the level of IAA labeling signifies the oxidation of the indicated Cys residues by cystamine. (C) Resistance of the C230S mutant of TG2 to cystamine-mediated activation. Cystamine treatment of TG2 in all panels was performed with a 10-fold cystamine:enzyme molar ratio for 30 min in 50 mM Tris and 1 mM EDTA at pH 7.6 and 22 °C.
To functionally validate the findings of our redox mapping experiment, we incubated the C230S mutant of human TG2, which retains full catalytic activity relative to that of the wild-type (WT) enzyme, in a 10-fold molar excess of cystamine for 30 min and assayed its deamidation activity. The C230S mutant retained >85% of its activity, whereas identically treated WT enzyme was completely inactivated (Figure 2C). This result confirms that the Cys230–Cys370–Cys371 allosteric redox triad is the target of cystamine.
Cystamine has been used to attenuate the activity of TG2 in a variety of animal models of disease.13 We therefore sought to identify an analogous Food and Drug Administration-approved oxidant that was able to inhibit TG2. For several reasons, we hypothesized that disulfiram, a drug used in the treatment of alcohol abuse, could have potential to serve as such a tool. First, disulfiram is a symmetric thiuram disulfide that is similar to cystamine in size (Chart 2). Second, on the basis of values reported for structurally related compounds,21 the redox potential of disulfiram should be sufficiently positive to provide adequate thermodynamic driving force for oxidation of the Cys370–Cys371 disulfide of TG2 (E°′ ≈ −184 mV).15 Last but not least, disulfiram is administered orally and relatively well tolerated at doses of 500 mg/day.22 Although it is rapidly reduced following intestinal absorption,23 its presystemic bioavailability could facilitate TG2 inactivation in the intestinal mucosa.
Chart 2.
Structure of Disulfiram
To evaluate whether disulfiram could irreversibly inhibit TG2, an aliquot of TG2 was incubated with a 10-fold molar excess of the compound for 2.5 min, disulfiram was quickly removed by centrifugal size exclusion, and TG2 activity was measured. Strikingly, after this short incubation, TG2 was approximately 90% inactivated (Figure 3A), suggesting that disulfiram is an inhibitor that is considerably more potent than cystamine (compare to Figure 1A).
Figure 3.
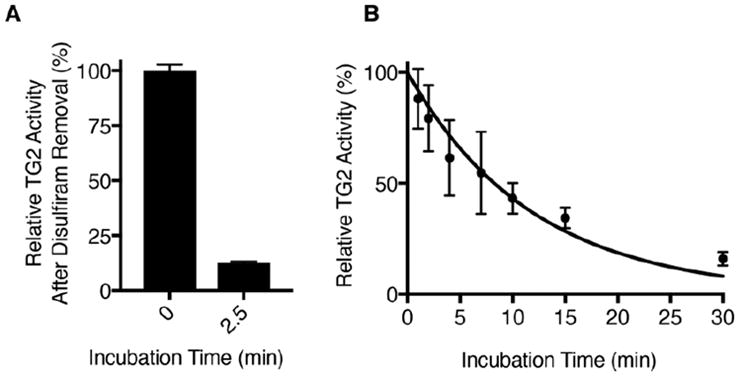
Disulfiram is an irreversible inhibitor of human TG2. (A) Residual TG2 deamidation activity after incubation of TG2 (10 μM) with disulfiram (100 μM) for 2.5 min, followed by removal of disulfiram by size exclusion. (B) Time-dependent inactivation of TG2 (50 nM) by disulfiram (10 μM). TG2 transamidation activity was measured relative to enzyme maintained in the absence of disulfiram. Data are fit to eq 1 (R2 = 0.94). All measurements were performed in 50 mM Tris, 1 mM EDTA, and 2.5% DMSO at pH 7.6 and 22 °C. Error bars show the standard deviation of triplicate or quadruplicate measurements.
The kinetics of disulfiram-mediated TG2 inactivation were too rapid to allow us to estimate kinh/Ki using the coupled deamidation activity assay. We therefore utilized the trans-amidation assay, which is sensitive enough to be performed with as little as 50 nM TG2. This allowed us to decrease the disulfiram concentration while still maintaining inhibitor in excess relative to the enzyme. In this assay, we estimate that the bimolecular rate constant for disulfiram-mediated TG2 inactivation is 8.3 mM−1 min−1 (Figure 3B).
In conclusion, we have demonstrated through several lines of evidence that the widely used TG2 inhibitor cystamine oxidatively inactivates TG2 by promoting formation of the Cys370–Cys371 allosteric disulfide bond. This inhibitory mechanism is fundamentally different from those previously proposed. Guided by this novel mechanistic insight, we have discovered that disulfiram, a commonly prescribed drug, is also an TG2 inhibitor. To the best of our knowledge, this is the first clinically approved compound that displays inhibitory activity toward human TG2.
Finally, we note that we have recently developed an acute model for activating TG2 in the small intestinal mucosa of living mice by reducing the Cys370–Cys371 disulfide bond via administration of exogenous thioredoxin.16 In this model, the active site-directed dihydroisoxazole compound ERW1041E was shown to effectively block TG2 activity. The potency of disulfiram (kinh/Ki = 8.3 mM−1 min−1) is comparable to that reported for ERW1041E (kinh/Ki = 16 mM−1 min−1).7 Thus, this physiologically relevant model system can be used to assess the efficacy of orally administered disulfiram as a TG2 inhibitor in vivo. If successful, trials of disulfiram to attenuate TG2 activity in disorders in which extracellular TG2 activity is elevated (such as celiac disease) may be warranted.
Supplementary Material
Acknowledgments
Funding
This work was supported by National Institutes of Health Grants R01 DK063158 and DK100619 to C.K.
The authors acknowledge Prof. Josh Elias and Dr. Lichao Zhang for assistance with mass spectrometry and thank Arek Melkonian and Nielson Weng for assistance with experiments.
Footnotes
Author Contributions
B.A.P. and C.K. conceived the project. B.A.P. designed and conducted the experiments and analyzed the data. B.A.P. and C.K. wrote the manuscript.
Notes
The authors declare the following competing financial interest(s): C.K. is a shareholder in and consultant to Sitari Pharmaceuticals.
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.bio-chem.8b00204.
References
- 1.Eckert RL, Kaartinen MT, Nurminskaya M, Belkin AM, Colak G, Johnson GVW, Mehta K. Transglutaminase regulation of cell function. Physiol Rev. 2014;94:383–417. doi: 10.1152/physrev.00019.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Klöck C, Khosla C. Regulation of the activities of the mammalian transglutaminase family of enzymes. Protein Sci. 2012;21:1781–1791. doi: 10.1002/pro.2162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gundemir S, Colak G, Tucholski J, Johnson GVW. Transglutaminase 2: A molecular Swiss army knife. Biochim Biophys Acta, Mol Cell Res. 2012;1823:406–419. doi: 10.1016/j.bbamcr.2011.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Klöck C, Diraimondo TR, Khosla C. Role of transglutaminase 2 in celiac disease pathogenesis. Semin Immunopathol. 2012;34:513–522. doi: 10.1007/s00281-012-0305-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Burhan I, Furini G, Lortat-Jacob H, Atobatele AG, Scarpellini A, Schroeder N, Atkinson J, Maamra M, Nutter FH, Watson P, Vinciguerra M, Johnson TS, Verderio EAM. Interplay between transglutaminases and heparan sulphate in progressive renal scarring. Sci Rep. 2016;6:31343. doi: 10.1038/srep31343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Keillor JW, Apperley KYP. Transglutaminase inhibitors: a patent review. Expert Opin Ther Pat. 2016;26:49–63. doi: 10.1517/13543776.2016.1115836. [DOI] [PubMed] [Google Scholar]
- 7.Klöck C, Herrera Z, Albertelli M, Khosla C. Discovery of Potent and Specific Dihydroisoxazole Inhibitors of Human Transglutaminase 2. J Med Chem. 2014;57:9042–9064. doi: 10.1021/jm501145a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Badarau E, Wang Z, Rathbone DL, Costanzi A, Thibault T, Murdoch CE, El Alaoui S, Bartkeviciute M, Griffin M. Development of Potent and Selective Tissue Transglutaminase Inhibitors. Chem Biol. 2015;22:1347–1361. doi: 10.1016/j.chembiol.2015.08.013. [DOI] [PubMed] [Google Scholar]
- 9.Klöck C, Jin X, Choi K, Khosla C, Madrid PB, Spencer A, Raimundo BC, Boardman P, Lanza G, Griffin JH. Acylideneoxoindoles: A new class of reversible inhibitors of human transglutaminase 2. Bioorg Med Chem Lett. 2011;21:2692–2696. doi: 10.1016/j.bmcl.2010.12.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jeitner TM, Delikatny EJ, Ahlqvist J, Capper H, Cooper AJL. Mechanism for the inhibition of trans-glutaminase 2 by cystamine. Biochem Pharmacol. 2005;69:961–970. doi: 10.1016/j.bcp.2004.12.011. [DOI] [PubMed] [Google Scholar]
- 11.Wu YW, Tsai YH. A Rapid Transglutaminase Assay for High-Throughput Screening Applications. J Biomol Screening. 2006;11:836–843. doi: 10.1177/1087057106291585. [DOI] [PubMed] [Google Scholar]
- 12.Lorand L, Conrad SM. Transglutaminases. Mol Cell Biochem. 1984;58:9–35. doi: 10.1007/BF00240602. [DOI] [PubMed] [Google Scholar]
- 13.Caccamo D, Currò M, Ientile R. Potential of transglutaminase 2 as a therapeutic target. Expert Opin Ther Targets. 2010;14:989–1003. doi: 10.1517/14728222.2010.510134. [DOI] [PubMed] [Google Scholar]
- 14.Stamnaes J, Pinkas DM, Fleckenstein B, Khosla C, Sollid LM. Redox regulation of transglutaminase 2 activity. J Biol Chem. 2010;285:25402–25409. doi: 10.1074/jbc.M109.097162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jin X, Stamnaes J, Klöck C, Diraimondo TR, Sollid LM, Khosla C. Activation of extracellular transglutaminase 2 by thioredoxin. J Biol Chem. 2011;286:37866–37873. doi: 10.1074/jbc.M111.287490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Plugis NM, Palanski BA, Weng CH, Albertelli M, Khosla C. Thioredoxin-1 Selectively Activates Transglutaminase 2 in the Extracellular Matrix of the Small Intestine. J Biol Chem. 2017;292:2000–2008. doi: 10.1074/jbc.M116.767988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yi MC, Melkonian AV, Ousey JA, Khosla C. Endoplasmic reticulum-resident protein 57 (ERp57) oxidatively inactivates human transglutaminase 2. J Biol Chem. 2018;293:2640–2649. doi: 10.1074/jbc.RA117.001382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yi MC, Palanski BA, Quintero SA, Plugis NM, Khosla C. An unprecedented dual antagonist and agonist of human transglutaminase 2. Bioorg Med Chem Lett. 2015;25:4922–4926. doi: 10.1016/j.bmcl.2015.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Day N, Keillor JW. A continuous spectrophotometric linked enzyme assay for transglutaminase activity. Anal Biochem. 1999;274:141–144. doi: 10.1006/abio.1999.4255. [DOI] [PubMed] [Google Scholar]
- 20.Piper JL, Gray GM, Khosla C. High Selectivity of Human Tissue Transglutaminase for Immunoactive Gliadin Peptides. Biochemistry. 2002;41:386–393. doi: 10.1021/bi011715x. [DOI] [PubMed] [Google Scholar]
- 21.Nichols PJ, Grant MW. Reduction potentials of thiuram disulfide/dithiocarbamate couples in acetone/water. Aust J Chem. 1982;35:2455–2463. [Google Scholar]
- 22.Malcolm R, Olive MF, Lechner W. The safety of disulfiram for the treatment of alcohol and cocaine dependence in randomized clinical trials: guidance for clinical practice. Expert Opin Drug Saf. 2008;7:459–472. doi: 10.1517/14740338.7.4.459. [DOI] [PubMed] [Google Scholar]
- 23.Johnson BA. Disulfiram. In: Stolerman IP, Price LH, editors. Encyclopedia of Psychopharmacology. Springer; Berlin: 2015. pp. 531–534. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.