

Abstract
As one of the most abundant elements on earth, sulfur is part of many small molecular metabolites and is key to their biological activities. Over the past few decades, some general strategies have been discovered for the incorporation of sulfur into natural products. In this review, we summarize recent efforts in elucidating the biosynthetic details for two sulfur-containing metabolites, ergothioneine and ovothiol. Their biosyntheses involve an unprecedented trans-sulfur strategy, a combination of a mononuclear non-heme iron enzyme-catalyzed oxidative C–S bond formation reaction and a PLP enzyme-mediated C–S lyase reaction.
Graphical Abstract
Sulfur is a component of both small molecular metabolites and macromolecules,1–3 and the roles of sulfur in primary metabolism are well-established.4–8 Over the past few decades, studies of sulfur-containing vitamins/cofactors (e.g., thiamin pyrophosphate,9,10 biotin,9,11–14 lipoic acid,15 molybdopterin,16 coenzyme B,17 and coenzyme M18,19) have greatly enriched our understanding on the biosynthetic C–S bond construction strategies. In recent years, an explosion of genetic information from genome sequencing efforts revealed many potential sulfur-containing natural product biosynthetic gene clusters.20–23 Currently known enzymatic C–S bond formation reactions can be roughly divided into two categories: ionic mechanisms and radical mechanisms.
Ionic Types of C–S Bond Formation Biosynthetic Pathways
The pKa of cysteine thiol is ~8.3, and under physiological conditions (e.g., pH 7.4), a portion of the cysteine side chain is present as a thiolate, which could function as a nucleophile directly. In nisin 1 biosynthesis (Figure 1A),24 an example of lantibiotic biosynthesis,25 thioether bond formation involves several steps. The serine and threonine residues are dehydrated by NisB, resulting in a 2,3-didehydroalanine (Dha) or 2,3-didehydrobutyrine (Dhb) moiety (Figure 1A). A Zn2+-containing cyclase, NisC, catalyzes the nucleophilic attacks on the β-carbon position of Dhb or Dha by cysteine residues, producing thioether linkages. In the NisC reaction, the coordination of a cysteine residue to Zn2+ decreases its pKa, facilitating thioether bond formation (Figure 1B).24 In the last step, a protease, NisP, cleaves off the leader peptide from the thioether-containing peptide to give nisin 1 as the final product (Figure 1A).
Figure 1.

Ionic type of C–S bond formation in nisin biosynthesis. (A) Overall biosynthetic pathway of nisin. (B) NisC activates the cysteine thiol by coordinating to a Zn2+ ion, allowing nucleophilic attacks of the Dhb or Dha β-carbon position to give a thioether linkage.
In some cases, electrophiles for the nucleophilic reactions are created by oxidative activation mechanisms. Several types of oxidases have been reported to be part of this type of activation mechanism, including P450 monooxygenases, non-heme iron enzymes, flavin-dependent enzymes, and copper-containing oxidases.20 This strategy is present in the biosyntheses of gliotoxin [12 (Figure 2)],26,27 leukotriene D4,28,29 and glucosinolate.30 In gliotoxin 12 biosynthesis, a nonribosomal peptide synthetase, GliP, catalyzes the condensation between L-Ser 2 and L-Phe 3 to cyclo-phenylalanyl-Ser 4 (Figure 2). Hydroxyl groups are incorporated into the α-positions of 4 by a P450-catalyzed hydroxylation (GliC catalysis). The subsequent dehydration reaction of 5 produces an electrophilic acyliminium intermediate 5a, which then reacts with glutathione to form the C–S bonds in bis-glutathionylated intermediate 6 (GliG catalysis). Then, a γ-glutamyl-transferase, GliK, cleaves the isopeptide bond, and the resulting Cys–Gly dipeptide bond in compound 7 is hydrolyzed by a metal-dependent dipeptidase, GliJ. A PLP-dependent C–S lyase, GliI, finishes the sulfur transfer process to produce the epidithiol 9. The disulfide bridge in gliotoxin 12 is formed by a flavin-dependent oxidase [GliT, 11 → 12 conversion (Figure 2)].27,31,32
Figure 2.

C–S bond formation through an ionic type of reaction in gliotoxin biosynthesis. In the gliotoxin 12 biosynthetic pathway, C–S bond formation mediated by GliG undergoes a nucleophilic attack by GSH on an acyliminium intermediate 5a, giving bis-glutathionylated intermediate 6.
In many other ionic sulfur transfer reactions, sulfur atoms are first activated into a persulfide (R-S-SH)33 or a thiocarboxylate intermediate (R-CO-SH).4,34,35 Biosyntheses of sulfur-containing cofactors (e.g., thiamine pyrophosphate10 and molybdopter-in16), and the tRNA modification reaction,9,36 provide mechanistic details about this type of transformation. These two intermediates were also proposed in several secondary metabolite biosynthetic pathways, including BE-7585A,37 thioquinolobactin,38,39 thiolactomycin,40–42 and 6-thiogua-nine.43,44 The recently reported biosynthetic details for a thiosugar [BE-7585A, 15 (Figure 3)] are consistent with the findings of studies of sulfur-containing cofactors, while some unique features were uncovered.37 In the BE-7585A biosynthetic gene cluster, the BexX gene is homologous to the thiazole synthase thiG gene in thiamine biosynthesis.9,10,45 The covalent adduct between BexX and the glucose 6-phosphate-derived 2-ketosugar has been confirmed biochemically45 [13a (Figure 3)], and the crystal structure of the BexX–substrate complex has also been reported.37 Results from biochemical and structural studies support the mechanistic similarities between BexX in BE-7585A biosynthesis and ThiG in thiamine biosynthesis.37,45
Figure 3.

Sulfur-delivery machinery in BE-7585A biosynthesis. MoeZ not only activates the C-terminus of a sulfur carrier protein (SCP) using ATP but also mediates the transfer of the sulfur to the activated SCP to form a thiocarboxylate. The activated SCP then incorporates sulfur into BexX–substrate complex 13a to produce 14.
Interestingly, the BE-7585A biosynthetic gene cluster does not contain any cysteine desulfurases, sulfur carrier proteins, or rhodanese-like proteins. These proteins are required for efficient transfer of the cysteine sulfur to BexX–substrate complex 13a, if the sulfur transfer strategy, described above, discovered in the biosynthesis of sulfur-containing cofactors is followed in BexX-catalysis. This observation raises the question of what sulfur-delivery machinery is employed by BexX. The genome of the BE-7585A-producing strain, Amycolatopsis orientalis, contains five cysteine desulfurase homologues, four sulfur carrier protein homologues (ThiS, MoaD, CysO, and MoaD2), and five rhodanese homologues. The thiS, moaD, and cysO genes are part of the thiamine, molybdopterin, and cysteine biosynthetic gene clusters, respectively, while moaD2 is a stand-alone gene without clear association with any biosynthetic pathway. Moreover, the thiamine, molybdopterin, and cysteine biosynthetic gene clusters do not encode the enzyme responsible for activating the sulfur carrier proteins (SCPs). In the genome, there is a MoeZ gene whose N-terminus is homologous to ThiF, an activating enzyme in thiamine biosynthesis. The C-terminus of MoeZ has a rhodanese domain. MoeZ can catalyze the activation of both MoaD2 and CysO to produce the thiocarboxylate intermediate at the C-termini of these SCPs (Figure 3). In addition, the C-terminal rhodanese domain of MoeZ mobilizes the sulfur atom into the activated SCP (e.g., CysO or MoaD2), from which sulfur is transferred to the BexX–substrate complex [13a → 14 conversions (Figure 3)].37 This study demonstrated that the sulfur-delivery system (e.g., desulfurases, sulfur carrier proteins, and their activating enzymes) may not be located in a particular secondary metabolite biosynthetic gene cluster. Instead, the sulfur transfer system can be used for multiple pathways, including both primary and secondary metabolism.
Radical Types of C–S Bond Formation Biosynthetic Pathways
Radical-type C–S bond formation chemistries include two major subcategories:18 anaerobic and aerobic reactions. Biotin synthase, lipoate synthase (anaerobic radical type), and isopenicillin N-synthase (IPNS) (aerobic radical type) are well-known examples. Biotin synthase is a radical SAM enzyme,46 and the reduction of S-adenosylmethionine (SAM, 16) by a [4Fe-4S] cluster leads to the formation of L-Met 17 and 5′-deoxyadenosyl radical (18, 5′-dAdo•), which abstracts a hydrogen from the substrate to produce a substrate-based radical [19a (Figure 4B)]; this is followed by a sulfur insertion using an auxiliary [2Fe-2S] cluster in BioB as the sulfur source [19b → 19c (Figure 4)].11,12,15,18 The process is repeated one more time to form the thioether bond in biotin [19d → 20 conversion (Figure 4B)].
Figure 4.

Radical type of sulfur insertion mechanism (BioB catalysis of biotin biosynthesis). (A) Reductive cleavage of SAM. (B) Proposed BioB catalytic mechanism in which thioether bond formation goes through two substrate-based radical intermediates, 19a and 19d.
A similar process is also observed in the Escherichia coli lipoic acid biosynthetic pathway (Figure 5). In this pathway, an octanoyltransferase (LipB) first transfers an n-octanoyl moiety 21 to a conserved lysine residue on lipoyl carrier protein [22 (Figure 5)]. Then, a lipoyl synthase, LipA, mediates the sulfur transfer. Like other radical SAM enzymes, the first [4Fe-4S] cluster of LipA undergoes reductive cleavage of SAM to produce 5′-dAdo• [16 → 18 conversion (Figure 4)], which then abstracts a hydrogen atom from the C6 position of the substrate to form a substrate-based radical 22a (Figure 5).46 The substrate-based radical reacts with the bridging μ-sulfido ion of the auxiliary [4Fe-4S] cluster, while one of the Fe3+ ions is reduced to Fe2+ and released from the iron–sulfur cluster, resulting in intermediate 22b. The cycle is repeated one more time to incorporate the other sulfur into the C8 position of the octanoyl moiety to produce 22d. In this mechanism, LipA also functions as the sulfur source and supports only a single turnover. Determining how the iron–sulfur cluster is regenerated to support catalytic turnovers has been a challenge for decades. Recently, Booker et al. demonstrated that in E. coli, when iron–sulfur cluster carrier protein IscU or Nfu is included in the reaction, LipA can catalyze multiple turnovers. In this case, Nfu or IscU replenishes the auxiliary [4Fe-4S] cluster of LipA to support multiple turnovers.47,48
Figure 5.

Proposed lipoic acid biosynthetic mechanism. In lipoic acid biosynthesis, a radical SAM enzyme LipA mediates the transfer of sulfurs from an auxiliary [4Fe-4S] cluster to octanoyl-LCP substrate 22. The auxiliary [4Fe-4S] cluster is then replenished for the next cycle by the iron–sulfur cluster maturation system, e.g., iron–sulfur cluster delivered by IscU or Nfu protein.
In addition to the aforementioned cases, another type of radical SAM enzyme-mediated C–S bond formation strategy has been reported recently (Figure 6). In sactipeptides, one subclass of ribosomally synthesized and post-translationally modified peptides (RiPP),20,49 there are also thioether linkages. However, different from lantibiotics discussed in Figure 1, the thioether linkage in sactipeptides is formed between a cysteine sulfur atom and the α-carbon, instead of the β-carbon of another amino acid.50–53 Some recently characterized examples include subtilosin A, subtilosin A1,49,51 thuricin CD (composed of two peptides, thuricin α and thuricin β),52 and thuricin H.53 In the biosynthesis of subtilosin A, AlbA is a radical SAM enzyme with two [4Fe-4S] clusters. The first cluster undergoes reductive cleavage of SAM to produce 5′-dAdo• [16 → 18 conversion (Figure 4)], which then abstracts a hydrogen from the α-position of the amino acid, where the thioether bond will be formed [23 → 23a conversion (Figure 6)]. The role of the auxiliary [4Fe-4S] cluster in AlbA catalysis is not yet well-defined, but two mechanisms have been proposed (Figure 6). In the first model [pathway I (Figure 6)],49 SAM binds to the unique iron site of the first [4Fe-4S] cluster, while the sulfur group from the peptide substrate coordinates to one of the irons of the auxiliary [4Fe-4S] cluster [23b (Figure 6)]. The substrate-based radical reacts with the cysteine sulfur to form the thioether bond [23a → 23b conversion (Figure 6)]. At the same time, one electron is transferred from the substrate to reduce the auxiliary [4Fe-4S] cluster. In this model, the auxiliary [4Fe-4S] cluster serves as an electron acceptor. After the product is released, the reduced auxiliary cluster relays the electron back to the first [4Fe-4S] cluster to initiate another catalytic cycle.
Figure 6.

Proposed mechanistic models of Alba, which catalyzes sactipeptide thioether linkage formation in the biosynthesis of subtilosin A. The first [4Fe-4S] cluster undergoes reductive cleavage of SAM. The role of the auxiliary cluster is not yet clear.
In the other model [pathway II (Figure 6)], substrate-based radical 23a is oxidized by the auxiliary [4Fe-4S]2+ cluster. The intermediate is then trapped by the sulfur from a cysteine residue in the substrate to form a thioether [23c → 24 conversion (Figure 6)]. In this pathway, the auxiliary [4Fe-4S] cluster is also key to the redox chemistry required for thioether formation, while a direct coordination between the substrate and the auxiliary [4Fe-4S] cluster is not necessary.54 Additional studies are required to differentiate between these two models.
In the biosynthesis of penicillin, isopenicillin N-synthase (IPNS) is responsible for installing β-lactam and thiazolidine rings through an aerobic radical type of sulfur insertion mechanism (Figure 7). IPNS is a mononuclear non-heme iron enzyme, which catalyzes a four-electron oxidation of a tripeptide δ-(L-α-aminoadipoyl)-L-cysteinyl-D-valine (LLD-ACV, 25) to isopenicillin N 26 (Figure 7).55,56 After binding and activation of oxygen by the FeII center, the FeIII–superoxo intermediate oxidizes the cysteine sulfur to generate a thiolaldehyde 25a, which is attacked by the valine amide to form the β-lactam ring. Heterolytic cleavage of the O–O bond of the hydroperoxo intermediate generates FeIV═O species 25b, which abstracts a hydrogen atom from the CVal,β position, resulting in a valinyl radical 25c. Subsequently, the thiazolidine ring is formed by combining a valinyl radical and a thiol radical to produce the final product, isopenicillin N 26.57
Figure 7.
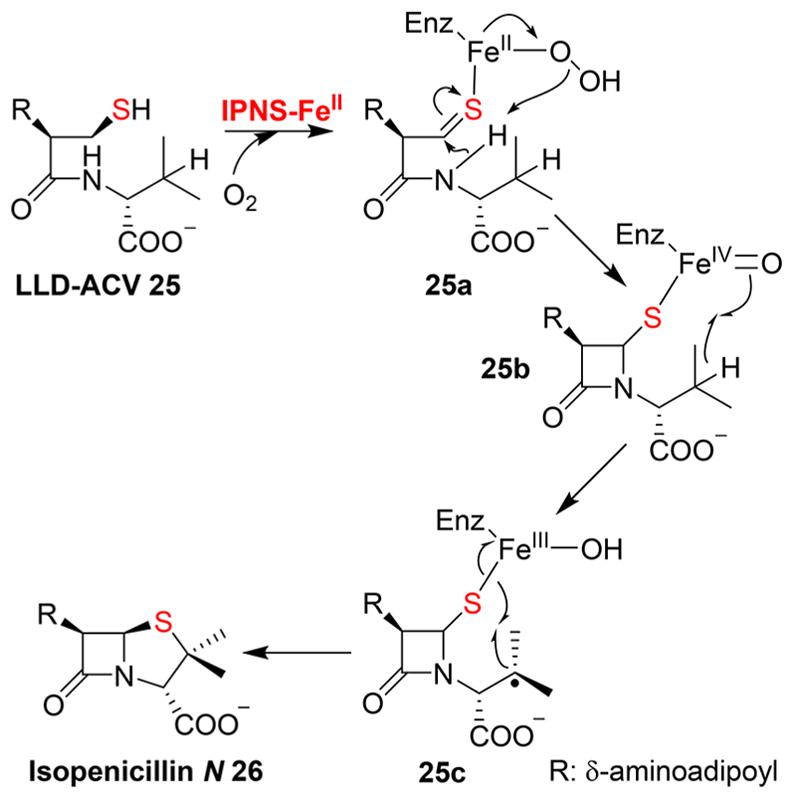
Proposed mechanistic model of IPNS that involves two stages:β-lactam 25b formation followed by thiazolidine ring formation (25c → 26) through an FeIV═O species 25b.
There is currently growing interest in the biosynthesis of sulfur-containing natural products. The examples mentioned above represent some of the general strategies. Many of the recently discovered biosynthetic pathways for sulfur-containing natural products have not yet been biochemically characterized. In the next section, we summarize the sulfur transfer strategy in ergothioneine and ovothiol biosynthesis in which sulfur is incorporated through a combination of a mononuclear non-heme iron enzyme-catalyzed oxidative C–S bond formation reaction and a PLP enzyme-mediated C–S lyase reaction. This sulfur transfer strategy differs from all other examples discussed in previous sections (Figures 1–7).
BIOSYNTHESIS OF ERGOTHIONEINE AND OVOTHIOL
Early Ergothioneine Biosynthetic Studies Using Isotopically Labeled Precursors
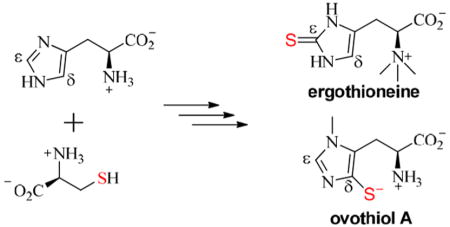
Ergothioneine 32 and ovothiol 36a–c are histidine derivatives with sulfur substitutions at the ε-and δ-carbons of the histidine imidazole side chain, respectively. Ergothioneine [32 (Figure 8A)] was isolated from ergot by Tanret in 1909.58 The biosynthetic community began the search for the ergothioneine biosynthetic details as early as the 1950s. At that time, ergothioneine had been isolated from various microorganisms.59–61 Feeding studies of ergothioneine-producing strains led to the structural characterization of some of the key intermediates. Culturing ergothioneine-producing strain Claviceps purpurea with [2-14C]acetate showed that L-His and ergothioneine have similar labeling distribution patterns, suggesting that L-His is an ergothioneine biosynthetic precursor.62 When [14C]His was fed to Neurospora crassa, it was efficiently incorporated into ergothioneine, whereas [14C]-thiohistidine was not.63 On the basis of these results, it was suggested that hercynine 28 is most likely an intermediate in ergothioneine biosynthesis. The source of sulfur was also examined, and feeding studies indicated that thiosulfate and some sulfur-containing amino acids (e.g., cysteine and cystine) were the sulfur sources for ergothioneine. Using [14C]methyl-labeled methionine resulted in ergothioneine with 14C-labeled methyl groups, suggesting that methionine is the methyl source for ergothioneine. Feeding experiments in C. purpurea led to similar conclusions.63–65
Figure 8.

Ergothioneine and ovothiol biosynthetic pathways. (A) Two aerobic ergothioneine biosynthetic pathways. (B) Proposed biosynthetic pathway of ovothiols. (C) OvoA reaction when hercynine 28 was used as the substrate, where OvoA changes its regioselectivity to the Egt1 type. (D) Anaerobic ergothioneine biosynthetic pathway, in which sulfur is incorporated at the ε-position of the hercynine imidazole ring by a rhodanese, EanB. (E) Dominant reaction of the EgtB Y377F mutant.
These early feeding studies suggested that ergothioneine biosynthesis most likely starts with the methylation of the L-His amino group to produce hercynine 28 (Figure 8A).66 This conclusion was further substantiated by using N. crassa cell extract, which catalyzes the conversion of L-His to hercynine using SAM as the methyl source.67,68 When [14C]hercynine and L-Cys were incubated with N. crassa cell extract, the production of ergothioneine was observed. Interestingly, in the presence of 200 μM hydroxylamine, S-(β-amino-β-carboxyethyl)ergothioneine sulfoxide 31 accumulated. In addition, adding S-(β-amino-β-carboxyethyl)ergothioneine sulfoxide 31 to the N. crassa cell extract resulted in ergothioneine production.69 Results from these studies suggested that sulfoxide 31 might be another ergothioneine biosynthetic intermediate (Figure 8A).
The exact enzymes responsible for these transformations in ergothioneine biosynthesis were not identified at that time. However, it was discovered that supplementing the N. crassa cell extract with Fe2+ and pyridoxamine phosphate (PLP) significantly improved the ergothioneine production yield. The identities of these enzymes were uncovered recently,70 and their biochemical properties correspond to the observations in the study of N. crassa cell extract.
Ovothiol Biosynthetic Studies Using Cell Extracts
Ovothiol A 36a is another thiohistidine isolated from eggs and ovaries of marine invertebrates71 and trypanosomatids (Figure 8B).72,73 Similar to the ergothioneine studies, the initial characterizations of ovothiol A biosynthetic studies were conducted using the cell extracts from ovothiol-producing organisms. Using Crithidia fasciculate, an ovothiol-producing organism, Steenkamp et al. demonstrated that ovothiol is derived from L-His and L-Cys and that the sulfur transfer occurs prior to methylation.74 Using partially purified proteins from Cr. fasciculate, Steenkamp and co-workers further demonstrated that ovothiol A biosynthesis also involves an iron enzyme-catalyzed oxidative coupling reaction and a PLP-catalyzed C–S lyase reaction.75 Thus far, only the identity of the non-heme iron enzyme is known.76
Ergothioneine and Ovothiol Biosynthetic Pathways
Recently, Seebeck and co-workers discovered the ergothioneine biosynthetic gene cluster using a comparative genomic approach (Figure 8A).77 In one of the ergothioneine-producing organisms, Mycobacterium aviu, there are 78 annotated methyltransferases, and among these genes, 29 of them have homologues in N. crassa, another ergothioneine-producing species. Eliminating homologues in ergothioneine nonproducing species narrows the list to 10 candidate methyltransferases. Interestingly, one of them is located next to a PLP-containing enzyme in the context of a five-gene cluster, designated egtABCDE (EgtA–EgtE genes). The recombinant EgtD indeed catalyzes the methylation of L-His 27 to hercynine 28, and the methylation is processive (Figure 8A), resulting in hercynine 28 with only small amounts of mono- or dimethylhistidine products. This processivity is attributed to the fact that EgtD binds to dimethyl-histidine 70-fold more tightly than to histidine.78
Feeding study results discussed previously implied that an Fe-dependent enzyme is responsible for oxidative C–S bond formation. Indeed, EgtB in the gene cluster reported by Seebeck is a non-heme iron enzyme. In addition, the presence of γ-glutamyl-cysteine ligase (EgtA) in the mycobacterium ergo-thioneine biosynthetic gene cluster suggests that γ-glutamyl-cysteine 29 is the sulfur source.77 As expected, EgtB catalyzes the oxidative coupling between hercynine and γ-glutamyl-cysteine to form a sulfoxide 30. Then, the glutamate group is removed by a glutamine amidotransamidase homologue (EgtC). The last step of ergothioneine biosynthesis is catalyzed by a PLP-dependent C–S lyase (EgtE), whose activity has also been demonstrated in vitro.79,80 Recently, OvoA, an EgtB homologue, was identified from one of the ovothiol A producers, Erwinia tasmaniensis.76 Recombinant OvoA catalyzes the oxidative coupling between L-His 27 and cysteine to a sulfoxide 33 in vitro (Figure 8B). However, the proposed C–S lyase and methyltransferase in ovothiol biosynthesis remain to be identified.
Biochemical Information about Ergothioneine and Ovothiol Biosynthesis
In ergothioneine and ovothiol biosyntheses, the trans-sulfuration method differs from those discussed previously (Figures 1–7). A combination of a non-heme iron enzyme-catalyzed oxidative C–S bond formation (EgtB, OvoA catalysis) and a reductive C–S lyase represents one of the most efficient sulfur transfer strategies in natural product biosyntheses. After the discovery of the mycobacterial EgtB gene, the functions of its homologues in fungi (N. crassa,81 Aspergillus fumigates,82 and the fission yeast, Schizosaccharomyces pombe83) have also been examined by genetic studies. A systematic analysis of all completed genomes in the NCBI database indicated that ergothioneine biosynthetic genes are widely distributed in actinobacteria,84 while EgtB and EgtD were found across proteobacterial and cyanobacterial species.85
EgtB and OvoA both mediate sulfoxide formation; however, they differ in their substrate selectivity and C–S bond formation regioselectivity (panel A vs panel B of Figure 8). In ergothioneine biosynthesis, EgtB makes use of hercynine and γ-glutamyl-cysteine as the substrates and the C–S bond is formed at the ε-position (Figure 8A). OvoA, on the other hand, uses L-His and L-Cys as the substrates, resulting in the C–S bond being formed at the δ-position (Figure 8B). Interestingly, with hercynine 28 as the substrate, OvoA catalyzes the oxidative coupling between hercynine and L-Cys to hercynyl-cysteine sulfoxide [31 (Figure 8C)].86,87 OvoA also mediates the oxidation of cysteine to cysteine sulfinic acid 34 (Figure 8C), an activity solely reported in cysteine dioxygenase (CDO).88 On the basis of the EgtB and OvoA biochemical information and guided by sequence similarity network analysis results, we identified and characterized the Egt1 gene from fungal N. crassa biochemically in vitro. The fungal Egt1 preferentially catalyzes the oxidative coupling between hercynine 28 and L-Cys to give hercyl-cysteine sulfoxide 31 (Figure 8A).70 These studies led to the elucidation of two aerobic ergothioneine biosynthetic pathways: the mycobacterial pathway77 [EgtA–EgtE (Figure 8A)] and the fungal N. crassa pathway70 [Egt1 and Egt2 (Figure 8A)].
The discoveries of these aerobic ergothioneine biosynthetic pathways are in line with the function of ergothioneine as a cellular antioxidant. Seebeck and co-workers analyzed the genomes of a green sulfur bacterium Chlorobium limicola DSM 245 and identified Clim_1148 (EanA) as an EgtD homologue.89 Ch. limicola grows in illuminated anoxic waters and conducts anaerobic photosynthesis using sulfides as the electron donor for carbon dioxide fixation. The recombinant EanA can catalyze the methylation of L-His 27 to hercynine 28. Analysis of the EanA gene neighborhood revealed a putative rhodanese-like sulfur transferase (EanB, Clim_1149). This pair of enzymes was found in at least 20 genomes from predominantly anaerobic bacteria and archaea. In the presence of cysteine desulfurase IscS, EanB can mediate the transfer of sulfur from cysteine to hercynine 28 to yield ergothioneine (Figure 8D). This surprising discovery contradicts the general hypothesis of having ergothioneine as an antioxidant, which removes reactive oxygen species (ROS) or reactive nitrogen species (RNS) generated under aerobic conditions.90 The presence of ergothioneine in anaerobic organisms raised the question of its biological functions. In addition, the trans-sulfuration catalyzed by EanB is intriguing because this transformation differs from other rhodanase-catalyzed sulfur transfer reactions (Figure 3). This discovery suggests that at least three different ergothioneine biosynthetic pathways exist in nature: two aerobic and one anaerobic (Figure 8A,D).91
Mechanistic Characterization of Oxidative C–S Bond Formation in Ergothioneine and Ovothiol Biosyntheses
The crystal structure of Mycobacterium thermoresistible EgtB has been reported.92 It has an N-terminal DinB-like four-helix bundle (residues 7–150) and a C-terminal C-type lectin fold. The EgtB structure contains few secondary structural elements and is rich in loops stabilized by buried ionic interactions (Figure 9A). The enzyme active site is in a 15 Å deep and 10 Å wide tunnel, and at the bottom of the tunnel, the catalytic iron is coordinated by three histidine residues (H51, H134, and H138). The Fe2+ center is also coordinated to three water ligands. When the Fe2+ center was replaced with Mn2+, the structure of the EgtB·dimethylhis-tidine·γ-glutamyl-cysteine tertiary complex was obtained (Figure 9B). Dimethylhistidine (DMH) coordinates to the Mn2+ center using its imidazole side chain, while γ-glutamyl-cysteine coordinates to the metallocenter using its side chain sulfur. The sixth ligand is a water molecule, which is most likely the site for oxygen binding and activation. In this structure, the water ligand is hydrogen-bonded to the hydroxyl group of an active site tyrosine residue (Y377). Upon mutation of Y377 to F377 in EgtB, the dominant activity of this mutant is the cysteine dioxygenase activity [kcat of 1.2 s−1 (Figure 8E)], instead of oxidative C–S bond formation in wild-type EgtB. This activity is at a level comparable to that of native CDO activity (kcat of 1.8 s−1).93,94 This discovery suggested that Y377 might serve as a general acid–base or be part of the redox chemistries in EgtB catalysis.
Figure 9.

M. thermoresistible EgtB structure. (A) Overall EgtB topology showing the N-terminus with a DinB-like helix bundle (blue) with the C-terminus containing a lectin fold (salmon/orange). EgtB contains a two-stranded β-sheet region (cyan). (B) EgtB tertiary complex with dimethylhistidine (DMH, green sticks) and γ-glutamyl-cysteine (γGC, magenta sticks) as the ligands of the Mn2+ center (purple sphere), which is coordinated by H51, H134, and H138. Notably, the water ligand (red sphere) also forms a hydrogen bond with active site Y377.
The kinetic parameters (kcat and Km values for hercynine and γ-glutamyl-cysteine) for EgtB Y377F are very similar to those of wild-type EgtB.93 As in the case of wild-type OvoA catalysis, the γ-glutamyl-cysteine dioxygenase activity depends on the presence of hercynine 28. In the absence of hercynine 28, γ-glutamyl-cysteine is oxidized to γ-glutamyl-cystine at a rate orders of magnitude lower than the γ-glutamyl-cysteine sulfinic acid or sulfoxide formation rates. Cysteine sulfinic acid formation and sulfoxide formation are possibly two pathways branching out from a common intermediate in both EgtB and OvoA catalysis (Figure 8).93,95
These two reactions have also been characterized by studies measuring the kinetic isotope effect (KIE). Using [ε-2H]-hercynine 28 as the substrate, no primary KIEs were observed. The solvent KIEs on wild-type EgtB and the EgtB Y377F mutant were measured for both the γ-glutamyl-cysteine dioxygenase activity and the sulfoxide synthase activity. The solvent KIEs for wild-type EgtB sulfoxide synthase and Y377F mutant γ-glutamyl-cysteine dioxygenase activity were 1.2 ± 0.2 and 0.9 ± 0.1, respectively. For the sulfoxide synthase activity of the EgtB Y377F mutant, the determined solvent KIE is 1.9 ± 0.1.93 The chemistries involved in the ergothioneine and ovothiol biosynthetic pathways are very similar (Figure 8) despite the differences in substrate selectivity and product regioselectivity between EgtB/Egt1 and OvoA.70,76,77,86,95 In addition, cysteine dioxygenase activity was discovered in OvoA (Figure 8C).95 Although wild-type EgtB does not show γ-glutamyl-cysteine dioxygenase activity, the dominant reaction in the EgtB Y377F mutant is γ-glutamyl-cysteine dioxygenase activity (Figure 8E).93 The discovery of sulfur oxidation88,96,97 in EgtB and OvoA adds another level of complexity to their mechanistic investigations. Both EgtB catalysis and OvoA catalysis are four-electron oxidations, in which molecular oxygen is fully reduced to water. In this process, oxidative C–S bond formation and formation of sulfoxide each provide two electrons. In EgtB and OvoA catalysis, it has yet to be determined whether C–S bond formation or sulfur oxidation occurs first. Several mechanistic options have been proposed for EgtB and OvoA reactions.76,95
Using EgtB catalysis as the example, the proposed mechanistic models and roles of Y377 are summarized (Figure 10). The EgtB mononuclear iron center has three histidine ligands and three exchangeable water ligands. In pathway I (Figure 10), the binding of substrates, γ-glutamyl-cysteine and hercynine, replaces two exchangeable H2O ligands. The remaining ligand site is for oxygen binding and activation to produce FeIII–superoxo species, which reacts with the thiolate to form a γ-glutamyl-cysteine sulfenic acid and an FeIV═O species [38b (Figure 10)]. After formation of the FeIV═O species, both two-electron chemistry and one-electron chemistries have been proposed for the subsequent steps [pathways IA–IC (Figure 10)].76,98–100 The FeIV═O species may abstract a hydrogen atom from the histidine imidazole ring δ-carbon to produce an imidazole-based radical [38c, pathway IA (Figure 10)]. Subsequently, the C–S bond forms by combining the thiol radical with the imidazole radical (38c → 30). Alternatively, one electron transfer from the imidazole ring to the FeIV═O species results in an imidazole cation radical [38d, pathway IB (Figure 10)]. After the C–S bond is formed (38d → 38e), Y377 is proposed to function as a base to deprotonate 38e to finish the catalytic cycle (38e → 30). It has also been suggested that direct nucleophilic attack by the imidazole on the sulfenic acid functional group leads to C–S bond formation [38b → 38e, pathway IC (Figure 10)]. Subsequent deprotonation of the imidazole ring in 38e by a base, potentially Y377, results in coupling product 30.
Figure 10.

Proposed mechanistic models for EgtB catalysis. (A) In pathway I, substrate binding and oxygen activation result in an Fe(III)–superoxo intermediate, which reacts with cysteine thiolate to form sulfenic acid and an FeIV═O species 38b. In this pathway, Y377 is proposed to function as a general base. In pathway II, after oxygen activation, FeIII–superoxo intermediate 38f oxidizes Y337 by a proton-coupled electron transfer process to generate a tyrosyl radical 38g. (B) Energy diagram of intermediates along pathway II in panel A based on quantum mechanics/molecular mechanics calculations.
Recently, the cysteine dioxygenase activity and the sulfoxide formation activity of EgtB have been examined through quantum mechanics/molecular mechanics (QM/MM)-based calculations (energy diagram in Figure 10B) using the M. thermoresistibile EgtB structure as a starting point.98 In the mechanistic model reported by Visser and co-workers, when oxygen binds to the iron center, the first intermediate is an FeIII–superoxo intermediate [38f, pathway II (Figure 10)]. In the subsequent step, the hydrogen bond network between Y377 and the active site water allows a proton-coupled electron transfer process to occur to produce FeIII–hydroperoxo species 38g along with a tyrosyl radical. Then, a nucleophilic or radical attack of the cysteine sulfur atom on the imidazole side chain of hercynine links the two substrates together through a thioether linkage (38g → 38h). Simultaneously with C–S bond formation, the hydrogen atom of hydroperoxo species is relayed back to the Y377-based radical to regenerate tyrosine and at the same time leads to an FeII–superoxo intermediate 38h. In the next step, the FeII–superoxo intermediate abstracts a hydrogen atom from the imidazole ring to regenerate the aromaticity and produces the FeII–hydroperoxo intermediate [38i (Figure 10)]. In this pathway, the C–S bond formation step, specifically, the formation of intermediate 38h is the rate-limiting step with an energy barrier of ~14.2 kcal/mol (Figure 10B). For 38h → 38i conversion, the energy barrier is ~4.4 kcal/mol and the transition from 38i to the product complex is energetically favorable (Figure 10B). The results from QM/MM-based mechanistic studies suggested that Y377 is involved in EgtB catalysis through a proton-coupled electron transfer process to reduce superoxo intermediate 38f to a hydroperoxo intermediate 38g. This model differs from the model proposed by Seebeck originally, in which the Y377 serves as a Lewis acid to provide a proton to peroxo species to produce a hydroperoxo intermediate.92 In addition, Visser and co-workers also proposed that the reduction of FeIII–superoxo intermediate 38f by a proton-coupled electron transfer process from Y337 plays the key role in suppressing thiol dioxygenase activity [38b → 37 (Figure 10A)].
Starting from the same M. thermoresistibile EgtB structure, Wei et al. also examined the EgtB catalytic mechanism using density functional theory.99 In this work, it is proposed that the first half of EgtB catalysis undergoes the sulfur oxidation to generate a sulfenic acid intermediate along with FeIV═O species 38b. Wei et al. proposed that the deprotonation of 38e is the rate-limiting step (pathway IB in Figure 10A), and a KIE as high as 5.7 was predicted. Thus far, KIEs measured from at least two reports were all close to unity, far from the predicted value of 5.7.87,100
There is currently no crystal structure for OvoA; therefore, we generated an OvoA structure model using I-TASSER.101 In this model, Y417 in OvoA is likely the EgtB Y377 counterpart.100 Wild-type OvoA has ~10% cysteine dioxygenase activity (Figure 8B). Replacing Y417 with a tyrosine analogue, 2-amino-3-[4-hydroxy-3-(methylthio)phenyl] propanoic acid [MtTyr, 39 (Figure 11A)], a mimic of the tyrosine–cysteine cross-link in cysteine dioxygenase,102–104 resulted in 30% cysteine dioxygenase activity (Figure 11A,B). In addition, for the OvoA Y417MtTyr variant, the amount of cysteine sulfinic acid further increases from 30% in H2O buffer to approximately 50% in D2O buffer. These results demonstrated that the two activities in OvoA catalysis can be modulated by replacing the active site tyrosine using a tyrosine analogue.100 Further systematic characterization using tyrosine analogues could provide mechanistic insights related to these novel transformations. Thus far, no mechanistic information for the anaerobic ergothioneine biosynthetic pathway is available (Figure 8D).89
Figure 11.
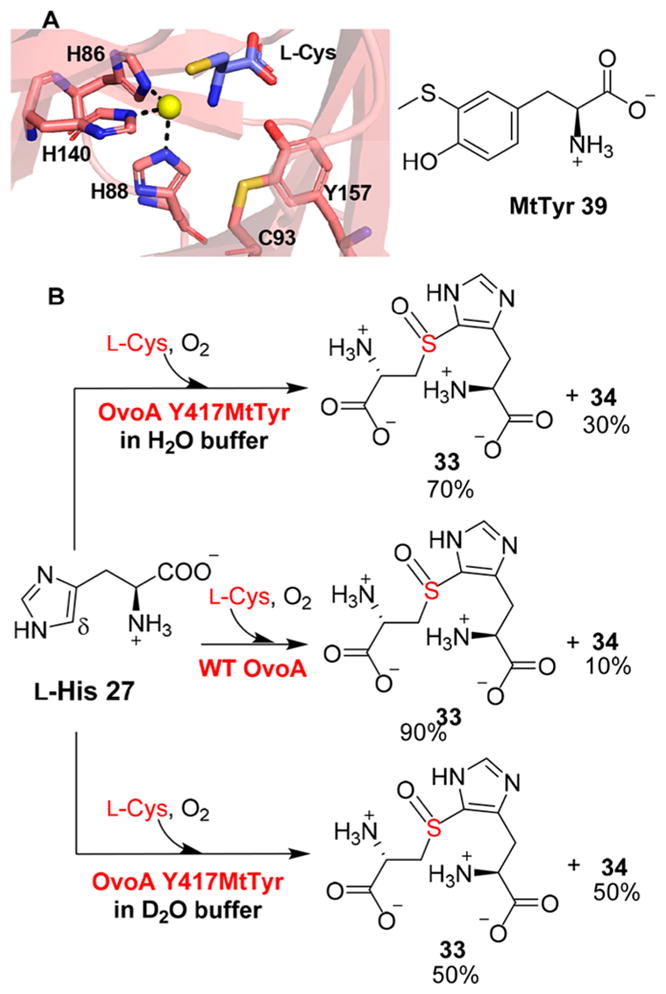
Modulating OvoA reactivity by replacing Y417 using a tyrosine analogue. (A) The active site of the cysteine dioxygenase crystal structure shows the tyrosine–cysteine cross-link, which is mimicked by a tyrosine analogue MtTyr in the OvoA Y417MtTyr variant. (B) Reaction results for wild-type OvoA and the Y417MtTyr variant, suggesting that the OvoA activities can be modulated by replacing Y417 using tyrosine analogues.
Mechanistic Studies of the C–S Lyase in Ergothioneine Biosynthesis
In ergothioneine and ovothiol biosynthesis, aside from the non-heme iron-dependent enzyme-catalyzed oxidative C–S bond formation (EgtB, Egt1, and OvoA catalysis), the other novel transformation is the EgtE/Egt2-catalyzed C–S lyase reaction. Most of the reported C–S lyases use thioethers as the substrates.105 The C–S lyase (EgtE/Egt2) in ergothioneine biosynthesis, however, is among the few lyases using sulfoxide substrates.79,106 Through a combination of biochemical and structural approaches, we have characterized how the reduction process is coupled with the C–S lyase reaction. Recombinant EgtE makes use of both thioether 40 and sulfoxide 31 as its substrate; pyruvate 41 and ammonia were produced as the side products (Figure 12A).79 Using thiol ether 40 as the substrate, ergothioneine was produced in a reductant-independent manner. When hercyl-cysteine sulfoxide 31 was used as the substrate in the absence of a reductant, ergothioneine 32 and ergothioneine-2-sulfinic acid 42b were isolated in a 1:1 ratio (Figure 12B). A plausible explanation for this result is that ergothioneine sulfenic acid 42 is the product of EgtE/Egt2 catalysis (Figure 12B). After intermediate 42 is released from the EgtE/Egt2 active site, a disproportionation reaction between two molecules of ergo-thioneine sulfenic acid 42 leads to the formation of a thio-sulfinic acid 42a, which is hydrolyzed to ergothioneine 32 and ergothioneine-2-sulfinic acid 42b (Figure 12B). The spontaneous decomposition of ergothioneine sulfinic acid 42b produces hercynine and SO2. In the presence of DTT, ergothioneine sulfenic acid 42 is reduced to ergothioneine (Figure 12A). The sulfenic acid intermediate 42 has also been trapped using 1,3-cyclohexanedione, and the adduct was isolated and characterized.80
Figure 12.

EgtE/Egt2 reactions. (A) C–S lyase EgtE reactions using sulfoxide 31 as the substrate in the presence of reductants or thioether 40 as the substrate in the absence of reductants. (B) Mechanistic model for the reaction using sulfoxide 31 as the substrate in the absence of reductants, in which ergothioneine 32 and hercynine 28 are produced in a 1:1 ratio.
Recently, we have successfully determined the crystal structure of Egt2 (the N. crossa ergothioneine C–S lyase).80 In its native structure, PLP is covalently linked to Egt2 protein through a Schiff base with K247. The pyridine ring of PLP interacts with the aromatic ring of Y134 via π–π stacking interactions. The Egt2 Y134F mutant is much less active than wild-type Egt2, while this Egt2 Y134F mutant still maintains some enzymatic activity [kcat/Km of (4.8 ± 0.1) × 10−3 μM−1 s−1 at pH 7.0, which is ~11.5-fold lower than that of wild-type Egt2]. The low activity of the Egt2 Y134F mutant allowed us to capture several intermediates in Egt2 catalysis (Figure 13). In one of the monomers, the active site electron density is consistent with the assignment of the Egt2 Y134F·substrate 31 binary complex prior to external aldimine intermediate formation (Figure 13B). When the crystals were transferred to a cryo buffer (pH 8.0), the electron density of the active site suggests that the chemical reaction has occurred, and a sulfenic acid intermediate in Egt2 catalysis was trapped (Figure 13C). In addition to the sulfenic acid intermediate, the positive density next to the PLP C4′ position in one of the EgtE monomers is consistent with an aminoacrylate geminal diamine intermediate (Figure 13D).
Figure 13.

Egt2 structure and our mechanistic model. (A) Overall structure of the Egt2 Y134F·substrate 31 binary complex in which the central catalytic domain is colored salmon. (B) Active site of the Egt2 Y134F·substrate 31 binary complex (sulfoxide 31 shown as magenta sticks and the PLP cofactor as yellow sticks). (C) Interactions between the active site residues and ergothioneine sulfenic acid intermediate 42 (shown as magenta sticks). (D) Aminoacrylate germinal diamine intermediate (shown as yellow sticks). (E) Proposed Egt2 mechanistic model. Sulfenic acid intermediate 42 could be trapped by C156 at the exit of the active site (pathway I) or be reduced directly (pathway II) to ergothioneine by reductants after it is released from the active site.
On the basis of these biochemical and structural studies, a mechanistic model was proposed (Figure 13E). Similar to other PLP-containing enzymes, the first step is the formation of external aldimine intermediate 31a between the PLP cofactor and sulfoxide substrate 31. After deprotonation of the Cys α-carbon to generate a quinonoid intermediate 31b, subsequent C–S bond cleavage produces ergothioneine sulfenic acid 42 and the PLP-based aminoacrylate intermediate, which decomposes to pyruvate 41 and ammonia. Egt2 structural analysis reveals a C156 residue located adjacent to the active site exit, implying that C156 might trap ergothioneine sulfenic acid 42 to produce a disulfide intermediate. This intermediate is then reduced to ergothioneine and regenerates C156 for the next cycle [pathway I (Figure 13E)]. Alternatively, ergothioneine sulfenic acid 42 formed might be released to the solution directly, which is then reduced to ergothioneine by either a small molecular reductant (e.g., glutathione) or a thiol reduction system [e.g., the thioredoxin/thioredoxin reductase pair, pathway II (Figure 13E)].
Ergothioneine Biological Functions
In humans, ergo-thioneine accumulates in many tissues with concentrations ranging from 100 μM to 2 mM, with the highest concentrations being in erythrocytes, bone marrow, liver, kidney, seminal fluid, and the lens and cornea of eyes,107–110 through an ergothioneine-specific transporter, OCTN1.110
The concentrations of ergothioneine in various tissues are correlated with the abundance of OCTN1 mRNA.111–113 In addition, when OCTN1 was deleted, ergothioneine accumulation is abolished, which suggests that OCTN1-mediated uptake is probably the sole ergothioneine uptake mechanism in animals.114,115 Ergothioneine has been suggested to play a role complementary to that of glutathione in vivo.90 The exact biological target of ergothioneine remains to be identified. Ergothioneine’s reduction potential (E0′ = −0.06 V3,116,117) is significantly higher than that of glutathione (E0′ = −0.25 V118). It has been suggested that the primary role of ergothioneine is as an antioxidant to eradicate reactive oxygen species (ROS) and reactive nitrogen species (RNS),90 including •OH, peroxyl radicals, peroxynitrite (ONOO−), nitrosoperoxycarbonate (ONOOCO2−), and carbonate radical. Several human diseases have been suggested to be related to ergothioneine, including rheumatoid arthritis,119,120 Crohn’s disease,121,122 neurodege-nerative diseases,123–126 cardiovascular disorders,127 and diabetes.128 Ergothioneine can pass through the blood–brain barrier123–125 and might serve as a protection mechanism against neurodegenerative diseases. Overstimulation of N-methyl-D-aspartate (NMDA) leads to neuronal cell death due to an increased level of production of free radicals.129 Intravitreal injection of NMDA in rats leads to a significant loss of retinal neurons. However, the NMDA excitotoxicity was relieved by intraperitoneal injection of ergothioneine.126 Ergothioneine treatment decreases the level of expression of adhesion molecules VACM-1, ICAM-1, and E-selectin. As a result, it inhibits the binding of monocytes to the endothelium,130 which might be the reason explaining ergothioneine’s protective effects against cardiovascular disorders.127
Because of the wide distribution of ergothioneine in both aerobic and anaerobic systems, it is tempting to assume that ergothioneine may have biological roles besides being a biological antioxidant.89,91 Recently, Zhao et al. reported the biochemical characterization of lincomycin A biosynthesis, in which ergothioneine plays a key role (Figure 14).131 Lincomycin 49 is an antibiotic produced from Streptomyces lincolnensis. It has an N-methyl-4-propyl-L-Pro (PPL) moiety and an unusual eight-carbon sugar (lincosamide) decorated with a C-1 position methylthiol group. In the licomycin A (49) biosynthetic gene cluster, there is a DinB-2-like domain-containing protein, LmbV. DinB-2 domain-containing proteins make up a large superfamily with more than 10000 members, which are involved in the transformations using small molecular thiols.132 LmbV was proposed to mediate the incorporation of the thiol group at the lincosamide C-1 position. In the ΔlmbV mutant, as anticipated, no lincomycin A was produced. Surprisingly, an ergothioneine S-conjugate 46 was isolated. To further characterize the function of LmbV, Zhao et al. overexpressed and isolated a LmbV homologue (CcbV, 57% identical to LmbV) because LmbV overexpression was challenging. When ergothioneine S-conjugate 46 and MSH were supplied as the CcbV substrates, MSH S-conjugate 47 was observed along with the formation of ergothioneine as the side product.131 This was the first discovery of ergothioneine being involved in sugar activation.
Figure 14.

Lincomycin A biosynthesis shows the involvement of two small molecular thiols, ergothioneine (EGT) and mycothiol (MSH).
Sequence analysis suggests that LmbT is a glycosyltransferase. In vitro characterization demonstrated that LmbT mediates the equilibrium between GDP-lincosamide and the lincosamide–ergothioneine conjugate [44 → 45 (Figure 14)]. The LmbT-catalyzed reaction exhibited an equilibrium constant Keq of 1.94, and the reaction slightly favors GDP-lincosamide 44.
Subsequent genetic and biochemical characterizations suggested that LmbC, LmbD, and LmbN work together to incorporate the PPL moiety into the lincosamide–ergothioneine conjugate to produce 46. LmbC encodes an adenylation protein, which activates PPL using ATP and transfers PPL onto the peptidyl carrier protein (PCP) domain of LmbN protein. LmbD then catalyzes the condensation between the activated PPL and EGT S-conjugate 45 to yield 46. In the lincomycin-producing organism, S. lincolnensis, LmbE is an amidase homologue. As predicted, a MSH-associated lincomycin analogue 47 accumulated in the ΔlmbE mutant strain. The recombinant LmbE rapidly converts 47 to 1-O-(2-N-acetyl)-glucosamine-D-myo-inositol-3-phosphate (GlcNAc-Ins-3-P) and 48. Thus far, most of the critical steps in licomycin A 49 biosynthesis have been established. This biosynthetic process involves two small molecular thiols, mycothiol (MSH) and ergothioneine (EGT).
Over the years, it has been suggested that small molecular thiols are involved in providing protection against oxidative stress or employed as tools to detoxify electrophilic toxins. The discovery of the key roles played by ergothioneine in lincomycin A biosynthesis is the first example of its involvement in a natural product biosynthetic pathway. Several biosynthetic pathways have been suggested to involve MSH S-conjugates. Given the presence of ergothioneine across biological systems, it is likely that many more roles of ergothioneine may be discovered in the future.133,134 S-Alk(en)yl-L-Cys sulfoxides have been discovered as metabolites in many plants, and some of them play important biological roles, e.g., suppression of hypercholesterolemia,135 stimulation of insulin secretion,136 or priming neutrophils for respiratory burst.137 In addition to an examination of the roles of ergothioneine and ovothiol, it may also be beneficial to characterize the biological function of ergothioneine and ovothiol biosynthetic intermediates (Figure 8).
In summary, ergothioneine and ovothiol biosynthetic studies not only provide an excellent system for characterizing new trans-sulfuration strategies in nature but also offer ample opportunities for future investigations of the biological functions of small molecular thiols.
Acknowledgments
Funding
This work is supported in part by grants from the National Institutes of Health (R01 GM093903) and the National Science Foundation (CHE-1309148) to P.L. and a grant from National Natural Science Foundation of China (31670030) to C.Z. L.C. and C.Z. are supported by fellowships from the China Scholarship Council. N.N. is supported by a Warren-McLoed fellowship from the Boston University Marine Program.
Footnotes
Notes
The authors declare no competing financial interest.
References
- 1.Kessler D. Enzymatic activation of sulfur for incorporation into biomolecules in prokaryotes. FEMS Microbiol Rev. 2006;30:825–840. doi: 10.1111/j.1574-6976.2006.00036.x. [DOI] [PubMed] [Google Scholar]
- 2.Hand CE, Honek JF. Biological chemistry of naturally occurring thiols of microbial and marine origin. J Nat Prod. 2005;68:293–308. doi: 10.1021/np049685x. [DOI] [PubMed] [Google Scholar]
- 3.Fahey RC. Novel thiols of prokaryotes. Annu Rev Microbiol. 2001;55:333–356. doi: 10.1146/annurev.micro.55.1.333. [DOI] [PubMed] [Google Scholar]
- 4.Mueller EG. Trafficking in persulfides: delivering sulfur in biosynthetic pathways. Nat Chem Biol. 2006;2:185–194. doi: 10.1038/nchembio779. [DOI] [PubMed] [Google Scholar]
- 5.Shigi N. Biosynthesis and functions of sulfur modifications in tRNA. Front Genet. 2014;5:67. doi: 10.3389/fgene.2014.00067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fontecave M, Atta M, Mulliez E. S-adenosylme-thionine: nothing goes to waste. Trends Biochem Sci. 2004;29:243–249. doi: 10.1016/j.tibs.2004.03.007. [DOI] [PubMed] [Google Scholar]
- 7.Struck AW, Thompson ML, Wong LS, Micklefield J. S-adenosyl-methionine-dependent methyltransferases: highly versatile enzymes in biocatalysis, biosynthesis and other biotechnological applications. ChemBioChem. 2012;13:2642–2655. doi: 10.1002/cbic.201200556. [DOI] [PubMed] [Google Scholar]
- 8.Beld J, Sonnenschein EC, Vickery CR, Noel JP, Burkart MD. The phosphopantetheinyl transferases: catalysis of a post-translational modification crucial for life. Nat Prod Rep. 2014;31:61–108. doi: 10.1039/c3np70054b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Begley TP, Xi J, Kinsland C, Taylor S, McLafferty F. The enzymology of sulfur activation during thiamin and biotin biosynthesis. Curr Opin Chem Biol. 1999;3:623–629. doi: 10.1016/s1367-5931(99)00018-6. [DOI] [PubMed] [Google Scholar]
- 10.Jurgenson CT, Begley TP, Ealick SE. The structural and biochemical foundations of thiamin biosynthesis. Annu Rev Biochem. 2009;78:569–603. doi: 10.1146/annurev.biochem.78.072407.102340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Berkovitch F, Nicolet Y, Wan JT, Jarrett JT, Drennan CL. Crystal structure of biotin synthase, an S-adenosylmethionine-dependent radical enzyme. Science. 2004;303:76–79. doi: 10.1126/science.1088493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ugulava NB, Frederick KK, Jarrett JT. Control of adenosylmethionine-dependent radical generation in biotin synthase: a kinetic and thermodynamic analysis of substrate binding to active and inactive forms of BioB. Biochemistry. 2003;42:2708–2719. doi: 10.1021/bi0261084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ugulava NB, Gibney BR, Jarrett JT. Biotin synthase contains two distinct iron-sulfur cluster binding sites: chemical and spectroelectrochemical analysis of iron-sulfur cluster interconversions. Biochemistry. 2001;40:8343–8351. doi: 10.1021/bi0104625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ugulava NB, Sacanell CJ, Jarrett JT. Spectroscopic changes during a single turnover of biotin synthase: destruction of a [2Fe-2S] cluster accompanies sulfur insertion. Biochemistry. 2001;40:8352–8358. doi: 10.1021/bi010463x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Booker SJ, Cicchillo RM, Grove TL. Self-sacrifice in radical S-adenosylmethionine proteins. Curr Opin Chem Biol. 2007;11:543–552. doi: 10.1016/j.cbpa.2007.08.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schwarz G, Mendel RR, Ribbe MW. Molybdenum cofactors, enzymes and pathways. Nature. 2009;460:839–847. doi: 10.1038/nature08302. [DOI] [PubMed] [Google Scholar]
- 17.Graham DE, White RH. Elucidation of methanogenic coenzyme biosyntheses: from spectroscopy to genomics. Nat Prod Rep. 2002;19:133–147. doi: 10.1039/b103714p. [DOI] [PubMed] [Google Scholar]
- 18.Fontecave M, Ollagnier-de-Choudens S, Mulliez E. Biological radical sulfur insertion reactions. Chem Rev. 2003;103:2149–2166. doi: 10.1021/cr020427j. [DOI] [PubMed] [Google Scholar]
- 19.Graham DE, Xu HM, White RH. Identification of coenzyme M biosynthetic phosphosulfolactate synthase - A new family of sulfonate-biosynthesizing enzymes. J Biol Chem. 2002;277:13421–13429. doi: 10.1074/jbc.M201011200. [DOI] [PubMed] [Google Scholar]
- 20.Dunbar KL, Scharf DH, Litomska A, Hertweck C. Enzymatic carbon-sulfur bond formation in natural product biosynthesis. Chem Rev. 2017;117:5521–5577. doi: 10.1021/acs.chemrev.6b00697. [DOI] [PubMed] [Google Scholar]
- 21.Repka LM, Chekan JR, Nair SK, van der Donk WA. Mechanistic understanding of lanthipeptide biosynthetic enzymes. Chem Rev. 2017;117:5457–5520. doi: 10.1021/acs.chemrev.6b00591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Waldman AJ, Ng TL, Wang P, Balskus EP. Heteroatom-heteroatom bond formation in natural product biosynthesis. Chem Rev. 2017;117:5784–5863. doi: 10.1021/acs.chemrev.6b00621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gilbert JA, Jansson JK, Knight R. The Earth Microbiome Project: successes and aspirations. BMC Biol. 2014;12:69. doi: 10.1186/s12915-014-0069-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li B, Yu JPJ, Brunzelle JS, Moll GN, van der Donk WA, Nair SK. Structure and mechanism of the lantibiotic cyclase involved in nisin biosynthesis. Science. 2006;311:1464–1467. doi: 10.1126/science.1121422. [DOI] [PubMed] [Google Scholar]
- 25.Willey JM, van der Donk WA. Lantibiotics: peptides of diverse structure and function. Annu Rev Microbiol. 2007;61:477–501. doi: 10.1146/annurev.micro.61.080706.093501. [DOI] [PubMed] [Google Scholar]
- 26.Davis C, Carberry S, Schrettl M, Singh I, Stephens JC, Barry SM, Kavanagh K, Challis GL, Brougham D, Doyle S. The role of glutathione S-transferase GliG in gliotoxin biosynthesis in Aspergillus fumigatus. Chem Biol. 2011;18:542–552. doi: 10.1016/j.chembiol.2010.12.022. [DOI] [PubMed] [Google Scholar]
- 27.Scharf DH, Chankhamjon P, Scherlach K, Heinekamp T, Willing K, Brakhage AA, Hertweck C. Epidithiodike-topiperazine biosynthesis: a four-enzyme cascade converts glutathione conjugates into transannular disulfide bridges. Angew Chem, Int Ed. 2013;52:11092–11095. doi: 10.1002/anie.201305059. [DOI] [PubMed] [Google Scholar]
- 28.Haeggstrom JZ, Funk CD. Lipoxygenase and leukotriene pathways: biochemistry, biology, and roles in disease. Chem Rev. 2011;111:5866–5898. doi: 10.1021/cr200246d. [DOI] [PubMed] [Google Scholar]
- 29.Hammerstrom S, Samuelsson B. Detection of leukotriene-A4 as an intermediate in the biosynthesis of leukotriene-C4 and leukotriene-D4. FEBS Lett. 1980;122:83–86. doi: 10.1016/0014-5793(80)80407-8. [DOI] [PubMed] [Google Scholar]
- 30.Hansen CH, Du L, Naur P, Olsen CE, Axelsen KB, Hick AJ, Pickett JA, Halkier BA. CYP83b1 is the oxime-metabolizing enzyme in the glucosinolate pathway in Arabidopsis. J Biol Chem. 2001;276:24790–24796. doi: 10.1074/jbc.M102637200. [DOI] [PubMed] [Google Scholar]
- 31.Dolan SK, O’Keeffe G, Jones GW, Doyle S. Resistance is not futile: gliotoxin biosynthesis, functionality and utility. Trends Microbiol. 2015;23:419–428. doi: 10.1016/j.tim.2015.02.005. [DOI] [PubMed] [Google Scholar]
- 32.Welch TR, Williams RM. Epidithiodioxopiperazines occurrence, synthesis and biogenesis. Nat Prod Rep. 2014;31:1376–1404. doi: 10.1039/c3np70097f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hidese R, Mihara H, Esaki N. Bacterial cysteine desulfurases: versatile key players in biosynthetic pathways of sulfur-containing biofactors. Appl Microbiol Biotechnol. 2011;91:47–61. doi: 10.1007/s00253-011-3336-x. [DOI] [PubMed] [Google Scholar]
- 34.Dahl JU, Urban A, Bolte A, Sriyabhaya P, Donahue JL, Nimtz M, Larson TJ, Leimkuhler S. The identification of a novel protein involved in molybdenum cofactor biosynthesis in Escherichia coli. J Biol Chem. 2011;286:35801–35812. doi: 10.1074/jbc.M111.282368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Webb ME, Marquet A, Mendel RR, Rebeille F, Smith AG. Elucidating biosynthetic pathways for vitamins and cofactors. Nat Prod Rep. 2007;24:988–1008. doi: 10.1039/b703105j. [DOI] [PubMed] [Google Scholar]
- 36.Cavuzic M, Liu Y. Biosynthesis of sulfur-containing tRNA modifications: A comparison of bacterial, archaeal, and eukaryotic pathways. Biomolecules. 2017;7:27. doi: 10.3390/biom7010027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sasaki E, Zhang X, Sun HG, Lu MY, Liu TL, Ou A, Li JY, Chen YH, Ealick SE, Liu HW. Co-opting sulphur-carrier proteins from primary metabolic pathways for 2-thiosugar biosynthesis. Nature. 2014;510:427–431. doi: 10.1038/nature13256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Matthijs S, Baysse C, Koedam N, Tehrani KA, Verheyden L, Budzikiewicz H, Schafer M, Hoorelbeke B, Meyer JM, De Greve H, Cornelis P. The Pseudomonas siderophore quinolobactin is synthesized from xanthurenic acid, an intermediate of the kynurenine pathway. Mol Microbiol. 2004;52:371–384. doi: 10.1111/j.1365-2958.2004.03999.x. [DOI] [PubMed] [Google Scholar]
- 39.Godert AM, Jin M, McLafferty FW, Begley TP. Biosynthesis of the thioquinolobactin siderophore: an interesting variation on sulfur transfer. J Bacteriol. 2007;189:2941–2944. doi: 10.1128/JB.01200-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tang X, Li J, Millan-Aguinaga N, Zhang JJ, O’Neill EC, Ugalde JA, Jensen PR, Mantovani SM, Moore BS. Identification of thiotetronic acid antibiotic biosynthetic pathways by target-directed genome mining. ACS Chem Biol. 2015;10:2841–2849. doi: 10.1021/acschembio.5b00658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yurkovich ME, Jenkins R, Sun Y, Tosin M, Leadlay PF. The polyketide backbone of thiolactomycin is assembled by an unusual iterative polyketide synthase. Chem Commun. 2017;53:2182–2185. doi: 10.1039/c6cc09934c. [DOI] [PubMed] [Google Scholar]
- 42.Tao W, Yurkovich ME, Wen S, Lebe KE, Samborskyy M, Liu Y, Yang A, Liu Y, Ju Y, Deng Z, Tosin M, Sun Y, Leadlay PF. A genomics-led approach to deciphering the mechanism of thiotetronate antibiotic biosynthesis. Chem Sci. 2016;7:376–385. doi: 10.1039/c5sc03059e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Coyne S, Chizzali C, Khalil MN, Litomska A, Richter K, Beerhues L, Hertweck C. Biosynthesis of the antimetabolite 6-thioguanine in Erwinia amylovora plays a key role in fire blight pathogenesis. Angew Chem, Int Ed. 2013;52:10564–10568. doi: 10.1002/anie.201305595. [DOI] [PubMed] [Google Scholar]
- 44.Numata T, Ikeuchi Y, Fukai S, Suzuki T, Nureki O. Snapshots of tRNA sulphuration via an adenylated intermediate. Nature. 2006;442:419–424. doi: 10.1038/nature04896. [DOI] [PubMed] [Google Scholar]
- 45.Sasaki E, Liu HW. Mechanistic studies of the biosynthesis of 2-thiosugar: evidence for the formation of an enzyme-bound 2-ketohexose intermediate in BexX-catalyzed reaction. J Am Chem Soc. 2010;132:15544–15546. doi: 10.1021/ja108061c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Broderick JB, Duffus BR, Duschene KS, Shepard EM. Radical S-adenosylmethionine enzymes. Chem Rev. 2014;114:4229–4317. doi: 10.1021/cr4004709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lanz ND, Booker SJ. Identification and function of auxiliary iron–sulfur clusters in radical SAM enzymes. Biochim Biophys Acta, Proteins Proteomics. 2012;1824:1196–1212. doi: 10.1016/j.bbapap.2012.07.009. [DOI] [PubMed] [Google Scholar]
- 48.McCarthy EL, Booker SJ. Destruction and reformation of an iron-sulfur cluster during catalysis by lipoyl synthase. Science. 2017;358:373–377. doi: 10.1126/science.aan4574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fluhe L, Marahiel MA. Radical S-adenosylmethionine enzyme catalyzed thioether bond formation in sactipeptide biosynthesis. Curr Opin Chem Biol. 2013;17:605–612. doi: 10.1016/j.cbpa.2013.06.031. [DOI] [PubMed] [Google Scholar]
- 50.Bruender NA, Wilcoxen J, Britt RD, Bandarian V. Biochemical and spectroscopic characterization of a radical S-adenosyl-L-methionine enzyme involved in the formation of a peptide thioether cross-link. Biochemistry. 2016;55:2122–2134. doi: 10.1021/acs.biochem.6b00145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fluhe L, Knappe TA, Gattner MJ, Schafer A, Burghaus O, Linne U, Marahiel MA. The radical SAM enzyme AlbA catalyzes thioether bond formation in subtilosin A. Nat Chem Biol. 2012;8:350–357. doi: 10.1038/nchembio.798. [DOI] [PubMed] [Google Scholar]
- 52.Fluhe L, Burghaus O, Wieckowski BM, Giessen TW, Linne U, Marahiel MA. Two [4Fe-4S] clusters containing radical SAM enzyme SkfB catalyze thioether bond formation during the maturation of the sporulation killing factor. J Am Chem Soc. 2013;135:959–962. doi: 10.1021/ja310542g. [DOI] [PubMed] [Google Scholar]
- 53.Wieckowski BM, Hegemann JD, Mielcarek A, Boss L, Burghaus O, Marahiel MA. The PqqD homologous domain of the radical SAM enzyme ThnB is required for thioether bond formation during thurincin H maturation. FEBS Lett. 2015;589:1802–1806. doi: 10.1016/j.febslet.2015.05.032. [DOI] [PubMed] [Google Scholar]
- 54.Benjdia A, Guillot A, Lefranc B, Vaudry H, Leprince J, Berteau O. Thioether bond formation by SPASM domain radical SAM enzymes: CαH-atom abstraction in subtilosin A biosynthesis. Chem Commun. 2016;52:6249–6252. doi: 10.1039/c6cc01317a. [DOI] [PubMed] [Google Scholar]
- 55.Baldwin JE, Bradley M. Isopenicillin-N synthase -mechanistic studies. Chem Rev. 1990;90:1079–1088. [Google Scholar]
- 56.Roach PL, Clifton IJ, Fulop V, Harlos K, Barton GJ, Hajdu J, Andersson I, Schofield CJ, Baldwin JE. Crystal structure of isopenicillin N synthase is the first from a new structural family of enzymes. Nature. 1995;375:700–704. doi: 10.1038/375700a0. [DOI] [PubMed] [Google Scholar]
- 57.Tamanaha E, Zhang B, Guo Y, Chang W-c, Barr EW, Xing G, St Clair J, Ye S, Neese F, Bollinger JM, Jr, Krebs C. Spectroscopic Evidence for the Two C–H-Cleaving Intermediates of Aspergillus nidulans Isopenicillin N Synthase. J Am Chem Soc. 2016;138:8862–8874. doi: 10.1021/jacs.6b04065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tanret C. Sur une base nouvelle retiree du seigle ergote, l’ergothioneine. C R Acad Sci. 1909;149:222–224. [Google Scholar]
- 59.Genghof DS, Inamine E, Kovalenko V, Melville DB. Ergothioneine in microorganisms. J Biol Chem. 1956;223:9–17. [PubMed] [Google Scholar]
- 60.Genghof DS, Vandamme O. Biosynthesis of ergothioneine and hercynine by mycobacteria. J Bacteriol. 1964;87:852–862. doi: 10.1128/jb.87.4.852-862.1964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Genghof DS, Van Damme O. Biosynthesis of ergothioneine from endogenous hercynine in Mycobacterium smegmatis. J Bacteriol. 1968;95:340–344. doi: 10.1128/jb.95.2.340-344.1968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Heath H, Wildy J. Biosynthesis of ergothioneine. Nature. 1957;179:196–197. doi: 10.1038/179196a0. [DOI] [PubMed] [Google Scholar]
- 63.Melville DB, Eich S, Ludwig ML. The biosynthesis of ergothioneine. J Biol Chem. 1957;224:871–877. [PubMed] [Google Scholar]
- 64.Heath H, Wildy J. The biosynthesis of ergothioneine and histidine by Claviceps purpurea I The incorporation of [2-14C]-acetate. Biochem J. 1956;64:612–620. doi: 10.1042/bj0640612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Heath H, Wildy J. Biosynthesis of ergothioneine by Claviceps purpurea III The incorporation of labelled histidine. Biochem J. 1958;68:407–410. doi: 10.1042/bj0680407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Askari A, Melville DB. The reaction sequence in ergothioneine biosynthesis: hercynine as an intermediate. J Biol Chem. 1962;237:1615–1618. [PubMed] [Google Scholar]
- 67.Reinhold VN, Ishikawa Y, Melville DB. Conversion of histidine to hercynine by Neurospora crassa. J Bacteriol. 1970;101:881–884. doi: 10.1128/jb.101.3.881-884.1970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ishikawa Y, Melville DB. The enzymatic a-N-methylation of histidine. J Biol Chem. 1970;245:5967–5973. [PubMed] [Google Scholar]
- 69.Ishikawa Y, Israel SE, Melville DB. Participation of an intermediate sulfoxide in the enzymatic thiolation of the imidazole ring of hercynine to form ergothioneine. J Biol Chem. 1974;249:4420–4427. [PubMed] [Google Scholar]
- 70.Hu W, Song H, Sae Her A, Bak DW, Naowarojna N, Elliott SJ, Qin L, Chen X, Liu P. Bioinformatic and biochemical characterizations of C-S bond formation and cleavage enzymes in the fungus Neurospora crassa ergothioneine biosynthetic pathway. Org Lett. 2014;16:5382–5385. doi: 10.1021/ol502596z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Shapiro BM, Hopkins PB. Ovothiols: biological and chemical perspectives. Adv Enzymol Relat Areas Mol Biol. 2006;64:291–316. doi: 10.1002/9780470123102.ch6. [DOI] [PubMed] [Google Scholar]
- 72.Spies HS, Steenkamp DJ. Thiols of intracellular pathogens. Identification of ovothiol A in Leishmania donovani and structural analysis of a novel thiol from Mycobacterium bovis. Eur J Biochem. 1994;224:203–213. doi: 10.1111/j.1432-1033.1994.tb20013.x. [DOI] [PubMed] [Google Scholar]
- 73.Steenkamp DJ, Spies HS. Identification of a major low-molecular-mass thiol of the trypanosomatid Crithidia fasciculata as ovothiol A. Facile isolation and structural analysis of the bimane derivative. Eur J Biochem. 1994;223:43–50. doi: 10.1111/j.1432-1033.1994.tb18964.x. [DOI] [PubMed] [Google Scholar]
- 74.Steenkamp DJ, Weldrick D, Spies HS. Studies on the biosynthesis of ovothiol A. Eur J Biochem. 1996;242:557–566. doi: 10.1111/j.1432-1033.1996.0557r.x. [DOI] [PubMed] [Google Scholar]
- 75.Vogt RN, Spies HS, Steenkamp DJ. The biosynthesis of ovothiol A (N-methyl-4-mercaptohistidine). Identification of S-(4′-L-histidyl)-L-cysteine sulfoxide as an intermediate and the products of the sulfoxide lyase reaction. Eur J Biochem. 2001;268:5229–5241. doi: 10.1046/j.0014-2956.2001.02444.x. [DOI] [PubMed] [Google Scholar]
- 76.Braunshausen A, Seebeck FP. Identification and characterization of the first ovothiol biosynthetic enzyme. J Am Chem Soc. 2011;133:1757–1759. doi: 10.1021/ja109378e. [DOI] [PubMed] [Google Scholar]
- 77.Seebeck FP. In vitro reconstitution of mycobacterial ergothioneine biosynthesis. J Am Chem Soc. 2010;132:6632–6633. doi: 10.1021/ja101721e. [DOI] [PubMed] [Google Scholar]
- 78.Vit A, Misson L, Blankenfeldt W, Seebeck FP. Ergothioneine biosynthetic methyltransferase EgtD reveals the structural basis of aromatic amino acid betaine biosynthesis. ChemBioChem. 2015;16:119–125. doi: 10.1002/cbic.201402522. [DOI] [PubMed] [Google Scholar]
- 79.Song H, Hu W, Naowarojna N, Her AS, Wang S, Desai R, Qin L, Chen X, Liu P. Mechanistic studies of a novel C-S lyase in ergothioneine biosynthesis: the involvement of a sulfenic acid intermediate. Sci Rep. 2015;5:11870. doi: 10.1038/srep11870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Irani S, Naowarojna N, Tang Y, Kathuria KR, Wang S, Dhembi A, Lee N, Yan W, Lyu H, Costello CE, Liu P, Zhang YJ. Snapshots of CS Cleavage in Egt2 Reveals Substrate Specificity and Reaction Mechanism. Cell Chem Biol. 2018 doi: 10.1016/j.chembiol.2018.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Bello MH, Barrera-Perez V, Morin D, Epstein L. The Neurospora crassa mutant NcΔEgt-1 identifies an ergothioneine biosynthetic gene and demonstrates that ergothioneine enhances conidial survival and protects against peroxide toxicity during conidial germination. Fungal Genet Biol. 2012;49:160–172. doi: 10.1016/j.fgb.2011.12.007. [DOI] [PubMed] [Google Scholar]
- 82.Sheridan KJ, Lechner BE, O’Keeffe G, Keller MA, Werner ER, Lindner H, Jones GW, Haas H, Doyle S. Ergothioneine biosynthesis and functionality in the opportunistic fungal pathogen, Aspergillus fumigatus. Sci Rep. 2016;6:35306. doi: 10.1038/srep35306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Pluskal T, Ueno M, Yanagida M. Genetic and metabolomic dissection of the ergothioneine and selenoneine biosynthetic pathway in the fission yeast, S. pombe, and construction of an overproduction system. PLoS One. 2014;9:e97774. doi: 10.1371/journal.pone.0097774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Jones GW, Doyle S, Fitzpatrick DA. The evolutionary history of the genes involved in the biosynthesis of the antioxidant ergothioneine. Gene. 2014;549:161–170. doi: 10.1016/j.gene.2014.07.065. [DOI] [PubMed] [Google Scholar]
- 85.Liao C, Seebeck FP. Convergent evolution of ergothioneine biosynthesis in cyanobacteria. ChemBioChem. 2017;18:2115–2118. doi: 10.1002/cbic.201700354. [DOI] [PubMed] [Google Scholar]
- 86.Song H, Leninger M, Lee N, Liu P. Regioselectivity of the oxidative C–S bond formation in ergothioneine and ovothiol biosyntheses. Org Lett. 2013;15:4854–4857. doi: 10.1021/ol402275t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Mashabela GT, Seebeck FP. Substrate specificity of an oxygen dependent sulfoxide synthase in ovothiol biosynthesis. Chem Commun. 2013;49:7714–7716. doi: 10.1039/c3cc42594k. [DOI] [PubMed] [Google Scholar]
- 88.Joseph CA, Maroney MJ. Cysteine dioxygenase: structure and mechanism. Chem Commun. 2007:3338–3349. doi: 10.1039/b702158e. [DOI] [PubMed] [Google Scholar]
- 89.Burn R, Misson L, Meury M, Seebeck FP. Anaerobic origin of ergothioneine. Angew Chem, Int Ed. 2017;56:12508–12511. doi: 10.1002/anie.201705932. [DOI] [PubMed] [Google Scholar]
- 90.Cheah IK, Halliwell B. Ergothioneine; antioxidant potential, physiological function and role in disease. Biochim Biophys Acta, Mol Basis Dis. 2012;1822:784–793. doi: 10.1016/j.bbadis.2011.09.017. [DOI] [PubMed] [Google Scholar]
- 91.Ruszczycky MW, Liu HW. The surprising history of an antioxidant. Nature. 2017;551:37–38. doi: 10.1038/551037a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Goncharenko KV, Vit A, Blankenfeldt W, Seebeck FP. Structure of the sulfoxide synthase EgtB from the ergothioneine biosynthetic pathway. Angew Chem, Int Ed. 2015;54:2821–2824. doi: 10.1002/anie.201410045. [DOI] [PubMed] [Google Scholar]
- 93.Goncharenko KV, Seebeck FP. Conversion of a non-heme iron-dependent sulfoxide synthase into a thiol dioxygenase by a single point mutation. Chem Commun. 2016;52:1945–1948. doi: 10.1039/c5cc07772a. [DOI] [PubMed] [Google Scholar]
- 94.Li W, Pierce BS. Steady-state substrate specificity and O2-coupling efficiency of mouse cysteine dioxygenase. Arch Biochem Biophys. 2015;565:49–56. doi: 10.1016/j.abb.2014.11.004. [DOI] [PubMed] [Google Scholar]
- 95.Song H, Her AS, Raso F, Zhen Z, Huo Y, Liu P. Cysteine oxidation reactions catalyzed by a mononuclear non-heme iron enzyme (OvoA) in ovothiol biosynthesis. Org Lett. 2014;16:2122–2125. doi: 10.1021/ol5005438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Kumar D, Thiel W, de Visser SP. Theoretical study on the mechanism of the oxygen activation process in cysteine dioxygenase enzymes. J Am Chem Soc. 2011;133:3869–3882. doi: 10.1021/ja107514f. [DOI] [PubMed] [Google Scholar]
- 97.Aluri S, de Visser SP. The mechanism of cysteine oxygenation by cysteine dioxygenase enzymes. J Am Chem Soc. 2007;129:14846–14847. doi: 10.1021/ja0758178. [DOI] [PubMed] [Google Scholar]
- 98.Faponle AS, Seebeck FP, de Visser SP. Sulfoxide synthase versus cysteine dioxygenase reactivity in a nonheme iron enzyme. J Am Chem Soc. 2017;139:9259–9270. doi: 10.1021/jacs.7b04251. [DOI] [PubMed] [Google Scholar]
- 99.Wei WJ, Siegbahn PE, Liao RZ. Theoretical study of the mechanism of the non-heme iron enzyme EgtB. Inorg Chem. 2017;56:3589–3599. doi: 10.1021/acs.inorgchem.6b03177. [DOI] [PubMed] [Google Scholar]
- 100.Chen L, Naowarojna N, Song H, Wang S, Wang J, Deng Z, Zhao C, Liu P. Use of a tyrosine analog to modulate the two activities of a non-heme iron enzyme OvoA in ovothiol biosynthesis, cysteine oxidation versus oxidative C-S bond formation. J Am Chem Soc. 2018:4604–4612. doi: 10.1021/jacs.7b13628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Yang J, Yan R, Roy A, Xu D, Poisson J, Zhang Y. The I-TASSER Suite: protein structure and function prediction. Nat Methods. 2015;12:7. doi: 10.1038/nmeth.3213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Simmons CR, Liu Q, Huang Q, Hao Q, Begley TP, Karplus PA, Stipanuk MH. Crystal structure of mammalian cysteine dioxygenase. A novel mononuclear iron center for cysteine thiol oxidation. J Biol Chem. 2006;281:18723–18733. doi: 10.1074/jbc.M601555200. [DOI] [PubMed] [Google Scholar]
- 103.McCoy JG, Bailey LJ, Bitto E, Bingman CA, Aceti DJ, Fox BG, Phillips GN. Structure and mechanism of mouse cysteine dioxygenase. Proc Natl Acad Sci U S A. 2006;103:3084–3089. doi: 10.1073/pnas.0509262103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Ye S, Wu Xa, Wei L, Tang D, Sun P, Bartlam M, Rao Z. An insight into the mechanism of human cysteine dioxygenase. Key roles of the thioether-bonded tyrosine-cysteine cofactor. J Biol Chem. 2007;282:3391–3402. doi: 10.1074/jbc.M609337200. [DOI] [PubMed] [Google Scholar]
- 105.Jacob C. A scent of therapy: pharmacological implications of natural products containing redox-active sulfur atoms. Nat Prod Rep. 2006;23:851–863. doi: 10.1039/b609523m. [DOI] [PubMed] [Google Scholar]
- 106.Kuettner EB, Hilgenfeld R, Weiss MS. The active principle of garlic at atomic resolution. J Biol Chem. 2002;277:46402–46407. doi: 10.1074/jbc.M208669200. [DOI] [PubMed] [Google Scholar]
- 107.Melville DB, Horner WH, Lubschez R. Tissue ergothioneine. J Biol Chem. 1954;206:221–228. [PubMed] [Google Scholar]
- 108.Shires TK, Brummel MC, Pulido JS, Stegink LD. Ergothioneine distribution in bovine and porcine ocular tissues. Comp Biochem Physiol, Part C: Pharmacol, Toxicol Endocrinol. 1997;117:117–120. doi: 10.1016/s0742-8413(96)00223-x. [DOI] [PubMed] [Google Scholar]
- 109.Leone E, Mann T. Ergothioneine in the seminal vesicle secretion. Nature. 1951;168:205. doi: 10.1038/168205b0. [DOI] [PubMed] [Google Scholar]
- 110.Grundemann D, Harlfinger S, Golz S, Geerts A, Lazar A, Berkels R, Jung N, Rubbert A, Schomig E. Discovery of the ergothioneine transporter. Proc Natl Acad Sci U S A. 2005;102:5256–5261. doi: 10.1073/pnas.0408624102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Nakamura T, Yoshida K, Yabuuchi H, Maeda T, Tamai I. Functional characterization of ergothioneine transport by rat organic cation/carnitine transporter Octn1 (slc22a4) Biol Pharm Bull. 2008;31:1580–1584. doi: 10.1248/bpb.31.1580. [DOI] [PubMed] [Google Scholar]
- 112.Taubert D, Jung N, Goeser T, Schömig E. Increased ergothioneine tissue concentrations in carriers of the Crohn’s disease risk-associated 503F variant of the organic cation transporter OCTN1. Gut. 2009;58:312–314. doi: 10.1136/gut.2008.164418. [DOI] [PubMed] [Google Scholar]
- 113.Grigat S, Harlfinger S, Pal S, Striebinger R, Golz S, Geerts A, Lazar A, Schömig E, Gründemann D. Probing the substrate specificity of the ergothioneine transporter with methimazole, hercynine, and organic cations. Biochem Pharmacol. 2007;74:309–316. doi: 10.1016/j.bcp.2007.04.015. [DOI] [PubMed] [Google Scholar]
- 114.Kato Y, Kubo Y, Iwata D, Kato S, Sudo T, Sugiura T, Kagaya T, Wakayama T, Hirayama A, Sugimoto M, Sugihara K, Kaneko S, Soga T, Asano M, Tomita M, Matsui T, Wada M, Tsuji A. Gene knockout and metabolome analysis of carnitine/organic cation transporter OCTN1. Pharm Res. 2010;27:832–840. doi: 10.1007/s11095-010-0076-z. [DOI] [PubMed] [Google Scholar]
- 115.Paul B, Snyder S. The unusual amino acid L-ergothioneine is a physiologic cytoprotectant. Cell Death Differ. 2010;17:1134. doi: 10.1038/cdd.2009.163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Hartman PE. Ergothioneine as antioxidant. Methods Enzymol. 1990;186:310–318. doi: 10.1016/0076-6879(90)86124-e. [DOI] [PubMed] [Google Scholar]
- 117.Hand CE, Taylor NJ, Honek JF. Ab initio studies of the properties of intracellular thiols ergothioneine and ovothiol. Bioorg Med Chem Lett. 2005;15:1357–1360. doi: 10.1016/j.bmcl.2005.01.014. [DOI] [PubMed] [Google Scholar]
- 118.Scott EM, Duncan IW, Ekstrand V. Purification and properties of glutathione reductase of human erythrocytes. J Biol Chem. 1963;238:3928–3933. [PubMed] [Google Scholar]
- 119.Taubert D, Lazar A, Grimberg G, Jung N, Rubbert A, Delank KS, Perniok A, Erdmann E, Schömig E. Association of rheumatoid arthritis with ergothioneine levels in red blood cells: a case control study. J Rheumatol. 2006;33:2139–2145. [PubMed] [Google Scholar]
- 120.Tokuhiro S, Yamada R, Chang X, Suzuki A, Kochi Y, Sawada T, Suzuki M, Nagasaki M, Ohtsuki M, Ono M, Furukawa H, Nagashima M, Yoshino S, Mabuchi A, Sekine A, Saito S, Takahashi A, Tsunoda T, Nakamura Y, Yamamoto K. An intronic SNP in a RUNX1 binding site of SLC22A4, encoding an organic cation transporter, is associated with rheumatoid arthritis. Nat Genet. 2003;35:341–348. doi: 10.1038/ng1267. [DOI] [PubMed] [Google Scholar]
- 121.Peltekova VD, Wintle RF, Rubin LA, Amos CI, Huang Q, Gu X, Newman B, Van Oene M, Cescon D, Greenberg G, Griffiths AM, St George-Hyslop PH, Siminovitch KA. Functional variants of OCTN cation transporter genes are associated with Crohn disease. Nat Genet. 2004;36:471–475. doi: 10.1038/ng1339. [DOI] [PubMed] [Google Scholar]
- 122.Leung E, Hong J, Fraser AG, Merriman TR, Vishnu P, Krissansen GW. Polymorphisms in the organic cation transporter genes SLC22A4 and SLC22A5 and Crohn’s disease in a New Zealand Caucasian cohort. Immunol Cell Biol. 2006;84:233–236. doi: 10.1111/j.1440-1711.2006.01423.x. [DOI] [PubMed] [Google Scholar]
- 123.Kaneko I, Takeuchi Y, Yamaoka Y, Tanaka Y, Fukuda T, Fukumori Y, Mayumi T, Hama T. Quantitative determination of ergothioneine in plasma and tissues by TLC-densitometry. Chem Pharm Bull. 1980;28:3093–3097. doi: 10.1248/cpb.28.3093. [DOI] [PubMed] [Google Scholar]
- 124.Briggs I. Ergothioneine in the central nervous system. J Neurochem. 1972;19:27–35. doi: 10.1111/j.1471-4159.1972.tb01250.x. [DOI] [PubMed] [Google Scholar]
- 125.Crossland J, Mitchell J, Woodruff GN. The presence of ergothioneine in the central nervous system and its probable identity with the cerebellar factor. J Physiol. 1966;182:427–438. doi: 10.1113/jphysiol.1966.sp007830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Moncaster JA, Walsh DT, Gentleman SM, Jen LS, Aruoma OI. Ergothioneine treatment protects neurons against N-methyl-D-aspartate excitotoxicity in an in vivo rat retinal model. Neurosci Lett. 2002;328:55–59. doi: 10.1016/s0304-3940(02)00427-5. [DOI] [PubMed] [Google Scholar]
- 127.Libby P, Ridker PM, Hansson GK. Progress and challenges in translating the biology of atherosclerosis. Nature. 2011;473:317–325. doi: 10.1038/nature10146. [DOI] [PubMed] [Google Scholar]
- 128.Bastard JP, Maachi M, Van Nhieu JT, Jardel C, Bruckert E, Grimaldi A, Robert JJ, Capeau J, Hainque B. Adipose tissue IL-6 content correlates with resistance to insulin activation of glucose uptake both in vivo and in vitro. J Clin Endocrinol Metab. 2002;87:2084–2089. doi: 10.1210/jcem.87.5.8450. [DOI] [PubMed] [Google Scholar]
- 129.Lafon-Cazal M, Pietri S, Culcasi M, Bockaert J. NMDA-dependent superoxide production and neurotoxicity. Nature. 1993;364:535. doi: 10.1038/364535a0. [DOI] [PubMed] [Google Scholar]
- 130.Martin KR. The bioactive agent ergothioneine, a key component of dietary mushrooms, inhibits monocyte binding to endothelial cells characteristic of early cardiovascular disease. J Med Food. 2010;13:1340–1346. doi: 10.1089/jmf.2009.0194. [DOI] [PubMed] [Google Scholar]
- 131.Zhao Q, Wang M, Xu D, Zhang Q, Liu W. Metabolic coupling of two small-molecule thiols programs the biosynthesis of lincomycin A. Nature. 2015;518:115. doi: 10.1038/nature14137. [DOI] [PubMed] [Google Scholar]
- 132.Newton GL, Leung SS, Wakabayashi JI, Rawat M, Fahey RC. The DinB superfamily includes novel mycothiol, bacillithiol, and glutathione S-transferases. Biochemistry. 2011;50:10751–10760. doi: 10.1021/bi201460j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Rawat M, Av-Gay Y. Mycothiol-dependent proteins in actinomycetes. FEMS Microbiol Rev. 2007;31:278–292. doi: 10.1111/j.1574-6976.2006.00062.x. [DOI] [PubMed] [Google Scholar]
- 134.Jothivasan VK, Hamilton CJ. Mycothiol: synthesis, biosynthesis and biological functions of the major low molecular weight thiol in actinomycetes. Nat Prod Rep. 2008;25:1091–1117. doi: 10.1039/b616489g. [DOI] [PubMed] [Google Scholar]
- 135.Komatsu W, Miura Y, Yagasaki K. Suppression of hypercholesterolemia in hepatoma-bearing rats by cabbage extract and its component, S-methyl-L-cysteine sulfoxide. Lipids. 1998;33:499–503. doi: 10.1007/s11745-998-0233-7. [DOI] [PubMed] [Google Scholar]
- 136.Augusti KT, Sheela CG. Antiperoxide effect of S-allyl cysteine sulfoxide, an insulin secretagogue, in diabetic rats. Experientia. 1996;52:115–119. doi: 10.1007/BF01923354. [DOI] [PubMed] [Google Scholar]
- 137.Kodama H, Zhang J, Sugahara K. Novel priming compounds of cystathionine metabolites on superoxide generation in human neutrophils. Biochem Biophys Res Commun. 2000;269:297–301. doi: 10.1006/bbrc.1999.1970. [DOI] [PubMed] [Google Scholar]