

Abstract
Polymer vesicles, i.e. polymersomes (PS), present unique nanostructures with an interior aqueous core that can encapsulate multiple independent cargos concurrently. However, the sequential release of such co-loaded actives remains a challenge. Here, we report the rational design and synthesis of oxidation-responsive shell-crosslinked PS with capability for the controlled, sequential release of encapsulated hydrophilic molecules and hydrogels. Amphiphilic brush block copolymers poly(oligo(ethylene glycol) methyl ether methacrylate)-b-poly(oligo(propylene sulfide) methacrylate) (POEGMA-POPSMA) were prepared to fabricate PS via self-assembly in aqueous solution. As a type of unique drug delivery vehicle, the interior of the PS was co-loaded with hydrophilic molecules and water-soluble poly(N-isopropylacrylamide) (PNIPAM) conjugates. Due to the thermosensitivity of PNIPAM, PNIPAM conjugates within the PS aqueous interior underwent a phase transition to form hydrogels in situ when the temperature was raised above the lower critical solution temperature (LCST) of PNIPAM. Via control of the overall shell permeability by oxidation, we realized the sequential release of two water-soluble payloads based on the assumption that hydrogels have much smaller membrane permeability than that of molecular cargos. The ability to control the timing of release of molecular dyes and PNIPAM-based hydrogels was also observed within live cell. Furthermore, leakage of hydrogels from the PS was effectively alleviated in comparison to molecular cargos, which would facilitate intracellular accumulation and prolonged retention of hydrogels within the cell cytoplasm. Thus, we demonstrate that the integration of responsive hydrogels into PS with crosslinkable membranes provides a facile and versatile technique to control the stability and release of water-soluble cargos for drug delivery purposes.
Keywords: Self-assembly, Polymersomes, Shell Crosslinking, Sequential release, Oxidation-responsive
Graphical Abstract
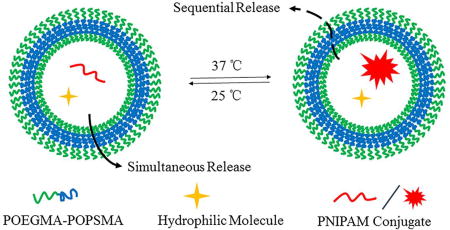
Introduction
Polymer vesicles, i.e. polymersomes (PS), fabricated via the self-assembly of amphiphilic block copolymers (BCP) in aqueous solution [1], are finding increasing applications in biomedical fields, including drug delivery, gene therapy, and diagnostic imaging [2–7]. Considering the high molecular weight of amphiphilic BCP, PS exhibit greatly enhanced robustness and stability in comparison to their structural analogues of liposomes, making PS attractive candidates as nanocarriers for extended circulation and prevention of premature drug release [8–11]. Moreover, the synthetic nature of BCP makes the resulting PS highly versatile materials with tunable structural and physicochemical properties [12, 13]. This flexibility in methods of synthesis permits almost infinite diversity of BCP in terms of nature, properties and composition, which can be rationally engineered to meet the requirements of specific applications [14–16]. For drug delivery purposes, it is possible to control the release behavior of PS by switching the membrane stability and permeability [17–20]. Through the incorporation of stimuli-responsive components, smart PS can be developed to recognize various external physical or internal biological environmental stimuli, thus realizing “on demand” release in dose-, spatial-, and temporal-controlled fashions [2, 4, 21, 22]. Also, surface functionalization of PS can be extensively modified and tailored using the high flexibility and customizability of BCP [23–25]. For example, the surface conjugation of various ligands can enable PS to function as targeted drug carriers that enhance the selective recognition of specific cells or tissues, thus optimizing their pharmacokinetics and biodistribution [26, 27].
The ability to controllably release different therapeutic drugs will play a pivotal role in improving patient care and simplifying treatment regimes in the clinic [28]. Traditional ways to concurrently unload multiple types of drugs remains limited, often leading to dissimilarities in pharmacokinetics, tissue distribution, and serious side effects [29]. The stimulus-responsive and controlled sequential release of different cargos allows the overcoming of these challenges. However, most reported nanocarriers to date can load only a single molecular payload within either their interior or shell for triggered release. To circumvent this limitation, next-generation drug carriers with multicompartmental polymeric structures have been suggested to deliver multiple cargo molecules in a preprogrammed and sequential manner [30, 31]. Based on this concept, a number of multicompartmentalized polymeric systems [31] have been developed using different types of polymeric components included layer-by-layer films [28, 32, 33], hydrogels [34, 35], vesosomes [36], and other capsules [37–40]. PS are of considerable interest due to the compartmentalized structure provided by their bilayer membrane and resulting aqueous interiors. These enable PS the capability of encapsulating diverse cargos within their aqueous cavities or the hydrophobic bilayer membranes depending on the characteristics of the cargo. However, mechanisms for sequential release from PS have been much less studied and are primarily limited to strategies employing multiple PS [29, 41] and vesosomes [42, 43] possessing complex or multi-layer membrane structures. The more commonly used single-layer PS have simple or homogenous structures and composition, and their sequential unloading remains an important challenge, particularly for PS co-loaded with different water-soluble cargos.
As drug delivery vehicles, PS typically encapsulate hydrophilic molecules within their central aqueous core. To realize a sequential release, the polymeric shell of PS must maintain integrity while allowing the diffusion of selective cargos through the membrane at different rates, otherwise simultaneous unloading is inevitable due to irreversible disruption or rupture. Conventional chemical crosslinking strategies have been used to address this stability issue [18, 44, 45]. Cargo size can strongly influence permeability through bilayer membranes [19], and we therefore hypothesized that PS with controllable membrane permeability could serve as sequential release capsules for differently sized co-encapsulated payloads. Furthermore, PS have the capacity to encapsulate a wide range of cargo sizes within their lumen, even including smaller nanoparticles [42, 43]. The loading of nanoparticles into PS can be facilitated by employing responsive molecules for in situ generation strategies. For example, thermosensitive poly(N-isopropylacrylamide) (PNIPAM) gel-containing PS (hydrosomes) were reported by Feijen Group [46, 47]. Previous studies demonstrated the application of these multicompartmental PS as a controlled delivery system, but the release behavior of the encapsulated smaller nanoparticles requires further investigation.
Herein, we synthesize a new type of amphiphilic BCP, poly(oligo(ethylene glycol) methyl ether methacrylate)-b-poly(oligo(propylene sulfide) methacrylate) (POEGMA-POPSMA, Figure 1a), for fabrication of PS with controllable membrane permeability to achieve sequential release of differently sized hydrophilic payloads. PS self-assembled from POEGMA-POPSMA were successfully formed by controlling the molecular weight ratio of the hydrophilic and hydrophobic blocks. Shell crosslinking approaches were used to endow the PS with stimuli-responsive membrane permeability. Upon oxidation, POEGMA-POPSMA PS membranes demonstrated increased permeabilization without affecting the overall vesicular architecture. The in situ generation of hydrogels within the PS lumen was achieved by facile conjugation of one cargo with thermo-sensitive PNIPAM (Figure 1b), allowing comparison of the release kinetics of differently sized molecular payloads. Through the use of a single crosslinkable BCP, we demonstrate concurrent loading and sequential unloading of two different water-soluble cargos from simple bilayer PS without the need for complex and hierarchical vesicle nanostructures.
Figure 1.

POEGMA-POPSMA block copolymers (BCP) self-assemble into polymersomes (PS) and support the size-dependent dual release of hydrophilic payloads. (a) Chemical structures of oxidation-responsive amphiphilic POEGMA-POPSMA BCP used for the fabrication of PS with crosslinkable membranes, and (b) schematic of sequential release from the shell crosslinked PS at 37 °C under a stimulus of oxidation.
Results and Discussion
Synthesis and Self-Assembly of Oxidation-Responsive Amphiphilic BCP
PS formed in water via the self-assembly of amphiphilic BCP are typically more robust than liposomes, presenting membranes with limited permeability. To synthesize PS with enhanced on-demand membrane sensitivity, we selected oxidation-responsive poly(propylene sulfide) (PPS) [48] as the hydrophobic block of an amphiphilic BCP to self-assemble vesicular nanostructures. Based on our previous work, poly(ethylene glycol)-poly(propylene sulfide) (PEG-PPS) can be engineered to assemble into stable PS in aqueous solutions reliably [9, 12, 13]. However, such linear BCP amenable to end-functionalization present insufficient reactive groups for further shell crosslinking, and the resulting PS would undergo a morphological transition from vesicle to smaller objects (worm-like and spherical micelles) upon exposure to oxidative conditions [48]. As sequential release vehicles, the PPS membrane must maintain mechanical integrity under oxidative stimuli, and hence we designed and synthesized the novel brush/comb amphiphilic POEGMA-POPSMA BCP for increased display of crosslinkable moieties relative to linear PEG-PPS. As shown in Scheme 1, the basic scaffold of POEGMA-POPSMA is a methacrylate-based backbone, which was prepared by reversible addition-fragmentation chain transfer (RAFT) polymerization.
Scheme 1.

Synthetic routes employed for the preparation of OPSMA monomers and POEGMA-POPSMA comb-shaped amphiphilic BCP.
The synthesis was started with the preparation of POPSMA monomer, which is a novel methacrylate-based macromolecular monomer containing highly reactive pyridyl disulfide groups and an oxidation-responsive oligo(propylene sulfide) (OPS) hydrophobic segment. Using a modified version of a published synthesis for PPS [49], hydroxyl and pyridyl disulfide heterobifunctional OPS (OPS-OH) was first synthesized via anionic ring-opening polymerization of propylene sulfide using 2-mercaptoethanol as the initiator and 2,2’-dipyridyl disulfide for termination. Then OPSMA monomer was prepared from the esterification of OPS-OH with methacryloyl chloride. We initially synthesized OPSMA monomers with varying OPS lengths (OPSmMA; m = 4, 7, 17, and 20), and molecular compositions were confirmed by their 1H NMR spectra (Figure S1). Higher molecular weight monomers displayed higher yields due to less loss during purification, but led to decreased polymerization reactivity and controllability [50]. RAFT polymerization of OPSMA was conducted in parallel using poly(oligo(ethylene glycol) methyl ether methacrylate) macro chain-transfer agent (POEGMA-CTA) [51], affording a series of amphiphilic POEGMA-POPSmMAn BCP differing in hydrophobic block lengths (representative data of POEGMA-POPS7MA8, shown in Figure S2 and Figure S3). To the best of our knowledge, the grafting through polymerization of OPS-based macromonomers has not been previously described in the literature to date. This strategy enables the design and syntheses of a series of well-defined oxidation-responsive PPS materials, which can subsequently endowed with multi-functionality via a rapid and quantitative modification through thiol-disulfide exchange under mild conditions [52].
Similar to PEG-PPS [48], POEGMA-POPSMA also presents as a viscous, oily material at room temperature. As shown in Figure S4, the glass transition temperature (Tg) of POEGMA-POPSMA is determined to be −36 °C by differential scanning calorimetry (DSC). Using a thin-film hydration method as previously described [8], the self-assembly of POEGMA-POPSMA was performed in phosphate-buffered saline (PBS) and PS could be obtained within specific molecular weight ratios of the hydrophilic and hydrophobic blocks. Both POEGMA-POPS7MA8 and POEGMA-POPS17MA3 can be engineered to assemble into vesicles as confirmed by cryogenic transmission electron microscopy (cryoTEM, Figure 2) and by the capability to retain the hydrophilic fluorophore calcein. The POEGMA weight fractions for POEGMA-POPS7MA8 and POEGMA-POPS17MA3 are calculated to be 0.29 and 0.32, respectively. It is worth mentioning that the POEGMA weight fractions required for vesicle formation of POEGMA-POPSMA are consistent with our previous data for linear PEG-PPS, where PS formed with PEG fractions of 0.19–0.31 [9, 12]. Figure 2a shows the representative size and morphology of the nanostructures of POEGMA-POPS17MA3 in PBS. Besides larger PS, smaller micelles were also formed via the self-assembly of the BCP, which was due to the high molecular weight distribution (Mw/Mn) of POEGMA-POPS17MA3. In general, Mw/Mn decreases with increasing degree of polymerization for a living radical polymerization up to intermediate conversion levels [53]. As an oligomeric product of OPS17MA, POEGMA-POPS17MA3 has a relatively high Mw/Mn of 1.74, determined by gel permeation chromatography (GPC). Moreover, low polymerization degree results limited number of pyridyl disulfide groups for POEGMA-POPS17MA3, which is marginal for subsequent shell crosslinking reaction even at 100% conversion. Therefore, we continued to use only POEGMA-POPS7MA8 to fabricate PS via controlled self-assembly.
Figure 2.

CryoTEM images of nanostructures fabricated by the self-assembly of POEGMA-POPS17MA3 (a) and POEGMA-POPS7MA8 (b–f) in PBS. (a) Coexistence of large size-distributed PS and smaller micelles in a POEGMA-POPS17MA3 dispersion, (b) Large size distributions were observed for uncrosslinked POEGMA-POPS7MA8 PS prior to extrusion, (c) monodisperse single-layer uncrosslinked PS obtained following extrusion, (d) shell crosslinked PS, (e) PNIPAM-RhB encapsulated shell crosslinked PS (cryo-fixed at 37 °C), and (f) PNIPAM-RhB encapsulated uncrosslinked PS (cryo-fixed at 37 °C displayed frequent vesicle rupture (red arrows) and PNIPAM-RhB hydrogels. Scale bar = 200 nm.
Analysis with cryoTEM revealed a wide range of size distributions for PS formed immediately after self-assembly of POEGMA-POPS7MA8 (Figure 2b). Most of the PS were unilamellar, consisting of spheres as small as 40 nm, typically 200–300 nm in diameter, and occasionally as large as 1000 nm. A subsequently extrusion process was performed to reduce those PS in size and lamellarity [8]. After passing through a 200 nm porous membrane, single-layer PS were obtained with diameters below 200 nm (Figure 2c). The hydrodynamic diameters and size distributions of PS were further characterized by dynamic light scattering (DLS), which showed that the Z-average diameter and polydispersity index (PDI) were 181.8 ± 2.4 nm and 0.223 ± 0.013, respectively (Figure S5). Due to the carboxyl terminal group on the hydrophilic POEGMA block, the resulting PS exhibited negative zeta-potential of −8.03 ± 1.35 mV (Table S1).
Shell Crosslinking
Shell crosslinked PS (C-PS) were prepared by in situ thiol-disulfide exchange of uncrosslinked PS (U-PS) with 1,2-ethanedithiol. Thiol-disulfide exchange reactions exhibit several advantages that make them ideal for membrane crosslinking. First, the resultant disulfide linkage is sensitive to redox environments and cleavable for biodegradation. Second, the reaction is efficient and quantitative with formation of an equimolar amount of 2-thiopyridone. Accordingly, the degree of crosslinking could be estimated indirectly from the result of 2-thiopyridone generation as determined by UV absorption (Figure S6) [54]. Third, thiol-disulfide exchange occurs conveniently under mild conditions without disruption of morphology. Figure 2d shows the typical morphology obtained for C-PS with a crosslinking degree (CD) of 44%. The cryoTEM image revealed the well-defined spherical nature of PS, with the occasional presence of large compound vesicles resulting from intermembrane crosslinking. As determined by DLS (Figure 3a, Table S1), the Z-average diameter of C-PS was 245.1 ± 3.6 nm, with a PDI of 0.204 ± 0.014. The slight increase in size compared with U-PS was likely owing to the presence of large compound vesicles. This shell crosslinking approach likely decreases the fluidity of the BCP within the PS membrane, and as a result, the extrusion method cannot be used to reduce C-PS in size and lamellarity. Further characterization of C-PS (CD = 44%) was performed using small angle X-ray scattering (SAXS). The scattering profile in Figure 3b was best fitted using a vesicle model with core radius of ~106 nm and shell thickness of ~14.3 nm. These results were in agreement with our DLS and cryo-TEM measurements.
Figure 3.

Physicochemical characteristics of C-PS in PBS at 25 °C by DLS (a) and SAXS (b).
The vesicular structure of U-PS is dictated by the ratio of the hydrophobic to hydrophilic composition of the BCP, and only a small amount of oxidation of OPS to the corresponding hydrophile is required to initiate restructuring of the larger vesicles into much smaller micelles [17, 55]. We expected crosslinking of the hydrophobic shells to maintain the overall architecture of resulting C-PS over a certain range of oxidative conditions. Since the vesicle-to-micelle transition results in an optical density decrease[48], we used turbidity measurements to evaluate the efficacy of shell crosslinking and assess the upper limits of resistance to oxidation-dependent degradation. Figure 4a compares the UV-Vis absorption of PS suspensions with varying CD at 600 nm on exposure to 5% H2O2 (1.63 M). After 1 h incubation, the UV-Vis absorptions for U-PS (CD of 0%) and C-PS of lower CD (17% and 32%) exhibited obvious decreases, suggesting the disassembly of PS into micellar structures. Though slight decreases in optical density were also detected for C-PS of higher CD (44% and 52%), the absorptions are significantly higher than that of U-PS under the same conditions. A more intuitive result, however, can be observed by 24 h incubation, where the U-PS suspension became complete clear whilst C-PS of higher CD (44% and 52%) still presented as a kind of micellar solution in PBS. Low extent of crosslinking is reported to be sufficient to maintain architecture of PS [18, 44], however, it was found that C-PS of lower CD (particularly 17%) exhibited no significant difference with U-PS. We attributed this to the fact that the average number of OPS7MA repeat units in POEGMA-POPS7MA8 is 8, which means that an effective shell crosslinking should be higher than 25% (at least two pyridyl disulfide groups of a polymer chain are reacted). Anyway, we were able to use a sufficiently high CD within the membrane to stabilize the hydrophobic membranes, and the resulting C-PS could serve as a unique drug delivery system with both oxidation-responsiveness and preserved nanoarchitecture. To simplify this investigation, a CD of 44% was selected for further study.
Figure 4.

Crosslinking of PS membranes increases stability and support in situ gelation of PNIPAM for retention of large encapsulated cargo. (a) Time-dependent evolution of normalized absorbance (turbidity) for PS with different degrees of crosslinking (0%, 17%, 32%, 44%, and 52%) in PBS with 1.63 M of H2O2 (5%). (b) Leakage profiles of calcein from U-PS and C-PS at 25 °C. (c) Intensity-average hydrodynamic diameter distributions recorded for U-PS and C-PS loaded with PNIPAM-RhB in PBS after incubation at 37 °C for 24 h. (d) Leakage profiles of calcein and PNIPAM-RhB hydrogels co-loaded C-PS in PBS at 37 °C. Data represent means ± standard deviations (n = 3). Statistical significance: *p ≤ 0.01, **p ≤ 0.005, ***p ≤ 0.0001.
Water-soluble molecules such as rhodamine B-labeled PNIPAM (PNIPAM-RhB) and calcein were co-encapsulated within PS during the BCP self-assembly (Figure S7). We expected further crosslinking of the hydrophobic POPSMA membranes to strengthen the vesicles and ensure drug retention for long-term storage [20, 56]. Leakage profiles of calcein were consistent with this, where a significant decrease in leakage from C-PS was observed in comparison to U-PS. As presented in Figure 4b, U-PS showed a fast leakage (> 15%) after 2 weeks of incubation in PBS at 25 °C, while < 5% of calcein leaked from C-PS under the same conditions. Shell crosslinking was further demonstrated to be essential for the stable retention of PNIPAM-RhB following lower critical solution temperature (LCST) phase transition at 37 °C. Regardless of shape, it is expected that PNIPAM-RhB hydrogels formed in situ within the lumen of PS should have a nanoscale size smaller than the PS diameter. However, aggregations as large as 1000 nm were observed by DLS after incubation of PNIPAM-RhB loaded U-PS dispersion for 24 h at 37 °C (Figure 4c). In the absence of BCP at 37 °C, PNIPAM-RhB initially formed nanogels that eventually ripened into micron-sized aggregates, both of which were confirmed by cryoTEM (Figure S8). Phase separation and formation of PNIPAM-RhB hydrogels within the lumen of U-PS were observed by cryoTEM to rupture these vesicles (Figure 2f). We hypothesized that permeation of PNIPAM hydrogels into the POPSMA domain of U-PS could cause irreversible disruption of membranes by altering the hydrophilic-to-lipophilic balance. In contrast, C-PS showed an enhanced ability to encapsulate PNIPAM-RhB at 37 °C, and hydrogel formation inside the vesicle lumen did not disrupt the C-PS membrane (Figure 2e). PNIPAM-RhB aggregates were observed to be stably retained within C-PS by cryoTEM and did not modulate the vesicle membrane thickness (Figure 2d, 2e inserts). These results demonstrate that membrane crosslinking is required for stable retention of phase separated PNIPAM nanogel payloads within PS assembled from POEGMA-POPSMA.
Ideally, nanocarriers will retain as much cargo as possible following in vivo administration until they reach the desired targeted tissues and cells. Conventional strategies related to crosslinking hydrophobic membranes have been used to reduce drug leakage during blood circulation [20, 56, 57]. However, premature drug leakage is still a common problem for PS, particularly those loaded with water-soluble cargos [22]. Figure 4d compares the leak profiles of calcein and PNIPAM-RhB hydrogels from C-PS at 37 °C. Loaded PS were incubated at 37 °C immediately after purification from unloaded dyes by size exclusion chromatography (SEC) with a Sepharose CL-6B size exclusion column. A temperature induced immediate release of ~10% was detected for both cargos. During this one-week study, extensive leakage of calcein from C-PS was observed, whereas significantly lower leakage was observed for PNIPAM-RhB at all time points after the temperature was raised to 37 °C. These results demonstrate that the in situ solgel transition of PNIPAM encapsulated within PS afforded a facile technique to overcome payload leakage. Reversibly decorating PNIPAM, or other responsive polymers, with hydrophilic chemotherapeutics may therefore decrease unwanted drug release following in vivo administration of nanocarriers and slow the rate of drug release at the target site. Such an ability may be beneficial for controlled delivery by prolonging systemic circulation time, increasing the percentage of drug reaching the desired target and minimizing side effects.
Oxidation-Triggered Sequential Release
Using calcein as a model small molecule cargo, we assessed the oxidation-triggered release kinetics of C-PS with varying physiologically relevant H2O2 concentrations (0, 0.1, 100, and 163 mM in PBS) at 37 °C (Figure 5a). It was demonstrated that the release could be accelerated on exposure to oxidative conditions, and the release rate was significantly dependent on the H2O2 concentration. Faster unloading could thus be achieved by incubation with higher concentrations of H2O2. The results therefore indicated that the vesicle bilayer permeability can be controlled via the extent of oxidation. It should be noted that H2O2 is one of the most abundant and stable forms of reactive oxygen species (ROS) persistently generated in living organisms with a typical level of 50–200 µM [16, 58], but is only one of many ROS present within cell lysosomes that contribute to the oxidation and intracellular release of PEG-PPS-based nanocarriers. As a model system and for vesicle characterization, we and others typically employ higher than physiological levels of H2O2 for more rapid in vitro investigations (Figure 4a) [13, 48].
Figure 5.

Crosslinked PS display reduced release rates for encapsulated PNIPAM hydrogels in comparison to small hydrophilic payloads. (a) Oxidation-triggered release of calcein at 37 °C from C-PS upon incubation with varying H2O2 concentrations (0, 0.1, 100, and 163 mM in PBS). (b) In vitro co-release profiles of calcein and PNIPAM-RhB at 37 °C from C-PS in the presence of H2O2 (163 mM, 0.5% in PBS). Data represent means ± standard deviations (n = 3). Statistical significance: *p ≤ 0.01, **p ≤ 0.005, ***p ≤ 0.0001.
According to our molecular design, we expected oxidation conversion of PPS-based shells to occur without loss of vesicle integrity due to the enhanced stability of crosslinking, thus the C-PS should exhibit slower release than U-PS. The results in Figure S9 were consistent with this, where a significantly slower release of PNIPAM-RhB is detected after shell crosslinking upon exposure to H2O2 at 25 °C. Since the 163 mM H2O2 concentration resulted in the most rapid release of calcein from C-PS, this concentration was selected to investigate co-release characteristics of calcein and PNIPAM-RhB hydrogels at 37 °C. As shown in Figure 5, ~90% of calcein was unloaded over 12 h from C-PS under these conditions. In contrast, a significant delay in release was observed for PNIPAM-RhB, and the unloading was significantly slower than calcein. Over a period of approximately 3 days, PNIPAM-RhB was released at a nearly constant rate. The co-release profiles revealed the ability of C-PS to control the timing of release of calcein and PNIPAM-RhB. In consideration of the molecular weight difference between PNIPAM-RhB and calcein, we also compared their release behavior at 25 °C (Figure S10). No significant difference in release rate was observed, suggesting that the delayed release of PNIPAM-RhB at 37 °C was due to the temperature-induced hydrogel formation. The mechanism for in situ hydrogel generation within PS makes use of the thermosensitivity of PNIPAM, decreasing the penetrability of the PNIPAM-RhB, even under oxidative conditions. These results demonstrate that changing the cargo size within the C-PS lumen enabled sequential release of two water-soluble payloads from a simple bilayer PS delivery system. The diffusion of payloads across the membrane of C-PS was driven by oxidation and the overall release rate of components from the interior depended upon incubation time and H2O2 concentration.
Confocal laser scanning microscopy (CLSM) was employed to further evaluate release rates of hydrophilic cargo as a function of H2O2 concentration from POEGMA-POPSMA C-PS. To enhance visualization during CLSM imaging, we used calcein and PNIPAM-RhB co-loaded U-PS with large size distributions (Figure 2b), which could be crosslinked to prepare samples containing giant C-PS. These giant microscale PS settled from PBS solutions during incubation for 24 h at 37 °C and became immobilized for CLSM observation and direct verification of the retention of the fluorescent cargos (Figure 6a). Since a self-quenching concentration (30 mM) of calcein was encapsulated within the C-PS, we were able to monitor the release via an increase in the fluorescence intensity within vesicles upon washing [17]. Numerous vesicles with quenched calcein appeared as red spheres due to encapsulated PNIPAM-RhB within their interiors (Figure 6a). Oxidation-induced release of calcein was initiated by H2O2 (163 mM), which increased the green fluorescence and induced colocalization of the green dequenched calcein and co-encapsulated red PNIPAM-RhB within vesicle lumens than PBS controls after washing (Figure 6b). This indicated that calcein was unloaded faster than passive diffusion in PBS. The use of higher levels of H2O2 (326 mM) resulted in further release of calcein upon the same 24-hour incubation, wherein most green fluorescent particles became dim and even disappeared from view (Figure 6c). Retention of PNIPAM-RhB was distinguishable with most particles exhibiting only red fluorescence. Several particles displayed red membranes, suggesting that the increased permeability caused PNIPAM-RhB hydrogels to aggregate at the membrane interface or become trapped within the membranes as they diffused out of the PS. Even with 652 mM of H2O2, red fluorescent particles were still observed after 24 h with complete absence of green fluorescence (Figure 6d). The relatively slow release of PNIPAM-RhB hydrogels can likely be attributed to poor diffusive transport through and limited permeability of the oxidized, crosslinked PPS shells.
Figure 6.

Confocal microscopy images of calcein (green channel) and PNIPAM-RhB (red channel) co-loaded C-PS upon incubation in PBS with varying H2O2 concentrations at 37 °C for 24 h. From left to right: (a) PBS, (b) 163 mM (0.5%) H2O2, (c) 326 mM (1.0%) H2O2, (c) 652 mM (2.0%) H2O2. Scale bar = 5 µm.
Intracellular Dual Release
The sequential release of two or more therapeutic molecules in live cells would be beneficial for a wide range of drug delivery applications and combination therapies [59]. Previously reported examples of sequential release vehicles involved the use of multicompartmentalized polymeric systems with micrometer scale sizes [29, 31, 39], which presents limited applications for intracellular and in vivo delivery. To further demonstrate the ability to differentially tune the release of two cargos from the C-PS in live cells, we chose human A2780 ovarian carcinoma cells for oxidation-induced intracellular release due to their elevated generation of ROS [16]. The cytotoxicity of C-PS to A2780 was first investigated by incubating A2780 cells with varying concentrations (0, 1, 10, 100, 200, and 500 µg/mL) of C-PS for 24 h. A 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay suggested that C-PS is nontoxic up to 0.5 mg/mL (Figure S11). Concentration-dependent endocytosis of C-PS in A2780 cells was also assessed using flow cytometry. Efficient cellular uptake was observed for C-PS (Figure S12), which indicated feasibility of intracellular delivery.
Nile blue chloride (NBC) and PNIPAM-RhB were co-loaded into C-PS to investigate intracellular delivery to A2780 cells. As a well-known plasma membrane dye, NBC was chosen because the intracellular release could be monitored by its staining of cell membranes. Moreover, a control endocytosis experiment showed that the dye is cell impermeant (Figure S13), which is important for eliminating the interference of any premature leakage. A2780 cells were incubated with NBC and PNIPAM-Rh B co-loaded C-PS for predetermined time and the intracellular distributions of the two dyes were imaged by CLSM (Figure 7a). The images of 1-hour incubation demonstrated the efficient cellular uptake of NBC using the C-PS as delivery vehicles. Since NBC and PNIPAM-RhB were co-encapsulated in C-PS, the majority of the two dyes was colocalized, resulting in an extensive amount of yellow overlay within cellular distributions. However, separate and distinct distributions of NBC and PNIPAM-RhB were found at the 24 h time point. Release of NBC at 24 h was demonstrated by the staining of cell membranes, whereas no obvious change in distribution was observed for PNIPAM-RhB, which remained within puncta characteristic of endosomal compartments. The continued retention of PNIPAM-RhB within cell lysosomes was confirmed in Figure 7b. A2780 cells were incubated with PNIPAM-RhB containing C-PS for 1 h. The culturing media were then replaced with fresh media and cells were incubated for another 24 h for CLSM observation. The images revealed that the majority of PNIPAM-RhB localized with the lysosomal stain LysoTracker. The above results indicated that the timing of intracellular release of two different water-soluble molecules could be achieved by the facile conjugation of one cargo with PNIPAM for size-dependent retention within C-PS.
Figure 7.

Confocal fluorescence images of A2780 cells. (a) Cells were incubated with Nile blue chloride (red channel) and PNIPAM-Rh B (green channel) co-loaded C-PS for 1 h and 24 h. Nucleus were stained by DAPI. (b) Cells were first treated with PNIPAM-RhB loaded C-PS for 1 h, and then incubated with fresh medium for 1 h and 24 h. Nucleus and lysosome were stained by Hoechst 33342 and LysoTracker Green, respectively. Scale bar = 10 µm.
Conclusion
In summary, we present a novel amphiphilic brush BCP that self-assembles into PS for size-dependent dual delivery of hydrophilic payloads. The BCP was synthesized via RAFT to contain oxidation-responsive oligo(propylene sulfide) and highly reactive pyridyl disulfide moieties to allow subsequent crosslinking of PS membranes. Due to the dense brush/comb architecture of the BCP, vesicles assembled at much shorter block lengths than typically observed for linear PEG-PPS BCPs. Shell-crosslinked PS (C-PS) supported the previously observed oxidation sensitivity of PEG-PPS PS, but crosslinking of the pyridyl disulfide moieties allowed preservation of the vesicular architecture. These oxidized PS presented increased permeability for small molecule payloads, but could retain larger encapsulated hydrogels both in the presence of H2O2 and within cell lysosomes. For the purposes of drug delivery, PNIPAM conjugated hydrophilic molecules could be co-encapsulated within the C-PS with other water-soluble cargos concurrently. The enhanced stability of the C-PS allowed in situ gelation of the PNIPAM within the vesicle lumen, which otherwise resulted in vesicle rupture in the absence of crosslinking. Release experiments showed that the release of PNIPAM hydrogels was remarkably delayed by comparison to small hydrophilic dyes at 37 °C, revealing a sequential release behavior for the vehicles. We further demonstrated the ability to control the release of two different cargos in live cells using this delivery system. Overall, this work developed a feasible strategy to realize the release of two different water-soluble cargos in a sequential manner from simple bilayer PS nanoarchitectures, which was facilitated by conjugation of one cargo to thermo-sensitive PNIPAM for in situ gelation. These nontoxic C-PS have immense potential for in vivo applications including vaccine delivery, combination chemotherapy, and theranostics, with utility being even further expanded through the use of nontoxic thermoresponsive polymers like poly(polyethylene glycol citrate-co-N-isopropylacrylamide) (PPCN) [60].
Materials and Methods
Materials
All chemical reagents and solvents were purchased from Sigma-Aldrich and used as received without further purification, unless otherwise stated. Oligo(ethylene glycol) methyl ether methacrylate (OEGMA, average Mn = 300) was purified by basic alumina column chromatography to remove inhibitors. 2,2’-Azobis(isobutyronitrile) (AIBN) was recrystallized from 95% ethanol and stored at −20 °C. Amine terminated poly(N-isopropylacrylamide) (PNIPAM-NH2, average Mn = 2500) was purchased from Sigma-Aldrich, and rhodamine B-labeled PNIPAM (PNIPAM-RhB) was synthesized by the conjugation of PNIPAM-NH2 with Rhodamine B isothiocyanate following similar procedure in Ref. [61].
Characterization
1H NMR spectra (400 MHz) were recorded on a Bruker-400 NMR spectrometer using tetramethylsilane as an internal standard. Molecular weights were determined by gel permeation chromatography (GPC) using Waters Styragel tetrahydrofuran (THF) columns with a THF mobile phase (0.6 mL/min) via both refractive index and UV/vis detectors (ThermoFisher Scientific), and a calibration plot constructed with poly(ethylene oxide) standards. UV-vis absorbance and fluorescence measurements were taken on a SpectraMax M3 microplate reader (Molecular Devices). The size distribution and zeta potential of nanostructures were determined by dynamic light scattering (DLS) using Zetasizer Nano (Malvern Instruments) with a 4 mW He-Ne 633 nm laser at 1 mg/mL in PBS. Specimens for cryogenic transmission electron microscopy (cryoTEM) were prepared by applying 4 µL of 5 mg/ml sample on a pretreated, holey carbon 400 mesh TEM grids and were plunge-frozen with a Gatan Cryoplunge freezer at room temperature or 37 °C. Images were collected using a JEOL 3200FSC transmission electron microscope. Small angle X-ray scattering (SAXS) experiments were performed at the DuPont-Northwestern-Dow Collaborative Access Team (DND-CAT) beamline at Argonne National Laboratory’s Advanced Photon Source (Argonne, IL, USA) with 10 keV (wavelength λ = 1.24 Å) collimated X-rays. All the samples were analyzed in the q-range 0.001 to 0.5 Å-1 at an exposure time of 1 s and the q-range was calibrated using the diffraction patterns of silver behenate. The momentum transfer vector q is defined as q = 4π sinθ/λ, where θ is the scattering angle. PRIMUS 2.8.2 software was utilized for data reduction, where solvent/buffer scattering was removed from the acquired sample scattering. Then the model fitting of the reduced data was completed using SasView 4.0.1 software package. Flow cytometry was performed with FACSDiva on a LSRII flow cytometer (BD Biosciences) and data were analyzed with FlowJo software. Fluorescence visualization images were taken with a Leica TCS SP5 confocal laser-scanning microscope (CLSM) and images were open and processed using the FIJI ImageJ package. Two-tail Student t tests were performed to determine statistical significance.
Synthesis of OPSMA
First, hydroxyl and pyridyl disulfide heterobifunctional oligo(propylene sulfide) (OPS-OH) was synthesized via anionic ring-opening polymerization of propylene sulfide using 2-mercaptoethanol as the initiator according to the modified literature procedure [49]. Briefly, 2-mercaptoethanol (1.05 mL, 15 mmol) was dissolved in dimethylformamide (DMF) (180 mL) and the solution was stirred under argon atmosphere at room temperature. 1.2 equiv. of sodium methylate (36 mL, 0.5 M in methanol) was added via syringe, and the mixture was stirred for 15 min. To the solution 7 equiv. of propylene sulfide (8.23 mL) was added slowly via syringe, and then the reagents were allowed to react for 50 min. An excess of 2,2’-dipyridyl disulfide (12 g, 54.5 mmol) in DMF (30 mL) was finally added, and the reaction was continued overnight. DMF was removed, and the residue was dissolved in dichloromethane (DCM) (150 mL). The DCM solution was washed with dilute hydrochloric acid solution (0.1 M, 100 mL × 3), saturated sodium bicarbonate solution (100 mL × 1), water (100 mL × 3), and dried over anhydrous sodium sulfate. After filtration, yellowish-brown and oily OPS-OH was obtained by complete removal of the solvent. Then OPSMA monomer was prepared from the esterification of OPS-OH with methacryloyl chloride. OPS-OH (7 g, 10 mmol) and triethylamine (4.2 mL, 30 mmol) were dissolved in 70 mL of dried DCM. To the ice-water cooled solution, methacryloyl chloride (2 mL, 20 mmol) in 10 mL of DCM was added dropwise, and the mixture was stirred at room temperature for 24 h. After the removal of triethylamine hydrochloride by filtration, the DCM solution was washed with dilute hydrochloric acid solution (0.1 M, 100 mL × 3), saturated sodium bicarbonate solution (100 mL × 3), water (100 mL × 1), and dried over anhydrous sodium sulfate. After filtration, the obtained DCM solution was concentrated, and further purified by basic alumina column chromatography. Then the solvent was removed, and the yellowish-brown and oily OPSMA (5.65 g, yield 73%) was dried under vacuum. 1H NMR (CDCl3, δ, ppm, Figure S1): 8.437 (d, 1H, pyridyl), 7.727 (m, 1H, pyridyl), 7.621 (d, 1H, pyridyl), 7.082 (m, 1H, pyridyl), 6.100 (s, 1H, H2C=C), 5.566 (s, 1H, H2C=C), 4.277 (t, 2H, −OCH2−), 3.112–2.743 (broad, 16H, −SCH2−), 2.605 (broad, 7H, −SCH(CH3)CH3−), 1.926 (s, 3H, CH2=C-CH3), 1.363 (broad, 21H, −SCH(CH3)CH3−). According to the 1H NMR result, the average number of propylene sulfide repeat units is 7, thus the monomer was denoted as OPS7MA.
Synthesis of POEGMA-CTA
OEGMA (21.4 mL, 75 mmol), 4-cyano-4-(phenylcarbonothioylthio)pentanoic acid (0.84 g, 3 mmol), AIBN (40 mg, 0.25 mmol), and DMF (40 mL) were combined in a Schlenk flask which was capped with a rubber septum. The flask was degassed with argon by three freeze-pump-thaw cycles. After thermostating at 70 °C in an oil bath and stirring for 8 h, POEGMA-CTA was isolated by precipitation into an excess of n-hexane for three times. Yield 7.4 g (32%). Mn (GPC) = 5.5 kg/mol, Mw/Mn (GPC) = 1.21. 1H NMR (CDCl3, δ, ppm): 7.831 (m, 2H, phenyl), 7.473 (t, 1H, phenyl), 7.314 (m, 2H, phenyl), 4.039 (broad, 18H, −C(=O)OCH2−), 3.711–3.422 (broad, 150H, OEG), 3.334 (s, 27H, OEG−CH3), 2.430 (m, 2H, −CH2COOH), 2.061–0.714 (m, 47H, backbone). According to the 1H NMR result, the average number of OEGMA repeat units is 9.
Synthesis of POEGMA-POPSMA
Typically, POEGMA-CTA (150 mg, 0.055 mmol), OPS7MA (1 g, 1.3 mmol), AIBN (0.9 mg, 0.0055 mmol), and DMF (5 mL) were charged into a Schlenk flask which was capped with a rubber septum. The flask was degassed with argon by three freeze-pump-thaw cycles. After thermostating at 75 °C in an oil bath and stirring for 31 h, the mixture was precipitated into an excess of diethyl ether for three times. The final product was dried under vacuum overnight at room temperature, yielding a yellowish-brown oily product (0.37 g, 32%). Mn (GPC) = 8.5 kg/mol, Mw/Mn (GPC) = 1.28. The average number of OPS7MA repeat units was determined to be 8 by 1H NMR analysis in CDCl3, thus the polymer was denoted as POEGMA-POPS7MA8.
Fabrication of PS and Loading with Dyes
POEGMA-POPS7MA8 was assembled using the thin-film hydration method as previously described [8]. Typical procedures employed for the co-encapsulation of calcein and PNIPAM-RhB within PS are as follows. 20 mg of POEGMA-POPS7MA8 was dissolved in 200 µL of DCM within 1.8 mL clear glass vials (ThermoFisher Scientific). After desiccation to completely remove the solvent, a solution of calcein (30 mM) and PNIPAM-RhB (10 mM) in PBS (1 mL) was added to hydrate the resulting thin films under shaking (1500 rpm) for 24 h. The PS dispersion was then transferred into a syringe and passed through a 200 nm nylon membrane. Loaded PS were purified from unloaded dyes by size exclusion chromatography (SEC) with a Sepharose CL-6B size exclusion column. The loading efficiencies of calcein and PNIPAM-RhB were determined to be 10.06% and 10.69%, respectively (Figure S7). The co-encapsulation of Nile blue chloride and PNIPAM-RhB within PS was performed by using similar experimental protocols described above.
Shell Crosslinking Reaction
C-PS were prepared by the reaction of U-PS dispersion with 1,2-ethanedithiol through thiol-disulfide exchange reactions. In a typical example, l mL of U-PS dispersion (10 mg/mL) was mixed with 18 µL of 1,2-ethanedithiol solution (0.15 M in ethanol). The mixture was allowed to react under shaking (1500 rpm, room temperature) overnight and was then purified by SEC with a Sepharose CL-6B size exclusion column. The degree of crosslinking was 44%, which was estimated indirectly from the result of 2-thiopyridone determination (Figure S6) [54].
Release Experiments
Typically, 100 µL of C-PS or U-PS in PBS (5 mg/mL) loaded with calcein (30 mM) and PNIPAM-RhB (10 mM) were transferred into Slide-A-Lyzer™ MINI Dialysis Cassettes (10K MWCO, ThermoFisher Scientific). Then the Cassettes were immersed in 1.1 mL of PBS or H2O2-containing PBS at 25 °C. After shaking for predetermined time, 100 µL of samples were withdrawn and replenished with 100 µL of same release media. Dye contents were analyzed by fluorescence intensity and the cumulative release percentage was calculated. To determine the release percentage of calcein and PNIPAM-RhB at 37 °C, 2 mL of C-PS in PBS (5 mg/mL) loaded with calcein (30 mM) and PNIPAM-RhB (10 mM) were incubated with or without H2O2 at 37 °C. At predetermined time points, 0.2 mL of samples were withdrawn. The samples were cooled down to room temperature immediately and passed through a 100 nm nylon membrane filter. Dye contents were analyzed by fluorescence intensity and the release percentage was calculated. All samples were analyzed in triplicate.
Fluorescence Imaging of C-PS Using CLSM
Without the extrusion of 200 nm nylon membrane, large size distributed U-PS was prepared for shell crosslinking. Calcein and PNIPAM-Rh B were co-loaded during self-assembly following the similar procedures as described above. To a µ-Slide (chambered coverslip with 8 wells) containing 400 µL/well of PBS with varying H2O2 concentrations (0%, 0.5%, 1%, and 2%), 20 µL/ well of the resulting C-PS (10 mg/mL) were added. The C-PS dispersions were then incubated at 37 °C for 24 h. The supernatant was carefully aspirated, and washed gently with PBS at room temperature. The samples were imaged using CLSM with a 100x oil-immersion objective.
Intracellular Release
A2780 cells were seeded on 10 mm2 glass coverslips placed in a 24-well plate and cultured with Nile blue chloride and PNIPAM-RhB co-loaded C-PS (50 µg/mL) for predetermined time. The cells were washed twice with PBS and fixed with 4% paraformaldehyde for 30 min, following by staining with DAPI for 5 min. Rinsed the samples 3 times in PBS, then cells were mounted on glass slides and visualized using CLSM.
Internalization of C-PS Observed under CLSM
A2780 cells were seeded in a FluoroDish™ cell culture dish (10000 cells, 2 mL) and cultured overnight. The cells were treated with PNIPAM-RhB loaded C-PS (50 µg/mL) for 1 h, following by incubation in new culture medium for 1 h or 24 h. After that, the cells were further treated with LysoTracker® Green DND-26 (2.5 µg/mL, 30 min at 37 °C) and washed twice with PBS. Cell nuclei were stained in PBS using Hoechst 33342 (Invitrogen™ R37605, two drops per mL, 20 min), and CLSM images were then taken.
Supplementary Material
Acknowledgments
We would like to thank Dr. Jisheng Xiao and Dr. Jessica Hornick for their generous aid and helpful suggestion for CLSM imaging. The assistance from Jonathan Remis for cryoTEM observation is acknowledged. We acknowledge staff and instrumentation support from the Integrated Molecular Structure Education and Research Center, Structural Biology Facility, NU Atomic and Nanoscale Characterization Experimental Center, Robert H. Lurie Comprehensive Cancer Center Flow Cytometry Core, and Biological Imaging Facility at Northwestern University. This work was supported by the National Institutes of Health Director’s New Innovator Award (grant no. 1DP2HL132390-01), the Louis A. Simpson & Kimberly K. Querrey Center for Regenerative Nanomedicine Regenerative Nanomedicine Catalyst Award. SAXS experiments were performed at the DuPont-Northwestern-Dow Collaborative Access Team (DND-CAT) located at Sector 5 of the Advanced Photon Source (APS). DND-CAT is supported by Northwestern University, E.I. DuPont de Nemours & Co., and The Dow Chemical Company. This research used resources of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02-06CH11357.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributions
F.D. and E.A.S. contributed to the conception and study design. F.D. prepared the polymers. F.D. and S.B. characterized the materials. F.D., S.B., S.Y., and E.A.S. contributed to the data analysis. F.D. and E.A.S. wrote the manuscript.
Conflict of Interest
F.D., S.B., S.Y., and E.A.S. declare that they have no conflicts of interest.
References
- 1.Discher DE, Eisenberg A. Polymer Vesicles. Science. 2002;297(5583):967–973. doi: 10.1126/science.1074972. [DOI] [PubMed] [Google Scholar]
- 2.Zhu Y, et al. Polymer vesicles: Mechanism, preparation, application, and responsive behavior. Progress in Polymer Science. 2017;64(Supplement C):1–22. [Google Scholar]
- 3.Vasdekis AE, et al. Vesicle Photonics. Annual Review of Materials Research. 2013;43(1):283–305. [Google Scholar]
- 4.Thambi T, Park JH, Lee DS. Stimuli-responsive polymersomes for cancer therapy. Biomaterials Science. 2016;4(1):55–69. doi: 10.1039/c5bm00268k. [DOI] [PubMed] [Google Scholar]
- 5.Scott EA, Karabin NB, Augsornworawat Overcoming Immune Dysregulation with Immunoengineered Nanobiomaterials. Annual Review of Biomedical Engineering. 2017;19(1):57–84. doi: 10.1146/annurev-bioeng-071516-044603. [DOI] [PubMed] [Google Scholar]
- 6.Lee JS, Feijen J. Polymersomes for drug delivery: Design, formation and characterization. Journal of Controlled Release. 2012;161(2):473–483. doi: 10.1016/j.jconrel.2011.10.005. [DOI] [PubMed] [Google Scholar]
- 7.Brinkhuis RP, Rutjes FPJT, van Hest JCM. Polymeric vesicles in biomedical applications. Polymer Chemistry. 2011;2(7):1449–1462. [Google Scholar]
- 8.Scott EA, et al. Dendritic cell activation and T cell priming with adjuvant- and antigen-loaded oxidation-sensitive polymersomes. Biomaterials. 2012;33(26):6211–6219. doi: 10.1016/j.biomaterials.2012.04.060. [DOI] [PubMed] [Google Scholar]
- 9.Yi S, et al. Tailoring Nanostructure Morphology for Enhanced Targeting of Dendritic Cells in Atherosclerosis. ACS Nano. 2016;10(12):11290–11303. doi: 10.1021/acsnano.6b06451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Stano A, et al. Tunable Tcell immunity towards a protein antigen using polymersomes vs solid-core nanoparticles. Biomaterials. 2013;34(17):4339–4346. doi: 10.1016/j.biomaterials.2013.02.024. [DOI] [PubMed] [Google Scholar]
- 11.Iyisan B, et al. Multifunctional and Dual-Responsive Polymersomes as Robust Nanocontainers: Design, Formation by Sequential Post-Conjugations, and pH-Controlled Drug Release. Chemistry of Materials. 2016;28(5):1513–1525. [Google Scholar]
- 12.Allen S, et al. Facile assembly and loading of theranostic polymersomes via multi-impingement flash nanoprecipitation. Journal of Controlled Release. 2017;262(Supplement C):91–103. doi: 10.1016/j.jconrel.2017.07.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Du F, Liu Y-G, Scott EA. Immunotheranostic Polymersomes Modularly Assembled from Tetrablock and Diblock Copolymers with Oxidation-Responsive Fluorescence. Cellular and Molecular Bioengineering. 2017;10(5):357–370. doi: 10.1007/s12195-017-0486-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dowling DJ, et al. Toll-like receptor 8 agonist nanoparticles mimic immunomodulating effects of the live BCG vaccine and enhance neonatal innate and adaptive immune responses. Journal of Allergy and Clinical Immunology. 2017;140(5):1339–1350. doi: 10.1016/j.jaci.2016.12.985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Allen S, Liu Y-G, Scott E. Engineering Nanomaterials to Address Cell-Mediated Inflammation in Atherosclerosis. Regenerative Engineering and Translational Medicine. 2016;2(1):37–50. doi: 10.1007/s40883-016-0012-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Deng Z, et al. Engineering Intracellular Delivery Nanocarriers and Nanoreactors from Oxidation-Responsive Polymersomes via Synchronized Bilayer Cross-Linking and Permeabilizing Inside Live Cells. Journal of the American Chemical Society. 2016;138(33):10452–10466. doi: 10.1021/jacs.6b04115. [DOI] [PubMed] [Google Scholar]
- 17.Vasdekis AE, et al. Precision Intracellular Delivery Based on Optofluidic Polymersome Rupture. ACS Nano. 2012;6(9):7850–7857. doi: 10.1021/nn302122h. [DOI] [PubMed] [Google Scholar]
- 18.Wang Z, et al. Tunable Permeability of Cross-Linked Microcapsules from pH-Responsive Amphiphilic Diblock Copolymers: A Dissipative Particle Dynamics Study. Langmuir. 2017;33(29):7288–7297. doi: 10.1021/acs.langmuir.7b01586. [DOI] [PubMed] [Google Scholar]
- 19.Larrañaga A, et al. Polymer capsules as micro-/nanoreactors for therapeutic applications: Current strategies to control membrane permeability. Progress in Materials Science. 2017;90(Supplement C):325–357. [Google Scholar]
- 20.Lai M-H, et al. Tailoring Polymersome Bilayer Permeability Improves Enhanced Permeability and Retention Effect for Bioimaging. ACS Applied Materials & Interfaces. 2014;6(13):10821–10829. doi: 10.1021/am502822n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hu X, et al. Stimuli-Responsive Polymersomes for Biomedical Applications. Biomacromolecules. 2017;18(3):649–673. doi: 10.1021/acs.biomac.6b01704. [DOI] [PubMed] [Google Scholar]
- 22.Fu J, Liang L, Qiu L. In Situ Generated Gold Nanoparticle Hybrid Polymersomes for Water-Soluble Chemotherapeutics: Inhibited Leakage and pH-Responsive Intracellular Release. Advanced Functional Materials. 2017;27(18):1604981-n/a. [Google Scholar]
- 23.Nicolas J, et al. Design, functionalization strategies and biomedical applications of targeted biodegradable/biocompatible polymer-based nanocarriers for drug delivery. Chemical Society Reviews. 2013;42(3):1147–1235. doi: 10.1039/c2cs35265f. [DOI] [PubMed] [Google Scholar]
- 24.Kanamala M, et al. Mechanisms and biomaterials in pH-responsive tumour targeted drug delivery: A review. Biomaterials. 2016;85(Supplement C):152–167. doi: 10.1016/j.biomaterials.2016.01.061. [DOI] [PubMed] [Google Scholar]
- 25.Ulbrich K, et al. Targeted Drug Delivery with Polymers and Magnetic Nanoparticles: Covalent and Noncovalent Approaches, Release Control, and Clinical Studies. Chemical Reviews. 2016;116(9):5338–5431. doi: 10.1021/acs.chemrev.5b00589. [DOI] [PubMed] [Google Scholar]
- 26.Mokhtarzadeh A, et al. Nano-delivery system targeting to cancer stem cell cluster of differentiation biomarkers. Journal of Controlled Release. 2017;266(Supplement C):166–186. doi: 10.1016/j.jconrel.2017.09.028. [DOI] [PubMed] [Google Scholar]
- 27.Ma X, et al. Future of nanotherapeutics: Targeting the cellular sub-organelles. Biomaterials. 2016;97(Supplement C):10–21. doi: 10.1016/j.biomaterials.2016.04.026. [DOI] [PubMed] [Google Scholar]
- 28.Hong J, et al. Graphene Multilayers as Gates for Multi-Week Sequential Release of Proteins from Surfaces. ACS Nano. 2012;6(1):81–88. doi: 10.1021/nn202607r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Xu W, et al. Hierarchical Assembly of Star Polymer Polymersomes into Responsive Multicompartmental Microcapsules. Chemistry of Materials. 2016;28(3):975–985. [Google Scholar]
- 30.Delcea M, et al. Multicompartmental Micro- and Nanocapsules: Hierarchy and Applications in Biosciences. Macromolecular Bioscience. 2010;10(5):465–474. doi: 10.1002/mabi.200900359. [DOI] [PubMed] [Google Scholar]
- 31.Marguet M, Bonduelle C, Lecommandoux S. Multicompartmentalized polymeric systems: towards biomimetic cellular structure and function. Chemical Society Reviews. 2013;42(2):512–529. doi: 10.1039/c2cs35312a. [DOI] [PubMed] [Google Scholar]
- 32.Driever CD, et al. Converging layer-by-layer polyelectrolyte microcapsule and cubic lyotropic liquid crystalline nanoparticle approaches for molecular encapsulation. Soft Matter. 2011;7(9):4257–4266. [Google Scholar]
- 33.De Geest BG, et al. Self-Exploding Beads Releasing Microcarriers. Advanced Materials. 2008;20(19):3687–3691. [Google Scholar]
- 34.Zou Y, et al. Development of an injectable two-phase drug delivery system for sequential release of antiresorptive and osteogenic drugs. Journal of Biomedical Materials Research Part B: Applied Biomaterials. 2012;100B(1):155–162. doi: 10.1002/jbm.b.31933. [DOI] [PubMed] [Google Scholar]
- 35.Kulygin O, et al. Subcompartmentalized Polymer Hydrogel Capsules with Selectively Degradable Carriers and Subunits. Small. 2010;6(14):1558–1564. doi: 10.1002/smll.201000453. [DOI] [PubMed] [Google Scholar]
- 36.Walker SA, Kennedy MT, Zasadzinski JA. Encapsulation of bilayer vesicles by self-assembly. Nature. 1997;387:61. doi: 10.1038/387061a0. [DOI] [PubMed] [Google Scholar]
- 37.Katagiri K, et al. Layer-by-Layer Self-Assembling of Liposomal Nanohybrid “Cerasome” on Substrates. Langmuir. 2002;18(17):6709–6711. [Google Scholar]
- 38.Katagiri K, et al. Layered Paving of Vesicular Nanoparticles Formed with Cerasome as a Bioinspired Organic-Inorganic Hybrid. Journal of the American Chemical Society. 2002;124(27):7892–7893. doi: 10.1021/ja0259281. [DOI] [PubMed] [Google Scholar]
- 39.Xu W, et al. Multicompartmental Microcapsules with Orthogonal Programmable Two-Way Sequencing of Hydrophobic and Hydrophilic Cargo Release. Angewandte Chemie International Edition. 2016;55(16):4908–4913. doi: 10.1002/anie.201600383. [DOI] [PubMed] [Google Scholar]
- 40.Bobbala S, Allen SD, Scott EA. Flash nanoprecipitation permits versatile assembly and loading of polymeric bicontinuous cubic nanospheres. Nanoscale. 2017 doi: 10.1039/c7nr06779h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kim S-H, et al. Multiple Polymersomes for Programmed Release of Multiple Components. Journal of the American Chemical Society. 2011;133(38):15165–15171. doi: 10.1021/ja205687k. [DOI] [PubMed] [Google Scholar]
- 42.Peyret A, et al. Liposomes in Polymersomes: Multicompartment System with Temperature-Triggered Release. Langmuir. 2017;33(28):7079–7085. doi: 10.1021/acs.langmuir.7b00655. [DOI] [PubMed] [Google Scholar]
- 43.Marguet M, Edembe L, Lecommandoux S. Polymersomes in Polymersomes: Multiple Loading and Permeability Control. Angewandte Chemie. 2012;124(5):1199–1202. doi: 10.1002/anie.201106410. [DOI] [PubMed] [Google Scholar]
- 44.Wang X, et al. Concurrent Block Copolymer Polymersome Stabilization and Bilayer Permeabilization by Stimuli-Regulated “Traceless” Crosslinking. Angewandte Chemie. 2014;126(12):3202–3206. doi: 10.1002/anie.201310589. [DOI] [PubMed] [Google Scholar]
- 45.Miksa B. Recent progress in designing shell cross-linked polymer capsules for drug delivery. RSC Advances. 2015;5(107):87781–87805. [Google Scholar]
- 46.Lee JS, et al. Hydrosomes, novel thermosensitive gel-containing polymersomes. Journal of Controlled Release. 2008;132(3):e28–e29. [Google Scholar]
- 47.Lee JS, et al. Thermosensitive hydrogel-containing polymersomes for controlled drug delivery. Journal of Controlled Release. 2010;146(3):400–408. doi: 10.1016/j.jconrel.2010.06.002. [DOI] [PubMed] [Google Scholar]
- 48.Napoli A, et al. Oxidation-responsive polymeric vesicles. Nature Materials. 2004;3:183. doi: 10.1038/nmat1081. [DOI] [PubMed] [Google Scholar]
- 49.Napoli A, et al. New Synthetic Methodologies for Amphiphilic Multiblock Copolymers of Ethylene Glycol and Propylene Sulfide. Macromolecules. 2001;34(26):8913–8917. [Google Scholar]
- 50.Krivorotova T, et al. Conventional free-radical and RAFT copolymerization of polyethylene oxide) containing macromonomers. European Polymer Journal. 2010;46(3):546–556. [Google Scholar]
- 51.Roth PJ, Davis T, Lowe AB. Comparison between the LCST and UCST Transitions of Double Thermoresponsive Diblock Copolymers: Insights into the Behavior of POEGMA in Alcohols. Macromolecules. 2012;45(7):3221–3230. [Google Scholar]
- 52.Liu X, et al. Multi-Stimuli-Responsive Amphiphilic Assemblies through Simple Postpolymerization Modifications. Macromolecules. 2016;49(17):6186–6192. doi: 10.1021/acs.macromol.6b01397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Goto A, Fukuda T. Kinetics of living radical polymerization. Progress in Polymer Science. 2004;29(4):329–385. [Google Scholar]
- 54.Glatz Z, Mašlaňová H. Specific thiol determination by micellar electrokinetic chromatography and on-column detection reaction with 2,2 ′-dipyridyldisulfide. Journal of Chromatography A. 2000;895(1):179–187. doi: 10.1016/s0021-9673(00)00475-1. [DOI] [PubMed] [Google Scholar]
- 55.Cerritelli S, et al. Aggregation Behavior of Polyethylene glycol-bl-propylene sulfide) Di- and Triblock Copolymers in Aqueous Solution. Langmuir. 2009;25(19):11328–11335. doi: 10.1021/la900649m. [DOI] [PubMed] [Google Scholar]
- 56.Wang G, et al. Enhanced Retention of Encapsulated Ions in Cross-Linked Polymersomes. The Journal of Physical Chemistry B. 2015;119(11):4300–4308. doi: 10.1021/jp5125316. [DOI] [PubMed] [Google Scholar]
- 57.Dai J, et al. Interlayer-Crosslinked Micelle with Partially Hydrated Core Showing Reduction and pH Dual Sensitivity for Pinpointed Intracellular Drug Release. Angewandte Chemie International Edition. 2011;50(40):9404–9408. doi: 10.1002/anie.201103806. [DOI] [PubMed] [Google Scholar]
- 58.Bandyopadhyay A, Gao J. Iminoboronate-Based Peptide Cyclization That Responds to pH, Oxidation, and Small Molecule Modulators. Journal of the American Chemical Society. 2016;138(7):2098–2101. doi: 10.1021/jacs.5b12301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.De Koker S, Hoogenboom R, De Geest BG. Polymeric multilayer capsules for drug delivery. Chemical Society Reviews. 2012;41(7):2867–2884. doi: 10.1039/c2cs15296g. [DOI] [PubMed] [Google Scholar]
- 60.Yang J, et al. A thermoresponsive biodegradable polymer with intrinsic antioxidant properties. Biomacromolecules. 2014;15(11):3942–52. doi: 10.1021/bm5010004. [DOI] [PubMed] [Google Scholar]
- 61.Gursahani H, et al. Absorption of polyethylene glycol (PEG) polymers: The effect of PEG size on permeability. Journal of Pharmaceutical Sciences. 2009;98(8):2847–2856. doi: 10.1002/jps.21635. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.