

Abstract
Myeloid-derived suppressor cells (MDSCs) are known suppressors of anti-tumor immunity, affecting amino acid metabolism and T cell function in the tumor microenvironment (TME). However, it is unknown if MDSCs regulate B cell responses during tumor progression. Using a syngeneic mouse model of lung cancer, we show reduction in percentages and absolute numbers of B cell subsets including Pro-, Pre- and mature B cells in the bone marrow (BM) of tumor-bearing mice. The kinetics of this impaired B cell response correlated with the progressive infiltration of MDSCs. We identified that IL-7 and downstream STAT5 signaling that play a critical role in B cell development and differentiation were also impaired during tumor progression. Global impairment of B cell function was indicated by reduced serum IgG levels. Importantly, we show that anti-Gr-1 antibody-mediated depletion of MDSCs not only rescued serum IgG and IL-7 levels, but also reduced TGF-β1, a known regulator of stromal IL-7, suggesting MDSC-mediated regulation of B cell responses. Further, blockade of IL-7 resulted in reduced phosphorylation of downstream STAT5 and B cell differentiation in tumor-bearing mice and administration of TGF-β blocking antibody rescued these IL-7 dependent B cell responses. Adoptive transfer of BM-derived MDSCs from tumor-bearing mice into congenic recipients resulted in significant reductions of B cell subsets in the BM and in circulation. MDSCs also suppressed B cell proliferation in vitro in an arginase-dependent manner that required cell-to-cell contact. Our results indicate that tumor-infiltrating MDSCs may suppress humoral immune responses and promote tumor escape from immune surveillance.
Introduction
Myeloid-derived suppressor cells (MDSCs) are heterogeneous immature myeloid cells that are drivers of tumor associated immune suppression (1–6). Broadly identified as Gr-1+CD11b+ cells in tumor-bearing mice, MDSCs segregate further into granulocytic and monocytic subsets (1–4). Accumulating evidence suggests that MDSCs modulate T cell responses in the tumor microenvironment (TME), by induction of multiple pathways that regulate oxidative and nitrative stress such as inducible nitric oxide synthase (iNOS), arginase 1 (ARG1), reactive oxygen species (ROS), and by the induction of regulatory T (Treg) cells (1–3, 5, 6). Additionally, recent reports of suppression of B cell responses in experimental autoimmune myasthenia gravis and a murine acquired immunodeficiency model (7, 8) have been attributed to MDSCs. But the potential role of MDSCs in regulation of B cell responses during tumor progression is currently unknown.
B cells can either positively or negatively regulate immune responses (9). B cells positively regulate cellular immune responses by producing antibodies (10), by serving as antigen presenting cells (APCs) (11), by secreting cytokines and chemokines, and by providing co-stimulatory signals to T cells (12, 13). Tumor-reactive B cells play a pivotal role in generating potent and long-term T cell responses against cancer (13, 14). Recently identified subset of regulatory B (Breg) cells also is known to promote tumor growth (15–18).
Interleukine-7 (IL-7), a cytokine which plays a pivotal role in B cell lineage commitment, regulation of B cell survival, proliferation and maturation (19, 20), is primarily produced by non-hematopoietic cells including fibroblastic stromal cells in the BM and in the TME (21). Stromal IL-7 can be regulated by TGF-β (22), one of the key immunoregulatory cytokines produced by MDSCs (3). IL-7/IL-7R axis regulates early B cell development by activation of downstream signal transducer and activator of transcription 5 (STAT5) (23). Additionally, suppressor of cytokine signaling 1 (SOCS1) inhibits IL-7 responses in developing B lineage cells (24). A significant contribution of IL-7 and STAT5 signaling in B cell responses has not been described during tumor progression.
In the present study, we show that B cell differentiation and function are impaired during tumor progression. We provide evidence that MDSCs directly suppress B cell responses by inhibiting IL-7 and downstream STAT5 signaling that are essential for B cell differentiation. Anti-Gr-1 antibody-mediated depletion of MDSCs reduced TGF-β1 levels and partially rescued serum IgG, IL-7, phosphorylation of STAT5 and B cell differentiation in tumor-bearing mice. These data show that MDSCs directly inhibit B cell responses to tumors and suggest that targeted deletion of MDSCs could have beneficial effect by enhancing B cell responses in cancer.
Materials and Methods
Syngeneic orthotopic mouse model of lung cancer
Female C57BL/6 mice and C57BL/6 congenic CD45.1+ mice at 6 to 8 week of age were purchased from The Jackson Laboratory (Bar Harbor, ME). Mice were kept in pathogen-free conditions and handled in accordance with the Guidelines for Animal Experiments at the University of Alabama at Birmingham. The murine Lewis Lung Carcinoma (LLC) cell line was purchased from American Type Culture Collection (ATCC; Manassas, VA). LLC cells were cultured in Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 10% FBS, 1mmol/L sodium pyruvate, 2 mmol/L L-glutamine, 10 μg/mL penicillin-streptomycin, and 0.1 mmol/L nonessential amino acids (Life Technologies; Waltham, MA). 106 LLC cells in 100 μl PBS were injected either intravenously (i.v.) via tail-vein injection, or via an intracardiac (i.c.) route (25). BM and spleens were collected for analyses on day 16, or at other specified time points, after injection of LLC cells.
Flow cytometry
BM from tibias and femurs, as well as spleens were harvested as previously described (25). Red blood cells were removed by ACK lysis buffer. Fc receptors were blocked with 3% BSA in PBS containing 2.4G2 antibody (anti-mouse CD16/CD32; BD Pharmingin), followed by staining with relevant antibodies. Fluorescein isothiocyanate (FITC)–conjugated anti-IgM (II/41), anti-CD21 (eBio4E3), phycoerythrin (PE)–conjugated anti-CD43 (eBioR2/60), anti-CD23 (B3B4), anti-Gr-1 (RB6-8C5), anti-CD25 (PC61.5), anti-IL-10 (JES5-16E3), allophycocyanin (APC) –conjugated anti-CD93 (AA4.1), anti-CD24 (M1/69), anti-CD45.2 (104), anti-F4/80 (BM8), anti-CD5 (53-7.3), eFluor 450–conjugated anti-IgM (II/41), anti-CD1d (1B1), PerCP-eFluor 710–conjugated anti-IgD (11-26C), PerCP-Cyanine (Cy) 5.5 conjugated anti-Ly6C (HK1.4), anti-Foxp3 (FJK-16s), PE-eFluor 610–conjugated anti-CD45.1 (A20), PE-Cy7 conjugated anti-CD19 (eBio1D3), anti-CD4 (GK1.5), APC-eFluor 780 conjugated anti-CD45R (B220; RA3-6B2) antibodies were purchased from Life Technologies (Grand Island, NY). FITC–conjugated anti-Ly6G (1A8), anti-Annexin V and APC-Cy7–conjugated CD11b antibodies were purchased from BD Bioscience (San Jose, CA). Data were collected with LSR-II flow cytometer (Becton Dickinson) and analyzed with FlowJo software (version 8.5.2; TreeStar, Ashland, OR).
In vivo treatment with anti-Gr-1 antibody
Anti-Gr-1 or IgG2b control antibody was administered intraperitoneally into tumor-bearing mice as previously described (25, 26). Mice transplanted with LLC cells i.v. were given intraperitoneal injections of anti-Gr1 antibody (250 μg/100μl/mouse; BioXcell, West Lebanon, NH) or immunoglobulin G2b (IgG2b) control (BioXcell, West Lebanon, NH) on days 4, 7 and 11. BM, serum, and spleen tissues were collected for analyses on day 16 after injection of LLC cells.
In vivo treatment with anti-TGF-β antibody
Mice transplanted with LLC i.c. were given intraperitoneal injections of anti-TGF-β antibody (clone 1D11.16.8, 300 μg/100μl/mouse; BioXcell, West Lebanon, NH) on days 4 and 7. BM, serum, and spleen tissues were collected for analyses at day 11 after injection of LLC cells.
In vivo treatment with anti-IL-7 antibody
Mice transplanted with LLC i.c. were given intraperitoneal injections of anti-IL-7 antibody (clone M25, 300 μg/100μl/mouse; BioXcell, West Lebanon, NH) or mouse IgG2b isotype control (Clone MPC-11, 300 μg/100μl/mouse; BioXcell, West Lebanon, NH) on days 4 and 7. BM, serum, spleen and lung tissues were collected for analyses at day 11 after injection of LLC cells.
Adoptive transfer of MDSCs via intratibial injection
MDSCs from BM or tumor tissues from CD45.2+ mice challenged with LLCs were purified by cell sorting on FACSAria cell sorter (BD Bioscience; San Jose, CA). 5 × 105 sorted CD11b+Gr-1+ MDSCs were injected into tibias of CD45.1+ recipients. Peripheral blood, spleen and BM samples were collected for analysis on day 7 after MDSC transfer.
In vitro B cell inhibitory assays
Splenocytes from naïve mice were labeled with CFSE (Molecular Probes; Eugene, OR) and cultured for 72 hours with 20 μg/ml LPS (Sigma-Aldrich; St. Louis, MO) and 10 ng/ml IL-4 (PeproTech; Rocky Hills, NJ). The pre-activated splenocytes then were co-cultured for 48 hours with MDSCs purified from the BM of tumor-bearing mice at a ratio of 1:1 in the absence or presence of 20 μM arginase inhibitor nor-NOHA (Cayman Chemical; Ann Arbor, MI), 500 nM iNOS inhibitor 1400W (Cayman Chemical; Ann Arbor, MI) or 1 mM IDO inhibitor 1-D-MT (Sigma-Aldrich; St. Louis, MO). The percentages of CD19+CFSElow cells (proliferating cells) were determined by FACS analysis. In some experiments, splenocytes were depleted of T cells by using Dynabeads FlowComp Mouse Pan T kit (Life Technology, Grand Island, NY). The same co-culture assay was performed with the T cell depleted splenocytes as described above.
In some experiments, splenocytes from naïve mice were stimulated with LPS (20 μg/ml) and IL-4 (10 ng/ml) for 24 hrs. To determine whether the inhibitory effect of MDSCs on B cells is T cell dependent or not, B220+CD19+ cells were sorted and labeled with CFSE. The sorted B cells were co-cultured with MDSCs purified from the BM of tumor-bearing mice or immature myeloid cells (IMCs) from naïve mice in the absence or presence of nor-NOHA, 1400W for 72 hrs. To determine whether suppression of B cells by MDSCs is T cell dependent, T cells from the spleens of tumor-bearing mice were isolated with Dynabeads FlowComp Mouse Pan T kit (Life Technology, Grand Island, NY). The purified T cells were then added to the B cells and MDSCs in the co-cultures described above. In blocking experiments, anti-TGFβ antibody (clone 1D11.16.8, BioXcell, West Lebanon, NH) or isotype control mouse IgG1(clone MOPC21, BioXcell, West Lebanon, NH) were added to the B cells and MDSCs co-culture assays. To determine whether MDSCs suppress B cell proliferation through direct cell-to-cell contact, MDSC-B cell co-culture experiments were established in the presence or absence of transwell inserts (Corning, NY).
In vitro co-culture of B cells and Treg cells
Treg cells were purified from tumor-bearing Foxp3-DTR-GFP mice (kindly provided by Dr. Troy Randall at University of Alabama at Birmingham). Splenocytes were stimulated with LPS (20 μg/ml) and IL-4 (10 ng/ml) for 24 hrs. B220+CD19+ cells were sorted and labeled with CFSE. These pre-activated B cells were co-cultured with CD4+GFP+ Treg cells or CD4+GFP− T cells purified from spleens of tumor-bearing mice. The percentages of CD19+CFSElow cells (proliferating cells) were determined by FACS analysis.
Quantitation of IgG, IgM, IL-7, IL-2 and TGF-β1 by Enzyme Linked Immunosorbent Assay (ELISA)
The mouse sera were harvested from naïve and tumor-bearing mice. The supernatants were collected from the in vitro B cell inhibitory assays as described above. The levels of IgG and IgM were determined by ELISA kits from Life Technologies (Grand Island, NY) in sera and B cell culture supernatants following the instructions of the manufacturer. The levels of IL-7 were measured by a mouse IL-7 ELISA kit from R & D Systems (Minneapolis, MN). The levels of IL-2 in sera were quantified by an ELISA kit from Abcam (Cambridge, MA). The levels of TGF-β1 in sera or supernatants were measured using a commercial ELISA kit purchased from Life Technologies (Grand Island, NY).
Immunoblotting
BM cells sorted CD19+B220+ cells from BM were collected in RIPA lysis buffer. A total of 15-20 μg protein was used for the immunoblotting, unless otherwise indicated. Immunoblotting for total STAT5, phospho-STAT5, SOCS1, phospho-Btk (Try223) or Btk (Cell Signaling; Danvers, MA) was performed as described previously (25). β-actin was purchased from Sigma-Aldrich (St. Louis, MO), and used for the loading control.
Statistical analysis
One-way ANOVA with Tukey’s multiple comparisons test was used for multiple groups. Unpaired t test was used for the statistical analyses between two groups by using GraphPad Prism 5. P < 0.05 was considered statistically significant.
Results
Increased MDSCs in the BM of tumor-bearing mice
We used an established syngeneic orthotopic mouse model of lung cancer to evaluate mechanisms by which MDSCs affect B cell responses during tumor progression (26). Increased infiltration of MDSCs has been associated with tumor progression in murine models of lung cancer (25, 26). The percentages and total numbers of BM-MDSCs were increased in tumor-bearing mice on day 16 after intravenous implantation of LLC cells and on day 11 after intracardiac transplantation, compared to naïve tumor-free mice (Fig. 1A). Infiltration of both granulocytic (CD11b+Ly6G+Ly6Clow) and monocytic (CD11b+Ly6G−Ly6Chigh) MDSC subsets were significantly increased (Supplemental Fig. 1A, 1B) in BM, and in the spleens of tumor-bearing mice (Fig. 1B, Supplemental Fig. 1C, 1D).
Figure 1.
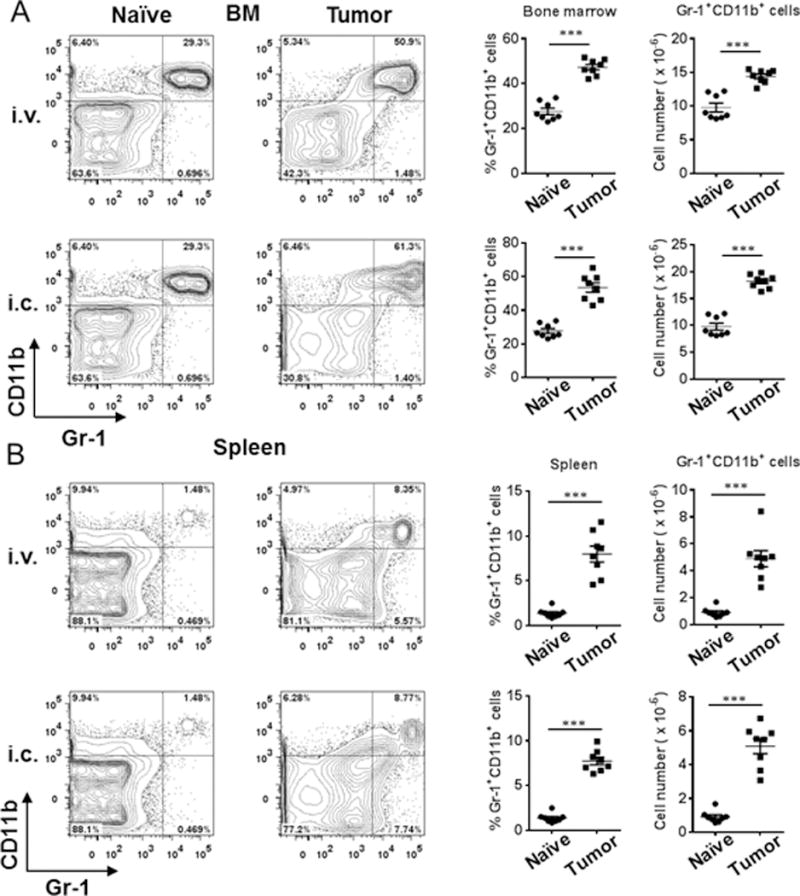
Increased MDSCs in the BM and spleens of tumor-bearing mice. (A) FACS plots, percentages and cell numbers of Gr-1+CD11b+ MDSCs in the BM from naïve mice and tumor-bearing mice on day 16 post-LLC intravenous injection and on day 11 post-LLC intra-cardiac injection (n = 8 mice/group). (B) FACS plots, percentages and cell numbers of Gr-1+CD11b+ MDSCs in the spleens from naïve mice and tumor-bearing mice on day 16 post-LLC intravenous injection and on day 11 post-LLC intra-cardiac injection (n = 8 mice/group). **, P < 0.01; ***, P < 0.001.
Impaired B cell differentiation in tumor-bearing mice
We then determined if B cell populations/numbers and/or their differentiation were impaired with increasing infiltration of MDSCs by enumerating B cell subsets in the BM and spleens of tumor-bearing mice (27, 28). The percentages of total B220+ cells, B220+IgD−IgM−CD24intCD43+ pro-B cells and B220+IgD+IgM+ mature B cells were decreased (Fig. 2A, i.v. model), while the absolute numbers of B220+IgD−IgM−CD24hiCD43− pre-B cells and B220+IgD−IgM+ immature B cells in addition to the above subsets of B cells were significantly decreased in the BM of tumor-bearing mice (Fig. 2B, i.v. model). To determine if modulation of B cell responses occurs with increasing tumor burden and correlates with increasing infiltration of tumor promoting MDSCs, we investigated the dynamics of this response. Time course studies revealed that this reduction in the numbers of B220+ cells, pro-B cells and pre-B cells in the BM occurred as early as 5 days post-tumor challenge (Supplemental Fig. 2A, 2B), whereas immature and mature B cells in the BM were reduced at day 11 (Supplemental Fig. 2C, 2D). Additionally, the dynamics of MDSC infiltration in BM coincided with the decline of B cell populations in the BM (Supplemental Fig. 3A, 3B). In the spleens of tumor-bearing mice, both the percentages and numbers of B220+CD93+ immature B cells were increased, while B220+CD93−CD21intCD23+ follicular B cells were decreased (Fig. 2C, 2D, i.v. challenged). Time course studies showed that the modulation of immature and follicular B cells was observed as early as 5 days after tumor challenge (Supplemental Fig. 3C-F). In the lungs of tumor-bearing mice, both the percentages and numbers of B cell populations were reduced in a time-dependent manner (Supplemental Fig. 3G, 3H). Similar results were obtained in intracardiac challenged tumor model (data not shown). These data suggest that impaired B cell differentiation was associated with MDSC infiltration and tumor progression.
Figure 2.
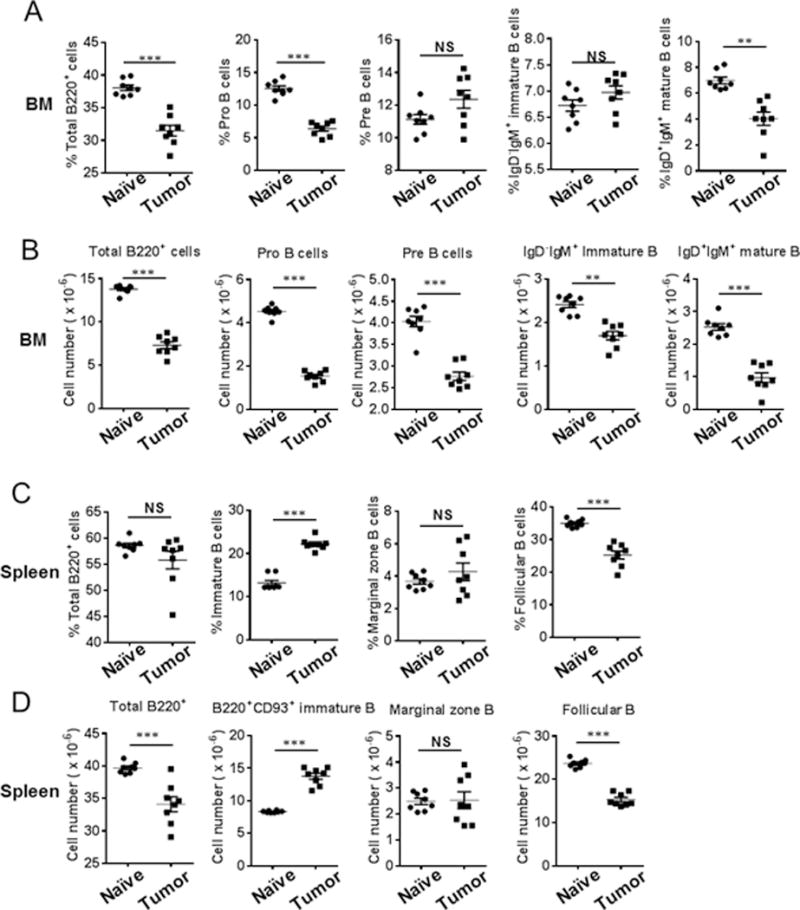
Impairment of B cell subsets in the BM and spleens of tumor-bearing mice. Percentages of Pro- and mature B cells were reduced whereas absolute numbers of Pro-, Pre-, immature and mature B cells decreased in bone marrow of tumor-bearing mice after intravenous challenge with LLC tumor cells. (A) The percentages of total B220+, Pro-, Pre-, immature, and mature B cells were determined by FACS analysis of cells harvested from the BM of naïve and tumor-bearing mice on day 16 post-LLC intravenous challenge (n = 8 mice/group). (B) Absolute numbers of total B220+, Pro-, Pre-, immature, and mature B cells in the BM were calculated. Percentages and absolute numbers of follicular B cells were reduced, while immature B cells were increased in the spleens of tumor-bearing mice on day 16 post-LLC intravenous challenge with LLC tumor cells (n = 8 mice/group). (C) The percentages of total B, immature B, marginal zone B, and follicular B cells were determined by FACS analysis of cells harvested from the spleens of naïve and tumor-bearing mice on day 16 post-LLC intravenous challenge (n = 8 mice/group). (D) Absolute numbers of total B, immature B, marginal zone B, and follicular B cells in the spleens are presented (n = 8 mice/group). **, P < 0.01; ***, P < 0.001.
Serum IgG and IL-7 are reduced in tumor-bearing mice
To determine whether B cell function is also impaired during tumor progression, we evaluated serum IgM and IgG levels in tumor-bearing mice. Serum IgG was decreased in tumor-bearing mice (Fig. 3A, 3B) as early as 5 days post tumor challenge (Fig. 3C) and correlated with the decline in B cell numbers (Fig. 3D, 3E); however, no difference was observed in IgM levels between tumor-bearing mice and tumor-free naïve mice (Fig. 3A, 3B).
Figure 3.
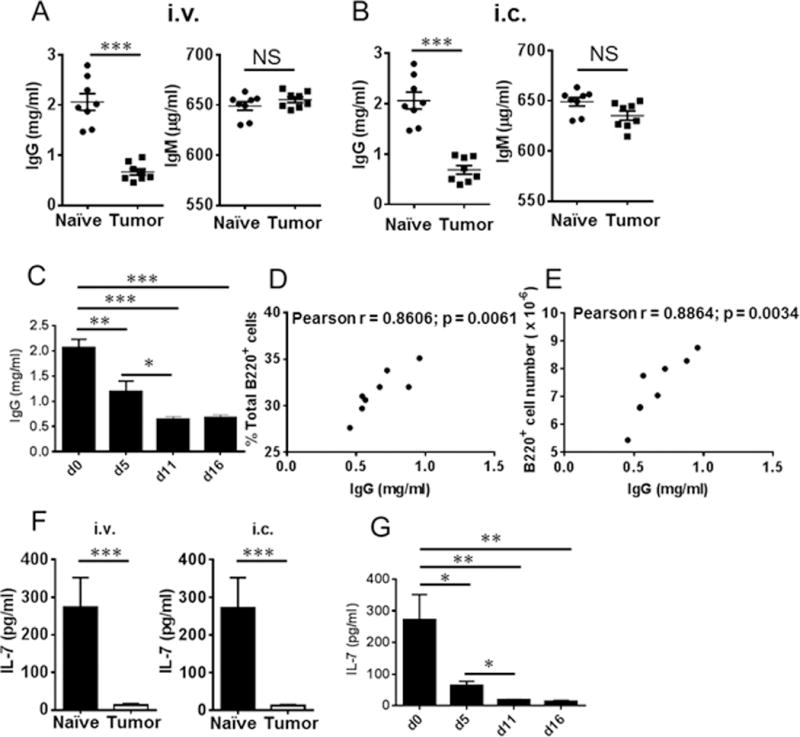
Reduced IgG and IL-7 levels in the serum of tumor-bearing mice. (A) IgG and IgM levels were detected in the serum of tumor-bearing mice on day 16 post-LLC intravenous challenge (n = 8 mice/group). (B) IgG and IgM levels were detected in the serum of tumor-bearing mice on day 11 post-LLC intracadiac challenge (n = 8 mice/group). (C) Time course of serum IgG at the indicated time points post-LLC intravenous injection (n = 4 mice/group). (D) Pearson correlation analysis of IgG levels with the percentages of total B cells in the BM of tumor-bearing mice (n = 8). (E) Pearson correlation analysis of IgG levels with the absolute numbers of B cells in the BM of tumor-bearing mice (n = 8). (F) IL-7 was reduced in the serum of tumor-bearing mice on 16 post-LLC intravenous challenge or on day 11 post-LLC intracadiac challenge (n = 6 mice/group). (G) Time course of serum IL-7 at the indicated time points after LLC intravenous challenge (n = 4 mice/group). *, P < 0.05; **, P < 0.01; ***, P < 0.001.
To further understand the regulatory mechanisms that may affect B cell differentiation and maturation during lung cancer progression, we focused on IL-7 signaling. B cell precursors are associated with stroma in the BM through expression of c-kit which interacts with stem cell factor (29). IL-7 plays a key role in initiating proliferation and differentiation of pro-B cells to pre-B cells (30, 31). We investigated whether modulation of IL-7 contributed to the impaired B cell differentiation observed in tumor-bearing mice. As shown in Figure 3F, IL-7 was significantly reduced in tumor-bearing mice, with reduction observed as early as 5 days after tumor challenge (Fig. 3G).
STAT5 activation plays a crucial role in directing IL-7-dependent B cell differentiation (23, 32). STAT5 signaling was impaired with reduced level of STAT5 phosphorylation in the BM of tumor-bearing mice on day 16 after LLC i.v. (Fig. 4A, 4B) or on day 11 after LLC i.c. challenge (Fig. 4C). Importantly, phosphorylation of STAT5 was reduced as early as day 5 (Fig. 4D), consistent with the decline of IL-7 and B cell numbers and infiltration of MDSCs, and remained reduced in the BM of tumor-bearing mice on day 11 (Supplemental Fig. S2, S3).
Figure 4.
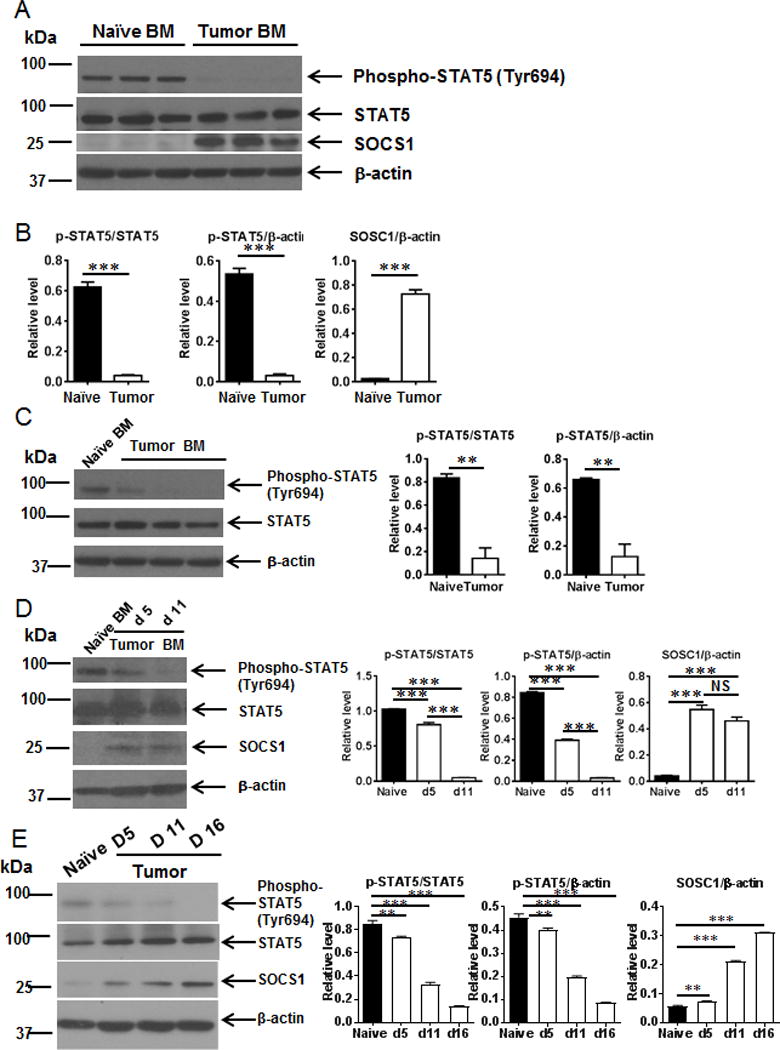
Impairment of STAT5 signaling in the BM of tumor-bearing mice. (A) and (B) Impairment of STAT5 signaling in the BM of tumor-bearing mice. BM lysates were collected from tumor-bearing mice on day 16 post-LLC intravenous challenge. Western blotting was performed with p-STAT5, STAT5 and SOCS1 antibodies. The relative expression of p-STAT5 or SOSC1 was normalized with STAT5, or β-actin, respectively. (C) Phosphorylation of STAT5 was reduced in the BM of tumor-bearing mice on day 11 post-LLC intra-cardiac challenge. The relative expression of p-STAT5 was normalized with STAT5, or β-actin, respectively. (D) Time course of impairment of STAT5 signaling in the BM of tumor-bearing mice at the indicated time points post-LLC intravenous challenge. The relative expression of p-STAT5 or SOSC1 was normalized with STAT5, or β-actin, respectively. Data are presented as mean ± s.e.m. of triplicates. (E) Time course of impairment of STAT5 signaling in the sorted CD19+B220+ cells from the BM of naïve or tumor-bearing mice at the indicated time points post-LLC intravenous challenge. The relative expression of p-STAT5 or SOSC1 was normalized with STAT5, or β-actin, respectively. **, P < 0.01; ***, P < 0.001.
SOCS1 is an important transcriptional regulator of cytokine signaling (24, 33). We investigated whether SOCS1 was upregulated during tumor progression and associated with the infiltration of tumor promoting MDSCs along with the inhibition of STAT5 activation. SOCS1 expression was increased in the BM tumor-bearing mice (Fig. 4A, 4B) as early as day 5 after tumor challenge (Fig. 4D).
Next, we sorted CD19+B220+ cells from BM of tumor-bearing mice. Phosphorylation of STAT5 was reduced while SOCS1 was elevated in a time-dependent manner (Fig. 4E). The results indicate that STAT5 signaling was impaired in B cells of BM from tumor-bearing mice.
Taken together, these results suggest that IL-7-mediated activation of downstream STAT5 signaling is disrupted during tumor progression, which contributes to the impairment of B cell development.
MDSC depletion rescued IL-7 signaling and B cell function and differentiation in tumor-bearing mice
Since the dynamics of MDSC infiltration during tumor progression was associated with suppression of B cell differentiation and function, we investigated if MDSCs would directly inhibit the B cell responses during tumor progression. TGF-β, an MDSC-associated cytokine that promotes MDSC-mediated Treg induction has been shown to negatively regulate stromal levels of IL-7 (22). Therefore, we first investigated whether levels of immunoregulatory TGF-β were altered during tumor progression. We observed a significant increase in serum TGF-β1 during tumor progression (Fig. 5A, 5B).
Figure 5.
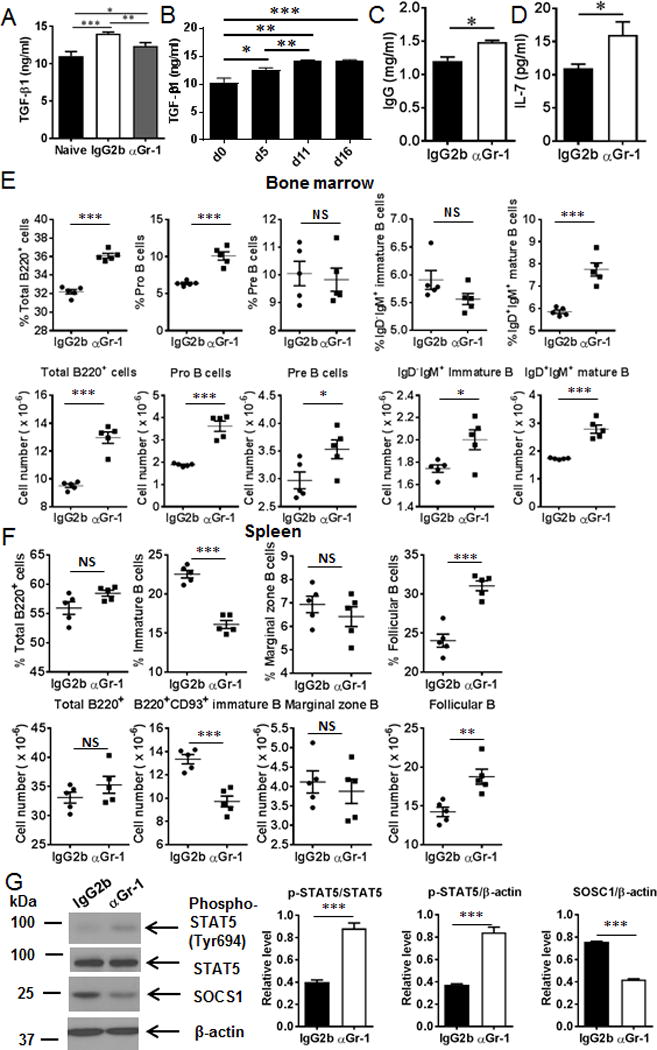
Anti-Gr-1 treatment partially rescued serum TGF-β1, IgG and IL-7 levels as well as B cell subsets and STAT5 signaling in tumor-bearing mice. (A) Serum TGF-β1 levels were elevated in tumor-bearing mice, and TGF-β1 levels were reduced after MDSC depletion by anti-Gr-1 treatment (n = 5 mice/group). (B) Time course of serum TGF-β1 at the indicated time points after LLC intravenous injection (n = 4 mice/group). (C) Serum IgG levels were elevated after anti-Gr-1 treatment (n = 5 mice/group). (D) Serum IL-7 levels were increased after anti-Gr-1 treatment (n = 5 mice/group). (E) Percentages and absolute numbers of total B220+, Pro-, Pre-, immature and mature B cells in the BM of tumor-bearing mice treated with anti-Gr-1 antibody (n = 5 mice/group). (F) The percentages and absolute numbers of immature B cells were decreased whereas the percentages and absolute numbers of follicular B cells were increased after anti-Gr-1 treatment (n = 5 mice/group). (G) Phospho-STAT5 was elevated whereas SOCS1 was reduced after anti-Gr-1 treatment in tumor-bearing mice. Densitometry data were quantified with ImageJ software. The relative expression of p-STAT5 or SOSC1 was normalized with STAT5, or β-actin, respectively. *, P < 0.05; .**, P < 0.01; ***, P < 0.001.
We then investigated whether MDSC depletion would reduce TGF-β1 levels which would then potentially rescue IL-7, which would then support B cell development and maturation and B cell function. As shown in Figure 5A, anti-Gr-1 antibody-mediated MDSC depletion reduced circulating TGF-β1 and partially rescued serum IL-7 and IgG levels in vivo (Fig. 5C, 5D).
Next, we tested whether dysregulation of B cell differentiation in the BM and spleens of tumor-bearing mice was reversed following MDSC depletion. The percentages of total B cells, pro-B cells, and mature B cells in the BM of tumor-bearing mice were significantly increased in anti-Gr-1 antibody treated group compared to the controls (Fig. 5E). Moreover, the absolute numbers of total B cells, pro-B cells, pre-B cells, immature B cells and mature B cells in the BM of tumor-bearing mice were also elevated after administration of anti-Gr-1 antibody (Fig. 5E). In the spleen, both the percentages and absolute numbers of immature B cells were reduced while both the percentages and absolute numbers of follicular B cells were increased by anti-Gr-1 treatment (Fig. 5F).
We then investigated whether rescue of levels of IL-7 following MDSC depletion would restore activation of STAT5. Phospho-STAT5 was increased in the BM of tumor-bearing mice after anti-Gr-1 treatment (Fig. 5G) compared to the control groups. In contrast, SOCS1 expression was reduced by MDSC depletion (Fig. 5G). Taken together, our studies show a direct effect of MDSCs on B cell differentiation and function mediated potentially by TGF-β-dependent regulation of IL-7 and its downstream effects on STAT-5 signaling.
TGF-β depletion rescued IL-7 signaling and B cell differentiation in tumor-bearing mice
We further tested whether TGF-β depletion would rescue IL-7 signaling and B cell differentiation in tumor-bearing mice. As shown in Figure 6A, anti-TGF-β depletion partially recued serum IL-7 level in vivo. Phospho-STAT5 was increased in the BM of tumor-bearing mice after anti-TGF-β treatment (Fig. 6B, 6C) compared to the control groups. In contrast, SOCS1 expression was reduced by TGF-β depletion (Fig. 6B, 6C). Both the percentages and absolute numbers of total B cells, pro-B cells, pre-B cells and mature B cells in the BM of tumor-bearing mice were significantly increased in anti-TGF-β antibody treated group compared to the controls (Fig. 6D, 6E). In the spleen, both the percentages and absolute numbers of immature B cells were reduced while both the percentages and absolute numbers of follicular B cells were increased by anti-TGF-β treatment (Fig. 6F, 6G). These results show that depletion of MDSC-associated cytokine TGF-β partially rescued serum IL-7 level and its downstream effects on STAT-5 signaling.
Figure 6.
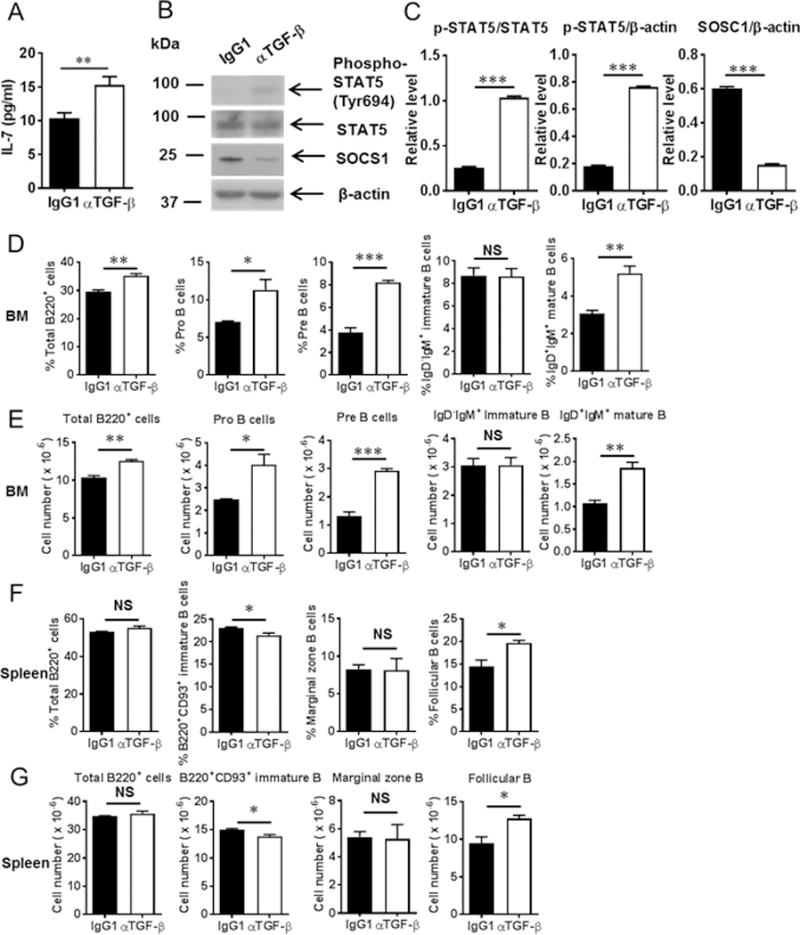
Anti-TGF-β treatment partially rescued serum IL-7 levels and STAT5 signaling as well as B cell subsets in tumor-bearing mice. (A) Serum IL-7 levels were increased after anti-TGF-β treatment (n = 3 mice/group). (B) Phospho-STAT5 was elevated whereas SOCS1 was reduced after anti-TGF-β treatment in tumor-bearing mice (n = 3 mice/group). (C) Densitometry data were quantified with ImageJ software. The relative expression of p-STAT5 or SOSC1 was normalized with STAT5, or β-actin, respectively. (D) and (E) Percentages and absolute numbers of total B220+, Pro-, Pre-, immature and mature B cells in the BM of tumor-bearing mice treated with anti-TGF-β antibody (n = 3 mice/group). (F) and (G) The percentages and absolute numbers of immature B cells were decreased whereas the percentages and absolute numbers of follicular B cells were increased in the spleens after anti-TGF-β treatment (n = 3 mice/group). *, P < 0.05; **, P < 0.01; ***, P < 0.001.
IL-7 blockade further impaired B cell differentiation in tumor-bearing mice
We next determined whether IL-7 blockade further impaired B cell differentiation in tumor-bearing mice. Serum levels of IL-7 were markedly reduced in the tumor-bearing mice by anti-IL-7 treatment (Fig. 7A). Phospho-STAT5 was further reduced in purified B cells from BM of tumor-bearing mice after anti-IL-7 treatment (Fig. 7B, 7C). The percentages and total numbers of MDSCs were increased the BM and lungs of tumor-bearing mice by anti-IL-7 treatment (Fig. 7D, 7F). These results indicate IL-7 blockade further impaired STAT5 signaling in tumor-bearing mice.
Figure 7.
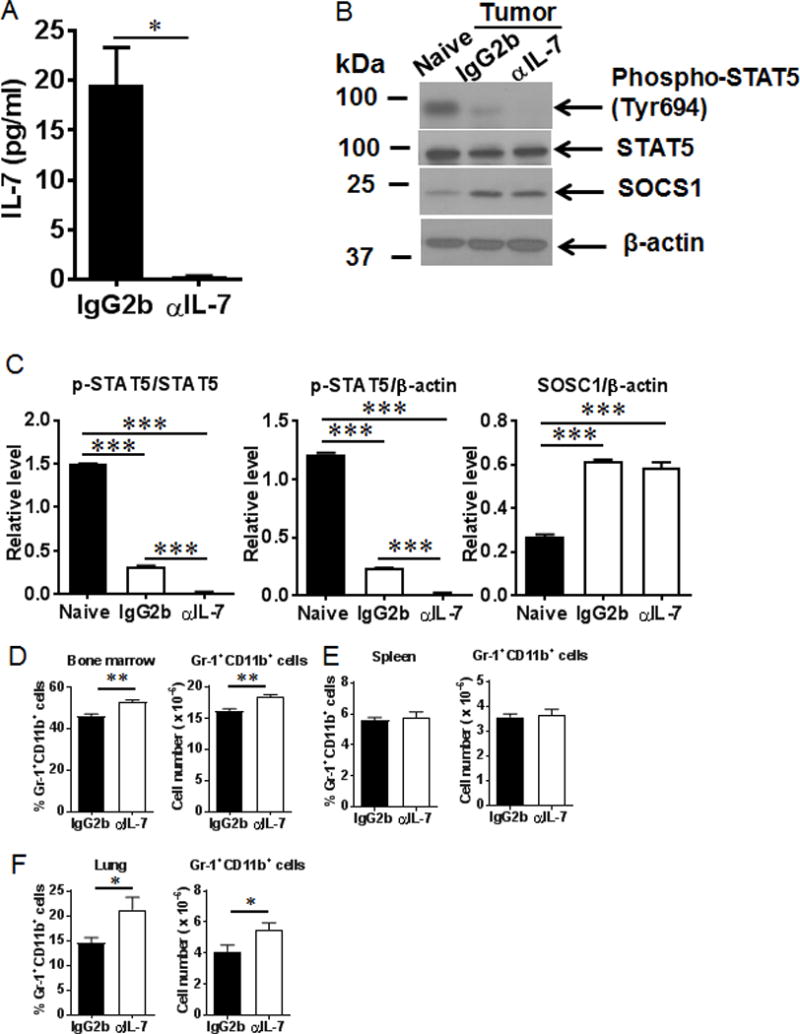
Serum IL-7 levels and STAT5 signaling were reduced after IL-7 blockade. (A) Serum IL-7 levels were decreased after anti-IL-7 treatment. (B) and (C) Phospho-STAT5 was reduced after anti-IL-7 treatment in tumor-bearing mice. The percentages and cell numbers of Gr-1+CD11b+ MDSCs were elevated in the bone marrow (D) and lungs (F) after anti-IL-7 treatment in tumor-bearing mice. No changes of MDSCs were observed in the spleens (E) of tumor-bearing mice after anti-IL-7 treatment (n = 5 mice/group). *, P < 0.05; **, P < 0.01; ***, P < 0.001.
As shown in Figure 8A and 8B, both the percentages and absolute numbers of total B cells, pro-B cells, pre-B cells, immature and mature B cells in the BM of tumor-bearing mice were significantly reduced by neutralizing of IL-7. In the spleen, both the percentages and absolute numbers of immature B cells were increased while both the percentages and absolute numbers of follicular B cells were reduced by anti-IL-7 treatment (Fig. 8C, 8D). Additionally, both the percentages and absolute numbers of B cell populations in the lung were reduced with IL-7 blockade (Fig. 8E and 8F). Taken together, these results indicated that IL-7 blockade further impaired B cell differentiation in tumor-bearing mice.
Figure 8.
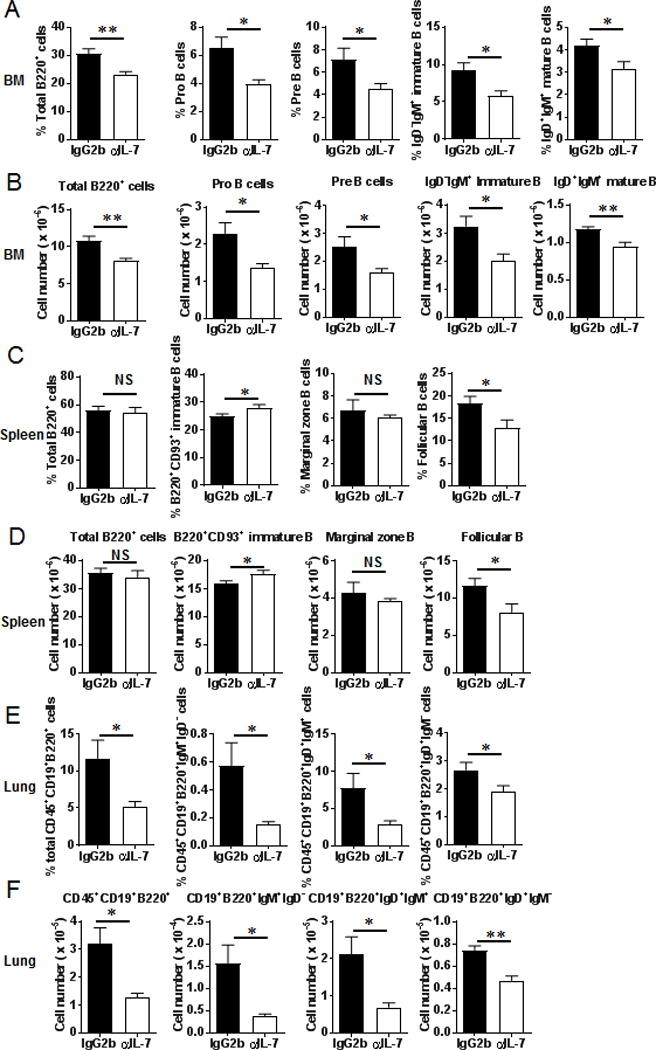
Anti-IL-7 treatment further reduced B cell subsets in the BM, spleens and lungs of tumor-bearing mice. (A) and (B) The percentages and absolute numbers of total B220+, Pro-, Pre-, immature and mature B cells in the BM of tumor-bearing mice treated with anti-IL-7 antibody (n = 5 mice/group). (C) and (D) The follicular B cells were reduced in the spleens of tumor-bearing mice after anti-IL-7 treatment (n = 5 mice/group). (E) and (F) B cell subsets were decreased in the lung of tumor-bearing mice after anti-IL-7 treatment (n = 5 mice/group). *, P < 0.05; **, P < 0.01.
Impaired B cell responses are independent of Tregs
MDSCs are known to suppress both CD4+ and CD8+ T cell responses in the TME (1, 3). Our studies also show suppression of tumor specific T cells in the TME coinciding with MDSC infiltration which was restored following MDSC depletion (25, 26). In parallel with MDSCs, Treg cells also contribute to the immunosuppression in tumor-bearing hosts (34). MDSCs are known to induce Treg cells in the TME (35). Since TGF-β1 levels were modulated upon MDSC depletion, we investigated whether this had an impact on Treg frequencies in the TME. As shown in Figure 9A-B, percentages of CD4+CD25highFoxP3+ Treg cells were elevated in both BM and spleen of tumor-bearing mice. Treg infiltration in the BM was suppressed upon MDSC depletion by anti-Gr-1 treatment (Fig. 9A) while Treg infiltration in the spleens remained unaffected (Fig. 9B).
Figure 9.
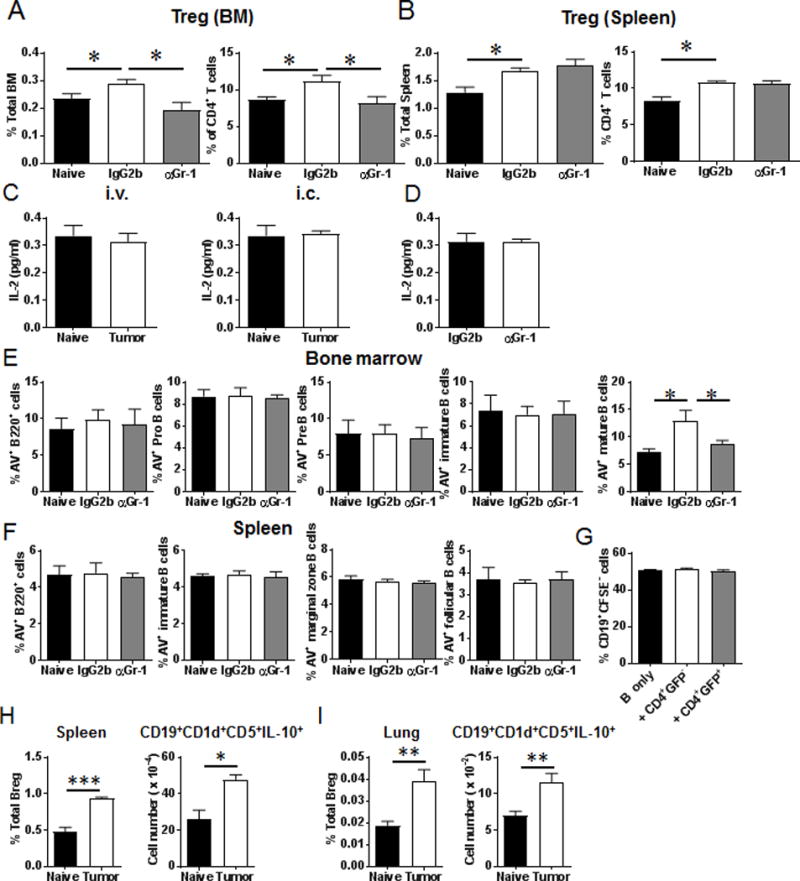
Treg cells and apoptosis of B cell subsets in the BM and spleens of tumor-bearing mice. (A) Percentages of Treg in total cells and CD4+ T cells in the BM (n = 4 mice/group). (B) Percentages of Treg in total cells and CD4+ T cells in the spleens (n = 4 mice/group). (C) Serum IL-2 levels were not altered in tumor-bearing mice (n = 4 mice/group). (D) Serum IL-2 levels were not changed after anti-Gr-1 treatment (n = 4 mice/group). (E) Percentages of Annexin V+ B220+ cells, Annexin V+ Pro B cells, Annexin V+ Pre B cells, Annexin V+ immature B cells and Annexin V+ mature B cells in the BM (n = 4 mice/group). (F) Percentages of Annexin V+ B220+ cells, Annexin V+ immature B cells, Annexin V+ marginal zone B cells and Annexin V+ follicular B cells in the spleens (n = 4 mice/group). Naïve: naïve tumor free mice; IgG2b: tumor-bearing mice treated with IgG2b control antibody; αGr-1: tumor-bearing mice treated with anti-Gr-1 antibody. (G) Splenocytes from naïve mice were stimulated with LPS (20 μg/ml) and IL-4 (10 ng/ml) for 24 hrs. B220+CD19+ cells were sorted and labeled with CFSE. The sorted B cells were co-cultured with CD4+GFP+ Treg cells or CD4+GFP− T cells purified from spleen of tumor-bearing Foxp3-DTR-GFP mice. The percentages of CD19+CFSElow cells were determined by FACS analysis. (H) Percentages and cell numbers of CD19+CD1d+CD5+IL-10+ Breg cells in the spleens of naïve and tumor-bearing mice on day 16 post-LLC intravenous injection (n = 4 mice/group). (I) Percentages and cell numbers of CD19+CD1d+CD5+IL-10+ Breg cells in the lungs of naïve and tumor-bearing mice on day 16 post-LLC intravenous injection (n = 4 mice/group). *, P < 0.05; **, P < 0.01; ***, P < 0.001.
Recent studies have demonstrated that IL-2 is involved in the differentiation and proliferation of B cells (36–39). In addition to IL-7, IL-2 can also regulate STAT-5 signaling (40). We hypothesized that increased infiltration of Treg in TME may lead to increased consumption of IL-2 which may then indirectly contribute to suppression of B cell differentiation in tumor-bearing hosts. As shown in Figure 9C, serum IL-2 levels did not change in tumor-bearing mice. Additionally, IL-2 was not altered by anti-Gr-1 treatment (Fig. 9D), suggesting an IL-2 independent mechanism for the impaired B cell responses.
To determine whether Treg cells directly affect the proliferation of B cells, pre-activated B cells were co-cultured with Treg cells purified from tumor-bearing Foxp3-DTR-GFP mice. Proliferation of B cells was not inhibited in the co-culture of Treg and B cells (Fig. 9G), indicating that impaired B cell responses are independent of Tregs.
Breg cells are elevated in the spleens and lungs of tumor-bearing mice
Breg cells are characterized as CD19+CD1d+CD5+IL-10+ subpopulation which play important roles in promoting tumor growth (12, 15–17). We next investigated whether Breg cells were increased in tumor-bearing mice. As shown in Figure 9H and 9I, the percentages and absolute numbers of CD19+CD1d+CD5+IL-10+ Breg cells were elevated in the spleens and lungs of tumor-bearing mice. These results suggested that while B cell differentiation was impaired in tumor-bearing mice, differentiation of immunosuppressive Breg cells were increased.
Apoptosis of mature B cells in BM are increased in tumor-bearing mice
We then determined whether B cell apoptosis accounted for the reduction in B cell subsets in tumor-bearing mice. As shown in Figure 9E, the percentage of Annexin V+ B220+IgD+IgM+ mature B cells in the BM was significantly increased in tumor-bearing mice, which was rescued by anti-Gr-1 treatment. However, no significant differences were found in the percentages of Pro B cells, Pre B cells, immature B cells in the BM, as well as splenic B cell subsets (Fig. 9E, 9F). These data suggest a role for MDSC mediated mechanisms in potentially modulating the apoptosis of mature B cells in the BM.
Adoptive transfer of MDSCs reduced B cell subsets
To further investigate the potential effects of MDSCs on B cell differentiation in vivo, we adoptively transferred MDSCs derived from the BM or tumors of CD45.2+ tumor-bearing mice by intratibial injection into congenic CD45.1+ mice. As shown in Supplemental Figure 4A, CD45.2+CD11b+ cells were detected in the peripheral blood of the CD45.1+ recipient mice 48 hours later after transfer. Both the percentages and absolute numbers of total B220+ cells, B220+IgD−IgM+ immature B cells and B220+IgD+IgM+ mature B cells were reduced in circulation of congenic CD45.1+ mice on day 7 after MDSC transfers (Fig. 10A). Additionally, both the percentages and cell numbers of pre-B cells and immature B cells were reduced in the BM of congenic CD45.1+ recipients of BM-MDSCs from CD45.2+ tumor-bearing mice (Fig. 10B). The percentages and absolute numbers of total B220+ cells and B220+CD93+ immature B cells were also reduced in the recipient spleens, whereas only the numbers of B220+CD93−CD21intCD23+ follicular B cells were reduced in these animals (Supplemental Fig. 4B). There was a trend toward reduction in serum IgG in the recipients of MDSC transfer, which did not reach statistical significance (Supplemental Fig. 4C).
Figure 10.
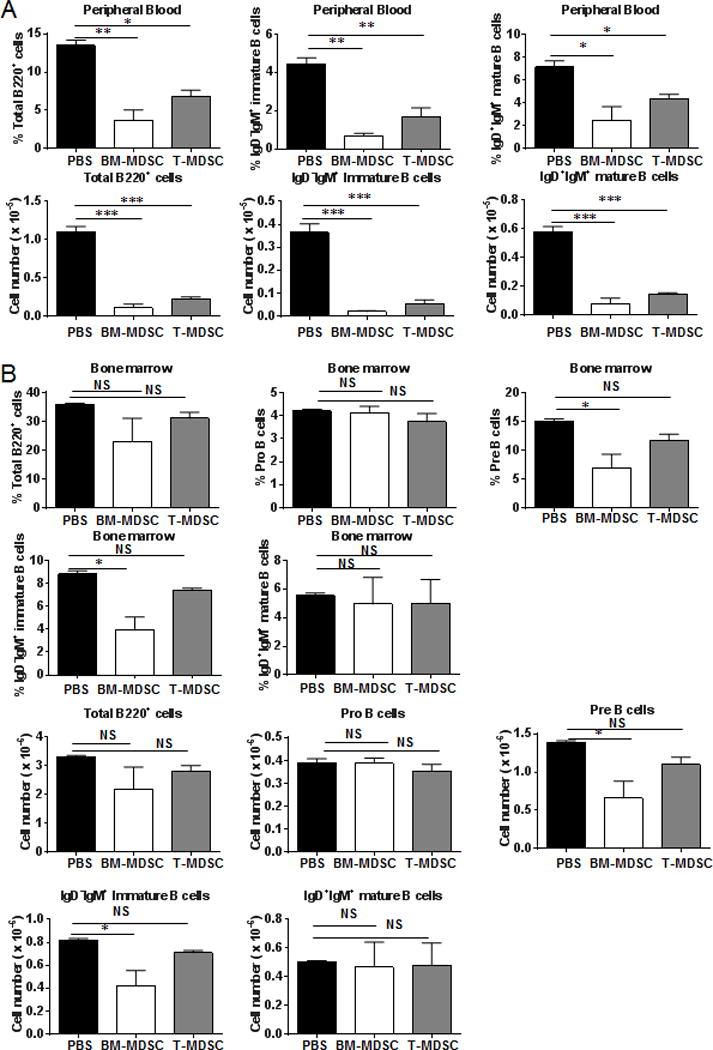
Adoptive intratibial transfer of MDSCs reduced circulating B cells as well as pre B and immature B cells in the BM. (A) Percentages and absolute numbers of total B, immature B, and mature B cells were reduced in peripheral blood of congenic CD45.1+ mice on day 7 after intra-tibial injection of BM-MDSCs and Tumor-MDSCs from CD45.2+ tumor-bearing mice (n = 4 mice/group). (B) Percentages and absolute numbers of Pre B and immature B cells were reduced in the BM of congenic CD45.1+ mice on day 7 after intratibial injection of BM-MDSCs from tumor-bearing mice (n = 4 mice/group). *, P < 0.05; **, P < 0.01; ***, P < 0.001.
MDSCs suppress B cell proliferation and function
We then investigated if MDSCs suppress B cell proliferation and IgG production in vitro. Pre-activated CFSE labeled splenocytes from naïve mice were co-cultured for 48 hours with BM-MDSCs purified from tumor-bearing mice. The percentages of CD19+CFSElow cells were reduced in these co-cultures, which were partially rescued by the addition of nor-NOHA or 1400W (Fig. 11A), inhibitors of arginase and iNOS, the regulatory pathways of MDSCs. IgG production in culture supernatants was decreased by BM-MDSCs, and this inhibition could also be partially reversed by nor-NOHA or 1400W, but not 1-D-MT (Fig. 11B), inhibitor of indoleamine 2,3 dioxygenase that regulates tryptophan metabolism. These results indicate that MDSCs suppress B cell proliferation and impair IgG production by B cells in an arginase- and iNOS-dependent manner but not by an IDO-dependent mechanism.
Figure 11.
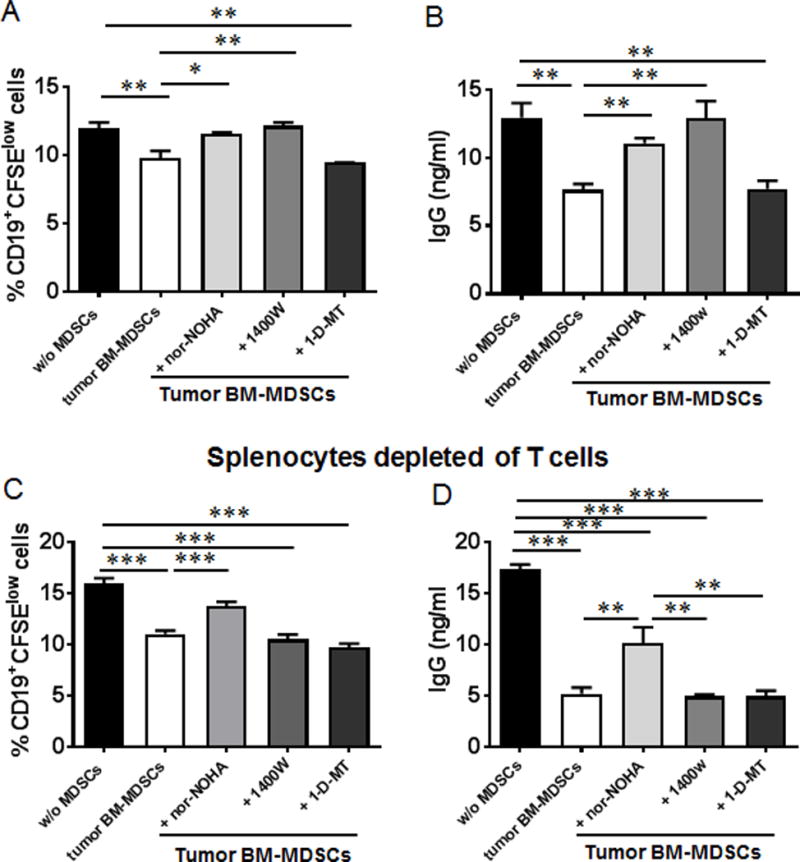
Suppression of B cell proliferation and function by MDSCs from tumor-bearing mice. (A) Splenocytes from naïve mice were labeled with CFSE and cultured with LPS (20 μg/ml) and IL-4 (10 ng/ml). 72 hours later, the pre-activated splenocytes were co-cultured with MDSCs purified from the BM of tumor-bearing mice in the absence or presence of arginase inhibitor nor-NOHA, iNOS inhibitor 1400W or IDO inhibitor 1-D-MT for 48 hrs. The percentages of CD19+CFSElow cells were determined by FACS analysis. (B) IgG detection from the supernatant collected from the co-culture. (C) and (D) Splenocytes depleted of T cells were labeled with CFSE and cultured with LPS and IL-4. The same experiments were performed as described in (A) and (B). *, P < 0.05; **, P < 0.01; ***, P < 0.001.
To address whether the suppression of B cells by MDSCs is T cell dependent, we examined the responses of B cells to MDSCs co-cultured with CFSE labeled splenocytes depleted of T cells. Both the percentages of CD19+CFSElow cells and IgG production were reduced in the co-cultures, which were partially recovered by the addition of arginase inhibitor, but not iNOS inhibitor or IDO inhibitor (Fig. 11C, 11D). These results suggest that the suppression of B cells by MDSCs through iNOS pathway is T cell dependent.
Next, we further determined if the suppression of B cells by MDSCs is T cell dependent. CD19+B220+ B cells were sorted from LPS- and IL-4-stimulated splenocytes, and then co-cultured with BM-MDSCs from tumor-bearing mice in the presence of nor-NOHA or 1400W. The percentages of CD19+CFSElow cells were reduced in the BM-MDSC co-cultures, which were partially rescued by treatment with arginase inhibitor, but not iNOS inhibitor (Fig. 12A). We then examined the effects of T cell addition to the co-culture of B cells with BM-MDSCs. The percentages of CD19+CFSElow cells were further reduced in the co-culture with the addition of T cells purified from the spleens of tumor-bearing mice, which was partially rescued by iNOS inhibitor (Fig. 12C). Both the T cell depletion (Fig 11C, 11D) and the T cell addition (Fig. 12C) experiments indicate that the suppression of B cells by MDSCs through iNOS pathways is T cell dependent. Co-cultures in transwells showed that suppression of B cell proliferation by BM-MDSCs was dependent on cell-cell contact (Fig. 12B).
Figure 12.
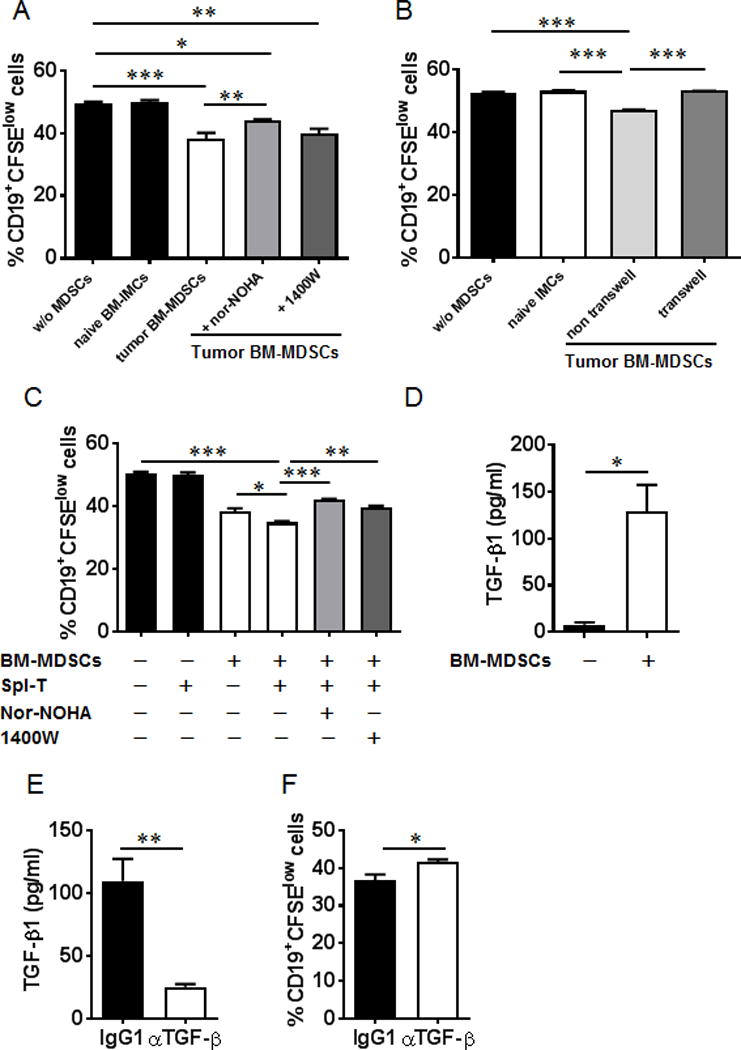
Suppression of B cell proliferation by MDSCs through iNOS is dependent on T cells. (A) Splenocytes from naïve mice were stimulated with LPS (20 μg/ml) and IL-4 (10 ng/ml) for 24 hrs. B220+CD19+ cells were sorted and labeled with CFSE. The sorted B cells were co-cultured with MDSCs purified from bone marrow of tumor-bearing mice in the absence or presence of arginase inhibitor nor-NOHA or iNOS inhibitor 1400W for 72 hrs. The percentages of CD19+CFSElow cells were determined by FACS analysis. (B) Purified B220+CD19+ cells were co-cultured with MDSCs from tumor-bearing mice in the absence or presence of a transwell system for 72 hrs. The percentages of CD19+CFSElow cells were determined by FACS analysis. (C) T cells purified from spleen of tumor-bearing mice were added to the co-cultures of B and MDSCs described in (A) in the absence or presence of arginase inhibitor nor-NOHA or iNOS inhibitor 1400W. The percentages of CD19+CFSElow cells were determined by FACS analysis. (D) TGF-β1 levels were elevated in the supernatants from the co-cultures of B cells and MDSCs. (E) TGF-β1 was reduced in the co-cultures of B cells and MDSCs in the presence of TGF-β neutralizing Ab. (F) The proliferation of B cells was increased in the co-cultures after TGF-β blockade. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
TGF-β1 levels were elevated in the co-culture of B cells and BM-MDSCs (Fig. 12D). The proliferation of B cells was also increased in the co-cultures with anti-TGF-β antibody (Fig. 12F). These results suggest that MDSCs suppressed B cell proliferation in an arginase- and TGF-β-dependent as well as in a cell-to-cell contact-dependent manner.
Discussion
B cells play a significant role in antitumor immune responses (41–44). In this report, we show that B cell differentiation and humoral immunity were impaired in vivo during tumor progression. We present evidence that immunosuppressive MDSCs directly impact B cell differentiation by TGF-β-mediated modulation of IL-7 and downstream STAT-5 signaling that are both essential for B cell differentiation and function. Our observation that B cell responses are impaired during tumor progression is consistent with studies by Richards et al. who have demonstrated that tumor growth decreases NK and B cells as well as common lymphoid progenitors (45). They observed a significant decrease in the percentage and absolute number of total B220+ cells in the BM of EL4 tumor-bearing mice (45). In humans, B cell dysfunction is correlated with loss of CD27+ B cells in patients with advanced melanomas and other solid tumors (46); however, the underlying mechanisms involved in the impairment of B cell subsets remain largely unknown.
Mice deficient in IL-7 or IL-7R display a severe block of early pro-B cell development (47, 48). IL-7 was significantly reduced in our tumor-bearing mice, along with reduced IgG and impairment of B cell differentiation. Total cell numbers of pro-, pre-, immature and mature B cell subsets were reduced in tumor-bearing mice, suggesting that B cell subsets were broadly suppressed in BM in tumor-bearing mice which might partially be mediated by the reduction of IL-7. In this regard, our observation that alterations in TGF-β levels correlating with tumor progression and the progressive decline in IL-7 levels suggests a role for immunoregulatory TGF-β in modulating IL-7. As TGF-β has been shown to regulate stromal IL-7 (22), and as MDSCs are significant contributors of TGF-β, the rescue of serum IL-7 levels following MDSC depletion suggests that MDSC-associated TGF-β-mediated mechanisms may account for the reduced IL-7 and downstream signaling and the impaired B cell responses. Additional evidence from our studies that treatment with TGF-β blocking antibody rescued IL-7 and downstream STAT-5 activation and B cell differentiation strongly support TGF-β-mediated regulation of IL-7 and STAT-5 and their impact on B cell differentiation and function. Targeting the recovery of IL-7 and B cell differentiation and function may provide new approaches for boosting humoral immunity against cancer.
MDSCs and Treg cells are major component of the immune suppressive TME. Several studies have shown that MDSCs support Treg cell development through TGF-β and IL-10 (49), ARG1 (50), or through CD40/CD40L interactions (51). These results suggest that the cross-talks between MDSCs and Treg cells are fine-tuned and dependent on the context of different tumor models. In our study, the elevation of Treg cells in the BM could be reduced by anti-Gr-1-mediated MDSC depletion, indicating that MDSCs contribute to the expansion of Treg cells in the BM of tumor-bearing mice. The reduction in TGF-β following the depletion of MDSCs thus not only regulated IL-7 levels, but also contributed to the reduction of Treg cells.
Although MDSCs have been shown to regulate B cell functions in different disease models, including experimental autoimmune myasthenia gravis (7), murine retrovirus-induced AIDS (8) and autoimmune arthritis (52), none of these studies have elucidated a direct mechanism involving TGF-β and IL-7 mediated signaling. IL-7/IL-7R signaling stimulates the JAK-STAT transcription factor pathway that leads to STAT5 phosphorylation (53). Our study identified that IL-7 and downstream STAT-5 signaling are impaired in tumor-bearing mice, which contributes to the disruption of B cell differentiation. Anti-Gr-1 treatment could partially rescue the phosphorylation of STAT-5, suggesting that MDSCs impact signaling pathways that regulate B cell differentiation. Moreover, anti-TGF-β depletion also partially restored IL-7 and downstream STAT5 signaling, as well as B cell differentiation indicating that MDSC-associated TGF-β may negatively influence B cell differentiation. IL-7 blockade further impaired B cell differentiation in tumor-bearing mice. MDSCs were increased in the BM and lung of tumor-bearing mice following anti-IL-7 antibody treatment. This finding is consistent with the report that radiofrequency thermal ablation combined with the administration of intralesional IL-7 and IL-15 reduced MDSCs in breast tumor models (54). Additionally, the contribution of apoptosis to the observed reduction in B cell differentiation may be mediated by other soluble mediators produced by MDSCs, such as reactive oxygen and nitrogen species. Thus MDSCs are central regulators of B cell differentiation and function.
It is unclear whether these suppressive responses of B cells mediated by MDSCs are T cell dependent. Our in vitro co-culture study in the presence of T cells suggest that MDSCs suppress B cell proliferation and IgG production by B cells in an arginase and iNOS-dependent manner but not an IDO-dependent manner. When splenocytes depleted of T cells were added to the co-culture, MDSCs suppressed B cell proliferation and IgG production by B cells in an arginase dependent manner. Further, when T cells were added to the co-culture of B cells with MDSCs, MDSCs suppressed B cell proliferation in an iNOS-dependent manner, indicating that the suppression of B cells by MDSCs through iNOS pathway is T cell dependent.
In the absence of T cells in the co-culture, MDSCs suppress B cell proliferation in an arginase- and TGF-β-dependent manner. It is possible that the induction of arginase expression and enzymatic activity in MDSCs may utilize all available L-Arginine (L-Arg) substrates and may override the availability of L-Arg for iNOS which shares this substrate with arginase. It has also been reported that arginine deficiency affects early B cell maturation and lymphoid organ development in transgenic mice overexpressing arginase in their enterocytes (55). It is intriguing to speculate whether L-Arg deficiency induced by the increased activity of arginase in MDSCs might affect B cell differentiation and maturation in vivo in tumor-bearing mice. Durante et al. reported that TGF-β1 stimulates L-Arg transport and metabolism in vascular smooth muscle cells (56). It has also been shown that TGF-β stimulates arginase activity in macrophages (57, 58). These evidences may account for the TGF-β- and arginase-dependent regulation of B cell proliferation. These combined results suggest that tumor-associated MDSCs may potentially regulate humoral immune responses in this murine lung cancer model.
Bruton tyrosine kinase (Btk) is a Tec family kinase with a well-defined role in B-cell antigen receptor (BCR) signaling (59, 60). Mutations in Btk result in X-linked agammaglobulinemia in humans. In the mouse, mutations in Btk cause X-linked immunodeficiency, characterized by blockage in B-cell development at multiple stages and impaired function of mature B cells (61). We investigated whether Btk signaling is involved in the impairment of B cell responses. There was no difference in the level of Btk phosphorylation in the purified B220+CD19+ BM-derived B cells between tumor-bearing mice and naïve mice (data not shown), suggesting the involvement of Btk-independent signaling pathways in the impairment of B cell subsets in the BM of tumor-bearing mice.
In summary, we found that B cell differentiation and function are impaired during tumor progression and that much of this is attributable to direct effects of MDSCs. They were correlated and seemingly mediated via a reduction of IL-7-mediated STAT-5 signaling and a dysfunctional B cell response. MDSC depletions and adoptive transfers show a direct role of these cells and their regulatory mechanisms in suppressing not only B cell differentiation but also their function. Additionally, we uncover TGF-β, a MDSC associated cytokine, as a central suppressive regulator of IL-7 signaling in B cells in the TME. Further, we demonstrate that the suppressive effects of MDSCs require cell-to-cell contact. Taken together, our findings suggest that tumor-infiltrating MDSCs may potentially regulate B cell response during tumor progression. Further efforts to investigate the potential mechanisms of B cell dysregulation during tumor progression may provide new insights into the immune incompetence in cancer, thereby suggesting novel approaches for targeted immunotherapies.
Supplementary Material
Acknowledgments
We acknowledge Marion Spell at the UAB Flow Cytometry Core Facilities for his technical assistance in acquiring sorted cell samples. We thank Dr. Chad Steele at UAB for use of his laboratory’s 96-well plate reader.
This work was supported by American Cancer Society – Institutional Research Grant Award IRG-60-001-53-IRG awarded to J.S.D, 1R01HL128502-01A1 awarded to J.S.D., R01-AI-083705 and the Lupus Research Institute Novel Research Award to H.-C.H., R01 AI14782 awarded to J.F.K., NIH P30 AR048311 and NIH P30 AI27667 for service and support provided by the UAB Comprehensive Flow Cytometry Core.
Abbreviations
- MDSCs
myeloid-derived suppressor cells
- TME
tumor microenvironment
- BM
bone marrow
- iNOS
inducible nitric oxide synthase
- ARG1
arginase 1
- ROS
reactive oxygen species
- Treg
regulatory T
- APCs
antigen presenting cells
- Breg
regulatory B
- SOCS1
suppressor of cytokine signaling 1
- IMCs
immature myeloid cells
Footnotes
Conflict of Interest: There are no financial conflicts of interest for any of the authors listed.
References
- 1.Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol. 2009;9:162–174. doi: 10.1038/nri2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Condamine T, Gabrilovich DI. Molecular mechanisms regulating myeloid-derived suppressor cell differentiation and function. Trends Immunol. 2011;32:19–25. doi: 10.1016/j.it.2010.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gabrilovich DI, Ostrand-Rosenberg S, Bronte V. Coordinated regulation of myeloid cells by tumours. Nat Rev Immunol. 2012;12:253–268. doi: 10.1038/nri3175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Youn JI, Nagaraj S, Collazo M, Gabrilovich DI. Subsets of myeloid-derived suppressor cells in tumor-bearing mice. J Immunol. 2008;181:5791–5802. doi: 10.4049/jimmunol.181.8.5791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Khaled YS, Ammori BJ, Elkord E. Myeloid-derived suppressor cells in cancer: recent progress and prospects. Immunol Cell Biol. 2013;91:493–502. doi: 10.1038/icb.2013.29. [DOI] [PubMed] [Google Scholar]
- 6.Ostrand-Rosenberg S, Sinha P. Myeloid-derived suppressor cells: linking inflammation and cancer. J Immunol. 2009;182:4499–4506. doi: 10.4049/jimmunol.0802740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Li Y, Tu Z, Qian S, Fung JJ, Markowitz SD, Kusner LL, Kaminski HJ, Lu L, Lin F. Myeloid-derived suppressor cells as a potential therapy for experimental autoimmune myasthenia gravis. J Immunol. 2014;193:2127–2134. doi: 10.4049/jimmunol.1400857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Green KA, Cook WJ, Green WR. Myeloid-derived suppressor cells in murine retrovirus-induced AIDS inhibit T- and B-cell responses in vitro that are used to define the immunodeficiency. J Virol. 2013;87:2058–2071. doi: 10.1128/JVI.01547-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zitvogel L, Kroemer G. Cancer: Antibodies regulate antitumour immunity. Nature. 2015;521:35–37. doi: 10.1038/nature14388. [DOI] [PubMed] [Google Scholar]
- 10.Carmi Y, Spitzer MH, Linde IL, Burt BM, Prestwood TR, Perlman N, Davidson MG, Kenkel JA, Segal E, Pusapati GV, Bhattacharya N, Engleman EG. Allogeneic IgG combined with dendritic cell stimuli induce antitumour T-cell immunity. Nature. 2015;521:99–104. doi: 10.1038/nature14424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schultze JL, Michalak S, Seamon MJ, Dranoff G, Jung K, Daley J, Delgado JC, Gribben JG, Nadler LM. CD40-activated human B cells: an alternative source of highly efficient antigen presenting cells to generate autologous antigen-specific T cells for adoptive immunotherapy. J Clin Invest. 1997;100:2757–2765. doi: 10.1172/JCI119822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fremd C, Schuetz F, Sohn C, Beckhove P, Domschke C. B cell-regulated immune responses in tumor models and cancer patients. Oncoimmunology. 2013;2:e25443. doi: 10.4161/onci.25443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nelson BH. CD20+ B cells: the other tumor-infiltrating lymphocytes. J Immunol. 2010;185:4977–4982. doi: 10.4049/jimmunol.1001323. [DOI] [PubMed] [Google Scholar]
- 14.DiLillo DJ, Yanaba K, Tedder TF. B cells are required for optimal CD4+ and CD8+ T cell tumor immunity: therapeutic B cell depletion enhances B16 melanoma growth in mice. J Immunol. 2010;184:4006–4016. doi: 10.4049/jimmunol.0903009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Balkwill F, Montfort A, Capasso M. B regulatory cells in cancer. Trends Immunol. 2013;34:169–173. doi: 10.1016/j.it.2012.10.007. [DOI] [PubMed] [Google Scholar]
- 16.Zhang Y, Gallastegui N, Rosenblatt JD. Regulatory B cells in anti-tumor immunity. Int Immunol. 2015;27:521–530. doi: 10.1093/intimm/dxv034. [DOI] [PubMed] [Google Scholar]
- 17.Mauri C, Menon M. Human regulatory B cells in health and disease: therapeutic potential. J Clin Invest. 2017;127:772–779. doi: 10.1172/JCI85113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mauri C, Bosma A. Immune regulatory function of B cells. Annu Rev Immunol. 2012;30:221–241. doi: 10.1146/annurev-immunol-020711-074934. [DOI] [PubMed] [Google Scholar]
- 19.Komschlies KL, Gregorio TA, Gruys ME, Back TC, Faltynek CR, Wiltrout RH. Administration of recombinant human IL-7 to mice alters the composition of B-lineage cells and T cell subsets, enhances T cell function, and induces regression of established metastases. J Immunol. 1994;152:5776–5784. [PubMed] [Google Scholar]
- 20.Grabstein KH, Waldschmidt TJ, Finkelman FD, Hess BW, Alpert AR, Boiani NE, Namen AE, Morrissey PJ. Inhibition of murine B and T lymphopoiesis in vivo by an anti-interleukin 7 monoclonal antibody. J Exp Med. 1993;178:257–264. doi: 10.1084/jem.178.1.257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mazzucchelli RI, Warming S, Lawrence SM, Ishii M, Abshari M, Washington AV, Feigenbaum L, Warner AC, Sims DJ, Li WQ, Hixon JA, Gray DH, Rich BE, Morrow M, Anver MR, Cherry J, Naf D, Sternberg LR, McVicar DW, Farr AG, Germain RN, Rogers K, Jenkins NA, Copeland NG, Durum SK. Visualization and identification of IL-7 producing cells in reporter mice. PLoS One. 2009;4:e7637. doi: 10.1371/journal.pone.0007637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tang J, Nuccie BL, Ritterman I, Liesveld JL, Abboud CN, Ryan DH. TGF-beta down-regulates stromal IL-7 secretion and inhibits proliferation of human B cell precursors. J Immunol. 1997;159:117–125. [PubMed] [Google Scholar]
- 23.Goetz CA, Harmon IR, O’Neil JJ, Burchill MA, Farrar MA. STAT5 activation underlies IL7 receptor-dependent B cell development. J Immunol. 2004;172:4770–4778. doi: 10.4049/jimmunol.172.8.4770. [DOI] [PubMed] [Google Scholar]
- 24.Corfe SA, Rottapel R, Paige CJ. Modulation of IL-7 thresholds by SOCS proteins in developing B lineage cells. J Immunol. 2011;187:3499–3510. doi: 10.4049/jimmunol.1100424. [DOI] [PubMed] [Google Scholar]
- 25.Schafer CC, Wang Y, Hough KP, Sawant A, Grant SC, Thannickal VJ, Zmijewski J, Ponnazhagan S, Deshane JS. Indoleamine 2,3-dioxygenase regulates anti-tumor immunity in lung cancer by metabolic reprogramming of immune cells in the tumor microenvironment. Oncotarget. 2016 doi: 10.18632/oncotarget.12249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sawant A, Schafer CC, Jin TH, Zmijewski J, Tse HM, Roth J, Sun Z, Siegal GP, Thannickal VJ, Grant SC, Ponnazhagan S, Deshane JS. Enhancement of antitumor immunity in lung cancer by targeting myeloid-derived suppressor cell pathways. Cancer Res. 2013;73:6609–6620. doi: 10.1158/0008-5472.CAN-13-0987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yabas M, Teh CE, Frankenreiter S, Lal D, Roots CM, Whittle B, Andrews DT, Zhang Y, Teoh NC, Sprent J, Tze LE, Kucharska EM, Kofler J, Farell GC, Broer S, Goodnow CC, Enders A. ATP11C is critical for the internalization of phosphatidylserine and differentiation of B lymphocytes. Nat Immunol. 2011;12:441–449. doi: 10.1038/ni.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Allman D, Lindsley RC, DeMuth W, Rudd K, Shinton SA, Hardy RR. Resolution of three nonproliferative immature splenic B cell subsets reveals multiple selection points during peripheral B cell maturation. J Immunol. 2001;167:6834–6840. doi: 10.4049/jimmunol.167.12.6834. [DOI] [PubMed] [Google Scholar]
- 29.Rico-Vargas SA, Weiskopf B, Nishikawa S, Osmond DG. c-kit expression by B cell precursors in mouse bone marrow. Stimulation of B cell genesis by in vivo treatment with anti-c-kit antibody. J Immunol. 1994;152:2845–2852. [PubMed] [Google Scholar]
- 30.Fry TJ, Mackall CL. Interleukin-7: from bench to clinic. Blood. 2002;99:3892–3904. doi: 10.1182/blood.v99.11.3892. [DOI] [PubMed] [Google Scholar]
- 31.Corfe SA, Paige CJ. The many roles of IL-7 in B cell development; mediator of survival, proliferation and differentiation. Semin Immunol. 2012;24:198–208. doi: 10.1016/j.smim.2012.02.001. [DOI] [PubMed] [Google Scholar]
- 32.Malin S, McManus S, Cobaleda C, Novatchkova M, Delogu A, Bouillet P, Strasser A, Busslinger M. Role of STAT5 in controlling cell survival and immunoglobulin gene recombination during pro-B cell development. Nat Immunol. 2010;11:171–179. doi: 10.1038/ni.1827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yoshimura A, Naka T, Kubo M. SOCS proteins, cytokine signalling and immune regulation. Nat Rev Immunol. 2007;7:454–465. doi: 10.1038/nri2093. [DOI] [PubMed] [Google Scholar]
- 34.Zou W. Regulatory T cells, tumour immunity and immunotherapy. Nat Rev Immunol. 2006;6:295–307. doi: 10.1038/nri1806. [DOI] [PubMed] [Google Scholar]
- 35.Lindau D, Gielen P, Kroesen M, Wesseling P, Adema GJ. The immunosuppressive tumour network: myeloid-derived suppressor cells, regulatory T cells and natural killer T cells. Immunology. 2013;138:105–115. doi: 10.1111/imm.12036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rolink A, Grawunder U, Winkler TH, Karasuyama H, Melchers F. IL-2 receptor alpha chain (CD25, TAC) expression defines a crucial stage in pre-B cell development. Int Immunol. 1994;6:1257–1264. doi: 10.1093/intimm/6.8.1257. [DOI] [PubMed] [Google Scholar]
- 37.Reya T, Yang-Snyder JA, Rothenberg EV, Carding SR. Regulated expression and function of CD122 (interleukin-2/interleukin-15R-beta) during lymphoid development. Blood. 1996;87:190–201. [PubMed] [Google Scholar]
- 38.Mingari MC, Gerosa F, Carra G, Accolla RS, Moretta A, Zubler RH, Waldmann TA, Moretta L. Human interleukin-2 promotes proliferation of activated B cells via surface receptors similar to those of activated T cells. Nature. 1984;312:641–643. doi: 10.1038/312641a0. [DOI] [PubMed] [Google Scholar]
- 39.Gearing A, Thorpe R, Bird C, Spitz M. Human B cell proliferation is stimulated by interleukin 2. Immunol Lett. 1985;9:105–108. doi: 10.1016/0165-2478(85)90019-7. [DOI] [PubMed] [Google Scholar]
- 40.Lin JX, Leonard WJ. The role of Stat5a and Stat5b in signaling by IL-2 family cytokines. Oncogene. 2000;19:2566–2576. doi: 10.1038/sj.onc.1203523. [DOI] [PubMed] [Google Scholar]
- 41.Sorrentino R, Morello S, Forte G, Montinaro A, De Vita G, Luciano A, Palma G, Arra C, Maiolino P, Adcock IM, Pinto A. B cells contribute to the antitumor activity of CpG-oligodeoxynucleotide in a mouse model of metastatic lung carcinoma. Am J Respir Crit Care Med. 2011;183:1369–1379. doi: 10.1164/rccm.201010-1738OC. [DOI] [PubMed] [Google Scholar]
- 42.Candolfi M, Curtin JF, Yagiz K, Assi H, Wibowo MK, Alzadeh GE, Foulad D, Muhammad AK, Salehi S, Keech N, Puntel M, Liu C, Sanderson NR, Kroeger KM, Dunn R, Martins G, Lowenstein PR, Castro MG. B cells are critical to T-cell-mediated antitumor immunity induced by a combined immune-stimulatory/conditionally cytotoxic therapy for glioblastoma. Neoplasia. 2011;13:947–960. doi: 10.1593/neo.11024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Forte G, Sorrentino R, Montinaro A, Luciano A, Adcock IM, Maiolino P, Arra C, Cicala C, Pinto A, Morello S. Inhibition of CD73 improves B cell-mediated anti-tumor immunity in a mouse model of melanoma. J Immunol. 2012;189:2226–2233. doi: 10.4049/jimmunol.1200744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Li Q, Teitz-Tennenbaum S, Donald EJ, Li M, Chang AE. In vivo sensitized and in vitro activated B cells mediate tumor regression in cancer adoptive immunotherapy. J Immunol. 2009;183:3195–3203. doi: 10.4049/jimmunol.0803773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Richards J, McNally B, Fang X, Caligiuri MA, Zheng P, Liu Y. Tumor growth decreases NK and B cells as well as common lymphoid progenitor. PLoS One. 2008;3:e3180. doi: 10.1371/journal.pone.0003180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Carpenter EL, Mick R, Rech AJ, Beatty GL, Colligon TA, Rosenfeld MR, Kaplan DE, Chang KM, Domchek SM, Kanetsky PA, Fecher LA, Flaherty KT, Schuchter LM, Vonderheide RH. Collapse of the CD27+ B-cell compartment associated with systemic plasmacytosis in patients with advanced melanoma and other cancers. Clin Cancer Res. 2009;15:4277–4287. doi: 10.1158/1078-0432.CCR-09-0537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Peschon JJ, Morrissey PJ, Grabstein KH, Ramsdell FJ, Maraskovsky E, Gliniak BC, Park LS, Ziegler SF, Williams DE, Ware CB, Meyer JD, Davison BL. Early lymphocyte expansion is severely impaired in interleukin 7 receptor-deficient mice. J Exp Med. 1994;180:1955–1960. doi: 10.1084/jem.180.5.1955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.von Freeden-Jeffry U, Vieira P, Lucian LA, McNeil T, Burdach SE, Murray R. Lymphopenia in interleukin (IL)-7 gene-deleted mice identifies IL-7 as a nonredundant cytokine. J Exp Med. 1995;181:1519–1526. doi: 10.1084/jem.181.4.1519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Huang B, Pan PY, Li Q, Sato AI, Levy DE, Bromberg J, Divino CM, Chen SH. Gr-1+CD115+ immature myeloid suppressor cells mediate the development of tumor-induced T regulatory cells and T-cell anergy in tumor-bearing host. Cancer Res. 2006;66:1123–1131. doi: 10.1158/0008-5472.CAN-05-1299. [DOI] [PubMed] [Google Scholar]
- 50.Serafini P, Mgebroff S, Noonan K, Borrello I. Myeloid-derived suppressor cells promote cross-tolerance in B-cell lymphoma by expanding regulatory T cells. Cancer Res. 2008;68:5439–5449. doi: 10.1158/0008-5472.CAN-07-6621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pan PY, Ma G, Weber KJ, Ozao-Choy J, Wang G, Yin B, Divino CM, Chen SH. Immune stimulatory receptor CD40 is required for T-cell suppression and T regulatory cell activation mediated by myeloid-derived suppressor cells in cancer. Cancer Res. 2010;70:99–108. doi: 10.1158/0008-5472.CAN-09-1882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Crook KR, Jin M, Weeks MF, Rampersad RR, Baldi RM, Glekas AS, Shen Y, Esserman DA, Little P, Schwartz TA, Liu P. Myeloid-derived suppressor cells regulate T cell and B cell responses during autoimmune disease. J Leukoc Biol. 2015;97:573–582. doi: 10.1189/jlb.4A0314-139R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hennighausen L, Robinson GW. Interpretation of cytokine signaling through the transcription factors STAT5A and STAT5B. Genes Dev. 2008;22:711–721. doi: 10.1101/gad.1643908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Habibi M, Kmieciak M, Graham L, Morales JK, Bear HD, Manjili MH. Radiofrequency thermal ablation of breast tumors combined with intralesional administration of IL-7 and IL-15 augments anti-tumor immune responses and inhibits tumor development and metastasis. Breast Cancer Res Treat. 2009;114:423–431. doi: 10.1007/s10549-008-0024-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.de Jonge WJ, Kwikkers KL, te Velde AA, van Deventer SJ, Nolte MA, Mebius RE, Ruijter JM, Lamers MC, Lamers WH. Arginine deficiency affects early B cell maturation and lymphoid organ development in transgenic mice. J Clin Invest. 2002;110:1539–1548. doi: 10.1172/JCI16143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Durante W, Liao L, Reyna SV, Peyton KJ, Schafer AI. Transforming growth factor-beta(1) stimulates L-arginine transport and metabolism in vascular smooth muscle cells: role in polyamine and collagen synthesis. Circulation. 2001;103:1121–1127. doi: 10.1161/01.cir.103.8.1121. [DOI] [PubMed] [Google Scholar]
- 57.Boutard V, Havouis R, Fouqueray B, Philippe C, Moulinoux JP, Baud L. Transforming growth factor-beta stimulates arginase activity in macrophages. Implications for the regulation of macrophage cytotoxicity. J Immunol. 1995;155:2077–2084. [PubMed] [Google Scholar]
- 58.Dzik JM. Evolutionary roots of arginase expression and regulation. Front Immunol. 2014;5:544. doi: 10.3389/fimmu.2014.00544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Niiro H, Clark EA. Regulation of B-cell fate by antigen-receptor signals. Nat Rev Immunol. 2002;2:945–956. doi: 10.1038/nri955. [DOI] [PubMed] [Google Scholar]
- 60.Satterthwaite AB, Witte ON. The role of Bruton’s tyrosine kinase in B-cell development and function: a genetic perspective. Immunol Rev. 2000;175:120–127. [PubMed] [Google Scholar]
- 61.Honigberg LA, Smith AM, Sirisawad M, Verner E, Loury D, Chang B, Li S, Pan Z, Thamm DH, Miller RA, Buggy JJ. The Bruton tyrosine kinase inhibitor PCI-32765 blocks B-cell activation and is efficacious in models of autoimmune disease and B-cell malignancy. Proc Natl Acad Sci U S A. 2010;107:13075–13080. doi: 10.1073/pnas.1004594107. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.