

Abstract
Tunable glutathione (GSH)-sensitive hollow mesoporous silica nanoparticles (HMSiO2 NPs) were developed using a structural difference-based selective etching strategy. These organosilica hollow nanoparticles contained disulfide linkages (S-S) in the outer shell which were degraded by GSH. The particles were compared with their nonGSH-sensitive tetraethyl orthosilicate (TEOS) HMSiO counterparts in terms of their synthesis method, characterization, DOX release profile, and in vitro cytotoxicity in MCF-7 breast cancer cells. Transmission electron microscopy (TEM) of the particles indicated that the fabricated HMSiO2NPs had an average diameter of 130 ± 5 nm. Thermogravimetric analysis (TGA) revealed that GSH-sensitive particles had approximately 5.3% more weight loss than TEOS HMSiO2 NPs. Zeta potential of these redox-responsive particles was −23 ± 1 mV at pH 6 in deionized (DI) water. Nitrogen adsorption-desorption isotherm revealed that the surface area of the hollow mesoporous nanoreservoirs was roughly 446 ± 6 m2 g−1 and the average diameter of the pores was 2.3 ± 0.5 nm. TEM images suggest that the nanoparticles started to lose mass integrity from Day 1. The particles showed a high loading capacity for DOX (8.9 ± 0.5%) as a model drug, due to the large voids existing in the hollow structures. Approximately 58% of the incorporated DOX released within 14 days in phosphate buffered saline (PBS) at pH 6 and in the presence of 10 mM GSH, mimicking intracellular tumor microenvironment while release from TEOS HMSiO2 NPs was only c.a. 18%. The uptake of these hollow nanospheres by MCF-7 cells and RAW 264.7 macrophages was evaluated using TEM and confocal microscopy. The nanospheres were shown to accumulate in the endolysosomal compartments after incubation for 24 h with the maximum uptake of c.a. 2.1 ± 0.3 % and 5.2 ± 0.4%, respectively. Cytotoxicity of the nanospheres was investigated using CCK-8 assay. Results indicate that intact hollow particles (both GSH-sensitive and TEOS HMSiO2 NPs) were nontoxic to MCF-7 cells after incubation for 24 h within the concentration range of 0–1000 μg ml−1. DOX-loaded GSH-sensitive nanospheres containing 6 μg ml−1 of DOX killed c.a. 51 % of MCF-7 cells after 24 h while TEOS HMSiO2 NPs killed c.a. 20% with the difference being statistically significant. Finally, cytotoxicity data in RAW 264.7 macrophages and NIH 3T3 fibroblasts shows that intact GSH-sensitive HMSiO2 NPs did not show any toxic effects on these cells with the concentrations equal or less than 125 μg ml−1.
Keywords: tunable, structural difference, hollow mesoporous silica nanoparticles, GSH-sensitive, degradable, high loading capacity
Graphical Abstract
Introduction
In recent years, various types of silica nanoparticles (SiO2 NPs) have attracted attention for delivery of bioactive agents due to their stability, robustness, ease of synthesis and scale up [1–3]. Silica nanoparticles provide the opportunity to protect active pharmaceutical ingredients, and to fabricate nanocomposite structures for theranostic applications (e.g. Iron Oxide/SiO2 composites) where imaging and delivery functionalities are combined into one system [4, 5]. However, there are still some challenges that limit the utility of SiO2 NPs as delivery vehicles. One problem is related to the limited control over loading and release of bioactive agents from porous structures. Although this limitation has to some extent been addressed by modifying the surface of SiO2 NPs with light-sensitive systems, pH-triggered ligands, or by capping of the pores [6–8], the complexity of such systems limits potential translation and scale up. Another drawback with majority of SiO2 NPs studied to date is the absence of controlled biodegradability under physiologic conditions. This can in turn result in unwanted accumulation in non-target tissues, potentially leading to acute and chronic inflammatory responses by inducing oxidative stress, lipid peroxidation, cell cycle arrest, apoptosis, and genotoxicity [9–11]. Hence it would be desirable to develop biodegradable SiO2 NPs with controlled loading and degradation profile.
To-date, several approaches have been investigated for fabricating degradable SiO2 NPs. These include silica nanospheres prepared by incorporation of biodegradable polymers such as poly(L-lactic acid) [12], dissolution of core-shell magnetic mesoporous SiO2 NPs immersed in phosphate buffered saline (PBS) [13], and the development of highly porous resorbable silica xerogels [14, 15]. In addition, stimuli-triggered SiO2 NPs have been designed by introducing breakable bonds into SiO2 NP structure using silane precursors containing such bonds which can be degraded intracellularly in response to enzymes [16], reducing environment [17], pH [18], etc. Degradable SiO2 NPs have also been prepared by condensation of an oxamide-bridged alkoxysilane (OBA) precursor which can be degraded in the presence of trypsin [19]. Redox-responsive degradable nanoparticles have been fabricated by incorporating disulfide (…S-S…) bonds within the SiO2 NP structure [20–23]. Since the concentration of glutathione (GSH) in intracellular microenvironment is ~100–1000 times higher than extracellular fluids, polysulfide-based compounds seem promising candidates for intracellular delivery as these carriers can be specifically degraded inside the cells but not outside [24]. Intratumoral GSH levels vary completely based on the type and the stage of the tumors [25]. For example, GSH levels in most brain tumors are usually lower in comparison to normal brain tissue except meningioma which has increased GSH levels [26–28]. In lung, colorectal, and breast tumors, GSH levels are found to be higher than the normal breast tissue [29, 30]. In head and neck tumors, higher levels of GSH are observed which rise with clinical stage. However, in esophagus, stomach, and liver tumors, decreased level of GSH has been shown [25].
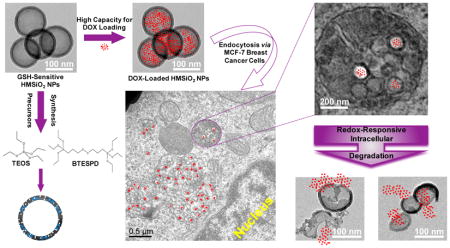
In our previous study, we fabricated porous and nonporous disulfide- and tetrasulfide-containing SiO2 NPs with a controlled degradation profile [31]. The degradation of these particles in the presence of GSH was predominantly governed by porosity and core composition in which mesoporous particles underwent surface and bulk degradation while nonporous particles underwent just surface erosion. A limitation of these systems however was their low loading capacity. In the present work, we report the design and development of GSH-sensitive biodegradable hollow mesoporous silica nanoparticles (HMSiO2 NPs) containing disulfide bonds. These nanoreservoirs were fabricated by controlling reaction conditions (e.g. vigorous stirring, high etchant concentration, etc.) and a unique structural difference-based selective etching technique [32] in which a core-shell structure is created with distinct structural and compositional differences between the inner dense silica core and the outer disulfide-based mesoporous silica shell (Scheme 1). The inner core was selectively etched by applying an appropriate etching agent (sodium carbonate; Na2CO3) while the outer shell remained roughly intact and ultimately a hollow nanoparticle was formed. This novel redox-triggered degradable system combines the advantages of degradability, low density, large surface area, high loading efficiency, ease of fabrication and scale up, stability, and the ability for surface modification/functionalization. Additionally, these nanostructures provide sustained drug release via tunable interior hollow cavity diameter (~80–250 nm) and variations in pore size and shell thickness (~10–30 nm). The fabricated nanoparticles were characterized by electron microscopy, nitrogen adsorption–desorption, X-ray photoelectron spectroscopy, X-ray diffraction, and thermogravimetric methods and compared to their nondegradable tetraethyl orthosilicate (TEOS) HMSiO2 NPs. In vitro degradation of these nanoreservoirs in the presence of GSH was assessed. Co-localization and cell toxicity in MCF-7 breast cancer epithelial cells as a model cancer cell were explored. Maximum cellular uptake in MCF-7 and RAW 264.7 macrophages was evaluated to understand the difference between nanoparticle uptake as a function of HMSiO2 NP concentration, incubation time, and cell type. Cytotoxicity in RAW 264.7 macrophages and NIH 3T3 fibroblasts was investigated for cell type, NP concentration-, and incubation time-dependence of toxicity. Finally, as a model drug, in vitro doxorubicin (DOX) loading, release profile, and cytotoxicity were explored in both GSH-sensitive and TEOS HMSiO2 NPs.
Scheme 1.

Top panel: schematic representation of GSH-sensitive HMSiO2 NPs fabrication steps: (1) Core Stöber SiO2 NP synthesized by TEOS precursor (hard template); (2) Disulfide-based mesoporous shell coated Stöber NP fabricated by TEOS and BTESPD precursors; (3) GSH-sensitive HMSiO2 NP formed via structural and compositional difference-based selective etching method using high concentration of Na2CO3 as the etching agent. (4) DOX-loaded HMSiO2 NP. These hollow NPs showed high loading capacity for DOX (up to c.a. 9% w/w). Bottom panel: representative microscopy images of the cell lines used for cell toxicity studies of GSH-sensitive HMSiO2 NPs; NIH 3T3 fibroblasts, MCF-7 breast cancer epithelial cells, and RAW 264.7 macrophages. These NPs can be degraded inside the cells by GSH as reducing agent via reduction of the disulfide bonds.
Material and methods
The following compounds were purchased from Sigma-Aldrich, Inc. (St. Louis, MO, USA): aminopropyltriethoxysilane (APTES, ≥ 98.0%), triethylamine (TEA, ≥99.0%), cetyltrimethylammonium bromide (CTAB, ≥99.0%), tetraethyl orthosilicate (TEOS, ≥99.0% GC), Triton™ X-100, bisbenzimide Hoechst No. 33342, fetal bovine serum (FBS), and glutathione (GSH). Dulbecco’s Modified Eagle Medium (DMEM), TrypLE™, fluorescein isothiocyanate (FITC), and LysoTracker™ Deep Red were received from Thermo Fisher Scientific (Grand Island, NY, USA). Bis[3-(triethoxysilyl)propyl] disulfide (BTESPD, 90.0%) was purchased from Gelest, Inc. (Morrisville, PA, USA). Doxorubicin hydrochloride salt (DOX, >99.0%) was acquired from LC Laboratories (Woburn, MA, USA). Sodium carbonate (Na2CO3, anhydrous, granular, ≥99.5%) was received from Mallinckrodt Chemicals (Phillipsburg, NJ, USA). Sodium Chloride High Purity Grade (NaCl, 99.9%) was purchased from AMRESCO® (Solon, OH, USA). Roswell Park Memorial Institute-1640 (RPMI-1640) medium, hydrochloric acid ACS Grade BDH (36.5–38.0%), and phosphate buffered saline (PBS) Biotechnology Grade tablets were received from VWR (Radnor, PA, USA). Absolute ethanol (200 proof) and ethanol 95% were purchased from Decon Labs, Inc. (King of Prussia, PA, USA) and Fisher Science Education (Nazareth, PA, USA), respectively. Trypan Blue Stain 0.4% was obtained from Invitrogen (Carlsbad, CA, USA). Hydrofluoric acid (HF, 48.0%) and ammonium hydroxide (NH4OH, 28.0–30.0% as NH3) were received from EMD Millipore Corporation (Billerica, MA, USA). RAW 264.7 macrophages (ATCC® TIB-71™), MCF-7 breast cancer cells (ATCC® HTB-22™), and NIH 3T3 (ATCC® CRL-1658™) fibroblasts were obtained from American Type Culture Collection (ATCC, Manassas, VA, USA). CCK-8 cytotoxicity assay kit was received from Dojindo (Rockville, MD, USA). All materials were used as received without further purification.
Synthesis of GSH-sensitive and TEOS HMSiO2 NPs
First, 100 nm dense Stöber core NPs were prepared as follows: 1700 mmol of absolute ethanol, 180 mmol of DI water, and 200 mmol of ammonium hydroxide were mixed in a flask under stirring rate of 400 RPM for 10 min. Then, 18 mmol of TEOS was added dropwise and the reaction was left under stirring for 24 h at room temperature. Next, the synthesized NPs (900 mg) were precipitated by centrifugation using Sorvall® RC-5B Refrigerated Superspeed Centrifuge (Du Pont Instruments Ltd., Wilmington, DE, USA) at 15,000 RPM for 20 min, washed thoroughly with DI water and ethanol 95%, and stored in 50 mL of DI water for further use (stock Stöber suspension: 18 mg mL−1).
Second, the synthesized Stöber NPs were coated with surfactant-based mesoporous silica shell (core-shell NPs). For this step, 1300 mmol of DI water, 60 mmol of absolute ethanol, 0.2 mmol of TEA, 0.16 mmol of CTAB, and 10 mL of the stock Stöber suspension were mixed in a 100 mL round bottom flask at 80 °C under stirring rate of 600 RPM for 50 min. Afterwards, for preparing GSH-sensitive shell, stirring rate was increased to 1400 RPM, and 0.42mmol of TEOS and 0.08 mmol of BTESPD were added simultaneously. Then, the reaction was allowed to stir for 4 h. For preparing TEOS shell, 0.65 mmol of TEOS was added to the suspension and the reaction was allowed to stir for 4 h at 80 °C and 1400 RPM. The hollow cavity size (~80–250 nm) and the shell thickness (~10–30 nm) were tunable depending on the amount of the reagents used in the formation of dense Stöber core and mesoporous coating procedure (the amount and ratio of TEOS to BTESPD), respectively. Next, the mesoporous coated Stöber NPs were precipitated by centrifugation at 15,000 RPM for 20 min and washed twice with DI water and ethanol 95%.
Third, HMSiO2 NPs were formed via etching with sodium carbonate due to structural/compositional differences between the core and the shell. For fabricating GSH-sensitive and TEOS HMSiO2 NPs, 15 and 12.5 mmol of sodium carbonate, respectively were dissolved in 10 mL of DI water in a 100 mL round bottom flask at 50 °C under stirring rate of 600 RPM for 30 min. The obtained core-shell NPs were dispersed in 10 mL of DI water and sonicated for 30 min. Then, stirring rate was increased to 1200 RPM and the NP suspension was added to sodium carbonate solution. The reaction was kept under stirring for 10 h. The product was washed thrice with water/ethanol 95% mixture, suspended in acidic ethanol (1 mL HCl 36.5% in 30 mL absolute ethanol), and heated to 80 °C under reflux for 6 h to remove the surfactant. Acidic ethanol washing step was repeated twice and HMSiO2 NPs were then stored in absolute ethanol for further use.
Physicochemical characterization of nanoparticles
Size and morphology of the nanoparticles were investigated by electron microscopy methods. Transmission electron microscopy (TEM) and scanning electron microscopy (SEM) images were acquired by FEI Tecnai™ 12 transmission electron microscope (Hillsboro, OR, USA) operating at 120 kV and FEI Quanta 600F scanning electron microscope (Hillsboro, OR, USA) operating at 20 kV, respectively. Hydrodynamic diameter and zeta potential measurements were determined by dynamic light scattering (DLS) in a Malvern Instruments Zetasizer Nano ZS (Malvern Instruments Ltd., Worcestershire, UK). Measurements were performed in triplicate. Thermogravimetric analyses (TGA) were conducted using a TA Instruments hi-res TGA 2950 Thermogravimetric Analyzer (New Castle, DE, USA). All TGA experiments were conducted under N2 atmosphere from 35 to 800 °C at a heating rate of 20 °C/min. Nitrogen adsorption–desorption isotherm analyses were conducted at −196 °C on a Micromeritics ASAP 2020 (Norcross, GA, USA) for measuring surface area and pore size. All samples were dried at 100 °C overnight prior to analysis. X-ray diffraction (XRD) patterns of all nanoparticles were investigated on a Bruker D2 Phaser X-ray diffractometer (Bruker AXS, Madison, WI, USA) using Cu Kα radiation (λ = 0.1542 nm) at 45 kV and 40 mA. The XRD spectra were recorded at a scanning speed of 0.01 deg/s, with a step size of 0.02° in a 2-theta scattering angle and in a range of 2–8. Scanning transmission electron microscopy (STEM) images and spectra of the nanoparticles were obtained on JEOL JEM-2800 (Akishima, Tokyo, Japan) scanning transmission electron microscope with dual energy dispersive X-ray spectrometer (EDS) detectors at an electron beam energy of 200 kV. Sample preparation was done by drop casting the nanoparticles on a carbon coated TEM grid. Pore volume and pore size distributions were acquired from an adsorption branch by using the Barrett-Joyner-Halenda (BJH) method. The Brunauer–Emmett–Teller (BET) specific surface areas were measured via adsorption data at P/P0= 0.05–0.20. X-ray photoelectron spectroscopy (XPS) analyses of the nanoparticles were performed by Axis Ultra DLD instrument (Kratos Analytical, Manchester, UK). For analyses, the samples were mounted on a C tape and pumped overnight in the load lock before introduction into the analysis chamber. A mono Al source was employed. Survey scans were collected with a pass energy of 160 eV, step size of 1 eV, and dwell time of 200 ms. High resolution region scans were collected with a pass energy of 40 eV, 0.1 eV step size, and 400 ms dwell time. Data were processed using CASA XPS software.
Degradation study of GSH-sensitive HMSiO2 NPs
Degradation of GSH-sensitive HMSiO2 NPs (50 μg mL−1) was evaluated mimicking intracellular and extracellular GSH concentrations (10 mM and 10 μM, respectively) in DI water at pH 6 and under constant shaking (160 RPM; Amerex Instruments Inc., Concord, CA, USA) at 37 °C. At predetermined time points (0, 6 h, 1, 3, and 7 days), samples were collected for TEM analysis. Next, water suspensions containing nanoparticles were drop-casted onto Formvar coated Cu grids and allowed to dry prior to visualization using FEI Tecnai T12 operating at 120 kV.
Cytotoxicity assays
Cytotoxicity of the intact GSH-sensitive HMSiO2 NPs, TEOS HMSiO2 NPs, DOX-loaded GSH-sensitive HMSiO2 NPs, DOX-loaded TEOS HMSiO2 NPs, and free DOX was evaluated in MCF-7 breast cancer epithelial cells. Cells were cultured at 37 °C in 5% CO2 in DMEM with 10% FBS. Then, the cells were stained using Trypan Blue Stain 0.4% and read by Invitrogen Countess™ automated cell counter (Thermo Fisher Scientific Corporation, Grand Island, NY, USA). For the cytotoxicity evaluation, cells were seeded onto 96-well plates with the density of 4,000 cells/well and incubated to grow for 48 h. After 48 h, the cells were washed with PBS. For GSH-sensitive HMSiO2 NPs and TEOS HMSiO2 NPs without DOX, fresh media containing 10% FBS was then added (120 μL) with varying nanoparticle concentrations ranging from 0 to 1000 μg mL−1. For DOX-loaded GSH-sensitive HMSiO2 NPs, DOX-loaded TEOS HMSiO2 NPs, and free DOX, samples were prepared based on DOX concentration ranging from 0 to 6 μg mL−1. Wells with media or Triton™ X-100 (without nanoparticles) were used as negative or positive controls, respectively. The cells were then incubated for another 24 and 48 h, the media was aspirated, and the cells were washed twice with PBS. Cell viability was determined using CCK-8 cytotoxicity assay kit according to an established protocol and absorbance was measured at 450 nm with SpectraMax® M2 microplate reader. Assays were performed at least in triplicate.
Cytotoxicity of the intact GSH-sensitive HMSiO2 NPs was also evaluated in RAW 264.7 macrophages and in NIH 3T3 fibroblasts. Cells were cultured at 37 °C in 5% CO2 in RPMI-1640 and DMEM (for RAW 264.7 macrophages and NIH 3T3, respectively) with 10% FBS and seeded onto 96-well plates with the density of 4,000 cells/well and incubated to grow for 48 h. Subsequently, the cells were washed with PBS. Fresh media containing 10% FBS was then added with varying nanoparticle concentrations ranging from 0 to 1000 μg mL−1. The cells were then incubated for another 24 and 48 h, the media was aspirated, and the cells were washed twice with PBS. Cell viability was measured using CCK-8 cytotoxicity assay kit at 450 nm. Assays were performed at least in triplicate.
Intracellular trafficking via confocal laser scanning microscopy (CLSM)
To explore intracellular trafficking and co-localization of the HMSiO2 NPs, CLSM was used and images were obtained by Olympus FluoView™ FV1000 confocal microscope (Olympus Corporation, Shinjuku, Tokyo, Japan) at 60x magnification. FITC-labeled HMSiO2 NPs were synthesized by the following procedure: 0.021 mmol of FITC and 0.021 mmol of APTES (1:1 molar ratio) were mixed in 10 mL of absolute ethanol and the reaction was kept under stirring (400 RPM) for 2 h at room temperature. Then, 5 mL of absolute ethanol containing 60 mg of HMSiO2 NPs was added to the previous mixture and the reaction was allowed to stir (400 RPM) for another 6 h at room temperature. Finally, FITC-labeled HMSiO2 NPs were centrifuged, washed 4 times with DI water and 95% ethanol to remove any unreacted species, and kept in absolute ethanol for further use. All the reactions were performed in dark to prevent FITC from bleaching.
Two chambered cover glasses (Lab-Tek® Chambered #1.0 Borosilicate Coverglass System) were used for this experiment. Cells were seeded in chambers (51,000 cells/chamber) and allowed to grow for 48 h at 37 °C in 5% CO2. Next, the media was removed, FITC-labeled HMSiO2 NPs (50 and 250 μg mL−1) were added to each chamber, and incubated for 4 and 24 h. Then, the cells were washed 3 times with PBS. LysoTracker™ Deep Red (50 nM) was then added to each chamber and the cells were incubated for 1 h. Afterwards, Hoechst 33342 (2 μg mL−1) was added to the chambers and the cells were incubated for another 10 minutes. Finally, the staining solutions were removed and the cells were washed 3 times with PBS. The excitation wavelength for FITC, Hoechst, and LysoTracker™ Deep Red were adjusted to 495, 350, and 647 nm, respectively.
Cell uptake
Uptake of GSH-sensitive HMSiO2 NPs by MCF-7 cells was explored using TEM. Cells were grown for 48 h on ACLAR® sheets in 12-well plates (48,000 cells/well) and treated with 50 and 250 μg mL−1 of HMSiO2 NPs. Controls were treated with media containing 10% FBS. After incubation for 24 h at 37 °C in 5% CO2, cells were washed 3 times with PBS and fixed with 1 mL of xing solution (2.5% glutaraldehyde + 1.0% paraformaldehyde). Then, the cells were post-fixed with osmium tetroxide (OsO4), stained with uranyl acetate, dehydrated, and embedded in an epoxy resin. Finally, ultrathin sections of the stained cells were prepared utilizing a diamond knife and imaged at room temperature by FEI Tecnai T12 operating at 120 kV.
HMSiO2 NPs uptake rate in MCF-7 cells and RAW 264.7 macrophages were also investigated via Inductively Coupled Plasma Mass Spectrometry (ICP-MS, Agilent 7500ce ICP-MS instrument, Santa Clara, CA, USA) using nontoxic NP concentrations in these cells. Cells were cultured in 6-well plates (120,000 cells/well) for 48 h. For MCF-7 cells, HMSiO2 NPs were added with the concentrations of 25, 50, 75, 100, 250, 500, 750, and 1000 μg mL−1 and the cells were incubated for 24 h. For time-dependent study, cells treated with 100 μg mL−1 of NPs were incubated for 0, 0.5, 1, 2, 4, 6, 12, 18, and 24 h. For RAW 264.7 macrophages, HMSiO2 NPs were added with the concentrations of 25, 50, 75, and 100 μg mL−1 and the cells were incubated for 24 h. For time-dependent study, cells treated with 100 μg mL−1 of NPs were incubated for 0, 4, 12, and 24 h. Cells without NP treatment were used as controls.
After incubation, the media was removed and the cells were washed thrice with 2.5 mL of PBS. Next, 1 mL of TrypLE™ was added to each well, incubated for 5–10 min, and the surface of the wells were scratched using 25 cm Cell Scraper (VWR, Radnor, PA, USA) to ensure that all the cells were detached. Then, the cells were collected in 1.5 mL microtubes, centrifuged (Centrifuge 5415 D Eppendorf, Hauppauge, NY, USA) for 5 min at 1500 RPM, supernatants were removed, 500 μLit of ultrapure water was added, and the samples were analyzed using ICP-MS for silicon (Si) content. Lastly, 20 μL cesium internal standard was added as an instrumental control. Silicon assays were performed in triplicate.
Drug loading and release profile
DOX as a model drug was incorporated in GSH-sensitive and TEOS HMSiO2 NPs according to the following procedure: 7.5 mg of DOX and 10 mg of HMSiO2 NPs were mixed in 7.5 mL of DI water. The reaction was kept under stirring (1000 RPM) for 24 h at room temperature. After 24 h, DOX-loaded HMSiO2 NPs were precipitated by centrifugation at 15,000 RPM for 20 min and washed five times with DI water to remove free DOX or the DOX adsorbed on the surface of these NPs. DOX loading capacity in HMSiO2 NPs was then calculated using UV-Vis spectroscopy at 480 nm based on DOX calibration curve plotted in the range of concentrations between 3.9 and 250 μg mL−1.
For release study, DOX-loaded GSH-sensitive HMSiO2 NPs (2 mg) were dispersed in 10 mL of PBS at pH 6 containing 2 and 10 mM GSH and in 10 mL of PBS at pH 7.2 containing 10 mM GSH. Vials with NPs in PBS at pH 6 (without GSH) were used as controls. For comparison study, DOX-loaded TEOS HMSiO2 NPs (2 mg) were dispersed in 10 mL of PBS pH 6. Subsequently, the suspension was kept under constant shaking (160 RPM) at 37 °C. Next, equal aliquots were taken out at specific time points (0, 2, 4, 6, 12, 24, 48, 72, 120, 168, and 366 h), centrifuged at 13,000 RPM for 20 min, and the amount of the released DOX in the supernatants was measured at excitation wavelength of 480 nm using a DOX standard curve. All measurements were performed at least in triplicate.
Results and discussion
Synthesis and characterization of HMSiO2 NPs
Different steps of the fabrication procedure for these hollow particles are outlined in Scheme 1. HMSiO2 NPs were fabricated using unique structural/compositional difference-based (between the core and the shell) selective etching strategy [33–35]. Three steps were involved for the preparation of these hollow nanostructures. First, dense Stöber SiO2 NPs were synthesized via modified Stöber method using TEOS precursor [36]. These Stöber cores are considered as hard template. Second, surfactant-based mesoporous shell containing …Si-O-Si-C-C-C-S-S-C-C-C-Si-O-Si… (in GSH-sensitive HMSiO2 NPs) and …Si-O-Si… (in TEOS HMSiO2 NPs) bonds were coated on the surface of these Stöber cores using TEOS and bis[3-(triethoxysilyl)propyl] disulfide (BTESPD) precursors. The size of the hollow cavity and the shell thickness of the particles was controlled by altering the size of the hard template and the amounts of the precursors used in the process of core-shell formation, respectively. Third, for forming the hollow structure, a high concentration of sodium carbonate was used. DOX-loaded HMSiO2 NPs was obtained by physical mixing of DOX with these particles. These hollow particles were reduced via intracellular GSH after endocytosis by NIH 3T3 fibroblasts, MCF-7 breast cancer epithelial cells, and RAW 264.7 macrophages.
Size, size distribution, and shape of the fabricated HMSiO2 NPs were characterized using TEM and SEM (Fig. 1). As shown (Fig. 1A), uniform Stöber particles were synthesized with an average diameter of c.a. 100 ± 5 nm, and coated with 15 nm mesoporous shell (Fig. 1B and G for GSH-sensitive and TEOS HMSiO2 NPs, respectively) using hydrolysis and co-condensation of silane precursors. These images confirm that the shell is porous. Fig. 1C and D indicate that uniform GSH-sensitive HMSiO2 NPs with an average diameter of c.a. 130 ± 5 nm were obtained by selective etching strategy. This occurs due to the structural (dense core vs. mesoporous shell) and compositional (TEOS in the core vs. TEOS and BTESPD in the shell) differences between the inner core and the outer shell, while TEOS HMSiO2 NPs were prepared via just structural difference between the dense core and the mesoporous shell (Fig. 1H and I). It has been shown that cationic surfactants can protect the outer shell of silica nanoparticles during hollow SiO2 NP synthesis [37]. It is postulated that CTAB plays a key role in the generation of HMSiO2 NPs from dense Stöber core by acting as a stabilizer to protect the silicate-CTAB shell from alkaline etching [38]. Fig. 1E and F are SEM images of GSH-sensitive HMSiO2 NPs and a particle with loss of mass integrity which confirms the existence of a large void in the hollow cavity. For obtaining hollow nanostructures, etching strategies were applied as follows: etching via hydrochloric acid/sodium chloride solution in which high temperature (150 °C) was applied to the core-shell particles but almost no etching was observed (This method is also known as hydrothermal treatment; Fig. S1A). In another experiment, hydrofluoric acid was used as the etchant in which the etching time and hydrofluoric acid concentration played major roles for the etching process. With high concentration of hydrofluoric acid (10% in water for 10 min at 50 °C) all the particles disintegrated (Fig. S1B). However, with lower concentration of hydrofluoric acid (2.5%) and longer etching times (4 and 10 h at 50 °C), rattle-type particles were formed instead of hollow NPs (Fig. S1C and D). Sodium carbonate was the best etchant for etching our NPs because when dissolved in water, it forms carbonic acid and sodium hydroxide. The latter acts as a strong base which can etch the inner Stöber core. In each experiment, different parameters where tested such as concentration of sodium carbonate, etching time, temperature, and stirring rate (Fig. S1E). For effective etching of HMSiO2 NPs, the optimal condition was to use high concentration of Na2CO3 (750 mM) at 50 °C under vigorous stirring (1200 RPM) for 10 h (Fig. 1). We demonstrated that by changing the precursors ratio, the nanoparticles could not completely be etched under the same optimal condition (Fig. S1F). After optimizing reaction parameters such as reaction time, stirring rate, ammonium hydroxide concentration, water/ethanol ratio, TEOS concentration, and TEOS/BTESPD ratio, several GSH-sensitive HMSiO2 NPs were fabricated with differences in size of the interior cavity and shell thickness (Fig. S2).
Fig. 1.

Electron microscopy images of the particles: TEM images of (A) Uniform c.a. 100 nm core Stöber NPs; (B) Disulfide-based mesoporous shell (15 nm) coated Stöber NPs synthesized by the addition of TEOS and BTESPD precursors; and (C and D) GSH-sensitive HMSiO2 NPs (c.a. 130 nm) under two magnifications. SEM images of (E) GSH-sensitive HMSiO2 NPs and (F) Broken NP showing voluminous (surface area: 446 m2 g−1) hollow interior. TEM images of (G) Mesoporous shell coated Stöber NPs synthesized by the addition of TEOS; (H and I) TEOS HMSiO2 NPs under two magnifications.
Fig. 2 displays further characterization of GSH-sensitive and TEOS HMSiO2 NPs in terms of nitrogen adsorption desorption isotherms, TGA, STEM, and XRD. Isotherm analyses were performed to explore BJH pore size distribution and BET specific surface area. As indicated in Fig. 2A, according to IUPAC classification, GSH-sensitive and TEOS HMSiO2 NPs (shown in green and red, respectively) exhibited sorption isotherms type IV ascribed to “mesoporous” NPs with average pore diameters of 2.3 and 3.1 nm, respectively. As shown in the isotherm plot of GSH-sensitive HMSiO2 NPs, there is a hysteresis loop which can be attributed to the capillary condensation in the mesopores and metastability of the gaseous phase at high relative pressures (P/P0) [31].
Fig. 2.

(A) Nitrogen adsorption desorption isotherms of the fabricated HMSiO2 NPs with/without hysteresis loop which show type IV isotherms according to IUPAC classification. Inset is the pore size distribution plots for each nanoparticle shown in angstrom; (B) STEM spectra for both HMSiO2 NPs. GSH-sensitive HMSiO2 NPs exhibit a small peak around 2.3 KeV which is ascribed to the presence of sulfur; (C) STEM images of GSH-sensitive HMSiO2 NPs demonstrate homogenous distribution of sulfur in the outer shell; (D) TGA graphs show c.a. 5.3% more weight loss in GSH-sensitive HMSiO2 NPs in comparison with TEOS HMSiO2 NPs attributed to the presence of organosilane matter in disulfide-based particles; (E) XRD plots of the synthesized HMSiO2 NPs confirm disordered pore structure in GSH-sensitive HMSiO2 NPs and well-ordered pore structure in TEOS HMSiO2 NPs due to the observation of two broad Bragg peaks. (Note: green lines and red lines/dash represent GSH-sensitive HMSiO2 NPs and TEOS HMSiO2 NPs, respectively).
Fig. 2B compares sulfur density between GSH-sensitive and TEOS HMSiO2 NPs. As shown in STEM spectra, sulfur peak can be observed in the binding energy around 2.3 KeV for GSH-sensitive particles. Fig. 2C indicates approximate atomic densities in GSH-sensitive HMSiO2 NP via EDS detector. This detector scans the surface of one NP in a raster pattern. The images confirm homogenous distribution of sulfur in the NP’s outer shell.
TGA in Fig. 2D demonstrates that GSH-sensitive HMSiO2 NPs had weight loss of c.a. 30.2% while for TEOS HMSiO2 NPs the weight loss was approximately 24.9%. This difference in weight loss is attributed to the presence of organosilane portion (…Si-C-C-C-S-S-C-C-C-Si…) in disulfide-based particles and calcination of these groups in the temperatures ranging from 600–800 °C. Loss of moisture existing within the NPs leads to weight loss less than 100 °C.
XRD plot (Fig. 2E) reveals that the mesopores existing in the shell of GSH-sensitive HMSiO2 NPs (green line) have a disordered structure since no typical Bragg peaks were observed at low 2θ areas. This phenomenon is often seen in periodic mesoporous organosilica NPs (PMO NPs) in which bissilylated precursor is used in the fabrication process [39]. Hence, in GSH-sensitive HMSiO2 NPs, the addition of BTESPD precursor in the shell coating step, made the formation of hexagonally-ordered pores difficult due to the presence of long silyl chains in comparison to TEOS HMSiO 2 NPs in which the shell is composed of only TEOS precursor. As shown in the XRD graph of TEOS HMSiO2 NPs (Fig. 2E; red line), two broad peaks can be observed around 2 and 4 degrees which suggest short-range ordering and a wormlike pore structure in these NPs [40].
Table 1 shows that surface area, total pore area, total pore volume, and pore diameter of GSH-sensitive HMSiO2 NPs were 446 ± 6 m2 g−1, 173 ± 8.7 m2 g−1, 0.9 ± 0.2 cm3 g−1, and 2.3 ± 0.5 nm, respectively; while the values for TEOS HMSiO2 NPs were: 523 ± 2 m2 g−1, 211 ± 11.3 m2 g−1, 1.1 ± 0.2 cm3 g−1, and 3.1 ± 0.7 nm, respectively (Table S1). This large surface area is directly related to the presence of interior hollow cavity of the particles which is c.a. 80 m2 g−1 higher than our previous disulfide-based degradable mesoporous SiO2 NPs (366 ± 9 m2 g−1) [31]. XPS, also known as electron spectroscopy for chemical analysis, is a quantitative technique that measures the elemental composition of the materials within the very top 1–10 nm of the NP’s surface. Table 1 indicates that the outer shell of GSH-sensitive HMSiO2 NPs contains c.a. 5.5% sulfur. In addition, XPS survey spectra indicate the presence and absence of S peaks in GSH-sensitive and TEOS HMSiO2 NPs, respectively (Fig. S3).
Table 1.
Composition percentages and nitrogen adsorption-desorption characteristics of GSH-sensitive HMSiO2 NPsa
| GSH-Sensitive HMSiO2 NPs | NP Composition Percentages Measured by XPS (% Mass Concentration) | BET-BJH Parameters | ||||||
| Si | O | S | C | Surface Area (m2 g−1) | Total Pore Area (m2 g−1) | Total Pore Volume (cm3 g−1) | Pore Diameter (nm) | |
| 39.4 | 40.8 | 5.5 | 14.3 | 446 ± 6 | 173 ± 8.7 | 0.9 ± 0.2 | 2.3 ± 0.5 | |
Data are mean ± SD (n = 3).
Table 2 shows hydrodynamic diameters of GSH-sensitive HMSiO2 NPs in various media. The average hydrodynamic diameters measured by DLS were 162 ± 10, 150 ± 3, and 189 ± 35 nm in DI water, DMEM + 10% fetal bovine serum (FBS), and RPMI + 10% FBS, respectively. Zeta potentials were −35 ± 1, −23 ± 1, −7 ± 1, and −7 ± 1 mV in DI water at pH 7.2, DI water at pH 6, DMEM + 10% FBS, and RPMI + 10% FBS, respectively. When GSH-sensitive HMSiO2 NPs were dispersed in DI water, their zeta potential values between −23 and −35 mV indicates the presence of surface silanol (Si-OH) groups. These groups can become deprotonated in aqueous environments and form Si-O− which lead to negative zeta potentials. These values decrease considerably by suspending the particles in media + 10% FBS (zeta potential −7 mV) due to high ionic strength of the media and the presence of serum proteins adsorbing on the surface of these NPs and forming protein corona [41]. For comparison, zeta potential values for TEOS HMSiO2 NPs were −30 ± 2, −21 ± 2, and −10 ± 1 mV in DI water at pH 7.2, DI water at pH 6, and DMEM + 10% FBS, respectively (Table S2).
Table 2.
Hydrodynamic diameter and zeta potential of GSH-sensitive HMSiO2 NPs in different mediaa
| GSH-Sensitive HMSiO2 NPs | Hydrodynamic Diameter (nm) Measured by DLS | Zeta Potential (mV) at 25 °C | |||||
| Deionized (DI) Water | DMEM + 10% FBS | RPMI + 10% FBS | DI Water pH 7.2 | DI Water pH 6 | DMEM + 10% FBS | RPMI + 10% FBS | |
| 162 ± 10 | 150 ± 3 | 189 ± 35 | −35 ± 1 | −23 ± 1 | −7 ± 1 | −7 ± 1 | |
Data are mean ± SD (n = 6).
Based on TEM images and the above-mentioned characterizations, it can be concluded that TEOS HMSiO2 NPs have similar features to GSH-sensitive HMSiO2 NPs except the presence of disulfide bonds.
In vitro degradation, DOX loading, and release
Fig. 3A illustrates in vitro degradation of GSH-sensitive HMSiO2 NPs (50 μg mL−1) in the presence of 10 mM of GSH at 37 °C and pH 6 for 7 days. This concentration was chosen to mimic intracellular concentration of GSH and pH 6 resembles pH of intracellular tumor microenvironment. GSH is a peptide composed of L-cysteine, glycine, and L-glutamic acid in which the L-cysteine amino acid residues have free S-H (thiol) group. These thiol groups can be oxidized to GSSG molecules and form an equilibrium. High intracellular concentrations of GSH (2–10 mM) facilitate degradation of disulfide bonds [24]. Extracellular concentrations of GSH are 100–1000 times lower (1–20 μM) than intracellular concentrations. Therefore such carriers can be applied in delivery systems where extracellular stability and intracellular release are desired [42]. We observed that these redox-responsive hollow particles start the degradation process from the first hours (Fig. 3A). As shown, many of the nanoparticles completely lost their mass integrity after day 7. Two mechanisms were observed for the degradation profile of these particles based on TEM images: first, majority of the NPs were broken down into smaller fragments. Second, some of the particles collapsed after day 7 which could result from loose structure of the shell when disulfide bonds started to break. In contrast, the particles in the control group after day 7, remained almost intact. It should be noted that the concentration of GSH is constant in vivo due to dynamic conditions and there is always an equilibrium between reduced glutathione (GSH) and oxidized (GSSG) via glutathione peroxidase/reductase enzymes [43]. Hence, it is likely that we may have higher in vivo degradation of these GSH-sensitive HMSiO2 NPs. Degradation of these particles at pH 7.2 and 10 mM of GSH was also performed. Results (Fig. S4) indicate that the change in pH did not alter degradation of GSH-sensitive HMSiO2 NPs. In addition, degradation was evaluated in the presence of 10 μM of GSH at pH 7.2. Fig. S5 shows that particles did not undergo any degradation at low GSH concentrations.
Fig. 3.

(A) TEM images for the degradation of GSH-sensitive HMSiO2 NPs in the presence of 10 mM of GSH in DI water at 37 °C and pH 6 for 7 days. Control samples are the NPs dispersed in DI water at 37 °C and pH 6 after day 7 (without GSH); (B) Digital image of DOX solution (100 μg mL−1) before (left) and after (right) interaction with HMSiO2 NPs. The image was taken after mixing, washing, and precipitating the NPs via centrifugation. High loading capacity 8.9 ± 0.5% was observed with GSH-sensitive HMSiO2 NPs due to their large interior voids; (C) DOX release profile from the GSH-sensitive and TEOS HMSiO2 NPs in solutions of different pH values at 37 °C for 14 days. Control is GSH-sensitive HMSiO2 NPs dispersed in PBS at pH 6 and without GSH. Data points are mean ± SD (n = 6). **Shows there was a statistically significant difference between the two study groups (Pvalue< 0.05).
HMSiO2 NPs can be used as delivery carriers due to their hollowness, low density, large surface area, and high drug loading capacity. For exploring drug loading capacity of GSH-sensitive and TEOS HMSiO2 NPs, DOX, was used as an anticancer drug model. DOX was physically incorporated in HMSiO2 NPs. In this process, electrostatic interactions facilitate incorporation of positively charged DOX within negatively charged NPs. After physical mixing for 24 h and washing the product thoroughly, loading capacity for GSH-sensitive and TEOS HMSiO2 NPs was 8.9 ± 0.5% (which means approximately 89 μg of DOX per 1 mg of GSH-sensitive HMSiO2 NPs) and 8.2 ± 0.4%, respectively. Drug loading efficiency for GSH-sensitive and TEOS HMSiO2 NPs was 12 ± 0.7% and 11 ± 0.5%, respectively. This high loading capacity is directly related to the size of the interior cavity that exists in these NPs and is proportional to the particle diameter which could be of great importance when we need to deliver two types of active agents simultaneously in the same carrier. Fig. 3B displays the GSH-sensitive hollow particles before and after interaction with DOX solution (100 μg mL−1). Next, DOX release profile from GSH-sensitive and TEOS HMSiO2 NPs in solutions of different pH values was evaluated for 14 days. Results (Fig. 3C) indicate redox-responsive hollow particles can release DOX approximately up to 60% in the presence of 10 mM of GSH at both pH 6 and pH 7.2. This release decreased when 2 mM of GSH was used at pH 6 and c.a. 45% of DOX released from the particles after 2 weeks. Release from control samples (GSH-sensitive HMSiO2 NPs dispersed in PBS at pH 6 and without GSH) and TEOS HMSiO2 NPs was approximately up to 14% and 18%, respectively. This suggests that DOX can be entrapped inside these hollow particles and release very slowly. In most samples, for the first 24 h, burst release was observed from the particles which results from the dissolution of DOX bound to the surface of the particles or entrapped in the pores of the shell.
In vitro cytotoxicity in MCF-7 cells
Cellular toxicity of GSH-sensitive and TEOS HMSiO2 NPs, DOX-loaded GSH-sensitive and TEOS HMSiO2 NPs, and free DOX was evaluated in MCF-7 breast cancer epithelial cells after incubation for 24 and 48 h. Results (Fig. 4A) reveal that both GSH-sensitive and TEOS HMSiO2 NPs were almost nontoxic to MCF-7 cells after incubation for 24 h in the concentration range between 3.9 to 1000 μg mL−1. However, cell viability decreased when the cells were treated with concentration of NPs higher than 125 μg mL−1 and incubated for 48 h (Fig. 4C). For GSH-sensitive HMSiO2 NPs, the toxic effects started from 250 μg mL−1 while for TEOS HMSiO2 NPs toxicity was observed with the concentrations equal or greater than 500 μg mL−1 which possibly results from the accumulation of particles inside the cells and disrupting cell membrane integrity.
Fig. 4.

MCF-7 cell toxicity of intact GSH-sensitive HMSiO2 NPs (green bars) and TEOS HMSiO2 NPs (red bars) after incubation for (A) 24 h and (C) 48 h. MCF-7 cytotoxicity of DOX-loaded GSH-sensitive HMSiO2 NPs (purple bars), DOX-loaded TEOS HMSiO2 NPs (blue bars), and free DOX (orange bars) after incubation for (B) 24 h and (D) 48 h. Purple, blue, and orange bars are plotted based on DOX concentration ranging from 0 to 6 μg mL−1. Assays confirm concentration- and time-dependent toxicity of the intact hollow particles and DOX-loaded ones. Data are mean ± SD (n = 3). **Shows there was a statistically significant difference between the two study groups (Pvalue< 0.05).
To demonstrate that DOX-loaded hollow particles can have toxic effects on MCF-7 cells, cytotoxicity of the loaded nanoparticles was tested and compared with toxicity of free DOX. As shown in Fig. 4B and D, cells were treated with DOX-loaded particles based on DOX concentration ranging from 0 to 6 μg mL−1. As expected free DOX had higher toxicity on MCF-7 cells than DOX-loaded particles with the maximum cell viability reduction of approximately 64% and 80% using 6 μg mL−1 of DOX after incubation for 24 and 48 h, respectively. Between two fabricated hollow NPs, a statistically significant difference was observed for the concentrations higher than 1.5 and 0.75 μg mL−1 of DOX after incubation for 24 and 48 h, respectively. DOX-loaded GSH-sensitive HMSiO2 NPs (containing 6 μg mL−1 of DOX) killed c.a. 50% of the cells while DOX-loaded TEOS HMSiO2 NPs with the same concentration killed 20% after 24 h incubation. This finding probably results from higher intracellular DOX release due degradation of disulfide-based particles. Cell viability reduction reached to 64% and 49% after incubation for 48 h with DOX-loaded GSH-sensitive HMSiO2 NPs and DOX-loaded TEOS HMSiO2 NPs, respectively (containing 6 μg mL−1 of DOX). This data suggests that there is an optimum concentration in which the intact hollow particles do not have toxic effects on the cells but DOX-loaded particles can effectively kill c.a. 50% of the cancer cells. In these experiments, the optimum concentration of GSH-sensitive HMSiO2 NPs is c.a. 66 μg mL−1 which can hold 6 μg mL−1 of DOX.
Cellular uptake and intracellular trafficking
Intracellular localization of GSH-sensitive HMSiO2 NPs was visualized using confocal laser scanning microscopy (CLSM). MCF-7 cells were co-cultured with 50 and 250 μg mL−1 of fluorescein isothiocyanate (FITC)-labeled GSH-sensitive HMSiO2 NPs and incubated for 4 and 24 h. Fig. 5 illustrates that most of the particles were entrapped in the endolysosomal compartments after they were internalized. It was observed that concentration of the particles and incubation time play major roles for internalization of hollow NPs. As shown in Fig. 5D, the number of FITC-labeled particles engulfed in the cells was much higher than the other conditions (Fig. 5A, B, and C). By increasing the concentration of the hollow particles and incubation time, cell uptake increased (green dots).
Fig. 5.

Intracellular trafficking of GSH-sensitive HMSiO2 NPs in MCF-7 cells: (A–D) Representative CLSM images of the cells treated with GSH-sensitive HMSiO2 NPs; (A) Cells were treated with 50 μg mL−1 of NPs and incubated for 4 h; (B) Cells were treated with 50 μg mL−1 of NPs and incubated for 24 h; (C) Cells were treated with 250 μg mL−1 of NPs and incubated for 4 h; and (D) Cells were treated with 250 μg mL−1 of NPs and incubated for 24 h. (A1-D1) Merge CLSM images under higher magnifications. Nuclei were stained in blue with Hoechst 33342 dye. NPs appeared with the green fluorescence of FITC. LysoTracker™ Deep Red dye was used to stain the lysosomes. As illustrated, NPs accumulated in the endolysosomal compartments and perinuclear regions. GSH-sensitive HMSiO2 NP uptake rate in MCF-7 cells measured by ICP-MS in terms of (E) NP concentration in the range of 0–1000 μg mL−1 with constant incubation time of 24 h and (F) incubation time in the range of 0–24 h with constant NP concentration of 100 μg mL−1. Results indicate that there is a threshold for the internalization of GSH-sensitive HMSiO2 NPs with the maximum uptake of c.a. 2.1% after incubation for 24 h. Cells without NP treatment were used as control. Data are mean ± SD (n = 3).
Previously we showed that there is a cellular toleration threshold for silica nanoconstructs in vitro [44]. Cell uptake of GSH-sensitive HMSiO2 NPs was evaluated in MCF-7 cells in terms of nanoparticle concentration (ranging from 0–1000 μg mL−1 with constant incubation time of 24 h) and incubation time (ranging from 0–24 h with constant nanoparticle concentration of 100 μg mL−1) to see when we reach to that cellular threshold. For this test, maximum chosen concentration was 1000 μg mL−1 since the nanoparticles did not show any toxic effect on MCF-7 cells at this concentration after incubation for 24 h. In this experiment, the amount of silicon (Si) was measured with Inductively Coupled Plasma Mass Spectrometry (ICP-MS). Results show (Fig. 5E) that c.a. 1.2% of the hollow particles were taken up when the cells were incubated with 25 μg mL−1 of NPs. Cell uptake increased by increasing the concentration of NPs and reached a plateau (c.a. 2.1%) at NP concentration of equal or greater than 250 μg mL−1. This 2.1% maximum uptake can be considered as a threshold of MCF-7 cells for internalizing GSH-sensitive HMSiO2 NPs. These observations were also consistent with the results of cell uptake obtained based on different incubation times in which the uptake of 100 μg mL−1 of hollow particles was approximately 1.7% after incubation for 24 h (Fig. 5F). In this experiment, MCF-7 cells without nanoparticle treatment were used as negative control.
In order to evaluate intracellular co-localization of GSH-sensitive HMSiO2 NPs in MCF-7 cells, uptake was also visualized by TEM (Fig. 6). The cells were treated with 50 (Fig. 6D) and 250 (Fig. 6E) μg mL−1 of the particles and incubated for 24 h. Cells without NP treatment were used as control (Fig. 6A). Fig. 6B illustrates that MCF-7 cells are internalizing the particles into the intracellular endosomal compartments. These NPs can be entrapped as an individual particle or as a group of particles (two or more) as indicated in Fig. 6C. It can be observed that some of the GSH-sensitive HMSiO2 NPs lost their hollow structure and underwent intracellular degradation in endocytic compartments (green arrows in Fig. 6D2 and E1). This degradation could result from the presence of gamma interferon-inducible lysosomal thiol-reductase (GILT) enzyme in the endocytic compartments which facilitates the degradation of disulfide-based materials with their optimal reductase activity at acidic pH values [45, 46].
Fig. 6.

Representative TEM images for intracellular co-localization of GSH-sensitive HMSiO2 NPs in MCF-7 cells: (A) Control (cells without NP treatment); (B) Cells phagocytosing the hollow particles; (C) Particles are internalized in the endocytic compartments as an individual particle or as a group of particles; (D) Cells were treated with 50 μg mL−1 of NPs and incubated for 24 h; (D1 and D2) TEM images under higher magnifications. (E) Cells were treated with 250 μg mL−1 of NPs and incubated for 24 h; (E1 and E2) TEM images under higher magnifications. Green arrows show degraded particles in endolysosomal compartments.
In vitro cytotoxicity and cell uptake in RAW 264.7 macrophages
The primary role of macrophages is early uptake of foreign material and their clearance. In this study, RAW 264.7 macrophages were chosen due to their high phagocytic activity. As shown in Fig. 7A and B, cell toxicity of GSH-sensitive HMSiO2 NPs was evaluated in RAW 264.7 macrophages after incubation for 24 and 48 h, respectively. Results (Fig. 7A) indicate that unlike MCF-7 cells, these hollow particles had toxic effects on RAW 264.7 macrophages when the NP concentration was equal or greater than 250 μg mL−1 and cell viability decreased to c.a. 70% when the cells were co-cultured with 1000 μg mL−1 of particles and incubated for 24 h. This toxicity was also time-dependent in which cell viability decreased c.a. 9% more under the same NP concentration and incubation for 48 h (Fig. 7B). Next, uptake of GSH-sensitive HMSiO2 NPs (25–100 μg mL−1 which is the maximum safe concentration of these hollow particles in RAW 264.7 macrophages after incubation for 24 h; Fig. 7C) was studied in RAW 264.7 macrophages. It was observed that after the dose of equal or greater than 75 μg mL−1, a plateau was observed with the maximum uptake of c.a. 5.2% which was approximately 2.5 times higher than the maximum uptake of the particles in MCF-7 breast cancer epithelial cells (2.1%). NP uptake in RAW 264.7 macrophages was also investigated with different incubation times (0–24 h and 100 μg mL−1 of NPs). Results (Fig. 7D) demonstrate that HMSiO2 NP uptake after incubation for 24 h was almost 3 times higher than when they were incubated for 4 h.
Fig. 7.

Cytotoxicity of GSH-sensitive HMSiO2 NPs in RAW 264.7 macrophages after incubation for (A) 24 h and (B) 48 h. Particles were toxic to the cells with the concentrations equal or greater than 250 μg mL−1 GSH-sensitive HMSiO2 NP uptake rate in RAW 264.7 macrophages measured by ICP-MS in terms of (C) NP concentration and (D) incubation time (in D, cells were treated with 100 μg mL−1 of NPs). Maximum toleration threshold (5.2%) was observed when the cells where co-cultured with 75–100 μg mL−1 of NPs and incubated for 24 h. Data are mean ± SD (n = 3).
In addition to these cytotoxicity studies conducted in MCF-7 and RAW 264.7 macrophages, we also performed cell toxicity in NIH 3T3 fibroblast cells to see the effect of GSH-sensitive HMSiO2 NPs on normal cells. Fig. 8A and B demonstrate that particles did not show any toxic effects on these cells with NP concentrations equal or less than 125 μg mL−1. However, with higher NP concentrations cell viability decreased in a time- and concentration-dependent fashion. c.a. 41% and 63% reduction were observed in cell viability when the cells were treated with 1000 μg mL−1 of HMSiO2 NPs and incubated for 24 and 48 h, respectively. In addition, c.a. 23% and 51% reduction were seen in cell viability when the cells were co-cultured with 500 μg mL−1 of HMSiO2 NPs and incubated for 24 and 48 h, respectively.
Fig. 8.

Cytotoxicity of GSH-sensitive HMSiO2 NPs in NIH 3T3 fibroblasts after incubation for (A) 24 h and (B) 48 h. Toxic effects of the particles started when the cells were treated with NPs in the concentrations between 250 and 1000 μg mL−1. Data are mean ± SD (n = 3).
Conclusions
In summary, we have fabricated and characterized novel GSH-sensitive biodegradable hollow mesoporous silica nanoparticles with the average diameter of 130 nm. These particles were size-tunable in terms of internal hollow cavity diameter and shell thickness and can incorporate substantial amount of active agents such as DOX (~8.9%). We have shown that these uniform nanoparticles can degrade from the outer shell containing disulfide bonds in the presence of intracellular concentrations of GSH (~10 mM). Approximately 60% of DOX released within 2 weeks in the presence of 10 mM of GSH at pH 6 resembling intratumoral microenvironment. These hollow nanoparticles were taken up by MCF-7 cells in a time- and concentration-dependent manner and entrapped in endocytic compartments. Cytotoxicity studies conducted in MCF-7 breast cancer cells, RAW 264.7 macrophages, and NIH 3T3 fibroblasts revealed that toxicity of the particles was proportional to nanoparticle concentration, incubation time, and cell type. Based on the results of cell toxicities after 24 h, these particles were nontoxic to MCF-7 cells in the concentration range of 0–1000 μg mL−1. However, they showed some toxic effects on RAW 264.7 macrophages and NIH 3T3 fibroblasts with the concentrations equal or greater than 250 μg mL−1. These cytotoxicity studies suggest that the optimal concentration of GSH-sensitive HMSiO2 NPs for drug delivery applications is approximately 100 μg mL−1 in which the particles can hold 9 μg mL−1 of DOX and effectively kill the cancerous cells. Cellular uptake measured via ICP-MS elucidate that there is a threshold for the uptake of these redox-responsive hollow particles in MCF-7 cells and RAW 264.7 macrophages with the maximum uptake of 2.1% and 5.2%, respectively. Together these results demonstrate the high loading capacity and controlled degradation of these nanoparticles which is an improvement over other SiO2 NPs systems. The next steps are accurate in vivo characterization of these GSH-sensitive HMSiO2 NPs and optimizing their degradation rate over a certain period of time for specific delivery applications.
Statistical analysis
Data points are mean ± standard deviations (SD) for at least three separate experiments. The difference between multiple groups was studied using analysis of variance (ANOVA). For comparison between two groups, t-test was applied. The difference compared to control was considered significant at Pvalue< 0.05.
Supplementary Material
Acknowledgments
We acknowledge financial support from the National Institute of Environmental Health Sciences of the NIH (R01ES024681), the University of Utah Nanotechnology Training Program Fellowship (Seyyed Pouya Hadipour Moghaddam), and the University of Utah College of Pharmacy Skaggs fellowship (Mostafa Yazdimamaghani). This work made use of the University of Utah shared facilities of the Micron Microscopy Suite and the University of Utah USTAR shared facilities supported in part by the MRSEC Program of the NSF under Award No. DMR-1121252.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Wu X, Wu M, Zhao JX. Recent development of silica nanoparticles as delivery vectors for cancer imaging and therapy. Nanomedicine: nanotechnology, biology, and medicine. 2014;10:297–312. doi: 10.1016/j.nano.2013.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lee JE, Lee N, Kim T, Kim J, Hyeon T. Multifunctional mesoporous silica nanocomposite nanoparticles for theranostic applications. Accounts of chemical research. 2011;44:893–902. doi: 10.1021/ar2000259. [DOI] [PubMed] [Google Scholar]
- 3.Šoltys M, Balouch M, ej Kašpar O, Lhotka M, Ulbrich P, Zadražil A, Kovaíik P, Št pánek F. Evaluation of scale-up strategies for the batch synthesis of dense and hollow mesoporous silica microspheres. Chemical Engineering Journal. 2017 [Google Scholar]
- 4.Lee JE, Lee N, Kim T, Kim J, Hyeon T. Multifunctional Mesoporous Silica Nanocomposite Nanoparticles for Theranostic Applications. Accounts of chemical research. 2011;44:893–902. doi: 10.1021/ar2000259. [DOI] [PubMed] [Google Scholar]
- 5.Zhao W, Gu J, Zhang L, Chen H, Shi J. Fabrication of Uniform Magnetic Nanocomposite Spheres with a Magnetic Core/Mesoporous Silica Shell Structure. Journal of the American Chemical Society. 2005;127:8916–8917. doi: 10.1021/ja051113r. [DOI] [PubMed] [Google Scholar]
- 6.Angelos S, Khashab NM, Yang YW, Trabolsi A, Khatib HA, Stoddart JF, Zink JI. pH Clock-Operated Mechanized Nanoparticles. Journal of the American Chemical Society. 2009;131:12912–12914. doi: 10.1021/ja9010157. [DOI] [PubMed] [Google Scholar]
- 7.Liu J, Luo Z, Zhang J, Luo T, Zhou J, Zhao X, Cai K. Hollow mesoporous silica nanoparticles facilitated drug delivery via cascade pH stimuli in tumor microenvironment for tumor therapy. Biomaterials. 2016;83:51–65. doi: 10.1016/j.biomaterials.2016.01.008. [DOI] [PubMed] [Google Scholar]
- 8.Luo Z, Ding X, Hu Y, Wu S, Xiang Y, Zeng Y, Zhang B, Yan H, Zhang H, Zhu L, Liu J, Li J, Cai K, Zhao Y. Engineering a Hollow Nanocontainer Platform with Multifunctional Molecular Machines for Tumor-Targeted Therapy in Vitro and in Vivo. ACS Nano. 2013;7:10271–10284. doi: 10.1021/nn404676w. [DOI] [PubMed] [Google Scholar]
- 9.Yu Y, Li Y, Wang W, Jin M, Du Z, Li Y, Duan J, Yu Y, Sun Z. Acute Toxicity of Amorphous Silica Nanoparticles in Intravenously Exposed ICR Mice. PLoS One. 2013;8:e61346. doi: 10.1371/journal.pone.0061346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ryu HJ, Seong N-w, So BJ, Seo H-s, Kim J-h, Hong J-S, Park M-k, Kim M-S, Kim Y-R, Cho K-B, Seo MY, Kim M-K, Maeng EH, Son SW. Evaluation of silica nanoparticle toxicity after topical exposure for 90 days. International Journal of Nanomedicine. 2014;9:127–136. doi: 10.2147/IJN.S57929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yu Y, Duan J, Li Y, Li Y, Jing L, Yang M, Wang J, Sun Z. Silica nanoparticles induce liver fibrosis via TGF-β(1)/Smad3 pathway in ICR mice. International Journal of Nanomedicine. 2017;12:6045–6057. doi: 10.2147/IJN.S132304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zalzberg L, Avnir D. Biocompatible hybrid particles of poly(l-lactic acid)@silica. Journal of Sol-Gel Science and Technology. 2008;48:47–50. [Google Scholar]
- 13.Chen K, Zhang J, Gu H. Dissolution from inside: a unique degradation behaviour of core-shell magnetic mesoporous silica nanoparticles and the effect of polyethyleneimine coating. Journal of Materials Chemistry. 2012;22:22005–22012. [Google Scholar]
- 14.Kortesuo P, Ahola M, Karlsson S, Kangasniemi I, Yli-Urpo A, Kiesvaara J. Silica xerogel as an implantable carrier for controlled drug delivery--evaluation of drug distribution and tissue effects after implantation. Biomaterials. 2000;21:193–198. doi: 10.1016/s0142-9612(99)00148-9. [DOI] [PubMed] [Google Scholar]
- 15.Radin S, El-Bassyouni G, Vresilovic EJ, Schepers E, Ducheyne P. In vivo tissue response to resorbable silica xerogels as controlled-release materials. Biomaterials. 2005;26:1043–1052. doi: 10.1016/j.biomaterials.2004.04.004. [DOI] [PubMed] [Google Scholar]
- 16.Croissant JG, Fatieiev Y, Julfakyan K, Lu J, Emwas AH, Anjum DH, Omar H, Tamanoi F, Zink JI, Khashab NM. Biodegradable Oxamide-Phenylene-Based Mesoporous Organosilica Nanoparticles with Unprecedented Drug Payloads for Delivery in Cells. Chemistry – A European Journal. 2016;22:14806–14811. doi: 10.1002/chem.201601714. [DOI] [PubMed] [Google Scholar]
- 17.Zhou M, Du X, Li W, Li X, Huang H, Liao Q, Shi B, Zhang X, Zhang M. One-pot synthesis of redox-triggered biodegradable hybrid nanocapsules with a disulfide-bridged silsesquioxane framework for promising drug delivery. Journal of Materials Chemistry B. 2017;5:4455–4469. doi: 10.1039/c6tb03368g. [DOI] [PubMed] [Google Scholar]
- 18.He Y, Zeng B, Liang S, Long M, Xu H. Synthesis of pH-Responsive Biodegradable Mesoporous Silica – Calcium Phosphate Hybrid Nanoparticles as a High Potential Drug Carrier. ACS Applied Materials & Interfaces. 2017 doi: 10.1021/acsami.7b16787. [DOI] [PubMed] [Google Scholar]
- 19.Fatieiev Y, Croissant JG, Julfakyan K, Deng L, Anjum DH, Gurinov A, Khashab NM. Enzymatically degradable hybrid organic-inorganic bridged silsesquioxane nanoparticles for in vitro imaging. Nanoscale. 2015;7:15046–15050. doi: 10.1039/c5nr03065j. [DOI] [PubMed] [Google Scholar]
- 20.Maggini L, Cabrera I, Ruiz-Carretero A, Prasetyanto EA, Robinet E, De Cola L. Breakable mesoporous silica nanoparticles for targeted drug delivery. Nanoscale. 2016;8:7240–7247. doi: 10.1039/c5nr09112h. [DOI] [PubMed] [Google Scholar]
- 21.Croissant J, Cattoen X, Man MW, Gallud A, Raehm L, Trens P, Maynadier M, Durand JO. Biodegradable ethylene-bis(propyl)disulfide-based periodic mesoporous organosilica nanorods and nanospheres for efficient in-vitro drug delivery. Advanced materials (Deerfield Beach, Fla) 2014;26:6174–6180. doi: 10.1002/adma.201401931. [DOI] [PubMed] [Google Scholar]
- 22.Croissant JG, Mauriello-Jimenez C, Maynadier M, Cattoen X, Wong Chi Man M, Raehm L, Mongin O, Blanchard-Desce M, Garcia M, Gary-Bobo M, Maillard P, Durand J-O. Synthesis of disulfide-based biodegradable bridged silsesquioxane nanoparticles for two-photon imaging and therapy of cancer cells. Chemical Communications. 2015;51:12324–12327. doi: 10.1039/c5cc03736k. [DOI] [PubMed] [Google Scholar]
- 23.Vivero-Escoto JL, Rieter WJ, Lau H, Huxford-Phillips RC, Lin W. Biodegradable Polysilsesquioxane Nanoparticles as Efficient Contrast Agents for Magnetic Resonance Imaging, Small (Weinheim an der Bergstrasse, Germany) 2013;9:3523–3531. doi: 10.1002/smll.201300198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cheng R, Feng F, Meng F, Deng C, Feijen J, Zhong Z. Glutathione-responsive nano-vehicles as a promising platform for targeted intracellular drug and gene delivery. Journal of controlled release: official journal of the Controlled Release Society. 2011;152:2–12. doi: 10.1016/j.jconrel.2011.01.030. [DOI] [PubMed] [Google Scholar]
- 25.Gamcsik MP, Kasibhatla MS, Teeter SD, Colvin OM. Glutathione Levels in Human Tumors. Biomarkers: biochemical indicators of exposure, response, and susceptibility to chemicals. 2012;17:671–691. doi: 10.3109/1354750X.2012.715672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Opstad KS, Provencher SW, Bell BA, Griffiths JR, Howe FA. Detection of elevated glutathione in meningiomas by quantitative in vivo 1H MRS. Magnetic resonance in medicine. 2003;49:632–637. doi: 10.1002/mrm.10416. [DOI] [PubMed] [Google Scholar]
- 27.Dudek H, Farbiszewski R, Rydzewska M, Michno T, Kozlowski A. Evaluation of antioxidant enzymes activity and concentration of non-enzymatic antioxidants in human brain tumours. Wiadomosci lekarskie (Warsaw, Poland: 1960) 2004;57:16–19. [PubMed] [Google Scholar]
- 28.Kudo H, Mio T, Kokunai T, Tamaki N, Sumino K, Matsumoto S. Quantitative analysis of glutathione in human brain tumors. Journal of neurosurgery. 1990;72:610–615. doi: 10.3171/jns.1990.72.4.0610. [DOI] [PubMed] [Google Scholar]
- 29.Barranco SC, Perry RR, Durm ME, Quraishi M, Werner AL, Gregorcyk SG, Kolm P. Relationship between colorectal cancer glutathione levels and patient survival: early results. Diseases of the colon and rectum. 2000;43:1133–1140. doi: 10.1007/BF02236562. [DOI] [PubMed] [Google Scholar]
- 30.Murray G, Burke M, Ewen S. Glutathione localisation in benign and malignant human breast lesions. British journal of cancer. 1987;55:605. doi: 10.1038/bjc.1987.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hadipour Moghaddam SP, Saikia J, Yazdimamaghani M, Ghandehari H. Redox-Responsive Polysulfide-Based Biodegradable Organosilica Nanoparticles for Delivery of Bioactive Agents. ACS Applied Materials & Interfaces. 2017;9:21133–21146. doi: 10.1021/acsami.7b04351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chen Y, Chen H, Guo L, He Q, Chen F, Zhou J, Feng J, Shi J. Hollow/Rattle-Type Mesoporous Nanostructures by a Structural Difference-Based Selective Etching Strategy. ACS Nano. 2010;4:529–539. doi: 10.1021/nn901398j. [DOI] [PubMed] [Google Scholar]
- 33.Wang X, Feng J, Bai Y, Zhang Q, Yin Y. Synthesis, Properties, and Applications of Hollow Micro-/Nanostructures. Chemical Reviews. 2016;116:10983–11060. doi: 10.1021/acs.chemrev.5b00731. [DOI] [PubMed] [Google Scholar]
- 34.Chen Y, Chen H, Guo L, He Q, Chen F, Zhou J, Feng J, Shi J. Hollow/Rattle-Type Mesoporous Nanostructures by a Structural Difference-Based Selective Etching Strategy. 2010 doi: 10.1021/nn901398j. [DOI] [PubMed] [Google Scholar]
- 35.Li Y, Shi J. Hollow-Structured Mesoporous Materials: Chemical Synthesis, Functionalization and Applications. Advanced Materials. 2014;26:3176–3205. doi: 10.1002/adma.201305319. [DOI] [PubMed] [Google Scholar]
- 36.Saikia J, Yazdimamaghani M, Hadipour Moghaddam SP, Ghandehari H. Differential Protein Adsorption and Cellular Uptake of Silica Nanoparticles Based on Size and Porosity. ACS Applied Materials & Interfaces. 2016;8:34820–34832. doi: 10.1021/acsami.6b09950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fang X, Chen C, Liu Z, Liu P, Zheng N. A cationic surfactant assisted selective etching strategy to hollow mesoporous silica spheres. Nanoscale. 2011;3:1632–1639. doi: 10.1039/c0nr00893a. [DOI] [PubMed] [Google Scholar]
- 38.Li Y, Shi J. Hollow-structured mesoporous materials: chemical synthesis, functionalization and applications. Advanced materials (Deerfield Beach, Fla) 2014;26:3176–3205. doi: 10.1002/adma.201305319. [DOI] [PubMed] [Google Scholar]
- 39.Van Der Voort P, Esquivel D, De Canck E, Goethals F, Van Driessche I, Romero-Salguero FJ. Periodic Mesoporous Organosilicas: from simple to complex bridges; a comprehensive overview of functions, morphologies and applications. Chemical Society Reviews. 2013;42:3913–3955. doi: 10.1039/c2cs35222b. [DOI] [PubMed] [Google Scholar]
- 40.Lin YS, Haynes CL. Impacts of Mesoporous Silica Nanoparticle Size, Pore Ordering, and Pore Integrity on Hemolytic Activity. Journal of the American Chemical Society. 2010;132:4834–4842. doi: 10.1021/ja910846q. [DOI] [PubMed] [Google Scholar]
- 41.Lee P, Knight R, Smit JM, Wilschut J, Griffin DE. A Single Mutation in the E2 Glycoprotein Important for Neurovirulence Influences Binding of Sindbis Virus to Neuroblastoma Cells. Journal of Virology. 2002;76:6302–6310. doi: 10.1128/JVI.76.12.6302-6310.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Quinn JF, Whittaker MR, Davis TP. Glutathione responsive polymers and their application in drug delivery systems. Polymer Chemistry. 2017;8:97–126. [Google Scholar]
- 43.Wu G, Fang YZ, Yang S, Lupton JR, Turner ND. Glutathione Metabolism and Its Implications for Health. The Journal of Nutrition. 2004;134:489–492. doi: 10.1093/jn/134.3.489. [DOI] [PubMed] [Google Scholar]
- 44.Gustafson H, Malugin A, Ghandehari H. Silica Nanoconstruct Cellular Toleration Threshold In Vitro. 2011 doi: 10.1016/j.jconrel.2011.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Arunachalam B, Phan UT, Geuze HJ, Cresswell P. Enzymatic reduction of disulfide bonds in lysosomes: characterization of a gamma-interferon-inducible lysosomal thiol reductase (GILT) Proceedings of the National Academy of Sciences. 2000;97:745–750. doi: 10.1073/pnas.97.2.745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hastings KT, Cresswell P. Disulfide Reduction in the Endocytic Pathway: Immunological Functions of Gamma-Interferon-Inducible Lysosomal Thiol Reductase. Antioxidants & Redox Signaling. 2011;15:657–668. doi: 10.1089/ars.2010.3684. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.