

Abstract
MicroRNAs (miRNA) are key modulators of gene expression and so act as putative fine-tuners of complex phenotypes. Here, we hypothesized that causal variants of complex traits are enriched in miRNAs and miRNA-target networks. First, we conducted a genome-wide association study (GWAS) for seven functional and milk production traits using imputed sequence variants (13~15 million) and >10,000 animals from three dairy cattle breeds, i.e., Holstein (HOL), Nordic red cattle (RDC) and Jersey (JER). Second, we analyzed for enrichments of association signals in miRNAs and their miRNA-target networks. Our results demonstrated that genomic regions harboring miRNA genes were significantly (P < 0.05) enriched with GWAS signals for milk production traits and mastitis, and that enrichments within miRNA-target gene networks were significantly higher than in random gene-sets for the majority of traits. Furthermore, most between-trait and across-breed correlations of enrichments with miRNA-target networks were significantly greater than with random gene-sets, suggesting pleiotropic effects of miRNAs. Intriguingly, genes that were differentially expressed in response to mammary gland infections were significantly enriched in the miRNA-target networks associated with mastitis. All these findings were consistent across three breeds. Collectively, our observations demonstrate the importance of miRNAs and their targets for the expression of complex traits.
Introduction
Understanding the genetic architecture, i.e., knowledge of causal genomic variants, their allele frequencies and effect sizes, underpinning complex phenotypes and diseases is a long ongoing quest in the field of genetics and genomics1–3. Complex phenotypes consist of contributions from multiple genomic loci, and accurate and reliable prediction of future complex traits and diseases based on genomic information is critical for human personalized medicine as well as for breeding of agricultural animal and plant species4–11. In the past decade, genome-wide association studies (GWAS) have been a productive way to achieve this goal5. Yet, big challenges remained in extending GWAS results to informative biological hypotheses underlying complex traits variation primarily due to 1) poor detection of many causal loci/genes with small effects by GWAS with limited sample sizes12,13, and 2) masking of true association signals by linkage disequilibrium (LD) between causal loci and neighboring markers4,14, especially in livestock with high degree of relatedness10,15. Recently, we have shown that the predictive ability of the genomic best linear unbiased prediction model increases in accuracy when the model utilizes and quantifies the combined contributions of markers in genomic regions associated with the genetic architecture of the underlying trait9,16–18. Also, biological knowledge has been incorporated to improve the understanding of GWAS results19–25. For instance, by integrating genome functional annotations with GWAS data, Finucane et al. (2015) revealed conserved genomic regions that were strongly enriched with genetic variation of many complex traits in human25. Holmans et al. (2009) provided insights into the biology of bipolar disorder through incorporation of Gene Ontology (GO) annotation into GWAS results23. Fang et al. (2017) revealed novel insights into the genetic and biological basis underpinning mastitis by incorporating transcriptome data22.
GWAS signals often map to non-coding regions of the genome, demonstrating that sequence variants in protein coding genes alone cannot explain most quantitative phenotypes. Indeed, conventional protein coding genes constitute only a very small percentage of the genome; yet ~75% of the genome generates RNA transcripts26. Thus, most transcripts in eukaryotic cells are non-coding RNA, and much of the regulatory capability and function of the genome are provided by non-coding RNA transcripts26–28. Among the many classes of non-coding RNA species are microRNAs, which act posttranscriptionally to fine-tune protein synthesis. The vertebrate genome encodes in the order of several hundred microRNAs comprising both evolutionary conserved and species-specific genes. The encoded miRNAs are ~22 nucleotide long RNA molecules that align to sequences in target mRNA molecules, leading to either deadenylation and decay of mRNAs or repression of protein translation29. In more rare cases, miRNAs can also activate and up-regulate gene expression in presence of distinct cofactors and under specific cellular conditions30. A single miRNA can target several different mRNAs, and moreover, mRNAs are often controlled by several different miRNAs29. Computational estimates suggest that more than 30% of all protein-coding genes in human are regulated by miRNAs31–34. This strongly indicates that miRNAs have widespread roles in vivo, and that all genetic networks and pathways are expected to be regulated to some degree by miRNAs. Genomic sequence variation can affect miRNAs functions in several ways: Variants can occur in sequences that are responsible for driving the expression of miRNA genes; variants can disrupt or create miRNA binding sites in target mRNAs; and the miRNAs themselves can exist as variants35. In this study, we aimed to investigate the joint effect of genetic variations in miRNA genes and in their targets-networks on complex traits of economic importance in dairy cattle.
Results
The overall study design is shown in Fig. 1. The quantitative traits in this study were body conformation (BC), mastitis, health, fertility and three milk production traits, including fat yield (FY), milk yield (MY) and protein yield (PY). Index for general health in dairy cattle describes genetic potential to resist reproductive, metabolic and feet-and-leg diseases, and this index is calculated based on data of veterinarians’ treatments in the first three lactations (http://www.nordicebv.info). We used imputed sequence variants (13~15 million single-nucleotide polymorphism (SNPs)) to ensure the coverage of most genes and miRNAs in the bovine genome (UMD 3.1). We performed within breed analyses for Holstein (HOL, n = 5,056), Nordic red (RDC, n = 4,310) and Jersey (JER, n = 1,231) cattle, to confirm our findings. Infection-induced transcriptome data were then integrated to validate the significant miRNA-target networks detected for mastitis, and to investigate the underlying genetic and biological basis of our findings.
Figure 1.
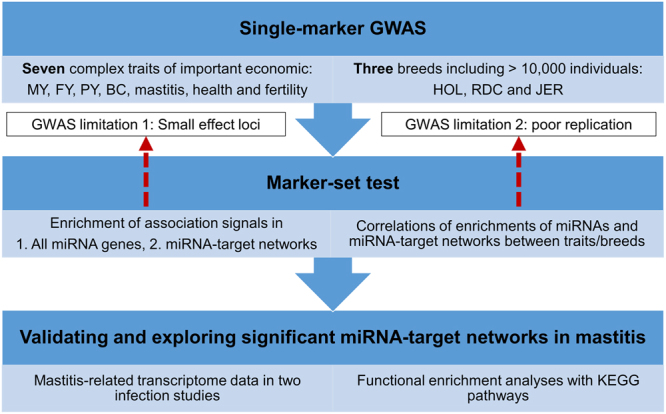
The overall study design. MY, FY, PY and BC are milk yield, fat yield, protein yield, and body conformation, respectively. HOL, RDC, and JER are Holstein, Nordic red, and Jersey cattle.
Single-marker GWAS for seven quantitative traits in three cattle breeds
The single-marker GWAS was performed to identify genomic variants associated with the traits being studied. The −log10P values of all tested sequence variants in the seven traits across HOL, RDC and JER are shown in the Manhattan plots, Supplementary Fig. S1, S2 and S3, respectively. At a nominal P-value < 3.3 × 10−9 (Bonferroni correction), a total of 6, 4, 12, 0, 5, 1 and 2 quantitative trait loci (QTL) were detected for MY, FY, PY, BC, mastitis, health and fertility, respectively in HOL, which jointly explained 24.1%, 22.3%, 19.3%, 0%, 9.7%, 8.2% and 5.3% of genomic variance in each corresponding trait, respectively. In RDC with the same P-value threshold as in HOL, 6, 6, 6, 4, 2, 0 and 4 QTLs were detected for MY, FY, PY, BC, mastitis, health and fertility respectively, which jointly explained 13.0%, 13.3%, 9.3%, 33.3%, 6.8%, 0% and 8.1% of genomic variance for each trait, respectively. In JER at P-value < 3.7 × 10−9, only 2 and 1 QTLs were detected for MY and FY, respectively, accounting for 3.9% and 3.1% of genomic variance for each trait, respectively. All the results demonstrated that the detected QTLs explained only a small fraction of genomic variance in complex traits, particularly in JER with a small sample size. The details of QTLs for all seven traits in the three breeds are summarized in Supplementary Table S1.
MiRNA genes are enriched with GWAS signals
In order to assess whether miRNAs influence the traits, we first mapped known miRNA genes in the bovine genome (UMD 3.1). A total of 750 bovine autosome miRNA genes expressed in different tissues from miRbase36 (http://www.mirbase.org/ftp.shtml) were observed to be distributed with an average of 26 (range: 7 to 77) miRNAs per chromosome (Fig. S4A and B). The average lengths of the miRNA precursor sequences were 77 base-pairs (bp) (range: 52 to 148 bp) (Fig. S4C). Since very few imputed SNPs were observed within the miRNA precursor regions due to their short lengths (see Supplementary Table S2), the analysis included the flanking ±3 kb, ±5 kb, ±0 kb, ±20 kb, ±50 kb sequences of all miRNA genes to capture proximal SNPs in the regulatory regions. Details of miRNAs and SNPs involved in the analyses under each extension are shown in Supplementary Table S1. Considering all miRNA genes together as a single set of markers, the marker-set test was carried out to analyze for enrichment of miRNA genes with association signals for each trait in all breeds. Analysis using the miRNA ±5 kb extended regions, significant (P < 0.05) enrichments were observed for most traits in the three breeds, especially for three milk production traits and mastitis (Fig. 2A), and the enrichments (i.e., −log10P values, marker-set test) were correlated between breeds (Fig. 2B–D). Of note, after removing all the four miRNAs (i.e., bta-miR-2309, bta-miR-1839, bta-miR-2308, and bta-miR-193a-2) that were in close proximity to the well-known milk/fat gene, DGAT1, on Bos taurus chromosome 14 (BTA 14)37, similar enrichments were still observed for the three production traits across the three breeds, particularly in RDC and JER (see Supplementary Table S3). In addition, similar enrichments were observed with other extensions for all seven traits in three breeds due to the extensive LD in bovine genome15 (see Supplementary Fig. S5). These results suggested that the phenotypic variations in the traits may be associated with the miRNA genes.
Figure 2.

The enrichments (i.e., −log10P) of association signals in miRNA genes with ±5 kb extension for seven traits across three breeds. (A) is the enrichments for seven traits among three breeds; the red dotted line corresponds to P = 0.05. (B–D) are correlations of enrichments for all seven traits in Holstein (HOL) vs. Jersey (JER), Nordic red cattle (RDC) vs. JER, and HOL vs. RDC, respectively. MY, FY, PY and BC are milk yield, fat yield, protein yield, and body conformation, respectively. −log10P is determined by marker-set test.
MiRNA-target networks provide new insights into the genetic architecture of complex traits
The targets of trait-associated miRNAs are likely to be enriched for GWAS signals. To test this, we employed the miRmap software to in silico predict targets of all miRNAs38 (http://mirmap.ezlab.org/). Computational target prediction remains a challenge as they predicted targets only rely on the physical properties of miRNA regulation (e.g., evolutionary conservation and secondary structures of 3′ UTRs), hence only the top 25% of predicted targets were analyzed in the downstream analysis, including 11,455 out of 24,616 bovine genes (UMD 3.1, http://www.ensembl.org/Bos_taurus/Info/Index). The average number of targets of miRNAs was 434 (range: 7 to 4,325) (Fig. 3A). To test for enrichment of GWAS signals, all targets of a single miRNA were considered as a miRNA target-network (i.e., as one marker set) and only SNPs located in the open reading frame (ORF) of targets were included. Furthermore, for comparison, 1,000 random target networks were generated for each miRNA, and 769 GO terms together with 916 Reactome terms were also analyzed (see Methods).
Figure 3.

Number of targets of miRNAs and the enrichments with miRNA-target networks for seven traits across three breeds. (A) Shows the number of target genes in each miRNA, and each blue point is a single miRNA. (B–D) are the difference in enrichments (i.e., −log10P) between the miRNA-target networks (miRNA-target) and random-target networks (Random-set) for seven traits in Holstein (HOL), Nordic red cattle (RDC), and Jersey (JER), respectively. Random-set is the random target-networks of miRNAs (replicates = 1000). −log10P is calculated using marker-set test. MY, FY, PY and BC are milk yield, fat yield, protein yield, and body conformation, respectively. The red dotted line corresponds to P = 0.05, and the blue line corresponds to P = 0.5.
Compared to the random-target networks, the enrichments with miRNA-target networks were significantly (P < 0.05, Wilcoxon-test) higher for all seven traits across three breeds, except for health and fertility in HOL (Fig. 3B–D). This was in agreement with the finding that fertility and health in HOL had the lowest enrichments with miRNA genes themselves as shown in Fig. 3A. Furthermore, the enrichments of fertility and health in both RDC and JER were significantly higher than in HOL, and the enrichment of BC in HOL was significantly higher than in RDC and JER, which were also in line with that the observations on the miRNA genes.
Correlations between traits
The enrichments with miRNA-target networks were highly significantly (P < 0.001) correlated between traits in HOL, and the highest correlations were observed among milk production traits (see Supplementary Fig. S6). Similar patterns were also observed in both RDC and JER, except for health and fertility in JER which might be due to their low statistical power (i.e., the combined influence of the small sample size and the small effect size) in GWAS (see Supplementary Figs S7 and S8). Assuming each correlation coefficient approximately follows a normal distribution, which was determined by 1,000 random-target networks (see Methods), a majority of between-traits correlations with miRNA-target networks were significantly (P < 0.05) greater than the random correlation coefficients, demonstrating that the genetic architecture of multiple complex traits shared certain similarities in the underlying miRNA-target networks. Only the significant between-traits correlations in HOL, RDC and JER are shown in Fig. 4A–C, respectively. The correlations among milk production traits were significant across three breeds. The correlations between BC with all three milk production traits were significant in HOL, and the correlations between health with other six traits (expect for BC) were significant in RDC. Of special note, mastitis was significantly correlated with the remaining six traits across all three breeds, except for mastitis vs. BC in RDC, and mastitis vs. health in JER. Additionally, the between-trait correlations with miRNA-target networks were typically higher than those with GO and Reactome terms, and the patterns were consistent across the three breeds (Fig. 4D–F). All the findings here provided genomic evidence that miRNAs may influence many complex phenotypes through regulation of their targets, and further suggests pleiotropic effects of miRNAs.
Figure 4.

The correlations for enrichments with miRNA-targets networks between traits in Holstein (HOL), Nordic red cattle (RDC) and Jersey (JER). (A–C) are the singificant (P < 0.05) between-trait correlations in HOL, RDC, and JER, respectively. The significant level was computed by assumping the correlation follows a normal distribution that was determined by random-target networks (replicates = 1,000). (D–F) are the difference in between-trait correlations among miRNA-target networks, Gene Ontology (GO) and Reactome terms for HOL, RDC, and JER, respectively.
Correlations between breeds
The correlations between breeds of enrichments with miRNA-target networks for the same trait were calculated. As shown in Table 1, a majority of between-breed correlations were significantly greater than the random correlation coefficients (P < 0.05). Furthermore, the between-breed correlations with miRNA-target networks tended to be higher than those with GO and Reactome terms (see Supplementary Fig. S9). These results indicated that the miRNA-target networks associated with complex traits shared certain similarities across breeds.
Table 1.
Between-breeds correlations based on enrichments with miRNA-target networks.
| FY | MY | PY | BC | Mastitis | Health | Fertility | |
|---|---|---|---|---|---|---|---|
| HOL vs. RDC | 0.45* | 0.41*** | 0.38*** | 0.11*** | 0.23** | 0.11 | 0.08 |
| HOL vs. JER | 0.11 | 0.22* | 0.02 | 0.39*** | 0.17* | 0.04 | 0.08˙ |
| RDC vs. JER | 0.12** | 0.23*** | 0.15*** | 0.07˙ | 0.02 | 0.24*** | 0.11˙ |
Note: FY, MY, PY and BC are fat yield, milk yield, protein yield and body conformation, respectively. The significant levels of correlations were determined acorrding to the 1000 correlations calculated using random-target networks, assuming the correlation approximately follows a normal distribution.
***is P < 0.001
**is P < 0.01
*is P < 0.05, and ˙is P < 0.1.
Significant miRNA target-networks
When a miRNA target-network had P-values < 0.05 (based on marker-set test) across all three breeds for a trait (P < 0.053 = 1.25e-04), this miRNA target-network was considered significant, suggesting that this miRNA and its target genes may participate in biological processes pertinent to this particular trait. A total of 55 significant miRNA-target networks were detected for seven traits and twelve miRNAs were involved in several traits (Fig. 5). The enriched (FDR < 0.1) KEGG pathways for the targets of these significant miRNAs are shown in Fig. 6. Thus, six miRNAs were shared by FY and MY, including bta-miR-188, bta-miR-2389, bta-miR-331, bta-miR-6526, bta-miR-670 and bta-miR-873, the targets of which were mainly engaged in MAPK signaling pathway, Ras signaling pathway, inositol phosphate and glycerophospholipid metabolism. Three miRNAs, bta-miR-10a/b and bta-miR-6525, were shared by MY and mastitis, and bta-miR-2366 was shared by FY and mastitis, the targets of which were engaged in MAPK signaling pathway, AMPK signaling pathway, Ras signaling pathway, salmonella infection, and fructose and mannose metabolism. Also, bta-miR-2306, was shared by MY, PY and fertility. Our results were in line with previous studies on miRNA transcriptomes in the mammary gland during lactating and infection39–42, suggesting it might be a promising way to detect candidate miRNAs for a trait through investigation of the enrichments of their targets for GWAS signals.
Figure 5.
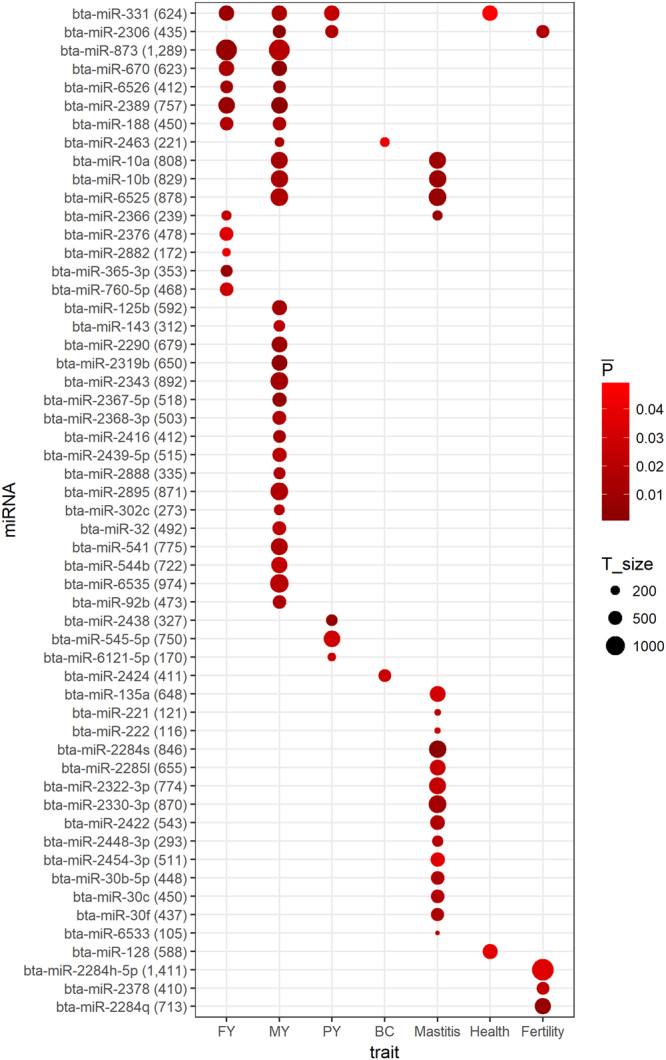
Significant miRNA-target networks for seven traits in three breeds. FY, MY, PY and BC are fat yield, milk yield, protein yield and body conformation, respectively. A miRNA target-network with P values < 0.05 across three breeds for the same trait was considered as significant for this particular trait. is the mean of P values (based on marker-set test) of a significant miRNA-target network in three breeds. T_size is the number of targets in a miRNA target-network.
Figure 6.

Enriched (p.adjust < 0.1) KEGG pathways for all significant miRNA-target networks. p.adjust is the corrected P-value using FDR methods. Values in the brackets are the number of targets that can be annotated in KEGG databases in the corresponding miRNAs, and GeneRatio is the proportion of enriched targets in a pathway over all the targets that were annotated in KEGG (values in the brackets).
Integrative analyses of infection-induced transcriptomes provide novel insights into the genetic and biological basis underlying mastitis
We detected eighteen miRNA-target networks that were associated with mastitis (Fig. 5). To provide new insight and to further improve prioritization of GWAS signals for mastitis, we integrated these associations with liver and mammary gland transcriptome data from two intra-mammary infection experiments performed in HOL cows. Differentially expressed genes (DEGs, FDR < 0.05) in the mastitis-related transcriptomes were identified at different time-points post infeciton relative to control groups (Fig. 7, see Methods). Enrichment analysis showed that the mastitis-associated miRNA-target networks were significantly enriched (P < 0.05, hypergeometric test) with DEGs detected in most of the conditions (Fig. 7A), supporting that these miRNAs could be involved in the response to mastitis. The top enriched miRNA in each condition is indicated in Fig. 7A. Targets of mastitis miRNAs that were observed differentially expressed in at least two infection conditions were considered as differentially expressed targets (DETs). The heat-map of log2 (fold change, FC) of DETs across all experimental conditions are shown in Fig. 7B. The numbers of DETs were 2,381 unique genes and the average number of DETs per miRNAs was 326 (range: 61 to 530). The results showed consistently across all three breeds that the mastitis GWAS signals were significantly more enriched in DETs than in non-DETs (P < 0.05, t-test) (Fig. 7C–E).
Figure 7.

Integrative analyses of infection-relevant transcriptome data with significant mastitis miRNA target-networks. (A) The enrichments for targets in mastitis miRNA target-networks with differently expressed genes (DEGs) that are detected in different infection conditions. Each point is a miRNA target-network. The significance (−log10P) is determined using hypergeometric test. The red line corresponds to P = 0.05. The red square, star and circle represent bta-miR-6525, bta-miR-10b and bta-miR-10a, respectively, which are shared between mastitis and milk yield. LL3, LL6, LL9, LL12 and LL48 represent the comparisons of 3, 6, 9, 12 and 48 h post intra-mammary infection with LPS to the control time-point (−22h), respectively in liver. LE12 and LE24 represent the comparisons of 12 and 24 h post intra-mammary infection with E. coli to the control time-point (−144h), respectively in liver. ME24 represents the comparison of the infected udder quarters to the control ones at 24 h post intra-mammary infection with E. coli. (B) The heatmap of log2(fold-change, FC) of differentially expressed target (DETs) of mastitis-associated miRNAs. (C–E) Represent the difference in the enrichments of association signals of mastitis between DETs and non-DE targets (N_DETs) of mastitis-associated miRNAs for HOL, RDC and JER, respectively. (F–H) Represent the difference in the enrichments of association signals of mastitis between up- (UP) and down- (DOWN) regulated DETs of mastitis-associated miRNAs for HOL, RDC and JER, respectively.
Next, the DETs of these miRNAs were separated into up- and down- regulated transcripts based on their log2FC > 0 (up) or <0 (down) across all infection conditions. In total, 1,002 unique up-regulated DETs were detected with an average of 142 (range: 31 to 231) for each miRNA, and 1,111 down-regulated DETs were detected with an average of 146 (range: 24 to 255) for each miRNA. The mastitis GWAS signals were significantly more enriched in up-regulated DETs of miRNAs than in down-regulated DETs across all three breeds (Fig. 7F–H). In agreement, the functional enrichment analyses based on KEGG pathways demonstrated that the up-regulated DETs of these mastitis miRNAs were mainly engaged in the innate immune system, infection defense and inflammatory response (Fig. 8).
Figure 8.

Functional enrichment analysis of up-regulated differentially expressed targets (DETs) of significant mastitis miRNAs. p.adjust is the corrected P-value using FDR methods. Values in the brackets are the number of up-regulated DETs that can be annotated in KEGG databases for the corresponding miRNAs, and GeneRatio is the proportion of enriched up-regulated DETs in a pathway over all the up-regulated DETs that were annotated in KEGG (values in the brackets).
Bta-miR-10a/b and bta-miR-6525 are associated with MY and mastitis
Three miRNA target networks, bta-miR-10a, bta-miR-10b and bta-miR-6525, were enriched for GWAS signals for both MY and mastitis. Integration with mastitis-related transcriptome data showed that transcripts that are up-regulated for all three miRNAs (Fig. 9A) were significantly more enriched with association signals for mastitis than for MY across the three breeds (Fig. 10C–E), while the down-regulated DETs (Fig. 9B) tended to be more associated with MY than with mastitis across all breeds (Fig. 9F–H). Consistently, functional analyses of up- and down-regulated DETs demonstrated that the up-regulated DETs of the three miRNAs were mainly engaged in inflammation and immune responses such as TNF signaling pathway and Epstein-Barr virus infection, while their down-regulated DETs were mainly engaged in amino acid metabolism, such as steroid biosynthesis and valine, leucine and isoleucine degradation (Fig. 10). These results were in agreement with previous reports showing involvement of bta-miR-10a and bta-miR-10b not only in innate immune responses but also in lipid and cholesterol metabolism43–49.
Figure 9.

Up- and down-regulated differentially expressed targets (DETs) of bta-miR-10a/b and bta-miR-6525. (A,B) are overlaps of up-/down- regulated DETs of bta-miR-10a/b and bta-miR-6525 respectively. (C–E) Are the difference in their up-regulated DETs in enrichments (−log10P) of association signals between mastitis and milk yield (MY) for HOL, RDC and JER, respectively, while (F–H) are the difference in the down-regulated DETs in enrichments of association signals between mastitis and MY for HOL, RDC, and JER, respectively. The red square, star and circle represent bta-miR-6525, bta-miR-10b and bta-miR-10a, respectively.
Figure 10.

Functional enrichment analyses of up-/down-regulated differentially expressed targets (DETs) of bta-miR-10a/b and bta-miR-6525. p.adjust is the corrected P-value using FDR methods. Values in the brackets are the number of DETs that can be annotated in KEGG databases, and GeneRatio is the proportion of enriched DETs in a pathway over all DETs that were annotated in KEGG (values in the brackets). UP_10a, UP_10b and UP_6525 correspond to up-regulated DETs of bta-miR-10a, bta-miR-10b, bta-miR-6525, respectively, while DOWN_10a, DOWN_10b and DOWN_6525 correspond to down-regulated DETs of bta-miR-10a, bta-miR-10b, bta-miR-6525, respectively.
Discussion
Knowledge-guided genotype-phenotype mapping and genomic prediction
Identifying and utilizing causal links between genotypes and quantitative phenotypes and diseases is a major goal in animal breeding as well as in clinical personalized medicine. Multiple statistical models have been used for genotype-phenotype mapping, and the performance of the models depends critically on the genetic architecture underlying complex traits. Single-marker regression models are suitable for detection of genomic variants with large effects but not for identifying variants with small effects50, and therefore typically accounts for only a fraction of the total genetic variance. Knowledge-guided gene-mapping approaches such as marker-set test based on single-marker statistics or genomic feature prediction models such as GFBLUP9 and BayesRC51 can contribute to the discovery of the so-called “missing heritability” in the standard GWAS. The sum-based marker-set test method applied in this study has been demonstrated to have higher power or at least equal to most commonly used marker-set test methods, particularly for the highly polygenic traits16,18. Provided the genomic features are enriched for causal variants, incorporating this type of biological information into the genomic prediction model can also improve the prediction accuracy9,17,18. Thus, previous studies demonstrated a positive correlation between association signal enrichment and the predictive ability of GFBLUP model that allowed us to differentially weight SNPs associated with that particular genomic feature11,18. In fact, any biological information that can be linked to specific genomic regions containing, or in LD with, SNPs, can be used to construct the genomic features. Important sources for defining trait-relevant features include the genome, epigenome, transcriptome, proteome and metabolome levels, or functional networks such as KEGG pathways, gene ontology terms and protein-protein interaction complexes. Information from several resources may be utilized simultaneously to capture the dynamic aspects of genome function. Clearly with the increasing availability of large-scale GWAS results and more comprehensive genome annotation, the marker-set test and the genomic feature based prediction models will be increasingly useful in the near future for identifying associations between a trait and a genomic feature and for improving genomic prediction of complex phenotypes.
MicroRNAs and their target networks contribute to genetic variations in complex traits
MicroRNAs are functional noncoding RNAs that modulate translation and decay of mRNA transcripts, thereby playing major roles as regulators of gene expression in a wide range of biological processes. Here we interrogated data from genome-wide association studies for evidence of overrepresentation of miRNA genes and their target genes. Our analysis revealed significant enrichment of GWAS signals in proximity to miRNA genes for complex traits in all three dairy cattle breeds. Overall, 55 significant miRNA-target networks were detected for the seven traits, out of which 12 were shared among multiple traits, indicating pleiotropic effects of miRNAs (Fig. 5). Our observations agree well with a number of other studies that addressed the roles of miRNAs in mammary gland development and lactation. Thus, a recent analysis of miRNA expression in milk during the entire lactation cycle detected a total 475 known and 348 novel miRNAs. Comparison between different cycle stages revealed 344, 366, and 209 differentially up- and down-regulated expression of miRNAs between lactogenesis and galactopoiesis, involution and galactopoiesis, and involution and lactogenesis, respectively52. Likewise, expression profiling of lactating versus non-lactating stages of mammary glands identified a total of 900 known and candidate microRNAs of which more than 60% were shared between the two periods53. The high diversity and abundance, as well as the differentially regulated expression patterns of miRNAs in mammary gland tissue clearly underscores that synthesis and secretion of milk as well as switches between lactation stage involves a high level of posttranscriptional regulation of gene expression by miRNAs.
Eighteen miRNA-target networks were associated with mastitis many of which have previously been observed in connection with mammary gland infections. Thus, Streptococcus agalactiae-induced mastitis of bovine mammary glands resulted in altered expression of thirty five miRNAs including a bta-miR-30 family member, bta-miR-135a, and bta-miR-2284 family members41. Challenge of bovine MAC-T mammary epithelial cells with pathogenic Staphylococcus aureus or Escherichia coli led to differential expression of seventeen miRNA genes among which were bta-miR-30b-5p and bta-miR-30c39. A total of 77 miRNAs that included bta-miR-2422, bta-miR-135a, a bta-miR-2285 member, and several bta-miR-2284 family members showed expression differences in Staphylococcus aureus infected mammary glands compared to control groups42. Furthermore, miRNA expression profiles of milk exosomes from cows prior and after infection of the mammary gland with Staphylococcus aureus showed significant changes in fourteen known genes including bta-miR-10a40. Also bta-miR-222 expression was significantly upregulated in mastitis-affected cows54. Co-expression network and pathway analysis revealed significant correlations between milk somatic cell count (SCC), an indicator trait for mastitis, and bta-miR-10a, bta-miR-2448, bta-miR-2284 family members and bta-miR-2285 family members55. Also in other species several of these eighteen miRNAs have been linked to mastitis and immune protection. In porcine mammary epithelial cells, coliform mastitis induced differential expression of miR-10a/10b and miR-30b/c/f56. Moreover, miR-221 was differentially expressed in colostrum and peak lactation periods in the caprine mammary gland57.
Finally we defined genomic features for mastitis by integrating both transcriptome and miRNA-target networks with GWAS data. The results showed consistently across all three breeds that the mastitis associated GWAS signals were significantly more enriched in DETs than in non-DETs. Additionally, partitioning of DETs showed that up-regulated targets had higher enrichment compared to down-regulated targets. Furthermore, three miRNA-target networks showed enrichment for GWAS signals for both MY and mastitis. Intriguingly, when integrated with the mastitis-related transcriptome data, the analysis revealed that up-regulated transcripts belonging the network targets of all three miRNAs were significantly more enriched with association signals for mastitis than for MY across the three breeds; and vice versa, the down-regulated transcripts were more associated with MY than with mastitis. High milk yield is often associated with risk of mastitis58. Our work showed a genetic link between the two traits mediated by miRNA-target networks, which may in part help understanding the unfavorable correlation between mastitis and MY.
Conclusion
In conclusion, the marker-set test of joint effects of SNPs linked to miRNAs enabled us to discover contributions from miRNAs as well as of their target networks to multiple complex traits in three cattle breeds. Overall, this work underscores that the use of biological priors such as genomic features defined by miRNAs and their targets enhances our insight into the genetic architecture underlying phenotypic diversity. With the rapid progress in the field of annotation of functional elements in livestock genomes, the genomic feature based analysis like marker-set test or GFBLUP will be increasingly useful for discovering small effect loci or for increasing the accuracy and reliability of prediction of consequences of genomic selection in animal and plant breeding.
Materials and Methods
Infection-induced transcriptome data
All experimental procedures involving animals have been approved by the Danish Animal Experiments Inspectorate and complied with Danish Ministry of Justice Laws concerning animal experimentation and care of experimental animals. All the experiments were performed in strict accordance with guidelines and regulations established by these committees. Members from these committees carried out inspections during the entire animal infection experiments.
All the transcriptome data used in this study came from two intra-mammary infection studies involving three and six healthy HOL animals, respectively at the very early stage of their first lactation. The details of the involved animals and the sample collection procedures were previously described by Jiang et al.59 and Jorgensen et al.60. Briefly, in the first study liver biopsies were sampled at −22, 3, 6, 9, 12, and 48 h relative to LPS intra-mammary treatment in all three studied animals (i.e., each group with three biological replicates). In the second study liver biopsies were collected from the six animals at −144, 12 and 24 h post intra-mammary infection with E. coli, and the mammary gland samples of the infected and control udder quarters were collected from the same animals at 24 h post infection (i.e., each group with six biological replicates). Finally, a total of 48 samples were applied with RNA sequencing using a 100 bp pair-end approach with llumina HiSeq 2000 by AROS Applied Biotechnology (Aarhus, Denmark). Time was considered as the only effect in the statistical model for analyzing liver samples data, while infection status was included in the model for mammary gland samples. The details of RNA-seq data analyses were previously described by Fang et al.22. FDR method was employed to control multiple testing, and genes with FDR < 0.05 were considered as DEGs.
Phenotypes and imputed sequence genotypes
The phenotypes currently analysed were de-regressed proofs (DRPs) of seven complex traits of economic importance in cattle, including three milk production traits (i.e., MY, FY and PY), BC, mastitis, health and fertility. These DRPs were obtained from a routine genetic evaluation by Nordic Cattle Genetic Evaluation (http://www.nordicebv.info/) and were available for 5,056 HOL, 4,310 RDC and 1,231 JER bulls. All known systematic effects have been corrected.
The whole genome sequence genotypes of these animals were obtained from the imputation of 50 K and High Density (HD) genotypes. The imputation was conducted using a two-step strategy as recommended by Brøndum et al.61, and the details has been previously described by Wu et al.62. Briefly, in the first step a 50 K was imputed into a HD genotype for each individual using IMPUTE2 v2.3.163 based on 3,383 reference animals (including 1,222 HOL, 1,326 RDC and 835 JER) that had been genotyped by Illumina BovineHD chips (Illumina, Inc., San Diego, CA). In the next step the imputed HD genotypes were imputed to the whole genome sequence level using Minimac264 based on the reference population of 1,228 animals from both Run4 of the 1,000 Bull Genomes Project65 and in-house whole genome sequence data62. In total, 22,751,039 biallelic variants were obtained after the imputation. The imputed sequence genotypes were then filtered to keep markers with deviation from Hardy-Weinberg proportions (HWP) > 10−6 and minor allele frequency (MAF) > 0.01. After the data editing, 15,355,382, 15,243,827 and 13,403,916 SNPs remained for the following analyses in HOL, RDC and JER, respectively with an average imputation accuracy of larger than 0.90. The imputation accuracy of a SNP increased with the increase of its MAF, and the details of the imputation accuracy were described by Wu et al. and Iso-Touru et al.62,66.
Single-marker GWAS using imputed sequence data
The single-marker GWAS for the imputed sequence genotypes was conducted using a two-step variance component-based method that was implemented in EMMAX67. The EMMAX was developed to take account for genetic relatedness and population structure as well as to improve the computational speed. In the first step, the polygenic and residual variances were estimated by
| 1 |
where is a vector of the phenotype (i.e., DRP); is the overall mean; is a vector of random polygenic effects, where ~ N(0, ), and is the genomic relationship matrix built using HD genotypes through excluding the chromosome containing the candidate SNP to control double fitting68, and is the additive genetic variance; is the vector of random residuals, where ~ N(0,), and is the identity matrix, and is the residual variance. 1 is a vector of ones. Z is a design matrix relating phenotypes to polygenic effects. In the second step, the individual association signal was assessed by a linear regression model
| 2 |
where y, μ and 1 are the same as described above, is a vector of genotype dosages (range: from 0 to 2), is the allele substitution effect, and is a vector of random residual deviates with (co)variance structure +. The genome-wide significance thresholds corresponding to an error rate of 0.05 (Bonferroni correction) were set at 3.3 × 10−9, 3.3 × 10−9, 3.7 × 10−9 for HOL, RDC and JER, respectively. Manhattan plots were generated using qqman v.0.1.2 implemented in the R package69.
Regions showing association signal could be very large due to high LD structure owing to small effective population sizes in dairy cattle breeds. Therefore, we demarcated a region around the top SNP, which most likely harbours the causal mutation. A QTL region boundaries were defined through extending the position of the top SNP up- and down- stream to include SNPs with −log10(P-values) higher than −log10(P-value) minus three of the top SNP70. The proportion of genomic variance explained by the top SNP within a QTL was calculated as , where and were allele frequencies, b was the allele substitution effect, and was the additive genomic variance.
Marker sets defined from GO and Reactome databases
Genes grouped into a specific GO or Reactome term were considered to be a single marker set (i.e., genomic feature). The Bioconductor package “org.Bt.eg.db” v. 3.3.0 (https://bioconductor.org/packages/release/data/annotation/html/org.Bt.eg.db.html) and “reactome.db” v 3.3.0 (https://bioconductor.org/packages/release/data/annotation/html/reactome.db.html) were used to link genes to the GO and Reactome terms, respectively. Here, we focused on the GO and Reactome terms consisting of at least 10 directly evidenced genes. In GO terms, only biological processes terms were considered. The imputed sequence variants were mapped to the bovine reference genome (UMD3.1). A SNP was assigned to a gene if its chromosome position was between the start and end positions of the gene (i.e., within the open reading frames, ORF).
Marker-set test
Since the genomic variance of the studied complex traits has been typically considered to be controlled by many loci of small to moderate effects, the following sum-based summary statistics for a genomic feature was chosen, and it has been demonstrated to have higher power or at least equal to most commonly used marker-set test methods, particularly for the highly polygenic traits16,18,21.
| 3 |
In which is the number of SNPs in a genomic feature, and is the square of t that was calculated as the SNP effect (b) divided by its standard error. It should be noticed that the LD patterns of SNPs among multiple genes in a genomic feature were typically low, especially for genes in different chromosomes. Although the LD patterns among SNPs and the sizes of marker sets were not directly taken into account in the summary statistics, they have been taken in account by the following cyclical permutation strategy as previously described21. Briefly, the test statistics (i.e., ) for all SNPs were ordered according to their chromosome positions (i.e., , , , ,). A random test statistic (i.e., ,) was chosen from this vector as the first, and the remaining test statistics were shifted to new positions, but retained their original orders (i.e., tk, tk+1 tm, t1 tk−1). Therefore, any association of SNPs with genomic features was uncoupled while maintaining the correlation structure among test statistics of SNPs. Afterwards, a new summary statistic of the genomic feature was computed according to its original chromosome position. The permutation procedure was repeated 1,000 times for each tested genomic feature, and an empirical P-value was computed according to one-tailed tests of the proportion of random summary statistics larger than that observed. This marker set-test method together with multiple quantitative genomic tools have been implemented in our QGG package (http://psoerensen.github.io/qgg/).
Significant testing for between-traits/breeds correlations using random-target networks
The between-trait correlations within a breed and the between-breed correlations for a same trait were computed based on the GWAS signal enrichments of miRNA target-networks. In order to determine the significant level of an observed correlation, we randomly generated targets with the same size as predicted for each miRNA, then a random correlation was calculated using the enrichments of random target-networks. We repeated this procedure for 1000 times, thereby 1000 random correlations were generated for each particular correlation. Assuming that a correlation approximately follows a normal distribution r ~ N(µ, σ2), where µ and σ2 can be approximately estimated as the mean and standard deviation (SD) of the 1000 randomly generated correlations. The significant level for an observed correlation based on miRNA target-networks was then determined by using one-sided test.
Functional enrichment analyses of gene lists
The functional enrichment analyses of a list of genes were conducted using R package clusterProfiler71, where a hypergeometric test, based on a KEGG pathway database, was applied, and the P-values for each pathway were adjusted using the FDR method. We used pathways with FDR < 0.1 to indicate the putative biological function of gene lists being studied.
Availability of data and materials
All genomic annotation data defining gene regions is available for download (ftp://ftp.ensembl.org/pub/release-84/gtf/bos_taurus). The miRNA annotation data is available for download (http://www.mirbase.org/ftp.shtml), and their best quantile predicted target genes is available in (http://mirmap.ezlab.org/). The GO annotation database can be publicly accessed (https://bioconductor.org/packages/release/data/annotation/html/org.Bt.eg.db.html). The Reactome annotation database can be publicly accessed (http://bioconductor.org/packages/release/data/annotation/html/reactome.db.html). The whole genome sequencing data from the 1000 Bull Genomes Project are publicly available from NBCI under SRA no. SRP039339 (http://www.ncbi.nlm.nih.gov/bioproject/PRJNA238491) and variations in dbSNP (http://www.ncbi.nlm.nih.gov/projects/SNP/). The genotype and phenotype data, and genome sequence data from Aarhus University are available only upon agreement with the commercial breeding organization (http://www.vikinggenetics.com/) and should be requested directly from the authors or the breeding organization.
Electronic supplementary material
Acknowledgements
This study was funded by the Danish Strategic Research Council (GenSAP: Centre for Genomic Selection in Animals and Plants, contract 12-132452) and the Aarhus University Research Foundation, contract AUFF-E-2015-FLS-9-16.
Author Contributions
L.F. performed the main statistical analyses, wrote the manuscript, and conceived and designed the study. P.S.and B.T. conceived the study and drafted the manuscript. GSahana performed the G.W.A.S. analyses. F.P., G.S.u., S.Z., Y.Y., B.L., L.M., G.L. and M.L. contributed to the study design. All authors read and approved the final manuscript.
Competing Interests
The authors declare no competing interests.
Footnotes
Electronic supplementary material
Supplementary information accompanies this paper at 10.1038/s41598-018-27729-y.
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Falconer, D. S & Mackay, T. F. Introduction To Quantitative Genetics (Longman, 1996).
- 2.Zeng ZB, Kao CH, Basten CJ. Estimating the genetic architecture of quantitative traits. Genet. Res. 1999;74:279–289. doi: 10.1017/S0016672399004255. [DOI] [PubMed] [Google Scholar]
- 3.Mackay TF. The genetic architecture of quantitative traits. Annu. Rev. Genet. 2001;35:303–339. doi: 10.1146/annurev.genet.35.102401.090633. [DOI] [PubMed] [Google Scholar]
- 4.Mackay TF, Huang W. Charting the genotype–phenotype map: lessons from the Drosophila melanogaster Genetic Reference Panel. Wiley Interdiscip. Rev. Dev. Biol. 2017;7:1. doi: 10.1002/wdev.289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Visscher PM, et al. 10 years of GWAS discovery: biology, function, and translation. Am. J. Hum. Genet. 2017;101:5–22. doi: 10.1016/j.ajhg.2017.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Daetwyler HD, Pong-Wong R, Villanueva B, Woolliams JA. The impact of genetic architecture on genome-wide evaluation methods. Genetics. 2010;185:1021–1031. doi: 10.1534/genetics.110.116855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Swami, M. Using genetic architecture to improve predictions. Nat. Rev. Genet. 11 (2010). [DOI] [PubMed]
- 8.Wientjes YC, Calus MP, Goddard ME, Hayes BJ. Impact of QTL properties on the accuracy of multi-breed genomic prediction. Genet. Sel. Evol. 2015;47:42. doi: 10.1186/s12711-015-0124-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Edwards SM, Sørensen IF, Sarup P, Mackay TF, Sørensen P. Genomic prediction for quantitative traits is improved by mapping variants to gene ontology categories in Drosophila melanogaster. Genetics. 2016;203:1871–1883. doi: 10.1534/genetics.116.187161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fang L, et al. Exploring the genetic architecture and improving genomic prediction accuracy for mastitis and milk production traits in dairy cattle by mapping variants to hepatic transcriptomic regions responsive to intra-mammary infection. Genet. Sel. Evol. 2017;49:44. doi: 10.1186/s12711-017-0319-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fang L, et al. Use of biological priors enhances understanding of genetic architecture and genomic prediction of complex traits within and between dairy cattle breeds. BMC Genomics. 2017;18:604. doi: 10.1186/s12864-017-4004-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Korte A, Farlow A. The advantages and limitations of trait analysis with GWAS: a review. Plant Methods. 2013;9:29. doi: 10.1186/1746-4811-9-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hou L, Zhao H. A review of post-GWAS prioritization approaches. Front. Genet. 2013;4:280. doi: 10.3389/fgene.2013.00280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schaub MA, Boyle AP, Kundaje A, Batzoglou S, Snyder M. Linking disease associations with regulatory information in the human genome. Genome Res. 2012;22:1748–1759. doi: 10.1101/gr.136127.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Farnir F, et al. Extensive genome-wide linkage disequilibrium in cattle. Genome Res. 2000;10:220–227. doi: 10.1101/gr.10.2.220. [DOI] [PubMed] [Google Scholar]
- 16.Sarup P, Jensen J, Ostersen T, Henryon M, Sørensen P. Increased prediction accuracy using a genomic feature model including prior information on quantitative trait locus regions in purebred Danish Duroc pigs. BMC Genet. 2016;17:11. doi: 10.1186/s12863-015-0322-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rohde PD, Gaertner B, Wards K, Sørensen P, Mackay TF. Genomic Analysis of Genotype by Social Environment Interaction for Drosophila Aggressive. Behavior. Genetics. 2017;206:1969–1984. doi: 10.1534/genetics.117.200642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sørensen IF, Edwards SM, Rohde PD, Sørensen P. Multiple Trait Covariance Association Test Identifies Gene Ontology Categories Associated with Chill Coma Recovery Time in Drosophila melanogaster. Sci. Rep. 2017;7:2413. doi: 10.1038/s41598-017-02281-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fridley BL, Biernacka JM. Gene set analysis of SNP data: benefits, challenges, and future directions. Eur J Hum Genet. 2011;19:837–43. doi: 10.1038/ejhg.2011.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jia P, Wang L, Meltzer HY, Zhao Z. Pathway-based analysis of GWAS datasets: effective but caution required. Int. J. Neuropsychopharmacol. 2011;14:567–572. doi: 10.1017/S1461145710001446. [DOI] [PubMed] [Google Scholar]
- 21.Rohde PD, Demontis D, Cuyabano BCD, Børglum AD, Sørensen P. Covariance Association Test (CVAT) Identifies Genetic Markers Associated with Schizophrenia in Functionally Associated Biological Processes. Genetics. 2016;203:1901–1913. doi: 10.1534/genetics.116.189498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fang L, et al. Integrating Sequence-based GWAS and RNA-Seq Provides Novel Insights into the Genetic Basis of Mastitis and Milk Production in Dairy Cattle. Sci. Rep. 2017;7:45560. doi: 10.1038/srep45560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Holmans P, et al. Gene Ontology Analysis of GWA Study Data Sets Provides Insights into the Biology of Bipolar Disorder. Am. J. Hum. Genet. 2009;85:13–24. doi: 10.1016/j.ajhg.2009.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Low TY, et al. Quantitative and qualitative proteome characteristics extracted from in-depth integrated genomics and proteomics analysis. Cell Rep. 2013;5:1469–1478. doi: 10.1016/j.celrep.2013.10.041. [DOI] [PubMed] [Google Scholar]
- 25.Finucane HK, et al. Partitioning heritability by functional annotation using genome-wide association summary statistics. Nat. Genet. 2015;47:1228. doi: 10.1038/ng.3404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Djebali S, et al. Landscape of transcription in human cells. Nature. 2012;489:101–108. doi: 10.1038/nature11233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ender C, Meister G. Argonaute proteins at a glance. J. Cell Sci. 2010;123:1819–1823. doi: 10.1242/jcs.055210. [DOI] [PubMed] [Google Scholar]
- 28.Hangauer MJ, McManus MT, Carpenter S. Discovering the complexity of the metazoan transcriptome. Genome Biol. 2014;15:112. doi: 10.1186/gb4172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. cell. 2004;116:281–297. doi: 10.1016/S0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- 30.Valinezhad-Orang, A., Safaralizadeh, R. & Kazemzadeh-Bavili, M. Mechanisms of miRNA-mediated gene regulation from common downregulation to mRNA-specific upregulation. Int. J. Genomics2014 (2014). [DOI] [PMC free article] [PubMed]
- 31.Urbich C, Kuehbacher A, Dimmeler S. Role of microRNAs in vascular diseases, inflammation, and angiogenesis. Cardiovasc. Res. 2008;79:581–588. doi: 10.1093/cvr/cvn156. [DOI] [PubMed] [Google Scholar]
- 32.Xiao F, et al. miRecords: an integrated resource for microRNA–target interactions. Nucleic Acids Res. 2008;37:D105–D110. doi: 10.1093/nar/gkn851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lewis BP, Burge CB, Bartel DP. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. cell. 2005;120:15–20. doi: 10.1016/j.cell.2004.12.035. [DOI] [PubMed] [Google Scholar]
- 34.Akhtar MM, Micolucci L, Islam MS, Olivieri F, Procopio AD. Bioinformatic tools for microRNA dissection. Nucleic Acids Res. 2015;44:24–44. doi: 10.1093/nar/gkv1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Moszyńska A, Gebert M, Collawn JF, Bartoszewski R. SNPs in microRNA target sites and their potential role in human disease. Open Biol. 2017;7:170019. doi: 10.1098/rsob.170019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kozomara A, Griffiths-Jones S. miRBase: annotating high confidence microRNAs using deep sequencing data. Nucleic Acids Res. 2013;42:D68–D73. doi: 10.1093/nar/gkt1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Grisart B, et al. Positional candidate cloning of a QTL in dairy cattle: identification of a missense mutation in the bovine DGAT1 gene with major effect on milk yield and composition. Genome Res. 2002;12:222–231. doi: 10.1101/gr.224202. [DOI] [PubMed] [Google Scholar]
- 38.Vejnar CE, Zdobnov EM. MiRmap: comprehensive prediction of microRNA target repression strength. Nucleic Acids Res. 2012;40:11673–11683. doi: 10.1093/nar/gks901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jin W, Ibeagha-Awemu EM, Liang G, Beaudoin F, Zhao X. Transcriptome microRNA profiling of bovine mammary epithelial cells challenged with Escherichia coli or Staphylococcus aureusbacteria reveals pathogen directed microRNA expression profiles. BMC Genomics. 2014;15:181. doi: 10.1186/1471-2164-15-181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sun J, et al. MicroRNA expression profiles of bovine milk exosomes in response to Staphylococcus aureus infection. BMC Genomics. 2015;16:806. doi: 10.1186/s12864-015-2044-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pu J, et al. Expression profiles of miRNAs from bovine mammary glands in response to Streptococcus agalactiae-induced mastitis. J. Dairy Res. 2017;84:300–308. doi: 10.1017/S0022029917000437. [DOI] [PubMed] [Google Scholar]
- 42.Li R, et al. Transcriptome microRNA profiling of bovine mammary glands infected with Staphylococcus aureus. Int. J. Mol. Sci. 2015;16:4997–5013. doi: 10.3390/ijms16034997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Al-Husseini W, et al. Characterization and profiling of liver microRNAs by RNA-sequencing in cattle divergently selected for residual feed intake. Asian-Australas J. Anim. Sci. 2016;29:1371. doi: 10.5713/ajas.15.0605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Madrigal-Matute J, Rotllan N, Aranda JF, Fernandez-Hernando C. MicroRNAs and atherosclerosis. Curr. Atheroscler Rep. 2013;15:322. doi: 10.1007/s11883-013-0322-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Quiat D, Olson EN. MicroRNAs in cardiovascular disease: from pathogenesis to prevention and treatment. J. Clin. Invest. 2013;123:11–8. doi: 10.1172/JCI62876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wang D, et al. Gut Microbiota Metabolism of Anthocyanin Promotes Reverse Cholesterol Transport in Mice Via Repressing miRNA-10bNovelty and Significance. Circulation Res. 2012;111:967–981. doi: 10.1161/CIRCRESAHA.112.266502. [DOI] [PubMed] [Google Scholar]
- 47.Wang M, et al. MicroRNA expression patterns in the bovine mammary gland are affected by stage of lactation. J. Dairy Res. 2012;95:6529–6535. doi: 10.3168/jds.2012-5748. [DOI] [PubMed] [Google Scholar]
- 48.Smolle E, Haybaeck J. Non-coding RNAs and lipid metabolism. Int. J. Mol. Sci. 2014;15:13494–13513. doi: 10.3390/ijms150813494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Li X, et al. Differential expression of micrornas in porcine parvovirus infected porcine cell line. Virol. J. 2015;12:128. doi: 10.1186/s12985-015-0359-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hirschhorn JN, Daly MJ. Genome-wide association studies for common diseases and complex traits. Nat. Rev. Genet. 2005;6:95. doi: 10.1038/nrg1521. [DOI] [PubMed] [Google Scholar]
- 51.MacLeod I, et al. Exploiting biological priors and sequence variants enhances QTL discovery and genomic prediction of complex traits. BMC Genomics. 2016;17:144. doi: 10.1186/s12864-016-2443-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Do DN, Li R, Dudemaine PL, Ibeagha-Awemu EM. MicroRNA roles in signalling during lactation: an insight from differential expression, time course and pathway analyses of deep sequence data. Sci. Rep. 2017;7:44605. doi: 10.1038/srep44605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Li Z, Liu H, Jin X, Lo L, Liu J. Expression profiles of microRNAs from lactating and non-lactating bovine mammary glands and identification of miRNA related to lactation. BMC Genomics. 2012;13:731. doi: 10.1186/1471-2164-13-731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lai YC, et al. Inflammation-related microRNA expression level in the bovine milk is affected by mastitis. PloS One. 2017;12:5. doi: 10.1371/journal.pone.0177182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Do DN, Dudemaine PL, Li R, Ibeagha-Awemu EM. Co-expression network and pathway analyses reveal important modules of miRNAs regulating milk yield and component traits. Int. J. Mol. Sci. 2017;18:1560. doi: 10.3390/ijms18071560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Jaeger A, et al. MicroRNA expression profiling of porcine mammary epithelial cells after challenge with Escherichia coli in vitro. BMC Genomics. 2017;18:660. doi: 10.1186/s12864-017-4070-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hou J, et al. Detection and comparison of microRNAs in the caprine mammary gland tissues of colostrum and common milk stages. BMC Genetics. 2017;18:38. doi: 10.1186/s12863-017-0498-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hogeveen, H. Mastitis In Dairy Production: Current Knowledge And Future Solutions (Wageningen Academic Pub, 2005).
- 59.Jiang L, Sørensen P, Røntved C, Vels L, Ingvartsen KL. Gene expression profiling of liver from dairy cows treated intra-mammary with lipopolysaccharide. BMC Genomics. 2008;9:443. doi: 10.1186/1471-2164-9-443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Jørgensen HB, et al. Transcriptional profiling of the bovine hepatic response to experimentally induced E. coli mastitis. Physiol. Genomics. 2012;44:595–606. doi: 10.1152/physiolgenomics.00084.2011. [DOI] [PubMed] [Google Scholar]
- 61.Brøndum RF, Guldbrandtsen B, Sahana G, Lund MS, Su G. Strategies for imputation to whole genome sequence using a single or multi-breed reference population in cattle. BMC Genomics. 2014;15:728. doi: 10.1186/1471-2164-15-728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wu X, Guldbrandtsen B, Lund MS, Sahana G. Association analysis for feet and legs disorders with whole-genome sequence variants in 3 dairy cattle breeds. J. Dairy. Sci. 2016;99:7221–7231. doi: 10.3168/jds.2015-10705. [DOI] [PubMed] [Google Scholar]
- 63.Howie B, Marchini J, Stephens M. Genotype imputation with thousands of genomes. 2011;G3(1):457–470. doi: 10.1534/g3.111.001198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Fuchsberger C, Abecasis GR, Hinds D. A. minimac2: faster genotype imputation. Bioinformatics. 2014;31:782–784. doi: 10.1093/bioinformatics/btu704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Daetwyler HD, et al. Whole-genome sequencing of 234 bulls facilitates mapping of monogenic and complex traits in cattle. Nat. Genet. 2014;46:858–865. doi: 10.1038/ng.3034. [DOI] [PubMed] [Google Scholar]
- 66.Iso-Touru T, Sahana G, Guldbrandtsen B, Lund M, Vilkki J. Genome-wide association analysis of milk yield traits in Nordic Red Cattle using imputed whole genome sequence variants. BMC Genet. 2016;17:55. doi: 10.1186/s12863-016-0363-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kang HM, et al. Variance component model to account for sample structure in genome-wide association studies. Nat. Genet. 2010;42:348–354. doi: 10.1038/ng.548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Listgarten J, et al. Improved linear mixed models for genome-wide association studies. Nat. Methods. 2012;9:525–526. doi: 10.1038/nmeth.2037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Turner, S. D. qqman: an R package for visualizing GWAS results using QQ and manhattan plots. BioRxiv, 005165 (2014).
- 70.Mao X, Sahana G, De Koning DJ, Guldbrandtsen B. Genome-wide association studies of growth traits in three dairy cattle breeds using whole-genome sequence data. J. Anim. Sci. 2016;94:1426–1437. doi: 10.2527/jas.2015-9838. [DOI] [PubMed] [Google Scholar]
- 71.Yu GC, Wang LG, Han YY, He Q. Y. clusterProfiler: an R Package for Comparing Biological Themes Among Gene Clusters. OMICS. 2012;16:284–287. doi: 10.1089/omi.2011.0118. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All genomic annotation data defining gene regions is available for download (ftp://ftp.ensembl.org/pub/release-84/gtf/bos_taurus). The miRNA annotation data is available for download (http://www.mirbase.org/ftp.shtml), and their best quantile predicted target genes is available in (http://mirmap.ezlab.org/). The GO annotation database can be publicly accessed (https://bioconductor.org/packages/release/data/annotation/html/org.Bt.eg.db.html). The Reactome annotation database can be publicly accessed (http://bioconductor.org/packages/release/data/annotation/html/reactome.db.html). The whole genome sequencing data from the 1000 Bull Genomes Project are publicly available from NBCI under SRA no. SRP039339 (http://www.ncbi.nlm.nih.gov/bioproject/PRJNA238491) and variations in dbSNP (http://www.ncbi.nlm.nih.gov/projects/SNP/). The genotype and phenotype data, and genome sequence data from Aarhus University are available only upon agreement with the commercial breeding organization (http://www.vikinggenetics.com/) and should be requested directly from the authors or the breeding organization.