

Abstract
Munc13-1 is a presynaptic active-zone protein essential for neurotransmitter release and presynaptic plasticity in the brain. This multidomain scaffold protein contains a C1 domain that binds to the activator diacylglycerol/phorbol ester. Although the C1 domain bears close structural homology with the C1 domains of protein kinase C (PKC), the tryptophan residue at position 22 (588 in the full-length Munc13-1) occludes the activator binding pocket, which is not the case for PKC. To elucidate the role of this tryptophan, we generated W22A, W22K, W22D, W22Y, and W22F substitutions in the full-length Munc13-1, expressed the GFP-tagged constructs in Neuro-2a cells, and measured their membrane translocation in response to phorbol ester treatment by imaging of the live cells using confocal microscopy. The extent of membrane translocation followed the order, wild-type > W22K > W22F > W22Y > W22A > W22D. The phorbol ester binding affinity of the wild-type Munc13-1C1 domain and its mutants was phosphatidylserine (PS)-dependent following the order, wild-type > W22K > W22A ≫ W22D in both 20% and 100% PS. Phorbol ester affinity was higher for Munc13-1 than the C1 domain. While Munc13-1 translocated to the plasma membrane, the C1 domain translocated to internal membranes in response to phorbol ester. Molecular dynamics (80 ns) studies reveal that Trp-22 is relatively less flexible than the homologous Trp-22 of PKCδ and PKCθ;. Results are discussed in terms of the overall negative charge state of the Munc13-1C1 domain and its possible interaction with the PS-rich plasma membrane. This study shows that Trp-588 is an important structural element for ligand binding and membrane translocation in Munc13-1.
Graphical Abstract
Munc13-1 is a presynaptic active-zone protein that stimulates SNARE complex formation. It is essential for synaptic vesicle priming1,2 and neurotransmitter release.3,4 Munc13-1 is expressed throughout the brain and modulates short-term presynaptic plasticity5,6 and long-term potentiation through its interactions with an active zone protein RIMα.7 The Munc13 family of proteins consists of four members, Munc13-1, Munc13-2, Munc13-3, and Munc13-4.8 Double-knockout of Munc13-1 and Munc13-2 in mice results in complete abolishment of neurotransmitter release.9–12 Munc13-1 has also been implicated in neurodegenerative disorders.13,14 It has been shown that Munc13-1 regulates Aβ-induced neurotoxicity in an AD model.15,16 A single-nucleotide polymorphism in Munc13-1 gene is associated with amyotrophic lateral sclerosis and frontotemporal dementia.17–20 Additionally, Munc13-1 is involved in insulin release21 and in alcohol addiction.22
Munc13-1 is a large peripheral membrane protein having multiple domains—three C2 domains, one C1 domain, and a MUN domain.23 The N-terminal C2A domain is followed by a high-affinity diacylglycerol (DAG)/phorbol ester binding C1 domain, a Ca2+ binding C2 domain (C2B), its characteristic MUN domain, and a C-terminal C2 domain (C2C).11,24 Residues 859–1531 between the C2B and C2C domains form an autonomously folded domain known as a MUN domain.25 The binding of DAG or phorbol ester to the Munc13-1 C1 domain stimulates synaptic vesicle priming as well as the translocation of Munc13-1 from the cytoplasm to the plasma membrane.26 There are strong sequence similarities among the C1 domains of Munc13, Dunc-13, Unc13, and the C1 domain of PKCs.27 The Unc13 C1 domain was shown to bind phorbol esters with high affinity, similar to that of PKCδ.28 Another study reported that the affinity of Munc13-1 was lower than that of PKC3 (While homologous, the C1 domains of Unc13 and Munc13 show a substantial difference in net charge, which may contribute to the difference in observed binding affinities.) The binding of DAG to the Munc13 C1 domain lowers the energy barrier for vesicle fusion and promotes neurotransmitter release.29 The H567K mutation in the C1 domain prevents the binding of phorbol esters, thereby inhibiting the activation of vesicle fusion.3,29
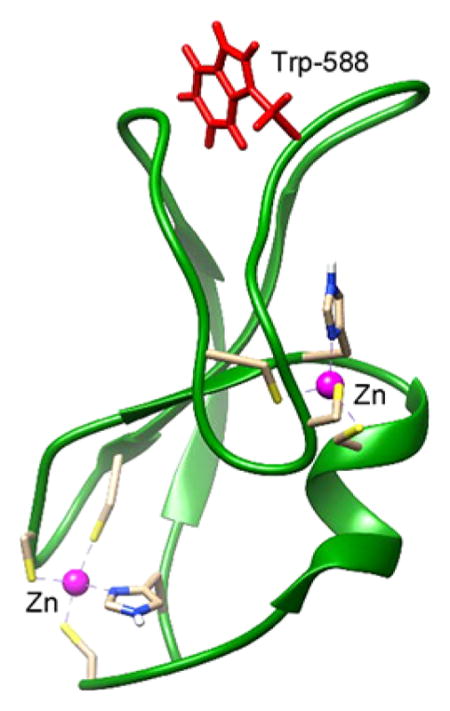
The C1 domain of Munc13-1 has close structural homology with the C1 domain of protein kinase C (PKC). A comparison of the PKCδ C1B and Munc13-1 C1 structures shows that both structures have two β-sheets, a short C-terminal α-helix, and two Zn2+ binding sites.30,31 DAG binds inside a groove formed by two loops. The NMR structure of Munc13-1 also revealed that the side chain of Trp-22 in the C1 domain (Trp-588 in the full-length Munc13-1) occludes the ligand binding site30 (Figure 1). The orientation of the tryptophan residue at the homologous position in the C1 domain of PKCδ31 and PKCθ32 is different than that of Munc13-1. Most of the phorbol ester-sensitive C1 domains contain Trp at this site with few exceptions (Tyr in C1B of PKC(α/β/γ); Val in MRCK α/β; Leu in MRCKγ; and Phe in PKD2C1A.27 Additionally, Leu is present at this position in RasGRP2, a RasGRP isoform with very low phorbol ester binding affinity.33 At this key homologous position, the switch between Trp and Tyr for the novel and conventional PKCs was found to influence their membrane affinity and cellular localization.34
Figure 1.

Orientation of Trp-588 (Trp-22 in the C1 domain) in the DAG/phorbol ester-binding C1 domain of Munc13-1.
To elucidate the role of Trp-22 in the ligand binding and subsequent function of the protein, we generated several mutants at this site and measured their efficiencies of membrane translocation in Neuro-2a cells in response to phorbol ester. We also compared the phorbol ester binding affinities and membrane translocation properties of the C1 domain and the full-length protein. Our results show that mutation of Trp to Asp at position 22 significantly reduces membrane translocation and phorbol ester binding, supporting the importance of Trp-588 in the ligand binding and membrane translocation of Munc13-1.
MATERIALS AND METHODS
Materials
[20-3H]Phorbol 12,13-dibutyrate ([3H]PDBu) (17.2 Ci/mmol) was obtained from PerkinElmer Life Sciences. PDBu and phorbol 12-myristate 13-acetate (PMA) were purchased from LC Laboratories (Woburn, MA). Phosphatidyl-L-serine (PS), phosphatidylcholine (PC), and sphingosine were from Avanti Polar Lipids (Alabaster, AL) and the isopropyl O-D-thiogalactopyranoside (IPTG) was from Sigma (St. Louis, MO). LNCaP human prostate cancer cells, Neuro-2a mouse neuroblastoma cells, HEK-293 cells, fetal bovine serum (FBS), RPMI 1640 medium, and L-glutamine were from the American Type Culture Collection (Manassas, VA). Reagents used for culturing bacteria (LB Broth, LB agar plates with ampicillin, ampicillin solution, etc.) were from K-D Medical, Inc. (Columbia, MD). The primers, the High Fidelity Polymerase, and the ligase used for cloning, the chemically competent MaxEfficiency DH5αT1R and BL-21(DE3) cells, the DMEM without phenol red used for confocal analysis, the Lipofactamine and Plus reagent, Lipofectamine 3000, and the GST spin purification kit were from Thermo Scientific (Pittsburgh, PA). The QIAquick PCR Purification Kit and the QIAprep Spin Miniprep Kit were from Qiagen (Germantown, MD). The pHTN HaloTag(R) CMV-neo vector and the kit for purification of Halo-tagged proteins were from Promega (Madison, WI).
Site Directed Mutagenesis of GFP-Labeled Full-Length Munc13-1
EGFP-tagged full-length Munc13-1 in pEGFP-N1 was provided by Dr. N. Brose of the Max Planck Institute for Experimental Medicine, Göttingen, Germany. The mutants W588A, W588D, W588F, W588K, and W588Y of Munc13-1 were constructed by site directed mutagenesis using the Quick-Change kit (Stratagene) using Munc13-1-pEGFP-N1 vector as template. EGFP-C1 was constructed between the BamH1 and Xho1 sites of the pEGFP-N1 vector. All mutations were confirmed by DNA sequencing for their correct sequences.
Construction of GST-Tagged C1 Domain of Munc13-1 WT and W22 Mutants
The C1 domain of rat Munc13-1 was cloned into the pGEX2T plasmid (GE Healthcare Life Sciences) using BamH1 and EcoR1 restriction sites. The appropriate sequences (50 amino acids of the C1 domain plus a 10 amino acid flanking region at both ends) were PCR amplified from the full-length cDNA clone of rat Munc13-1 WT and its W588 mutants using High Fidelity Polymerase and the following primers: forward primer: CATGGATCCTTA-ATCTACCCCATCTCCTGCACCA; reverse primer: CAT-GAATTCCACTTAGAACTCTTCTCAGCCGCC. The purified PCR products (QIAquick PCR purification kit) were ligated into the plasmid after digestion with BamH1 and EcoR1 (New England Biolabs, Beverly, MA) as recommended by the manufacturer. The ligation reaction was transformed into MaxEfficiency DH5αT1R cells to produce and purify the new constructs that were verified by sequencing conducted at the Genomics Core, Center for Cancer Research, NCI, National Institutes of Health.
Expression and Purification of GST-Tagged C1 Domains from Escherichia coli
pGEX2T1 plasmids containing the C1 domains of Munc13-1 WT and its W22 mutants were transformed into BL-21(DE3) chemically competent E. coli. For expression of the desired proteins, transformants were grown in LB medium at 37 °C until the optical density of the bacterial suspension reached 0.6–0.8 and they were then treated with 0.3 mM IPTG for 4–5 h at 37 °C. The bacteria were pelleted, lysed by sonication in B-PER bacterial protein extraction reagent, and the expressed C1 domains were purified using the B-PER GST spin purification kit according to the manufacturer’s instructions. The purification efficiency was evaluated by SDS-PAGE analysis followed by Coomassie staining (Instant Blue, Expedeon, San Diego, CA). The purified proteins were stored in 30% glycerol at –80 °C.
Expression and Purification of Full-Length Munc13-1
Rat Munc13-1 WT and its W588 mutants were cloned into the pHTN HaloTag CMV-neo vector (Promega), a mammalian expression vector that adds a Halo tag to the N-terminus of the protein, by conventional PCR based cloning using EcoRI and XbaI restriction sites. The constructs were verified by sequencing conducted at the Genomics Core, Center for Cancer Research, NCI, National Institutes of Health.
The constructs containing the rat-Munc13-1 WT or its W588 mutants were transfected into HEK-293 cells (ATCC) (grown on 10 cm plates to 70–80% confluency) using Lipofectamine and Plus reagent following the manufacturer’s instructions (Invitrogen). The purification of Halo-tagged proteins was performed based on the protocol suggested by the manufacturer (Promega). One day after transfection the cells were collected, lysed in purification buffer (PBS + 1 mM DTT + 0.005% Igepal CA-630) (2.5 mL for pellets from 16 dishes) containing protease inhibitors (Promega) by sonication (6 × 10 s at power level 3), and centrifuged for 15 min at 3000 rpm in a cooled tabletop centrifuge (Beckman). The supernatant was applied to equilibrated (in purification buffer) Halo-link resin (800 μL pelleted resin) (Promega) and gently rotated overnight at 4 °C followed by centrifugation, removal of supernatant, and washing of the resin (3× with 3–4 mL purification buffer). The purified protein was released from the resin by overnight treatment with TEV protease and stored in 30% glycerol at –80 °C (500 μL purification buffer +9 μL TEV protease). The full-length protein purified from 20 10 cm dishes was generally enough to perform about 3–5 binding assays. The purity of the protein was checked on 10–20% SDS-polyacrylamide gels stained with Instant Blue (Expedeon). In a single experiment, the purification procedure was performed in parallel on lysates of nontransfected HEK-293 cells and lysates from Munc13-1 transfected HEK-293 cells as well as on a preparation in which no lysate was included in the procedure but in which the TEV protease was still included in the “release” step.
In Vitro [3H]PDBu Binding Assays
The phorbol ester binding affinities of the WT and mutant C1 domains of Munc13-1 were determined by the in vitro [3H]PDBu binding assay developed in our laboratory and described earlier.35 Binding curves were measured at 18 °C for 10 min or 37 °C for 5 min in the presence of 100 μg/mL lipid of the indicated compositions (phosphatidylserine:phosphatidylcholine, w:w) and 0.1 mM Ca2+ to obtain the individual dissociation constants (Kd values). The Kd values are reported as the mean ± SEM of 3–6 experiments, where each data point in each experiment represents triplicate determinations. A single experiment was performed to test the PDBu binding in parallel of eluents from lysates of nontransfected HEK-293 cells and Munc13-1 transfected HEK-293 cells as well as from the preparation with the TEV protease alone to exclude the possibility that PDBu bound to a copurified protein present in the eluates in low amounts.
Confocal Microscopy and Quantification
LNCaP cells (ATCC) or Neuro-2a cells (ATCC) (between passage 2 and 15) were plated on Ibidi dishes (Ibidi, LLC) in RPMI-1640 medium containing FBS (10%) and L-glutamine (2 mM) and grown to 75% confluency at 37 °C. LNCaP cells were transfected using Lipofectamine in combination with Plus reagent (Thermo Scientific) and Neuro-2a cells were transfected using Lipofectamine 3000 (Thermo Scientific) per the manufacturer’s instructions. Twenty-four hours after transfection, cells were transferred to confocal medium (DMEM without phenol red +1% FBS) and translocation in response to the indicated PMA treatments was visualized using Zeiss LSM 510 NLO or Zeiss LSM 710 NLO confocal microscopes (Carl Zeiss, Inc.) equipped with a 63 × 1.4 NA oil objective. GFP was excited with an argon laser at 488 nm and filters of 500–530 nm were used for detecting emission. PMA (1 μM or as otherwise indicated) was added at time 0 and translocation was monitored every 20 s. For quantitation of GFP translocation to the membrane, 3 regions of about 5 μm2 were selected in each cell over the plasma membrane and cytoplasmic regions at each time point (as indicated in the figures), the signal intensities determined using Zeiss Zen software, the intensities averaged for each region, and the ratio of the mean intensities of the membrane/cytoplasm determined. Imaging was conducted in the Center for Cancer Research Confocal Microscopy Core facility, National Cancer Institute.
Molecular Dynamics (MD) Simulation
Molecular dynamics (MD) simulations were carried out on the three simulating systems composed of the Munc13-1 C1, PKCδ C1B, and PKCθ C1B using the GROMACS 4.6.5 package of programs36 with Amber99sb force field.37 The NMR structure of the Munc13-1 C1 (PDB: 1Y8F) and the X-ray crystal structures of PKCδ C1B (PDB: 1PTR) and PKCθ C1B (PDB: 4FKD) were selected for the MD simulation. The models were solvated by TIP3P water molecules38 with a dodecahedron box type and a box distance of 2.0 nm. The models were neutralized by adding Cl–counterions. Between 9,534 and 10,850 water molecules were used to solvate the systems. In order to remove steric clashes generated while solvating the systems, energy minimization was conducted using the steepest descent method until the maximum force (Fmax) was below 1000 kJ/(mol nm). After energy minimization, the systems were equilibrated for 80 ps by position-restrained MD simulation in order to maintain temperature and pressure of the systems and relax the solvent. The equilibration was performed in two phases. NVT optimization with 300 K was conducted in the first phase, and the second phase was conducted for NPT optimization with 1 bar. Following the equilibration, the MD production run was conducted using the Berendsen coupling method39 with 300 K and 1 bar for 80 ns for all three systems. The bond lengths were constrained by LINCS algorithm40 allowing a time step of 2 fs. The Particle Mesh Ewald (PME) method41 was used to compute electrostatic interactions. The van der Waals, electrostatic, and Coulombic interactions were calculated with a 1 nm cutoff. For data analysis, the atomic coordinates were saved every 10 ps during the MD simulation. The MD trajectories of the three systems were analyzed by GROMACS analysis tools, including g_energy and g_dist. The graphs were plotted by GraphPad Prism 5. The trajectories and structures were visualized using PyMol v1.7 (Schrodinger, LLC.) and Discovery Studio Visualizer 4.5 (Biovia Inc.).
Statistical Analysis
Significance was determined with Graph Pad Prism 6 software, using the Student’s t test. A value less than 0.05 was deemed significant.
RESULTS
Effect of Mutation of Trp-588 on the Membrane Translocation of Munc13-1
To explore the role of Trp-588 on the translocation of Munc13-1, we expressed the full-length protein and its mutated constructs in the Neuro-2a cell line, which was chosen because the physiological role of Munc13-1 is in vesicle priming in neural cells. Using the Neuro-2a cell line, we evaluated the impact on PMA-induced translocation of replacing W588 (numbering corresponding to W22 in the C1 domain) with a range of residues. It was previously reported that W22 in the C1 domain of PKCs makes an important contribution to ligand binding.34 As seen in the images and the quantitation of the translocation (Figure 2), the W588K and W588F mutations did not significantly influence the translocation in response to PMA, while the W588A mutation significantly decreased translocation and the W599D mutation totally abolished it. The translocation of the W588Y mutant was delayed compared to that of the wild-type Munc13-1 protein. The efficiency of the PMA-induced membrane translocation of full-length Munc13-1 thus followed the order, wild-type > W22K > W22F > W22Y > W22A > W22D.
Figure 2.

Translocation of full-length Munc13-1 WT and its mutants after PMA treatment in Neuro-2a cells. Full-length GFP-Munc13-1 and its mutants were transfected into Neuro-2a cells 24 h after plating. Twenty-four hours after transfection 1000 nM PMA was added and cells were imaged by confocal microscopy. A. Images shown are representative of those from the 3 independent experiments performed. B. The quantitation of translocation was performed as described in Methods. For each construct, the bars represent the results for translocation measured at 0, 2, 4, and 8 min after PMA addition (left to right). The values represent the mean ± SEM calculated for 3 individual cells from each of 3 independent experiments. * p < 0.5; **p < 0.1; ***p < 0.01.
Effect of Mutation of Trp-588 on Phorbol Ester Binding
GST-tagged C1 domains, containing the 50 amino acids of the rat Munc13-1 WT C1 domain or variants mutated at residue Trp-22 (Trp-588 in the full-length protein) and all including the endogenous flanking 10 amino acids at each end, were expressed in bacteria and affinity purified. Their affinities for [3H]PDBu were measured using a modification of the usual binding assay and included 0.1 mM Ca2+.35 At a concentration of 100 μg/mL phospholipid, the C1b domain of PKCδ was relatively insensitive to the proportion of the anionic PS in PS:PC mixtures over the range of 5–100% PS. In contrast, for the WT C1 domain of Munc13-1 as little as 2.5% PS supported almost maximal binding and PS proportions ≥60% decreased binding (Figure 3A). Indeed, the amount of [3H]PDBu bound by the WT Munc13-1 C1 domain at 100% PS was less than 15% of that at 20% PS. In contrast, the W22K mutant C1 domain, which is less negatively charged, showed a greater dependency on PS for optimal binding than even the PKCδ C1b domain; low binding was measured at PS proportions of ≤20% whereas PS proportions of 40% and above gave almost full binding. The binding of the W22A mutant of C1 domain was overall low but mostly resembled the PS dependence of that of WT C1 domain. The binding of the W22D mutant of the Munc13-1 C1 domain under any of these conditions was undetectable. In all of these in vitro binding assays, 0.1 mM Ca2+ was included. The rationale was that the Ca2+ might reduce repulsion between the negatively charged phospholipid and the negatively charged C1 domain, and preliminary experiments (data not shown) confirmed that binding affinities were modestly enhanced in its presence.
Figure 3.

PDBu binding of the isolated C1 domain of Munc13-1. A. Lipid requirement of different C1 domains. In vitro PDBu binding was measured at 5 nM [3H]PDBu in the presence of different ratios of phosphatidylserine (PS) to phosphatidylcholine (PC) as described in the Methods section. The amount of specifically bound PDBu is expressed relative to the amount bound in 100% PS. Data represent the mean ± SEM of three independent experiments. B. Saturation binding curve and Scatchard plot (inset) for [3H]PDBu binding to the Munc13-1 C1 domain. Binding was performed in the presence of 100 μg/mL lipid (20:80% PS/PC), 0.1 mM Ca2+ and increasing concentrations of radioactive PDBu. Data shown are from one representative experiment. Three additional experiments gave similar results.
The binding affinities (Kd) of the Munc13-1 WT C1 domain and its different W22 mutants varied with the lipid conditions (Table 1). The Kd of the WT C1 domain of Munc13-1 was 32.5 ± 2.6 and 98 ± 36 nM in 20% PS and 100% PS, respectively, indicating a 30- and 100-fold weaker binding affinity of the Munc13-1 C1 domain than that of the C1b domain of PKCδ. The Kd values for the W22K mutant were 420 ± 120 and 160 ± 20 nM in 20% PS and 100% PS, respectively, reflecting the higher PS requirement of this mutant for binding. The behavior of the W22A mutant not only showed a big loss in the specific binding but also induced a big loss in binding potency, especially at lower PS concentrations. The Kd for the W22A mutant in 100% PS was 460 ± 260 nM while in 20% PS it was >1000 nM. A representative experiment for binding of [3H]PDBu to the Munc13-1 WT C1 domain under conditions of 20% PS:80% PC is shown in Figure 3B.
Table 1.
Binding of [3H]PDBu by the Munc13-1 C1 Domain and Its Mutantsa
| Protein | Lipid (20:80 PS:PC) | Lipid (100:0 PS:PC) | ||
|---|---|---|---|---|
|
|
|
|||
| Kd (nM) | Kd(mutant)/Kd(WT) | Kd (nM) | Kd(mutant)/Kd(WT) | |
| Munc13-1 WT | 32.5 ± 2.6 | 98 ± 36 | ||
| Munc13-1 W22A mutant | >1000 nM | >31 | 460 ± 260 | 4.7 |
| Munc13-1 W22K mutant | 420 ± 120 | 13 | 160 ± 20 | 1.6 |
PDBu binding affinity (Kd) was determined for the C1 domain of Munc13-1 and its mutants under two different lipid conditions. Values represent the mean ± SEM from three independent experiments.
The Munc13-1 C1 domain was easily expressed in quantity in bacterial cells. For the full- length Munc13-1 protein, we expressed the Halo-tagged protein in HEK-293 cells and isolated the protein after TEV protease cleavage (Figure 4A). Although yields of protein did not compare with those for the C1 domain, they were still sufficient for measurement of binding by the constructs with higher [3H]PDBu binding affinity. It should be noted that, in addition to the major band of full-length Munc13-1 protein, some minor bands were also present. Some but not all of these could be attributed to the TEV protease used in the purification and/or proteins from the HEK lysates (SI Figure 1).
Figure 4.

PDBu binding to the wild-type full-length Munc13-1 protein. (A) Halo-tagged wild-type full-length Munc13-1 protein was expressed in HEK-293 cells, bound to Halo-link resin, and released with TEV protease. Aliquots of the column eluents were subjected to SDS gel electrophoresis and visualized with Coomassie Blue staining, as described in Methods. Results are shown for two representative batches of wild-type full-length Munc13-1, of five batches assayed. (B) Saturation binding curve and Scatchard plot (inset) for [3H]PDBu binding by the wild-type full-length Munc13-1 protein. Binding was performed in the presence of 100 μg/mL lipid (100% PS), 0.1 mM Ca2+, and increasing concentrations of [3H]PDBu. Symbols on the saturation curve represent the mean ± SE of triplicate measurements in one representative experiment. Where error bars are not visible, they fell within the size of the symbol. Three additional experiments gave similar results.
In contrast to the Munc13-1 C1 domain, the full-length Munc13-1 wild-type protein bound [3H]PDBu with high affinity under our in vitro binding conditions, with a Kd of 3.7 ± 0.32 nM (n = 3) (Figure 4B). Similarly, the full-length Munc13-1 protein incorporating the W22K mutation (W588 K using the numbering for the full-length protein) bound with a Kd of 3.3 ± 1.2 nM. Because of low overall protein expression of the Halo-tagged proteins, we were unable to characterize the binding of the W22A and W22D mutants of the full-length Munc13-1 protein. In a single experiment, we verified that there was no measurable specific [3H]PDBu binding in samples purified from nontransfected HEK-293 cells or TEV protease alone.
Our data argue that the full-length Munc13-1 protein binds phorbol ester with appreciably better affinity than does its isolated C1 domain. A potential rationale for this enhanced affinity is that positively charged residues on the C2B domain of Munc13-1 (R754, K752, and R750) help neutralize the negative charges (E584 and E582) of the C1 domain. These intramolecular interactions further could potentially help orient the C1 domain DAG/phorbol ester binding cleft relative to the membrane.42 A cautionary note, however, is imposed by the presence of minor protein bands in the Munc13-1 protein preparations. An alternative interpretation is that the full-length protein fails to bind [3H]PDBu with high affinity and that the high affinity binding instead represents a fragment of the Munc13-1. This model would be that the C1 domain in the intact protein is inaccessible to the phorbol ester because it is involved in intramolecular interactions. Rather, the binding would reflect a protein fragment of Munc13-1, in which these intramolecular interactions have been disrupted but in which other elements in the postulated fragment such as the C2B domain would provide the enhanced affinity relative to that of the isolated C1 domain.
We can rule out contaminating proteins from the HEK cells accounting for the binding since no binding was found when we carried out a parallel purification with nontransfected HEK cells. Likewise, we did not see any binding with cells expressing the full-length Munc13-1 mutated at W588D. Since this mutation in the C1 domain had abolished phorbol ester binding, the cells expressing the full-length Munc13-1 with this mutation should likewise provide a control for binding by extraneous proteins. Finally, we note that the translocation results, described below, show high sensitivity of the full-length Munc13-1 protein to phorbol ester, consistent with our [3H]PDBu binding data showing high affinity binding.
Munc13-1 and C1 Translocate to Different Location
The difference in phorbol ester binding of the isolated Munc13-1 C1 domain and the full-length protein was also reflected in the difference in their behavior in the context of the living cells. For these studies, we used LNCaP prostate cancer cells, which have been extensively used to characterize the translocation of C1 domains in response to phorbol esters and related derivatives. The choice of these cells also provided the opportunity to compare the response of the full-length Munc13-1 protein in a second cell type. The GFP-tagged C1 domain was localized to the nucleus and the cytoplasm while the full-length protein was solely localized to the cytoplasm in the great majority of the cells (Figure 5). After addition of 1000 nM PMA, the GFP-C1 domain translocated within 10 min from the nucleus to internal membranes but not to the plasma membrane. Translocation was not observed at 100 nM PMA. In contrast, the full-length Munc13-1 translocated to the plasma membrane.
Figure 5.

Translocation of Munc13-1 after PMA treatment in LNCaP cells. GFP tagged Munc13-1 C1 domain (left panel) or GFP-tagged full-length Munc13-1 (right panel) were transfected into LNCaP cells 24 h after plating. Twenty-four hours after transfection PMA at the indicated concentrations was added and cells were imaged by confocal microscopy. Images shown are representative of those from the 3 independent experiments performed.
The greater sensitivity of the full-length Munc13-1 to translocation in response to PMA is consistent with its greater in vitro phorbol ester binding affinity. Several factors may additionally contribute to the localization of the full-length Munc13-1 protein to the plasma membrane. The plasma membrane is more negatively charged, reflecting its higher content of PS. The neutralization of several of the negative charges on the C1 domain through intramolecular interactions with the C2B domain of the full-length Munc13-1 may reduce the effect of the negatively charged plasma membrane, and interactions between the C2B domain of the full-length Munc13-1 with phosphatidylinositol phosphates may further direct the plasma membrane localization of the full-length protein.23 Finally, it is possible that protein–protein interactions with other membrane proteins may make yet an additional contribution to the plasma membrane localization of the full-length Munc13-1 protein.
Flexibility of Trp-588 in Munc13-1C1
To understand flexibility of the side chain of Trp-22 in the C1 domain we performed molecular dynamics simulation of the Munc13-1 C1 domain during 80 ns and compared the data with that for the homologous Trp of PKCδ C1B and PKCθ C1B. Trp-22 in Munc13-1 C1 maintained its position between the two loops during 80 ns (Figure 6). In contrast, the Trp-22 in PKCδ C1B showed great flexibility during the MD simulation. Unlike in the case of the Munc13-1 C1, it was far from the center of the two loops at 0 ns, but within 30 ns it oriented itself in the opposite direction, toward the center of the two loops, before rotating away again by 80 ns (Figure 6). The Trp-22 of PKCθ showed intermediate behavior, with different orientations over the duration of MD simulation but with more residence time between the loops than was the case for Trp-22 of PKCδ C1B.
Figure 6.

Orientiation of Trp22 during MD simulation. Snapshots were taken at 10 ns time intervals during the MD simulation. The C1 domain ribbon structure is shown in gray. The line structure of Trp-22 is shown in different colors at every 10 ns interval. The structures at 0 ns were taken after the systems were equilibrated before MD production.
To measure the movement of the Trp in a quantitative manner, we measured the distance of the C6 of tryptophan and the Cα of one of the loop residues (Ser/Thr10). The distance between the Cα of Ser/Thr10 and the C6 of Trp-22, which is the farthest atom from Cα of Trp-22, was measured over the duration of MD simulation. The plot of PKCδ C1B showed high fluctuation during 80 ns. In the case of PKCθ C1B, it showed moderate fluctuation and kept a distance of about 7.8 Å from 20 to 55 ns. This result indicates that Trp-22 preferred a particular orientation, maintaining similar poses over this time interval (Figure 6). In contrast, the plot of Munc13-1 C1 showed little fluctuation and maintained a similar distance during the MD simulation. The distances between the loop Ser/Thr and the C6 of the tryptophan for these three C1 domains are shown in Figure 7. While for Munc13-1 the maximum range of this movement is 3.61 Å, for PKCδ and PKCθ it is 15.0 and 14.49 Å, respectively
Figure 7.

Fluctuation of the Trp-22 in Munc13-1 C1, PKCδ C1B, and PKCθ C1B during MD simulation. Top: The distance between the C6 of Trp-22 and the Cα of Thr-10/Ser-10 of the C1B domain was measured during the MD simulation. The red double headed arrow indicates the points of distance measurement. Ala-8 and Pro-11 are involved in hydrophobic interactions with Trp-22, as shown by the purple and black dashed lines. The hydrophobic interactions were identified using Discovery Studio Visualizer 4.5 program. Bottom: Plot showing the distance between Thr-10/Ser-10 and the Trp-22 for Munc13-1 C1, PKCδ C1B, and PKCθ C1B. The distance was calculated over the intervals of the MD simulation.
DISCUSSION
The objective of the present study is to understand the role of Trp-588 in the phorbol ester binding and membrane translocation of Munc13-1. Munc13-1 is a key regulator of vesicle priming, neurotransmitter release, and presynaptic plasticity.3,4,7 It is a peripheral membrane protein that acts as a scaffold by interacting with other SNARE complex proteins and the plasma membrane.42,43 Experiments using micro-injection of Munc13-1 and Munc13-1H567K (a C1 domain mutant that does not bind to phorbol ester) mRNA into Xenopus laevis embryos revealed that enhancement of neurotransmitter release by phorbol ester was dependent on a functional Munc13-1 C1 domain.3 Additionally, knock-in mice bearing Munc13-1H567K die shortly after birth even though the mutation causes no impairment of evoked release, suggesting that binding of DAG/phorbol ester to the C1 domain is essential for the synaptic efficacy and survival.44 Although the Munc13-1 C1 domain has close structural similarity with the C1 domains of PKC, the orientation of the side chain of Trp-22 occludes the activator binding site in Munc13-1 (Figure 1) unlike in the case of the homologous tryptophan in PKCs. In the present study, we generated several mutants at position 22 and characterized these mutants in terms of their phorbol ester binding and phorbol ester-induced membrane translocation. The efficiency of membrane translocation of the full-length Munc13-1 followed the order, WT > W22K > W22F > W22Y > W22A > W22D. The binding affinities of the three Munc13-1 C1 domains that we selected for phorbol ester binding studies followed the order WT> W22K > W22A ≫ W22D at both 20% and 100% PS.
Ligand binding to C1 domains depends both on the fit of the ligand to the C1 domain binding cleft as well as on the combination of elements—ligand, C1 domain, other lipid interacting domains, and lipid composition—which promote membrane association.27 This dependence on the other elements of the binding complex might be of particular importance for Munc13-1, in that its C1 domain differs from the other typical C1 domains in possessing a strong net negative charge, whereas the typical C1 domains possess a positive charge45 (Figure 8). Thus, the C1 domain of Munc13-1 might be subject to electrostatic repulsion by the plasma membrane, which is negatively charged because of the presence of phosphatidylserine and phosphatidylinositol phosphates.
Figure 8.

Sequence of the C1b domain of mouse PKCδ and of the C1 domain of rat Munc13-1 WT and its Trp-22 mutants. Capital letters indicate the amino acids of the C1 domain; small letters indicate the 10 amino acid flanking regions around the C1 domain; the black arrow points to Trp-22 mutated to A, K, and D in Munc13-1; the negatively charged acidic amino acids are colored blue while the positively charged ones are colored red; the net charge of the C1 domain/C1 domain with the 10 amino acid flanking regions are calculated from the sum of +1 and –1 charges for the K, R, and E and D amino acids, respectively.
Consistent with this expectation, the C1 domain of Munc13-1 with net negative charge shows maximum PDBu binding at 20% PS and not at 100% PS, while the PKCδ C1B and PKCθ C1B domains with net positive charge show maximum PDBu binding at 100% PS.32
Our binding data fit with previous observations. Irie et al.45 were unable to quantitate phorbol ester binding to the C1 domain of Munc13-1, which they prepared by chemical synthesis. In contrast, we had found strong binding affinity (1.1 nM) to the C1 domain of Unc13, the Munc13 ortholog from Caenorabditis elegans, which differs from Munc13 in that it is not negatively charged.28 Additionally, whereas Irie et al. were unable to detect appreciable phorbol ester binding to the C. elegans Munc13 C1 domain ortholog under their conditions, binding affinity was recovered in a synthetic construct to which they had added 5 arginine residues, conferring strong positive charge.45
Is hydrophobicity of Trp-22 important for phorbol ester binding? According to the White-Wimley hydrophobicity scale, Trp is the most hydrophobic amino acid.46,47 Besides, with its fused aromatic ring system Trp can undergo cation-π interactions48 with positively charged choline groups of the lipids and detergents49 and can also form hydrogen bonds, π-stacking, N–H···π, and C–H···π bonds.50 These diverse interactions of tryptophan allow it to serve as a strong anchoring point for membrane and peripheral membrane proteins. To understand the role of Trp on phorbol ester binding in the truncated C1 domain, we generated three mutants, W22A, W22K, and W22D, in which tryptophan was replaced with a small nonpolar residue, with a positively charged residue, and with a negatively charged residue, respectively. Trp, Ala, Lys, and Asp have ΔGbilayer to water values of +1.85 kcal/mol, –0.7 kcal/mol, –0.99 kcal/mol, and –1.23 kcal/mol, respectively, meaning that the hydrophobic effect (i.e., solvent exclusion) will make the largest favorable contribution to the free energy of binding in WT and the least in W22D following the order, WT > W22A > W22K > W22D. We, however, found that the order of the binding affinity was WT > W22K > W22A ≫ W22D at both 20% and 100% PS. The reverse order of W22A and W22K for the predicted and experimental binding affinity suggests that the positive charge of lysine in W22K outweighs the higher hydrophobic contribution from alanine in W22A. Similarly, replacement of tryptophan with lysine at position 22 in the C1b domain of PKCδ had negligible effect on phorbol ester binding, arguing that this behavior was not unique to the C1 domain on Munc13-1.51 Additionally, in Munc13-1, when tryptophan was replaced by alanine in W22A, only a ~5-fold reduction in binding affinity at 100% PS was observed as compared to >30-fold reduction of binding affinity at 20% PS. Our explanation is that at 100% PS the other factors that contribute to the phorbol ester binding would tend to be obscured by the negative charge of PS. However, when the PS was reduced to 20%, the hydrophobicity factor could take precedence over charge, showing higher fold reductions in binding affinity. For the W22K mutant, a 1.6-fold decrease in affinity at 100% PS and a 13-fold decrease in 20% PS again suggested the role of hydrophobicity of tryptophan, and possibly a compensatory charge effect for lysine, in phorbol ester binding. Detection of no binding for the W22D mutant at both 20% and 100% PS is indicative of electrostatic repulsion between the C1 domain and the membrane containing anionic PS. In the case of the W22D of PKCδC1B, a 1070-fold reduction of PDBu affinity was observed at 100% PS.52 In this PKC isoform, W22Y, W22T, W22K, and W22R showed modest effects on phorbol ester binding because of the overwhelming influence of 100% PS.52 Again, when the PS concentration was reduced to 10%, W22R, the only substitution for which the data is available, showed a 9-fold reduced of phorbol ester affinity as compared to the WT.52 This suggests that hydrophobicity of Trp-22 is also important for the phorbol ester binding in PKCδ. A closely related mutant W22G of PKCδ C1B28 and PKCθ C1B32 showed 31- and 8-fold reductions in phorbol ester affinity as compared to WT when measured in 100% PS, suggesting the role of hydrophobicity of Trp-22 in phorbol ester binding for these PKC isoforms. It is important to note that Trp-22 has been found to have greater influence on DAG binding than on binding of phorbol ester.34
The C1, C2B, and MUN domains (C1C2BMUN) of Munc13-1 form a 19.5-nm-long multihelical structure with the C1 and C2B domains packed at one end.42 It has been proposed that after binding of the C1 domain to DAG and of the C2B domain to Ca2+, Munc13-1 could orient itself in such a way that the plasma membrane and the vesicular membrane come closer to each other facilitating the SNARE complex formation. Although the surface residues, Glu-584, Glu-582, and Tyr-581 of the C1 domain are in close-proximity with Arg-754, Lys-752, and Arg-750 surface residues of the C2B domain, Trp-588 is oriented toward the plasma membrane. The pertinent question is whether the side chain of the Trp-588 still occludes the activator binding site. A simple MD simulation of the C1 domains suggests that during the 80 ns time scale the movement of Trp-588 is relatively restricted in the Munc13-1 C1 as compared to the homologous Trp-22 of PKCδ and PKCθ (Figure 6). The lower flexibility of Trp-22 in Munc13-1 as compared PKCδ and PKCθ can be explained by inspection of the residues with which the Trp-22 interacts in these three domains. In Munc13-1, Ala-8 and Pro-11 are involved in strong hydrophobic interactions in all the structures during 10 ns intervals, thereby restricting its movements (Figure 7). On the other hand, for PKCδ and PKCθ C1 domains, homologous Tyr does not interact with Trp-22 at all and interaction with Pro-11 was observed only in the 30 ns structure for PKCδ and the 70 ns structure for PKCθ. While the presence of DAG and the membrane is expected to affect the flexibility and the orientation of the Trp-22, our simulation data show the contribution of the surrounding amino acids in determining the flexibility of the tryptophan.
An additional objective of the present study was to compare the phorbol ester binding affinity and the phorbol ester-induced membrane translocation of the isolated C1 and the full-length Munc13-1. Our result that the affinity of Munc13-1 (Kd = 3.7 nM) is higher than the C1 domain (Kd= 98 nM) suggests that C2 and/or MUN domains also contribute to the phorbol ester affinity of Munc13-1. This pattern is thus analogous to that for the classic PKC isoforms, where membrane association is driven both by DAG binding to the C1 domains and by Ca2+ binding to their C2 domains. Interestingly, whereas the binding cleft of the C1 domains of PKCβII,53 β2-chimaerin,54 and RasGRP155 have all been shown to engage in intramolecular interactions in their unliganded state, this is not evident for Munc13-1. Rotation of Trp-22 into the binding cleft may provide an alternative solution to provide occupancy of the cleft in the absence of DAG/phorbol ester.
Our findings that the C1 domain translocates to the internal membranes instead of to the plasma membrane, which is relatively rich in PS and other anionic lipids as compared to the internal membranes,56,57 whereas Munc13-1 translocates to the plasma membrane, again highlights the contribution of charge and domain–domain interactions in dictating the specific localization of the full-length protein and its subsequent biological function. Unlike Munc13-1, both the C1A and C1B domains of PKCδ as well as the full-length PKCδ translocated to the plasma membrane in response to 1 μM PMA, albeit with varied kinetics.58
In summary, we show here that the phorbol ester affinity and membrane translocation properties of the isolated C1 domain of Munc13-1 are distinct from other typical C1 domains and that Trp-588 in Munc13-1 plays a critical role in phorbol ester binding and its membrane translocation. Given the role of Munc13-1 in glutamatergic neurotransmission9 and involvement in the neurodegenerative diseases,15,16,59 development of small molecule inhibitors targeting the C1 domain would be of great interest. The unique properties of the Munc13-1 C1 domain could be exploited for increasing the selectivity among the C1 domain containing proteins.
Supplementary Material
Acknowledgments
Funding
This research has been supported in part by funding from National Institutes of Health Grant 1R01 AA022414–01A1 to J.D. and in part by the Intramural Research Program, Center for Cancer Research, National Cancer Institute, NIH (Project Z1A BC 005270) to P.M.B.
We thank Dr. N. Brose of Max Planck Institute for Experimental Medicine, Göttingen, Germany for providing us with the constructs for Munc13-1. MD simulations were performed using the server at the Center for Advanced Computing and Data Systems (CACDS) of the University of Houston.
ABBREVIATIONS
- AD
Alzheimer’s disease
- DAG
diacylglycerol
- DMEM
Dulbecco’s modified eagle medium
- EGFP
enhanced green fluorescent protein
- FBS
fetal bovine serum
- GFP
green fluorescent protein
- GST
glutathione S-transferase
- HEK
Human embryonic kidney
- IPTG
isopropyl β-D-1-thiogalacto-pyranoside
- MHD
Munc13 homology domain
- MD
molecular dynamics
- PDBu
phorbol 12,13-dibutyrate
- PKC
protein kinase C
- PMA
phorbol 12-myristate 13-acetate
- PC
phosphatidylcholine
- PS
phosphatidylserine
- SDS-PAGE
sodium dodecyl sulfate polyacrylamide gel electrophoresis
- SNARE
Soluble NSF (N-Ethylmaleimide-Sensitive Factor) Attachment Protein Receptor
- WT
wild-type
Footnotes
Notes
The authors declare no competing financial interest.
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.biochem.7b00764.
Purification of full-length Munc13-1 (PDF)
References
- 1.Betz A, Okamoto M, Benseler F, Brose N. Direct interaction of the rat unc-13 homologue Munc13-1 with the N terminus of syntaxin. J Biol Chem. 1997;272:2520–2526. doi: 10.1074/jbc.272.4.2520. [DOI] [PubMed] [Google Scholar]
- 2.Sassa T, Harada S, Ogawa H, Rand JB, Maruyama IN, Hosono R. Regulation of the UNC-18-Caenorhabditis elegans syntaxin complex by UNC-13. J Neurosci. 1999;19:4772–4777. doi: 10.1523/JNEUROSCI.19-12-04772.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Betz A, Ashery U, Rickmann M, Augustin I, Neher E, Sudhof TC, Rettig J, Brose N. Munc13-1 is a presynaptic phorbol ester receptor that enhances neurotransmitter release. Neuron. 1998;21:123–136. doi: 10.1016/s0896-6273(00)80520-6. [DOI] [PubMed] [Google Scholar]
- 4.Brose N, Rosenmund C, Rettig J. Regulation of transmitter release by Unc-13 and its homologues. Curr Opin Neurobiol. 2000;10:303–311. doi: 10.1016/s0959-4388(00)00105-7. [DOI] [PubMed] [Google Scholar]
- 5.Rosenmund C, Sigler A, Augustin I, Reim K, Brose N, Rhee JS. Differential control of vesicle priming and short-term plasticity by Munc13 isoforms. Neuron. 2002;33:411–424. doi: 10.1016/s0896-6273(02)00568-8. [DOI] [PubMed] [Google Scholar]
- 6.Lipstein N, Sakaba T, Cooper BH, Lin KH, Strenzke N, Ashery U, Rhee JS, Taschenberger H, Neher E, Brose N. Dynamic control of synaptic vesicle replenishment and short-term plasticity by Ca(2+)-calmodulin-Munc13-1 signaling. Neuron. 2013;79:82–96. doi: 10.1016/j.neuron.2013.05.011. [DOI] [PubMed] [Google Scholar]
- 7.Yang Y, Calakos N. Munc13-1 is required for presynaptic long-term potentiation. J Neurosci. 2011;31:12053–12057. doi: 10.1523/JNEUROSCI.2276-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen Z, Cooper B, Kalla S, Varoqueaux F, Young SM., Jr The Munc13 proteins differentially regulate readily releasable pool dynamics and calcium-dependent recovery at a central synapse. J Neurosci. 2013;33:8336–8351. doi: 10.1523/JNEUROSCI.5128-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Augustin I, Rosenmund C, Sudhof TC, Brose N. Munc13-1 is essential for fusion competence of glutamatergic synaptic vesicles. Nature. 1999;400:457–461. doi: 10.1038/22768. [DOI] [PubMed] [Google Scholar]
- 10.Varoqueaux F, Sigler A, Rhee JS, Brose N, Enk C, Reim K, Rosenmund C. Total arrest of spontaneous and evoked synaptic transmission but normal synaptogenesis in the absence of Munc13-mediated vesicle priming. Proc Natl Acad Sci U S A. 2002;99:9037–9042. doi: 10.1073/pnas.122623799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Aravamudan B, Fergestad T, Davis WS, Rodesch CK, Broadie K. Drosophila UNC-13 is essential for synaptic transmission. Nat Neurosci. 1999;2:965–971. doi: 10.1038/14764. [DOI] [PubMed] [Google Scholar]
- 12.Richmond JE, Davis WS, Jorgensen EM. UNC-13 is required for synaptic vesicle fusion in C. elegans. Nat Neurosci. 1999;2:959–964. doi: 10.1038/14755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lipstein N, Verhoeven-Duif NM, Michelassi FE, Calloway N, van Hasselt PM, Pienkowska K, van Haaften G, van Haelst MM, van Empelen R, Cuppen I, van Teeseling HC, Evelein AM, Vorstman JA, Thoms S, Jahn O, Duran KJ, Monroe GR, Ryan TA, Taschenberger H, Dittman JS, Rhee JS, Visser G, Jans JJ, Brose N. Synaptic UNC13A protein variant causes increased neurotransmission and dyskinetic movement disorder. J Clin Invest. 2017;127:1005–1018. doi: 10.1172/JCI90259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rossner S. New players in old amyloid precursor protein-processing pathways. Int J Dev Neurosci. 2004;22:467–474. doi: 10.1016/j.ijdevneu.2004.07.004. [DOI] [PubMed] [Google Scholar]
- 15.Ikin AF, Causevic M, Pedrini S, Benson LS, Buxbaum JD, Suzuki T, Lovestone S, Higashiyama S, Mustelin T, Burgoyne RD, Gandy S. Evidence against roles for phorbol binding protein Munc13-1, ADAM adaptor Eve-1, or vesicle trafficking phosphoproteins Munc18 or NSF as phosphostate-sensitive modulators of phorbol/PKC-activated Alzheimer APP ectodomain shedding. Mol Neurodegener. 2007;2:23. doi: 10.1186/1750-1326-2-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hartlage-Rubsamen M, Waniek A, Rossner S. Munc13 genotype regulates secretory amyloid precursor protein processing via postsynaptic glutamate receptors. Int J Dev Neurosci. 2013;31:36–45. doi: 10.1016/j.ijdevneu.2012.10.001. [DOI] [PubMed] [Google Scholar]
- 17.Bosco DA, Landers JE. Genetic determinants of amyotrophic lateral sclerosis as therapeutic targets. CNS Neurol Disord: Drug Targets. 2010;9:779–790. doi: 10.2174/187152710793237494. [DOI] [PubMed] [Google Scholar]
- 18.Su XW, Broach JR, Connor JR, Gerhard GS, Simmons Z. Genetic heterogeneity of amyotrophic lateral sclerosis: implications for clinical practice and research. Muscle Nerve. 2014;49:786–803. doi: 10.1002/mus.24198. [DOI] [PubMed] [Google Scholar]
- 19.Finsterer J, Burgunder JM. Recent progress in the genetics of motor neuron disease. Eur J Med Genet. 2014;57:103–112. doi: 10.1016/j.ejmg.2014.01.002. [DOI] [PubMed] [Google Scholar]
- 20.Diekstra FP, Van Deerlin VM, van Swieten JC, Al-Chalabi A, Ludolph AC, Weishaupt JH, Hardiman O, Landers JE, Brown RH, Jr, van Es MA, Pasterkamp RJ, Koppers M, Andersen PM, Estrada K, Rivadeneira F, Hofman A, Uitterlinden AG, van Damme P, Melki J, Meininger V, Shatunov A, Shaw CE, Leigh PN, Shaw PJ, Morrison KE, Fogh I, Chio A, Traynor BJ, Czell D, Weber M, Heutink P, de Bakker PI, Silani V, Robberecht W, van den Berg LH, Veldink JH. C9orf72 and UNC13A are shared risk loci for amyotrophic lateral sclerosis and frontotemporal dementia: a genome-wide meta-analysis. Ann Neurol. 2014;76:120–133. doi: 10.1002/ana.24198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kwan EP, Xie L, Sheu L, Nolan CJ, Prentki M, Betz A, Brose N, Gaisano HY. Munc13-1 deficiency reduces insulin secretion and causes abnormal glucose tolerance. Diabetes. 2006;55:1421–1429. doi: 10.2337/db05-1263. [DOI] [PubMed] [Google Scholar]
- 22.Das J, Xu S, Pany S, Guillory A, Shah V, Roman GW. The pre-synaptic Munc13-1 binds alcohol and modulates alcohol self-administration in Drosophila. J Neurochem. 2013;126:715–726. doi: 10.1111/jnc.12315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shin OH, Lu J, Rhee JS, Tomchick DR, Pang ZP, Wojcik SM, Camacho-Perez M, Brose N, Machius M, Rizo J, Rosenmund C, Sudhof TC. Munc13 C2B domain is an activity-dependent Ca2+ regulator of synaptic exocytosis. Nat Struct Mol Biol. 2010;17:280–288. doi: 10.1038/nsmb.1758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ma C, Li W, Xu Y, Rizo J. Munc13 mediates the transition from the closed syntaxin-Munc18 complex to the SNARE complex. Nat Struct Mol Biol. 2011;18:542–549. doi: 10.1038/nsmb.2047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Basu J, Shen N, Dulubova I, Lu J, Guan R, Guryev O, Grishin NV, Rosenmund C, Rizo J. A minimal domain responsible for Munc13 activity. Nat Struct Mol Biol. 2005;12:1017–1018. doi: 10.1038/nsmb1001. [DOI] [PubMed] [Google Scholar]
- 26.Andrews-Zwilling YS, Kawabe H, Reim K, Varoqueaux F, Brose N. Binding to Rab3A-interacting molecule RIM regulates the presynaptic recruitment of Munc13-1 and ubMunc13–2. J Biol Chem. 2006;281:19720–19731. doi: 10.1074/jbc.M601421200. [DOI] [PubMed] [Google Scholar]
- 27.Das J, Rahman GM. C1 domains: structure and ligand-binding properties. Chem Rev. 2014;114:12108–12131. doi: 10.1021/cr300481j. [DOI] [PubMed] [Google Scholar]
- 28.Kazanietz MG, Lewin NE, Bruns JD, Blumberg PM. Characterization of the cysteine-rich region of the Caenorhabditis elegans protein Unc-13 as a high affinity phorbol ester receptor. Analysis of ligand-binding interactions, lipid cofactor requirements, and inhibitor sensitivity. J Biol Chem. 1995;270:10777–10783. doi: 10.1074/jbc.270.18.10777. [DOI] [PubMed] [Google Scholar]
- 29.Basu J, Betz A, Brose N, Rosenmund C. Munc13-1 C1 domain activation lowers the energy barrier for synaptic vesicle fusion. J Neurosci. 2007;27:1200–1210. doi: 10.1523/JNEUROSCI.4908-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shen N, Guryev O, Rizo J. Intramolecular occlusion of the diacylglycerol-binding site in the C1 domain of Munc13-1. Biochemistry. 2005;44:1089–1096. doi: 10.1021/bi0476127. [DOI] [PubMed] [Google Scholar]
- 31.Zhang G, Kazanietz MG, Blumberg PM, Hurley JH. Crystal structure of the cys2 activator-binding domain of protein kinase C delta in complex with phorbol ester. Cell. 1995;81:917–924. doi: 10.1016/0092-8674(95)90011-x. [DOI] [PubMed] [Google Scholar]
- 32.Rahman GM, Shanker S, Lewin NE, Kedei N, Hill CS, Prasad BV, Blumberg PM, Das J. Identification of the activator-binding residues in the second cysteine-rich regulatory domain of protein kinase Ctheta (PKCtheta) Biochem J. 2013;451:33–44. doi: 10.1042/BJ20121307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Czikora A, Lundberg DJ, Abramovitz A, Lewin NE, Kedei N, Peach ML, Zhou X, Merritt RC, Craft EA, Braun DC. Structural basis for the failure of the C1 domain of Ras guanine nucleotide releasing protein 2 (RasGRP2) to bind phorbol ester with high affinity. J Biol Chem. 2016;291:11133–11147. doi: 10.1074/jbc.M116.725333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dries DR, Gallegos LL, Newton AC. A single residue in the C1 domain sensitizes novel protein kinase C isoforms to cellular diacylglycerol production. J Biol Chem. 2007;282:826–830. doi: 10.1074/jbc.C600268200. [DOI] [PubMed] [Google Scholar]
- 35.Lewin NE, Blumberg PM. [3H]Phorbol 12,13-dibutyrate binding assay for protein kinase C and related proteins. Methods Mol Biol. 2003;233:129–156. doi: 10.1385/1-59259-397-6:129. [DOI] [PubMed] [Google Scholar]
- 36.Hess B, Kutzner C, van der Spoel D, Lindahl E. GROMACS 4: Algorithms for Highly Efficient, Load-Balanced, and Scalable Molecular Simulation. J Chem Theory Comput. 2008;4:435–447. doi: 10.1021/ct700301q. [DOI] [PubMed] [Google Scholar]
- 37.Hornak V, Abel R, Okur A, Strockbine B, Roitberg A, Simmerling C. Comparison of multiple Amber force fields and development of improved protein backbone parameters. Proteins: Struct, Funct, Genet. 2006;65:712–725. doi: 10.1002/prot.21123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML. Comparison of simple potential functions for simulating liquid water. J Chem Phys. 1983;79:926–935. [Google Scholar]
- 39.Berendsen HJ, Postma Jv, van Gunsteren WF, DiNola A, Haak J. Molecular dynamics with coupling to an external bath. J Chem Phys. 1984;81:3684–3690. [Google Scholar]
- 40.Hess B, Bekker H, Berendsen HJC, Fraaije JGEM. LINCS: A linear constraint solver for molecular simulations. J Comput Chem. 1997;18:1463–1472. [Google Scholar]
- 41.Essmann U, Perera L, Berkowitz ML, Darden T, Lee H, Pedersen LG. A Smooth Particle Mesh Ewald Method. J Chem Phys. 1995;103:8577–8593. [Google Scholar]
- 42.Xu J, Camacho M, Xu Y, Esser V, Liu X, Trimbuch T, Pan YZ, Ma C, Tomchick DR, Rosenmund C. Mechanistic insights into neurotransmitter release and presynaptic plasticity from the crystal structure of Munc13-1 C1C2BMUN. eLife. 2017;6:e22567. doi: 10.7554/eLife.22567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rizo J, Rosenmund C. Synaptic vesicle fusion. Nat Struct Mol Biol. 2008;15:665–674. doi: 10.1038/nsmb.1450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rhee JS, Betz A, Pyott S, Reim K, Varoqueaux F, Augustin I, Hesse D, Sudhof TC, Takahashi M, Rosenmund C, Brose N. Beta phorbol ester- and diacylglycerol-induced augmentation of transmitter release is mediated by Munc13s and not by PKCs. Cell. 2002;108:121–133. doi: 10.1016/s0092-8674(01)00635-3. [DOI] [PubMed] [Google Scholar]
- 45.Irie K, Masuda A, Shindo M, Nakagawa Y, Ohigashi H. Tumor promoter binding of the protein kinase C C1 homology domain peptides of RasGRPs, chimaerins, and Unc13s. Bioorg Med Chem. 2004;12:4575–4583. doi: 10.1016/j.bmc.2004.07.008. [DOI] [PubMed] [Google Scholar]
- 46.Wimley WC, Creamer TP, White SH. Solvation energies of amino acid side chains and backbone in a family of host-guest pentapeptides. Biochemistry. 1996;35:5109–5124. doi: 10.1021/bi9600153. [DOI] [PubMed] [Google Scholar]
- 47.Wimley WC, White SH. Experimentally determined hydrophobicity scale for proteins at membrane interfaces. Nat Struct Mol Biol. 1996;3:842–848. doi: 10.1038/nsb1096-842. [DOI] [PubMed] [Google Scholar]
- 48.Dougherty DA. Cation-pi interactions involving aromatic amino acids. J Nutr. 2007;137:1504S–1508S. doi: 10.1093/jn/137.6.1504S. discussion 1516S–1517S. [DOI] [PubMed] [Google Scholar]
- 49.de Jesus AJ, Allen TW. The role of tryptophan side chains in membrane protein anchoring and hydrophobic mismatch. Biochim Biophys Acta, Biomembr. 2013;1828:864–876. doi: 10.1016/j.bbamem.2012.09.009. [DOI] [PubMed] [Google Scholar]
- 50.Madhusudan Makwana K, Mahalakshmi R. Implications of aromatic-aromatic interactions: From protein structures to peptide models. Protein Sci. 2015;24:1920–1933. doi: 10.1002/pro.2814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wang QJ, Fang TW, Nacro K, Marquez VE, Wang S, Blumberg PM. Role of hydrophobic residues in the C1b domain of protein kinase C delta on ligand and phospholipid interactions. J Biol Chem. 2001;276:19580–19587. doi: 10.1074/jbc.M010089200. [DOI] [PubMed] [Google Scholar]
- 52.Kelsey JS, Geczy T, Lewin NE, Kedei N, Hill CS, Selezneva JS, Valle CJ, Woo W, Gorshkova I, Blumberg PM. Charge density influences C1 domain ligand affinity and membrane interactions. ChemBioChem. 2014;15:1131–1144. doi: 10.1002/cbic.201400041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Leonard TA, Różycki B, Saidi LF, Hummer G, Hurley JH. Crystal structure and allosteric activation of protein kinase C βII. Cell. 2011;144:55–66. doi: 10.1016/j.cell.2010.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Canagarajah B, Leskow FC, Ho JYS, Mischak H, Saidi LF, Kazanietz MG, Hurley JH. Structural mechanism for lipid activation of the Rac-specific GAP, β2-chimaerin. Cell. 2004;119:407–418. doi: 10.1016/j.cell.2004.10.012. [DOI] [PubMed] [Google Scholar]
- 55.Iwig JS, Vercoulen Y, Das R, Barros T, Limnander A, Che Y, Pelton JG, Wemmer DE, Roose JP, Kuriyan J. Structural analysis of autoinhibition in the Ras-specific exchange factor RasGRP1. eLife. 2013;2:e00813. doi: 10.7554/eLife.00813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Goldenberg NM, Steinberg BE. Surface charge: a key determinant of protein localization and function. Cancer Res. 2010;70:1277–1280. doi: 10.1158/0008-5472.CAN-09-2905. [DOI] [PubMed] [Google Scholar]
- 57.van Meer G, Voelker DR, Feigenson GW. Membrane lipids: where they are and how they behave. Nat Rev Mol Cell Biol. 2008;9:112–124. doi: 10.1038/nrm2330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Pu Y, Garfield SH, Kedei N, Blumberg PM. Characterization of the differential roles of the twin C1a and C1b domains of protein kinase C-delta. J Biol Chem. 2009;284:1302–1312. doi: 10.1074/jbc.M804796200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Rossner S, Fuchsbrunner K, Lange-Dohna C, Hartlage-Rubsamen M, Bigl V, Betz A, Reim K, Brose N. Munc13-1-mediated vesicle priming contributes to secretory amyloid precursor protein processing. J Biol Chem. 2004;279:27841–27844. doi: 10.1074/jbc.C400122200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.