

Abstract
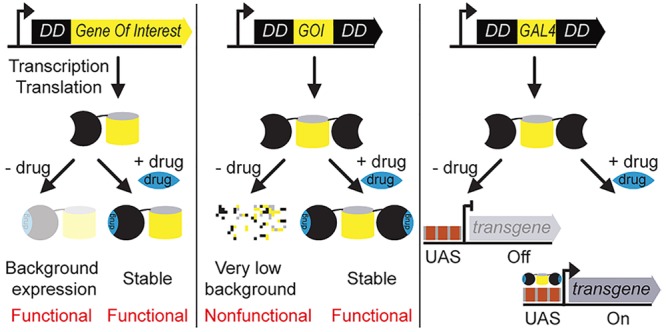
Destabilizing domains (DDs) are genetic tags that conditionally control the level of abundance of proteins-of-interest (POI) with specific stabilizing small-molecule drugs, rapidly and reversibly, in a wide variety of organisms. The amount of the DD-tagged fusion protein directly impacts its molecular function. Hence, it is important that the background levels be tightly regulated in the absence of any drug. This is especially true for classes of proteins that function at extremely low levels, such as lethality genes involved in tissue development and certain transcriptional activator proteins. Here, we establish the uninduced background and induction levels for two widely used DDs (FKBP and DHFR) by developing an accurate quantification method. We show that both DDs exhibit functional background levels in the absence of a drug, but each to a different degree. To overcome this limitation, we systematically test a double architecture for these DDs (DD-POI-DD) that completely suppresses the protein’s function in an uninduced state, while allowing tunable functional levels upon adding a drug. As an example, we generate a drug-stabilizable Gal4 transcriptional activator with extremely low background levels. We show that this functions in vivo in the widely used Gal4-UAS bipartite expression system in Drosophila melanogaster. By regulating a cell death gene, we demonstrate that only the low background double architecture enables tight regulation of the lethal phenotype in vivo. These improved tools will enable applications requiring exceptionally tight control of protein function in living cells and organisms.
Keywords: protein design, gene function, gene expression, gene toxicity, lethality gene
Biological systems have been classically explored by perturbing their genetic components and then determining the phenotypic consequences. These perturbations are typically achieved either through DNA mutagenesis, including random and targeted gene disruption via various techniques, or by RNA interference.1,2 However, such alterations are typically either irreversible or incomplete. This limits the characterization of pathways with toxic or conditionally lethal outputs.
Generic molecular tools that regulate protein stability synthetically at a post-translational level, and in a reversible fashion, are vital for the detailed understanding of conditional functions.3 To this end, various methods have been developed that directly control target protein levels inside living cells.4−12 These include protein degron systems induced by auxin, light, or destabilizing domains (DDs).11−14 Here, we mainly focus on the DD-based degron system.13,14 The DD strategy involves genetically fusing the protein of interest to a small unstable protein domain. This DD-fusion protein is recognized by the cellular protein quality control machinery, which will then degrade the whole fusion protein. However, in the presence of a DD-specific small molecule drug, the DD assumes a folded state and becomes stable, allowing the target protein to carry out its normal biochemical function.15 This methodology therefore allows the possibility to study both loss- and gain-of-function phenotypes of any protein of interest (POI) from a single POI-DD genetic construct.16
Recently, two orthogonal streamlined versions of these DD methods were developed that control target protein levels in a rapid, reversible and tunable fashion.13,14 The first engineered DD is based on a human FK506-rapamycin-binding protein (FKBP) with 107-amino acid residues. Point mutations in FKBP (F36V and L106P) confer instability to fusion partners, which can be rescued by the cell-permeable high affinity small molecule, Shield-1 (Shld-1).13 The second orthogonal DD is engineered from an Escherichia coli dihydrofolate reductase (DHFR) protein with 159-amino acid residues. Similarly, a few key mutations confer instability to DHFR, which can be rescued by the highly permeable small molecule drug, Trimethoprim (TMP).14 These systems have been demonstrated to function in a variety of contexts, including mammalian cell cultures, live mice, viral infections, and in pathogens like Plasmodium and Toxoplasma.13,14,17−22 However, both FKBP and DHFR DDs display high background levels in the absence of any drug.22,23 This basal level of expression is often sufficient for the target fusion protein to carry out its normal biochemical function and thereby precludes the observation of any loss-of-function phenotype.22,23 The double architecture for these DDs has recently been shown to minimize the background expression levels,22,23 but a systematic exploration should further expand the uses of this powerful methodology.
In this study, we establish the background levels of the original FKBP and DHFR DDs by developing an accurate quantification method. Further, by systematically testing double architectures for these original DDs, we show a reduction in the background levels to a very low level compared to the original DDs, in the absence of any drug. We demonstrate the applicability of the least-background double architecture by developing a drug-stabilizable Gal4 expression system for D. melanogaster. We show the functioning of the new drug-inducible Gal4 system in the format of the widely used Gal4-UAS bipartite expression system in vivo. Finally, we demonstrate the tightness of the regulation provided by the least background architecture, by regulating the expression of a highly toxic cell death-inducing transgene in vivo. This proof-of-concept application demonstrates the broader applicability of the double architecture constructs.
Results
A Ratiometric Quantification Method for DD Characterization
The original FKBP and DHFR DD studies compared the destabilization effects relative to the uninduced conditions.13,14 This approach basically omits the background levels as it baselines the destabilization effects by rescaling to the uninduced condition, and further normalizes the resulting data to the induced condition. Hence, we needed an alternative quantification method to directly compare the different DD-fusion constructs in terms of background and inducibility. To this end, we repurposed a commonly used method for expressing multiple genes under the same promoter regulation in eukaryotes.24 This method involves the coexpression of two different fluorescent proteins, mCherry and enhanced Green Fluorescent Protein (eGFP), under the same constitutive action5C promoter, making use of the highly efficient self-cleaving 2A peptide sequence from Thosea asigna virus (T2A).12,24,25 This design ensures the ratiometric expression of both upstream mCherry and downstream eGFP.24 We fuse the individual DDs to eGFP and use the mCherry as a reference to measure the amount of fusion protein synthesized (Figure 1a). By first normalizing the eGFP fluorescence intensity to mCherry intensity, this eliminates any cell-to-cell variability resulting from plasmid vector dosage due to transient transfection, or inherent stochastic gene expression. Finally, normalizing the resulting ratiometric score to a wild-type control eGFP (without DD),26 allows straightforward comparisons of DD-tagged fusion protein abundance at a percentage level.
Figure 1.

Quantitative assessment of the background levels and inducibility of drug-controllable destabilizing domains (DDs) engineered from bacterial DHFR and human FKBP proteins, by flow cytometry. (a) Overview of the quantification method developed to quantify the DD fusion proteins. The gene construct shows a multigene operon constitutively transcribing one mRNA. After translation, three independent proteins (mCherry, DD-eGFP and Puromycin selection marker) are derived from the self-cleaving 2A peptide (T2A). The DD-eGFPs are intrinsically unfolded and are hence degraded; they are only stabilized by adding a small molecule drug (blue diamond) that is specific to each DD. Hence, GFP fluorescence increases relative to the mCherry control upon induction with a drug. (b) Drosophila S2R+ cells with transient expression of these constructs were treated with and without their respective inducer drugs, and the mCherry and eGFP fluorescence was measured by flow cytometry. The histogram shows the normalized mean eGFP fluorescence in the mock-treatments with DMSO or Ethanol (−) and the presence (+) of 10 μM drug (TMP for DHFR; Shld-1 for FKBP). Control eGFP without DD: DMSO and Ethanol and 10 μM TMP and Shld-1. The fold-induction and statistical significance resulting from a t test are summarized with multiple asterisk marks representing the level of significance (**** = P-value ≤ 0.0001 and n.s. = P-value > 0.05). (c) Titration curve of DHFR-eGFP and FKBP-eGFP with various concentrations of TMP and Shld-1 drugs, respectively. Triangles and filled circles are experimental results, whereas the green line is a fitted Hill function. (d) Same as in (b) but for the human embryonic kidneys cell line (HEK293T). The error bars represent the standard deviation over the mean across the n biological replicates (b and d, n = 5 and c, n = 3). N.B., the maximum s.d. observed for DHFR-eGFP is ±0.38%, hence most error bars are invisible in (c).
To quantify the destabilizing effects of the original DDs, we have cloned the individual DDs in frame with a C-terminal eGFP (DHFR-eGFP and FKBP-eGFP) (see Methods). Since fusing DDs to the N-terminus is known to exert a stronger destabilizing effect compared to C-terminal fusions, we therefore chose the former configuration for quantifying the original DDs.13 We transiently transfected these constructs into D. melanogaster S2R+ cells, and measured the resulting fluorescence intensities from mCherry and eGFP proteins after 3 days, by flow cytometry. The DHFR-eGFP showed ∼18% background in the absence of TMP, but could only be stabilized to ∼28% of the wild-type control eGFP without DD, upon induction with TMP (Figure 1b). By contrast, for the FKBP-eGFP fusion, the background level was found to be higher at ∼38% without a drug, while the Shld-1 drug stabilized the level fully (Figure 1b). As expected, the wild-type control eGFP without DD, remained 100% in both the absence and the presence of TMP and Shld-1 drug molecules (Figure 1b). This suggests that the organic solvents used to dissolve the drug molecules, and the drugs themselves have no effect on the control eGFP fluorescence intensity, indicating of no apparent off-target effects (Supplementary Figure S1). Further titrations with various concentrations of the drug molecules showed that the DD-fusion protein levels can be tuned to a desired range using appropriate concentrations of the drugs, albeit to a low fold-induction (Figure 1c).
To further validate the fluorescence-based ratiometric quantification method by an orthogonal method, we directly quantified the protein abundance levels of mCherry and eGFP or FKBP-eGFP, in the absence and presence of the stabilizing drug, by performing a Western blot (Supplementary Figure S2a). This quantification method also involved the same data processing steps as the fluorescence-based quantification method, but instead utilized the protein band intensities quantified by an immunoblot (see Methods). Interestingly, this quantification method is consistent with the fluorescence-based quantification (Supplementary Figure S2b). In particular, no significant difference (P-value = 0.4) in the background levels are observed between the two orthogonal methods, in the absence of drug. Moreover, in the presence of drug, both methods confirm the level of abundance of FKBP-eGFP back to that of wild-type levels, which is within the measurement error. This confirms that the differences observed among the different DD-fusion constructs in the fluorescence based ratiometric quantification method are indeed reliable. Since the fluorescence-based quantification method involves the high-throughput collection of single-cell data by a flow cytometry, we chose this method for the subsequent quantification of the DD constructs.
The high background levels observed for DHFR-eGFP (∼18%) and FKBP-eGFP (∼38%) DD constructs could be due to transient transfection: the multiple copies of the DD constructs in individual cells, resulting from transient transfection, would result in more mRNA and this might ultimately produce more fusion protein, that could overload the proteasome machinery.13,22 To test this, we created stable D. melanogaster S2R+ cell lines of DD constructs by utilizing the coexpressed Puromycin selection marker (see Methods and Figure 1a). We measured the fluorescence intensities of mCherry and eGFP proteins after 3 days of the drug treatment in the stable lines, using flow cytometry (Supplementary Figure S3). Both in the absence and the presence of drug molecules, the DHFR- and FKBP-DD constructs displayed low background and low stabilization levels. However, this resulted in almost exactly the same level of inducibility to that of the transient transfection-based quantification. Based on this, we chose to characterize subsequent constructs in transient state.
The background levels observed for DHFR-eGFP (∼18%) and FKBP-eGFP (∼38%) are rather high in D. melanogaster S2R+ cells. Given that these systems have been widely used in the several organisms and cell types,13,14,17−22 they may have been performing better than in our observations. This suggests that there may be differential degradation of the unfolded protein by the ubiquitin–proteasome system in different organisms.27 To test this, we subcloned the DHFR-eGFP and FKBP-eGFP constructs into a mammalian expression vector (see Methods). We further quantified the resulting constructs in human embryonic kidney 293T cell line (HEK293T). Interestingly, we observed only ∼0.9% and ∼10% background levels for DHFR-eGFP and FKBP-eGFP, respectively (Figure 1d). This is a large reduction in the background levels for these constructs in HEK293T versus S2R+ cells. However, in the presence of their respective drug molecules, DHFR-eGFP could only be partially stabilized (∼8% of the wild-type abundance), while FKBP-eGFP stabilized to the wild-type level of abundance. As a result, these original DDs show 9- and 11-fold inducibility in mammalian cells (Figure 1d). However, qualitatively, their properties remain the same as in D. melanogaster S2R+ cells: DHFR-DD displays low background in the absence of a drug, while FKBP-DD reaches the wild-type level of abundance (Figure 1d).
To further assess the accuracy of ratiometric quantification method, we compared it to a simpler relative normalization by omitting the mCherry fluorescence signal, on the data obtained for the DHFR-eGFP and FKBP-eGFP constructs in D. melanogaster S2R+ cells (Figure 1b) and mammalian HEK293T cells (Figure 1d). This revealed a ∼13- and ∼26-fold induction in D. melanogaster S2R+ cells (Supplementary Figure S4a), whereas there was a ∼79 and ∼153-fold induction in mammalian HEK293T cells (Supplementary Figure S4b). This suggests that simple relative normalization overestimates the fold-induction by omitting the actual background levels. Furthermore, there is an increase in the variability of eGFP fluorescence observed particularly for the uninduced condition (Supplementary Figure S4). This can be attributed to the noise resulting from the plasmid vector dosage of the transient transfection or inherent stochastic gene expression, which could not be filtered-out due to the omission of the reference mCherry fluorescence intensity. Overall, the quantification revealed that the DHFR-DD displays lower background levels than the FKBP-DD in an uninduced state. However, the FKBP-DD could be stabilized fully upon induction with a drug, whereas the DHFR-DD could not. Notably, the ratiometric quantification method allows a highly reproducible comparison between DD constructs.
A Double DD Architecture Reduces the Background Levels and Improves the Fold-Induction
An ideal DD should have 0% background level in the absence of a drug, and should be stabilized back to 100% upon induction with the drug. Each of the two original DDs possesses one good property that is close to an ideal DD: DHFR displays a low background levels, while FKBP can be stabilized back to 100% with the drug (Figure 1b and 1d). We therefore reasoned that creating chimeric DDs by combining the individual DDs might capture their best qualities. Consequently, we fused the gene coding for the DHFR and FKBP in frame to the N-terminus of eGFP to create a chimeric DD architecture, DHFR-FKBP-eGFP. This architecture resulted in a relatively stable fusion protein, which showed a high background of ∼29% in the absence of both drug molecules. Moreover, this could only be stabilized back to ∼38%, resulting in a rather poor inducibility of 1.3-fold (Supplementary Figure S5). However, the introduction of a single zinc finger domain as a structured linker (zfln; 33-amino acid residues) in between the DHFR- and FKBP-DD, reduced the background level from ∼29% to ∼18% (Supplementary Figure S5). This is same as the background level of the parent DHFR-DD (Figure 1b), thus demonstrating the acquisition of desired property from the parent DHFR-DD. However, this chimeric architecture could only be stabilized back to ∼39% upon induction with both drug molecules. This percentage of stabilization is higher than the parent DHFR-DD (28%, Figure 1b), but does not reach anywhere near the 100% of the other parent FKBP-DD.
Because of the promising results with the structured linker separating the DDs, we next fused the gene coding for the FKBP-DD to the C-terminus of the DHFR-eGFP construct, to make a DHFR-eGFP-FKBP chimeric DD architecture. The quantitative assessment of this design revealed a much lower background level of ∼10% in the absence of drugs, while stabilization with both TMP and Shld-1 could be achieved up to ∼42% (Figure 2a). In the absence of drugs, the chimera degraded ∼2- and ∼4-times more efficiently than the parent DHFR- and FKBP-DD, respectively, hence drastically reducing the background levels (Figure 2a). Additionally, the stability of DHFR-eGFP-FKBP was increased by a factor of ∼2, when compared to the parent DHFR-DD. Overall, this increased the inducibility range to a factor of ∼4-fold, which is an improvement on the parent DDs. In the presence of just one drug, DHFR-eGFP-FKBP could only be partially stabilized to ∼21% (Supplementary Figure S6). The maximum stabilization was achieved (∼42%) only in the presence of both TMP and Shld-1 drug molecules, demonstrating the specificity of the two orthogonally acting drugs (Supplementary Figure S6).
Figure 2.

Quantitative assessment of the background levels and inducibility of the double architectures derived from the parent DHFR- and FKBP-DD in Drosophila S2R+ cells. (a) Histogram showing the normalized mean eGFP fluorescence in mock-treatments with DMSO and/or Ethanol (−) and the presence (+) of 5 μM Shld-1 and/or 5 or 10 μM TMP drug inducer molecules. The horizontal lines indicate the mean eGFP fluorescence of parent DHFR (dot) and FKBP (hyphen) DDs in the absence of drug. (b) Titration curve for chimeric DDs with TMP and/or Shld-1. Other labels are as for Figure 1.
Since the DHFR-DD has a higher propensity to degrade than FKBP-DD, this might account for the incomplete stabilization of the DHFR-eGFP-FKBP chimeric DD. We therefore made a double DD architecture, FKBP-eGFP-FKBP. Indeed, the quantitative assessment revealed a strong degradation without drug (∼18% background level), and a full stabilization was achieved with Shld-1. This design also degraded ∼2-times more efficiently than the parent FKBP-DD, in the absence of the drug, hence also reducing the background level (Figure 2a). Overall, this resulted in a ∼7-fold inducibility, hence widening the tunability (Figure 2a and b).
Although the DHFR-eGFP-FKBP and FKBP-eGFP-FKBP DD architectures degraded much better than the parent DDs in the absence of drug, there was still considerable background levels (10–18% background expression). This could be attributed to the lower propensity of FKBP-DD to degrade in the absence of a drug, compared with DHFR-DD. We therefore made a double DHFR-DD architecture, DHFR-eGFP-DHFR. This variant revealed a very strong degradation in the absence of drug (∼1% background level), and was stabilized back to 16% in the presence of TMP. Since this design degrades ∼17-times more efficiently in the absence of drug than the parent DHFR-DD, it was the lowest background DD system tested up to that point (Figure 2a). However, with TMP, this architecture was stabilized ∼2-fold less than the parent DHFR-DD. Nevertheless, this resulted overall in a ∼15-fold inducibility, making it the most tunable DD architecture (Figure 2a and b).
Recently, new DHFR-DD variants were engineered that are described as performing better in organisms that grow optimally at room temperature.28 We therefore further quantified the background levels and inducibility of these DHFR-DD variants. Our quantitation revealed that the DHFR-DD variant 07 (DHFR07) displayed a high background level of ∼13% (Figure 3a). Despite this, TMP could stabilize the level back to ∼51%, which is higher than DHFR and results in a ∼4-fold induction. By contrast the other construct, DHFR-DD variant 22 (DHFR22), displayed only ∼3% background expression, which is the lowest background among the parent DDs. Upon induction with TMP, this variant could be stabilized back to ∼37%, resulting in an overall induction of ∼11-fold, which is the highest among the parent DDs (Figure 3a).
Figure 3.

Quantitative assessment of the background levels and inducibility of DHFR-DD variants in Drosophila S2R+ cells that are optimized for room temperature.28 (a) Histogram showing the normalized mean eGFP fluorescence for the DHFR-DD variants 07 and 22 in mock-treatments with DMSO (−) and the presence (+) of 100 μM TMP. (b) Histogram showing the normalized mean eGFP fluorescence for the double architectures derived from the parent DHFR-DD variants 07 and 22, and with FKBP-DD. The horizontal lines indicate the mean eGFP fluorescence of parent DHFR-22 (dot) and DHFR-07 (hyphen) DDs in the absence of drug. The error bars represent the standard deviation over the mean across the 5 biological replicates. Other labels are as for Figure 1.
Since these new DDs appeared promising, we systematically created the double architectures for DHFR07, DHFR22 and FKBP DDs. We quantified the resulting constructs for their background levels and inducibility (Figure 3b). As before, there were trade-offs between background and induction levels, resulting in fold-inductions between ∼5- and ∼30-fold. Most interestingly, the double architecture DHFR22-eGFP-DHFR22 displayed as low as ∼0.5% background, making it the least background DD architecture described so far. Upon induction with TMP, the level could be stabilized back to ∼12%, which is lower than the parent DHFR22-DD (37%), but still results in a ∼25-fold induction.
In both single and double architectures, the DD sequences occur immediately downstream and upstream of the self-cleaving T2A peptide sequence, respectively. Since the cleavage of polypeptides occurs during translation, DD sequences might influence the processing efficiency of T2A sequences to produce independent polypeptides. To verify this, we performed a Western blot analysis on both single and double DD architecture constructs (Supplementary Figure S7). This confirms the correct processing of the T2A sequences.
We finally chose the least-background DHFR22 variant to explore further the single and double DD architectures in vivo. Because for classes of proteins that function at extremely low levels such as transcriptional activators,29 it is preferable to have low background levels, rather than the maximum possible stability, in order to completely suppress the function in an uninduced state.22
The Least-Background DD Architecture Tightly Regulates a Lethal Phenotype in Drosophila
One of the most challenging applications of the least-background DD architecture is to apply it to signal-amplifying transcriptional activators.29 These proteins often function at extremely low levels to initiate the transcription of target genes.30 The Gal4 transcriptional activator regulates transgene expression under upstream activating sequence (UAS) promoter, which is a widely used expression system in Drosophila.31−33 Despite much progress with the existing Gal4-UAS system, there is still a need to improve its functionality, especially to temporarily express genes that result in lethality upon constitutive expression.34−38 This is challenging because of the issue of “leakiness”: even low background expression cannot be tolerated due to the lethal phenotype.
We sought to compare the background levels of the original and double architecture DDs in vivo. Hence, we constructed both DHFR22-Gal4 (1xDHFR) and DHFR22-Gal4-DHFR22 (2xDHFR) architectures, by fusing the DHFR22-DD in frame with the transcriptional activator Gal4VP16 (Figure 4a). To facilitate the easy monitoring of the drug-induced phenotype, we spatially restricted the expression of these constructs to the Drosophila eye. This was achieved by placing the expression of the constructs under the control of the glass multiple reporter (GMR) eye-specific enhancer (Figure 4a). We created transgenic flies from these two constructs, by inserting them into the same genomic locus, to minimize the host chromatin context influence on their expression.39
Figure 4.

The drug-stabilizable Gal4 variants 1xDHFR22 and 2xDHFR22 function in vivo in the format of the widely used Gal4-UAS bipartite expression system for Drosophila. (a) Schematic representation of the constructs used to create drug-inducible Gal4 driver lines. 1xDHFR22 encodes the single DHFR-DD architecture with the DHFR variant 22,28 as a fusion to the Gal4VP16 transcription factor. A nuclear localization signal (NLS) is added N-terminally and expression is driven by the eye-specific enhancer, glass multiple reporter (GMR). Similarly, 2xDHFR22 encodes the double architecture of DHFR22-DD. (b) A population of F1 progenies from 1xDHFR22 and 2xDHFR22 genetic crosses with UAS-eGFP reporter line was allowed to feed on standard fly food supplemented with DMSO (mock-treatment) or various concentrations of TMP for 5-days. A negative control population was derived from the Curly wings phenotype resulting from a dominant CyO marker from the heterozygote 1xDHFR or 2xDHFR driver line. Samples of the population were imaged by fluorescence microscopy. Representative images of adult fly eyes display an increase in eGFP fluorescence intensity as a function of the inducer TMP. The upper, middle and lower panels display, respectively, F1 progenies with genotypes 1xDHFR22;UAS-eGFP, 2xDHFR22;UAS-eGFP and CyO;UAS-eGFP. Scale bar: 1 mm. (c) Quantification of eGFP fluorescence intensity in the Drosophila adult eyes either mock-treated with DMSO or with various concentrations of TMP. Data are presented as the mean fluorescence detected per eye. The statistical significance resulting from a one-way ANOVA and Tukey’s post hoc test is summarized with asterisk marks representing the level of significance (n.s.= P-value > 0.05, * = P-value ≤ 0.05, *** = P-value ≤ 0. 001, and **** = P-value ≤ 0.0001) on the indicated data set. The error bars represent the standard deviation over the mean across the biological replicates (n = 8–76 individual eyes per dose).
To test the inducibility of the DHFR22-DD architectures in vivo, we genetically crossed the driver transgenic fly lines to a reporter line that encodes eGFP under the UAS promoter. Feeding the F1 adult flies on standard fly food, supplemented with various concentrations of TMP, resulted in the induction of eGFP reporter expression. The level of eGFP fluorescence observed in the Drosophila eye increased with the amount of TMP in the food (Figure 4b and c). However, the mock-treated population from the original DHFR22-DD architecture (1xDHFR) showed no significant difference in the level of eGFP fluorescence compared to the double (2xDHFR) architecture. This population in turn did not show a significant difference in eGFP expression when compared with that of the negative control population (Figure 4b and c, Curly of Oster, CyO;UAS-eGFP). The latter negative control population encodes dominant Curly wings phenotype CyO marker instead of the 1xDHFR or 2xDHFR driver transgenes. As expected, this negative control population further showed no significant difference in eGFP expression among the various TMP drug conditions. The observation that there was no significant eGFP fluorescence difference between the mock-treated 1xDHFR, 2xDHFR and a negative control population indicates that the fluorescent microscopy used to quantify the eGFP fluorescence intensity is not able to differentiate the weak background eGFP signal from that of the cellular autofluorescence.40 Because of this reason, we could not assess the background levels at this stage (but see below). Overall, the induction experiment demonstrated that the DHFR-DD-Gal4 system is functional in a whole animal.
To further assess the background levels of both the original and double DHFR22-DD architectures, we applied the driver lines in regulating a lethal phenotype, induced by the expression of a pro-apoptotic gene. For this, we chose head involution defective gene, hid, which executes a cell death pathway in Drosophila.41 The F1 larvae from 1xDHFR and 2xDHFR crosses with UAS-hid line, were exposed to mock- and drug-treated conditions to test for background and drug-induced expression of hid in the eye. This resulted in the expected phenotype of structural defects in the eye in a drug-dependent manner (Figure 5). However, the original single DHFR22-DD architecture (1xDHFR) showed a mild phenotype even in the absence of TMP. The mild phenotype observed in 1xDHFR22;UAS-hid genotype progenies shows complete penetrance (Figure 5b). Also, in the in vitro data, the original architecture displayed a higher undegraded background level compare to the double architecture (Figure 3). This can be attributed to the background expression of 1xDHFR22 that could activate the transcription of hid even in the absence of TMP. This is the main limitation of single DDs. By contrast, the double DHFR22-DD architecture (2xDHFR) had a normal wild-type eye phenotype in the absence of TMP (Figure 5). Particularly, we observed no progenies with the 2xDHFR22;UAS-hid genotype that displayed eye defects in absence of TMP (Figure 5b). This suggests that 2xDHFR is completely silent in the absence of TMP.
Figure 5.
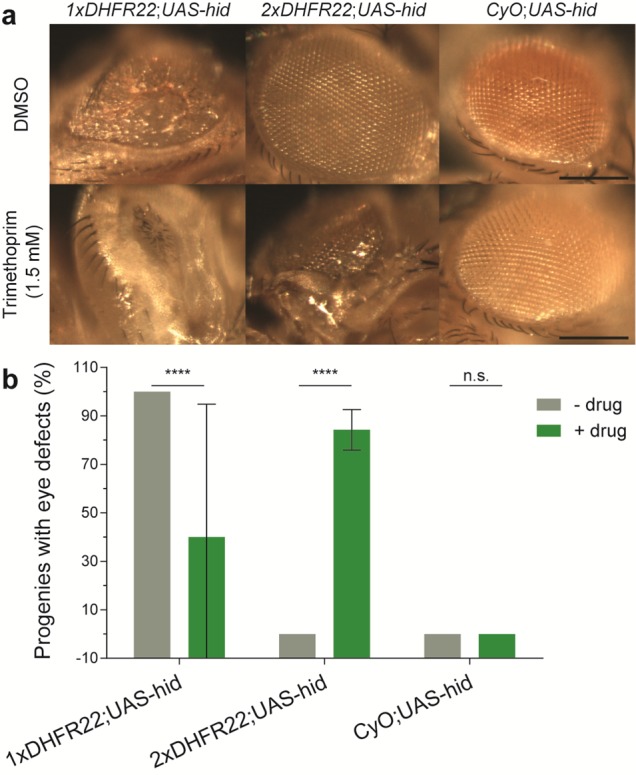
A drug-stabilizable Gal4 driver with double DHFR22-DD architecture regulates hid cell death gene expression tightly without any observable background. Genetic crosses were set up between a Gal4-driven reporter line (UAS-hid, encoding a pro-apoptotic gene hid under the UAS enhancer) and drug-inducible Gal4VP16 variants: 1xDHFR22 and 2xDHFR22 under a GMR enhancer. The resulting F1 population of third instar larvae were allowed to feed on standard fly food supplemented with DMSO (mock-treatment) or 1.5 mM of TMP. Eyes from emerged adult flies were imaged by bright-field microscopy. A negative control population was derived from the Curly wings phenotype resulting from a dominant CyO marker from the heterozygote 1xDHFR or 2xDHFR driver line. (a) Representative images showing structural defects in the adult eyes are displayed. Scale bar: 30 μm. (b) Quantification of the structural defects observed in the Drosophila adult eyes. The statistical significance of t tests are summarized with multiple asterisk marks representing the level of significance (n.s. = P-value > 0.05, and **** = P-value ≤ 0.0001), on each indicated data set. The error bars represent the standard deviation over the mean across the five independent experiments (n = 2–48 flies). N.B. several conditions produced consistent results across the individual experiments (i.e., zero s.d. and hence no error bars are displayed).
In the presence of 1.5 mM TMP, we observed complete loss of eye in both single and double architecture DHFR22 constructs. However, we observed no or very few emergent adult flies with the 1xDHFR22;UAS-hid genotype, in proportion to the CyO;UAS-hid genotype (expected 50% each according to Mendelian inheritance principles, given the crosses between heterozygote 1xDHFR22;CyO driver line and homozygote UAS-hid;UAS-hid reporter line). These progenies show a normal development until the pupal stage, but no or very few adult flies emerge from the pupal case. This is indicative of the lethality associated with higher expression of hid. This result is consistent with the constitutively active Gal4 driver line. The F1 larvae from GMR-Gal4 (Gal4 without DHFR22) and UAS-hid crosses also show a complete penetrance of lethality (data not shown). However, in the case of 1xDHFR22;UAS-hid genotype flies, we do rarely observe the eclosion of a few adult flies from the pupal case (Figure 5b, high error bar). This could be due to the low exposure of these larvae to TMP. The larvae that are in the later stage of the third instar stop feeding and climb away from their food for pupariation. Such larvae eventually eat less TMP compared to the first and second instar larvae, and hence appear to have sublethal levels of hid expression. This manages to rescue the lethality associated with high expression of hid. These larvae show complete loss of eyes upon maturation in to adult stage. On the other hand, flies with 2xDHFR22;UAS-hid genotype did not show any lethality in the 1.5 mM TMP condition. This suggests that the expression level of hid achieved with the 2xDHFR22 construct in 1.5 mM TMP is below the lethal level. Importantly, this is sufficient to induce eye defects in adult flies (Figure 5a). However, with TMP, we observed that on average 84% of progenies with the 2xDHFR22;UAS-hid genotype displayed the eye defects phenotype (Figure 5b). This is because the larvae that are in the later stage of the third instar stop feeding and climb away from their food for pupariation immediately upon inoculation of the fly food with TMP. This effect may be tuned to attain 100% by excluding the third instar larvae during inoculation. Moreover, a negative control encoding a CyO dominant marker showed a wild-type phenotype both in mock- and drug-treated conditions, suggesting that the TMP drug has no apparent off-target effects. Taken together, the in vivo data also support the improvement of the double DHFR22-DD architecture over the original in regulating a developmental lethal phenotype.
Discussion
In this study, we have developed an accurate quantification method to compare different DD constructs more directly, and found that the original DHFR-DD displays less background, whereas the FKBP-DD can be stabilized fully. We took advantage of these properties to make double architectures that drastically reduce background levels, albeit with some trade-off with respect to the maximal levels of stabilization achieved with drug. Nonetheless, the double architectures are suitable for applications requiring extremely low background levels over the maximum stability trait.22
We demonstrated the possible use of the least-background DD architecture by constructing a new drug-inducible transgene expression system for the widely used model organism, D. melanogaster. By implementing a drug-inducible control of a cell death gene, we demonstrated the advantage of the double architecture in tight regulation of transgene expression. This new drug-inducible Gal4-UAS expression system offers the potential for tighter spatial and temporal control of transgene expression. For example, here we used an eye-specific enhancer to drive expression of the DD-Gal4, but this can be easily adapted to generate further tissue-specific driver lines, to allow for wider spatial–temporal control of UAS-transgenes.42
The regulation of a transcription factor activity in a drug-dependent manner provides a direct ability to control genetic program of a cell at the time of one’s choosing. A plethora of well-characterized transcription factors is known, but small-molecule regulation is still not widely used. Our study demonstrates the use of DD elements to build these kinds of useful tools. Thus, the low-background DD architectures developed here should be generally applicable. Furthermore, the drug-mediated creation of tunable and reversible protein aggregates using DDs serves as a good model system to understand protein aggregation associated pathologies.43 Low background DD architectures could help in discovering the fundamental cellular mechanisms involved in unfolded protein clearance and unfolded protein response mounted cellular stress.44 These could also be used to explore the differential degradation of unfolded proteins by the ubiquitin–proteasome system in different organisms.
The low background expression that is achieved by the double architecture can be attributed to the specific configuration of the individual DDs in the architectures. The low background effect is clearly not resulting from a simple dosage effect of the multiple DD copies. For instance, the chimeric DD architectures DHFR-FKBP-eGFP and DHFR-eGFP-FKBP show different background expression and inducibility (Supplementary Figure S5 and Figure 2, respectively), indicating that the configuration of the individual DDs in the architectures matters. Furthermore, the concatenation architecture resulting from two or three copies of FKBP-DD in a row at the N- and/or C-terminus of the protein also showed no significant improvement over the single and double copy architecture.22,45 Taken together, these observations indicate that there may exist a context-dependent recognition of the unfolded protein domains in a protein, by the cell degradation system. In other words, given the differences in the degradation abilities of the fusion proteins with multiple unfolded domains, either in the N- or C-termini, or in both the N- and C-termini, this indicates independent mechanisms of recognition or degradation.13,46,47
In conclusion, these improved DD architectures should further widen the broad range of applications that currently rely on the control of protein function with small-molecules13,14,17−21 and may also reopen the studies that have been previously hindered by the high-background levels of the parent DDs.
Methods
DNA Constructs
The DHFR- and FKBP12-based destabilization domains used in this study were originally developed by the Wandless lab.13,14,28 The following DD variants were used in this study: DHFR (Addgene plasmid #29325), DHFR07 (#47080), DHFR22 (#47076) and FKBP (#31763). Corresponding DNA sequences were fused either 5′ or 3′ of the eGFP gene by overlap PCR extension and were cloned into the pAc5-STABLE2-puro24 multicistronic vector, using XbaI and HindIII restriction enzymes, and were verified by sequencing. The selected constructs were further cloned into the mammalian expression vector pTargeT by TA cloning, for measuring in human cell lines.
To create constructs for use in Drosophila in vivo, first a basic vector backbone was constructed by cloning the overlap PCR product from glass multiple reporter, HSP70 basal promoter, and simian virus 40 polyA sequences, with appropriate restriction sites, into the pCR4-TOPO vector (Invitrogen). The cloned cassette in this vector was flanked by short 40 base pairs (bps) attB recombinase sites,48 allowing site specific integration into the genome via ΦC31 recombinase mediated cassette exchange.
The DHFR22-DD DNA sequence was fused either to 5′ or 3′ of the gal4VP16 gene by overlap PCR extension, and was further fused to mCherry sequence, in frame, via the self-cleaving peptide sequence T2A, derived from Thosea asigna.24 The full-length PCR products were cloned into the basic vector backbone using AgeI and HindIII restriction enzymes.
Cell Culture, Transfection and Antibiotic Treatment
Drosophila S2R+ cells49 were obtained from the Drosophila Genome Resource Center (DGRC). Cells were cultured in Drosophila Schneider’s medium (Gibco), supplemented with 10% of fetal bovine serum (FBS), 1% of penicillin/streptomycin, at room temperature, in a humidified chamber. The human embryonic kidney 293T cell line (HEK293T) was obtained from American Type Culture Collection (ATCC), and was cultured in DMEM (Gibco) supplemented with 10% FBS, 1% of penicillin/streptomycin at 37 °C in a humidified incubator with 5% CO2.
Transfections were performed using Effectene reagent (Qiagen), following the manufacturer’s instructions, using 0.1 μg of DNA with a DNA:Enhancer ratio of 1:8 and DNA:Effectene ratio of 1:10. 50 μL of the transfection complexes were added to each well in a 96-well plate seeded with 1 × 105 (S2R+) cells or 1 × 104 (HEK293T) in 100 μL of medium. All transfections were performed in triplicates to quintuplicates.
For stable cell line creation, transfections were performed in Drosophila S2R+ cells using 0.4 μg of DNA with the same DNA:Enhancer and DNA:Effectene ratio, in a 6-well plate seeded with 4 × 106 cells. At 72 h post-transfection, the cells were selected in 10 μg/mL puromycin for a further 11 days.
Drug Treatment and Flow Cytometry
Cell cultures were titrated with various concentrations of the appropriate orthogonal drugs. 1.5 μL of different concentrations of 100× concentrated drug solution (TMP in Dimethyl sulfoxide (DMSO) and/or Shld-1 in absolute Ethanol) was added to the wells to achieve the final concentration. For cultures without any drug, corresponding volumes of solvents (DMSO and/or Ethanol) were added. The plates were incubated for 72 h before harvesting for measuring the fluorescence by flow cytometry: fluorescence measurements were performed on a BD LSRFortessa cell analyzer flow cytometer. The eGFP fluorescence was measured using a 488 nm excitation laser and a 515–545 nm emission filter, while mCherry fluorescence used 561 nm excitation and 600–620 nm emission. A minimum of 10 000 cells was measured from each sample. From these single-cell fluorescence intensities, we further computed the mean fluorescence intensity per cell representing the population average for both mCherry and eGFP separately using the FlowJo software (Treestar, Inc., San Carlos, CA). The mean eGFP fluorescence values were normalized to mCherry fluorescence intensities after subtracting for autofluorescence derived from mock-transfected cells. The resulting ratiometric scores were further converted to %, based on the ratiometric score of the control eGFP, without DDs or drug, but with the respective solvents of the drugs.
Western Blot and Quantitative Analysis
D. melanogaster S2R+ cells were harvested in 1x Laemmli sample buffer 72 h post-transfection. The lysates were resolved on a AnykD Criterion TGX Stain-Free protein gel (BioRad). The separated protein bands were subsequently transferred onto a Nitrocellulose membrane using an iBlot gel transfer device (Invitrogen). The FLAG-tagged mCherry and eGFP were detected using the primary antibodies anti-FLAG (F3165; Sigma), anti-α-Tubulin (T5168; Sigma) and anti-eGFP (Roche), respectively, at a 1:5000 dilution, and a peroxidase-conjugated sheep antimouse secondary antibody (Jackson ImmunoResearch) at a 1:50 000 dilution. Signals were detected using SuperSignal West Pico PLUS chemiluminescent substrate (Thermo Scientific) and a LAS-3000 imaging system (Fujifilm).
The mean protein band intensity values were extracted using freely available Fiji software.50 The eGFP intensity values were normalized to mCherry protein band intensities after subtracting background. The resulting ratiometric scores were further converted to %, based on the resulting ratiometric score of the control eGFP (without DDs or drug, but with Ethanol).
Transgenesis
In vivo demonstration constructs, 1xDHFR22 and 2xDHFR22, were inserted into the P{attP.w[+].attP}JB38FP51 (Bloomington Drosophila Stock Center, BDSC#27388) landing site on the second chromosome.51 This locus contains the mini-white gene, flanked by inverted attP sites. Insertion was done via ΦC31 recombinase mediated cassette exchange at BestGene Inc. (Chino Hills, CA, USA).
Protein Induction in Flies
Flies were reared at room temperature and raised on standard food. In addition to the transgenic flies created in this study, we used two published reporter lines: w[*]; P{w[+mC] = 10XUAS-IVS-GFP-WPRE}attP2 (BDSC#32202)52 and w[*], UAS-hid/FM6B.
For experiments involving TMP treatment, standard fly food was mixed with different concentrations of 100× concentrated TMP, after liquefying the food in the microwave. Fluorescence experiments were performed using 1 to 5 day-old adult progenies, obtained from the genetic crosses between heterozygote 1xDHFR22;CyO and 2xDHFR22;CyO driver lines with a homozygote UAS-eGFP reporter line. After 5 days in the food vials with various concentrations of TMP, fly heads were imaged for fluorescence. For experiments involving eye structural defects, larvae (from heterozygote 1xDHFR22;CyO and 2xDHFR22;CyO with homozygote UAS-hid reporter line crosses) were inoculated into food vials with DMSO or 1.5 mM TMP. Larvae were incubated in these vials until the eclosion of adult flies. Emerged adult fly eyes were imaged.
Imaging and Quantitative Analysis
All the microscopy images were acquired using either Leica MZ16 F or Zeiss Axio Zoom V16 fluorescence stereo microscope, mounted with a DFC300 FX or Axiocam 506 mono digital cameras, respectively. For representative eGFP fluorescence images, background intensities were subtracted and false colored, further linearly adjusted for levels using freely available Fiji software.50 Images were finally assembled using Adobe Illustrator version CS6.
From all the acquired eGFP fluorescence images, quantitative fluorescence intensities were extracted as follows: The region of interest was drawn around the fly eye and the mean fluorescence intensity of a pixel in this region was calculated for each eye. The background intensities were subtracted, by calculating mean pixel intensities from the areas around the Drosophila head object, and plotted on a graph, by calculating the mean over all the data for each condition.
Acknowledgments
MK was funded by FP7 ERC 201249 ZINC-HUBS and MI is funded by New Investigator Award No. WT102944 from the Wellcome Trust U.K. We are grateful to Tony Southall, Giorgio Gilestro and Gloria Rudenko for critical reading of the manuscript.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acssynbio.7b00302.
Figures S1–S7; Supplementary DNA sequences (PDF)
Author Contributions
MK conceived the study, performed the experiments and wrote the manuscript. MI supervised the work and wrote the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Fire A.; Xu S.; Montgomery M. K.; Kostas S. A.; Driver S. E.; Mello C. C. (1998) Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature 391, 806–811. 10.1038/35888. [DOI] [PubMed] [Google Scholar]
- St Johnston D. (2002) The art and design of genetic screens: Drosophila melanogaster. Nat. Rev. Genet. 3, 176–188. 10.1038/nrg751. [DOI] [PubMed] [Google Scholar]
- Campbell A. E.; Bennett D. (2016) Targeting protein function: the expanding toolkit for conditional disruption. Biochem. J. 473, 2573–2589. 10.1042/BCJ20160240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy F.; Johnston J. A.; Varshavsky A. (1999) Analysis of a conditional degradation signal in yeast and mammalian cells. Eur. J. Biochem. 259, 244–252. 10.1046/j.1432-1327.1999.00024.x. [DOI] [PubMed] [Google Scholar]
- Dohmen R. J.; Wu P.; Varshavsky A. (1994) Heat-inducible degron: a method for constructing temperature-sensitive mutants. Science 263, 1273–1276. 10.1126/science.8122109. [DOI] [PubMed] [Google Scholar]
- Banaszynski L. A.; Wandless T. J. (2006) Conditional control of protein function. Chem. Biol. 13, 11–21. 10.1016/j.chembiol.2005.10.010. [DOI] [PubMed] [Google Scholar]
- Kohler J. J.; Bertozzi C. R. (2003) Regulating cell surface glycosylation by small molecule control of enzyme localization. Chem. Biol. 10, 1303–1311. 10.1016/j.chembiol.2003.11.018. [DOI] [PubMed] [Google Scholar]
- Inoue T.; Heo W. D.; Grimley J. S.; Wandless T. J.; Meyer T. (2005) An inducible translocation strategy to rapidly activate and inhibit small GTPase signaling pathways. Nat. Methods 2, 415–418. 10.1038/nmeth763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneekloth J. S. Jr.; Fonseca F. N.; Koldobskiy M.; Mandal A.; Deshaies R.; Sakamoto K.; Crews C. M. (2004) Chemical genetic control of protein levels: selective in vivo targeted degradation. J. Am. Chem. Soc. 126, 3748–3754. 10.1021/ja039025z. [DOI] [PubMed] [Google Scholar]
- Janse D. M.; Crosas B.; Finley D.; Church G. M. (2004) Localization to the proteasome is sufficient for degradation. J. Biol. Chem. 279, 21415–21420. 10.1074/jbc.M402954200. [DOI] [PubMed] [Google Scholar]
- Nishimura K.; Fukagawa T.; Takisawa H.; Kakimoto T.; Kanemaki M. (2009) An auxin-based degron system for the rapid depletion of proteins in nonplant cells. Nat. Methods 6, 917–922. 10.1038/nmeth.1401. [DOI] [PubMed] [Google Scholar]
- Hermann A.; Liewald J. F.; Gottschalk A. (2015) A photosensitive degron enables acute light-induced protein degradation in the nervous system. Curr. Biol. 25, R749–750. 10.1016/j.cub.2015.07.040. [DOI] [PubMed] [Google Scholar]
- Banaszynski L. A.; Chen L. C.; Maynard-Smith L. A.; Ooi A. G.; Wandless T. J. (2006) A rapid, reversible, and tunable method to regulate protein function in living cells using synthetic small molecules. Cell 126, 995–1004. 10.1016/j.cell.2006.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwamoto M.; Bjorklund T.; Lundberg C.; Kirik D.; Wandless T. J. (2010) A general chemical method to regulate protein stability in the mammalian central nervous system. Chem. Biol. 17, 981–988. 10.1016/j.chembiol.2010.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egeler E. L.; Urner L. M.; Rakhit R.; Liu C. W.; Wandless T. J. (2011) Ligand-switchable substrates for a ubiquitin-proteasome system. J. Biol. Chem. 286, 31328–31336. 10.1074/jbc.M111.264101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Housden B. E.; Muhar M.; Gemberling M.; Gersbach C. A.; Stainier D. Y.; Seydoux G.; Mohr S. E.; Zuber J.; Perrimon N. (2017) Loss-of-function genetic tools for animal models: cross-species and cross-platform differences. Nat. Rev. Genet. 18, 24–40. 10.1038/nrg.2016.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banaszynski L. A.; Sellmyer M. A.; Contag C. H.; Wandless T. J.; Thorne S. H. (2008) Chemical control of protein stability and function in living mice. Nat. Med. 14, 1123–1127. 10.1038/nm.1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herm-Gotz A.; Agop-Nersesian C.; Munter S.; Grimley J. S.; Wandless T. J.; Frischknecht F.; Meissner M. (2007) Rapid control of protein level in the apicomplexan Toxoplasma gondii. Nat. Methods 4, 1003–1005. 10.1038/nmeth1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armstrong C. M.; Goldberg D. E. (2007) An FKBP destabilization domain modulates protein levels in Plasmodium falciparum. Nat. Methods 4, 1007–1009. 10.1038/nmeth1132. [DOI] [PubMed] [Google Scholar]
- Muralidharan V.; Oksman A.; Iwamoto M.; Wandless T. J.; Goldberg D. E. (2011) Asparagine repeat function in a Plasmodium falciparum protein assessed via a regulatable fluorescent affinity tag. Proc. Natl. Acad. Sci. U. S. A. 108, 4411–4416. 10.1073/pnas.1018449108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glass M.; Busche A.; Wagner K.; Messerle M.; Borst E. M. (2009) Conditional and reversible disruption of essential herpesvirus proteins. Nat. Methods 6, 577–579. 10.1038/nmeth.1346. [DOI] [PubMed] [Google Scholar]
- Qi Z.; Wilkinson M. N.; Chen X.; Sankararaman S.; Mayhew D.; Mitra R. D. (2017) An optimized, broadly applicable piggyBac transposon induction system. Nucleic Acids Res. 45, gkw1290. 10.1093/nar/gkw1290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maji B.; Moore C. L.; Zetsche B.; Volz S. E.; Zhang F.; Shoulders M. D.; Choudhary A. (2017) Multidimensional chemical control of CRISPR-Cas9. Nat. Chem. Biol. 13, 9–11. 10.1038/nchembio.2224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez M.; Martin-Ruiz I.; Jimenez S.; Pirone L.; Barrio R.; Sutherland J. D. (2011) Generation of stable Drosophila cell lines using multicistronic vectors. Sci. Rep. 1, 75. 10.1038/srep00075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J. H.; Lee S. R.; Li L. H.; Park H. J.; Park J. H.; Lee K. Y.; Kim M. K.; Shin B. A.; Choi S. Y. (2011) High cleavage efficiency of a 2A peptide derived from porcine teschovirus-1 in human cell lines, zebrafish and mice. PLoS One 6, e18556. 10.1371/journal.pone.0018556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z.; Chen O.; Wall J. B. J.; Zheng M.; Zhou Y.; Wang L.; Ruth Vaseghi H.; Qian L.; Liu J. (2017) Systematic comparison of 2A peptides for cloning multi-genes in a polycistronic vector. Sci. Rep. 7, 2193. 10.1038/s41598-017-02460-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt M.; Finley D. (2014) Regulation of proteasome activity in health and disease. Biochim. Biophys. Acta, Mol. Cell Res. 1843, 13–25. 10.1016/j.bbamcr.2013.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho U.; Zimmerman S. M.; Chen L. C.; Owen E.; Kim J. V.; Kim S. K.; Wandless T. J. (2013) Rapid and Tunable Control of Protein Stability in Caenorhabditis elegans Using a Small Molecule. PLoS One 8, e72393. 10.1371/journal.pone.0072393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanenbaum M. E.; Gilbert L. A.; Qi L. S.; Weissman J. S.; Vale R. D. (2014) A protein-tagging system for signal amplification in gene expression and fluorescence imaging. Cell 159, 635–646. 10.1016/j.cell.2014.09.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biggin M. D. (2011) Animal transcription networks as highly connected, quantitative continua. Dev. Cell 21, 611–626. 10.1016/j.devcel.2011.09.008. [DOI] [PubMed] [Google Scholar]
- Brand A. H.; Perrimon N. (1993) Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Development 118, 401–415. [DOI] [PubMed] [Google Scholar]
- Southall T. D.; Elliott D. A.; Brand A. H. (2008) The GAL4 System: A Versatile Toolkit for Gene Expression in Drosophila. Cold Spring Harb. Protoc. 2008, pdb.top49. 10.1101/pdb.top49. [DOI] [PubMed] [Google Scholar]
- Duffy J. B. (2002) GAL4 system in Drosophila: a fly geneticist’s Swiss army knife. Genesis 34, 1–15. 10.1002/gene.10150. [DOI] [PubMed] [Google Scholar]
- Roman G.; Endo K.; Zong L.; Davis R. L. (2001) P[Switch], a system for spatial and temporal control of gene expression in Drosophila melanogaster. Proc. Natl. Acad. Sci. U. S. A. 98, 12602–12607. 10.1073/pnas.221303998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osterwalder T.; Yoon K. S.; White B. H.; Keshishian H. (2001) A conditional tissue-specific transgene expression system using inducible GAL4. Proc. Natl. Acad. Sci. U. S. A. 98, 12596–12601. 10.1073/pnas.221303298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholson L.; Singh G. K.; Osterwalder T.; Roman G. W.; Davis R. L.; Keshishian H. (2008) Spatial and temporal control of gene expression in Drosophila using the inducible GeneSwitch GAL4 system. I. Screen for larval nervous system drivers. Genetics 178, 215–234. 10.1534/genetics.107.081968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGuire S. E.; Le P. T.; Osborn A. J.; Matsumoto K.; Davis R. L. (2003) Spatiotemporal rescue of memory dysfunction in Drosophila. Science 302, 1765–1768. 10.1126/science.1089035. [DOI] [PubMed] [Google Scholar]
- Zeidler M. P.; Tan C.; Bellaiche Y.; Cherry S.; Hader S.; Gayko U.; Perrimon N. (2004) Temperature-sensitive control of protein activity by conditionally splicing inteins. Nat. Biotechnol. 22, 871–876. 10.1038/nbt979. [DOI] [PubMed] [Google Scholar]
- Markstein M.; Pitsouli C.; Villalta C.; Celniker S. E.; Perrimon N. (2008) Exploiting position effects and the gypsy retrovirus insulator to engineer precisely expressed transgenes. Nat. Genet. 40, 476–483. 10.1038/ng.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niswender K. D.; Blackman S. M.; Rohde L.; Magnuson M. A.; Piston D. W. (1995) Quantitative imaging of green fluorescent protein in cultured cells: comparison of microscopic techniques, use in fusion proteins and detection limits. J. Microsc. 180, 109–116. 10.1111/j.1365-2818.1995.tb03665.x. [DOI] [PubMed] [Google Scholar]
- Grether M. E.; Abrams J. M.; Agapite J.; White K.; Steller H. (1995) The head involution defective gene of Drosophila melanogaster functions in programmed cell death. Genes Dev. 9, 1694–1708. 10.1101/gad.9.14.1694. [DOI] [PubMed] [Google Scholar]
- Lin C. C.; Potter C. J. (2016) Editing Transgenic DNA Components by Inducible Gene Replacement in Drosophila melanogaster. Genetics 203, 1613–1628. 10.1534/genetics.116.191783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyazaki Y.; Mizumoto K.; Dey G.; Kudo T.; Perrino J.; Chen L. C.; Meyer T.; Wandless T. J. (2016) A method to rapidly create protein aggregates in living cells. Nat. Commun. 7, 11689. 10.1038/ncomms11689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai Y. C.; Weissman A. M. (2010) The Unfolded Protein Response, Degradation from Endoplasmic Reticulum and Cancer. Genes Cancer 1, 764–778. 10.1177/1947601910383011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu B. W.; Banaszynski L. A.; Chen L. C.; Wandless T. J. (2008) Recent progress with FKBP-derived destabilizing domains. Bioorg. Med. Chem. Lett. 18, 5941–5944. 10.1016/j.bmcl.2008.09.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyazaki Y.; Chen L. C.; Chu B. W.; Swigut T.; Wandless T. J. (2015) Distinct transcriptional responses elicited by unfolded nuclear or cytoplasmic protein in mammalian cells. eLife 10.7554/eLife.07687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu B. W.; Kovary K. M.; Guillaume J.; Chen L. C.; Teruel M. N.; Wandless T. J. (2013) The E3 ubiquitin ligase UBE3C enhances proteasome processivity by ubiquitinating partially proteolyzed substrates. J. Biol. Chem. 288, 34575–34587. 10.1074/jbc.M113.499350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bateman J. R.; Wu C. T. (2008) A simple polymerase chain reaction-based method for the construction of recombinase-mediated cassette exchange donor vectors. Genetics 180, 1763–1766. 10.1534/genetics.108.094508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanagawa S.; Lee J. S.; Ishimoto A. (1998) Identification and characterization of a novel line of Drosophila Schneider S2 cells that respond to wingless signaling. J. Biol. Chem. 273, 32353–32359. 10.1074/jbc.273.48.32353. [DOI] [PubMed] [Google Scholar]
- Schindelin J.; Arganda-Carreras I.; Frise E.; Kaynig V.; Longair M.; Pietzsch T.; Preibisch S.; Rueden C.; Saalfeld S.; Schmid B.; Tinevez J. Y.; White D. J.; Hartenstein V.; Eliceiri K.; Tomancak P.; Cardona A. (2012) Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676–682. 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bateman J. R.; Lee A. M.; Wu C. T. (2006) Site-specific transformation of Drosophila via phiC31 integrase-mediated cassette exchange. Genetics 173, 769–777. 10.1534/genetics.106.056945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeiffer B. D.; Ngo T. T.; Hibbard K. L.; Murphy C.; Jenett A.; Truman J. W.; Rubin G. M. (2010) Refinement of tools for targeted gene expression in Drosophila. Genetics 186, 735–755. 10.1534/genetics.110.119917. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.