

Abstract
Atherosclerotic cardiovascular diseases (ASCVDs) are associated with a substantial mortality, physical morbidity, and mental disability. Elevated plasma low-density lipoprotein cholesterol (LDL-C) levels play a major role in the pathophysiology of ASCVDs. Statins have been shown to reduce ASCVD risk and associated events and are recommended as first-line therapy for treatment of hypercholesterolemia by current international guidelines. The key issue is to attain guideline-recommended LDL-C levels (below 70 mg/dl) for patients at very high cardiovascular risk. However, many high-risk and very-high-risk patients on statin therapy remain beyond treatment goals despite lifestyle modification and statins, and are exposed to a high risk of future cardiovascular events including myocardial infarction (MI), stroke, revascularization procedures, and death. This clearly emphasizes the urgent need for additional LDL-C reduction with new therapeutic strategies to target these highly atherogenic particles and to further reduce the burden of ASCVDs. Proprotein convertase subtilisin/kexin type 9 (PCSK9) plays a major role as a key regulator of the hepatic LDL receptor recycling process. Developments over the past 15 years have demonstrated PCSK9 inhibition to be a novel therapeutic strategy to manage increased LDL-C levels. A number of clinical studies using humanized monoclonal antibody technology against PCSK9 have shown profound reductions of LDL-C levels when used either alone or in combination with statin therapy. Recently, the first cardiovascular outcome study demonstrated a significant reduction of ASCV events when evolocumab was added to a statin therapy. This review will discuss current knowledge about antibody-mediated PCSK9 inhibition as add-on therapy to statin and the clinical potential that may be expected.
Keywords: alirocumab, cholesterol metabolism, genetics, proprotein convertase subtilisin/kexin type 9 inhibition, statins
Introduction
Statins are recommended by most current guidelines as a therapeutic strategy of first choice for reducing major adverse cardiovascular events (MACEs) in patients with atherosclerotic cardiovascular disease (ASCVD) risk.1–4 Furthermore, as suggested by such guidelines, patients categorized in high-risk and very-high-risk groups should be treated to achieve low-density lipoprotein cholesterol (LDL-C) levels below 100 mg/dl and below 70 mg/dl, respectively.2–4 However, many high-risk patients on statin therapy do not reach treatment goals for LDL-C. It has been suggested that only one in four high-risk patients treated with statins for more than 3 months had LDL-C levels below 70 mg/dl.5 Several European epidemiological studies confirmed these findings6–8 and additional data could demonstrate that most patients (about 80%) with heterozygous familial hypercholesterolemia treated with statins do not reach LDL-C levels below 100 mg/dl.9,10 Real world data suggest that most patients are usually started with a moderate-intensity statin with the potential to lower LDL-C levels by about 30%.3,11 However, both US and European guidelines recommend high-intensity statin therapy to reduce LDL-C by approximately 50% for higher risk patients, including those with diabetes with target organ damage, ASCVD, and for patients with LDL-C of more than190 mg/dl.1,4 In patients not at their LDL-C-goal with the initial statin therapy, further therapeutic strategies include increasing the statin dose, adding a nonstatin LDL-C-lowering drug, or changing to a higher-intensity statin such as atorvastatin or rosuvastatin.12,13 If higher doses of moderate-intensity statins or high-intensity statins are not tolerated, a nonstatin LDL-C-lowering drug (in most cases ezetimibe) can be added to further reduce LDL-C.1,12,13
Considering the high number of patients at risk whose condition remains poorly controlled with maximally tolerated doses of statins (and ezetimibe), there might be a benefit from more intensive cholesterol-lowering therapy beyond currently used strategies.1,5,13,14
Statins are competitive inhibitors of the 3-hydroxy-3-methylglutaryl coenzyme A reductase activity (HMG-CoA-R), which is the rate-limiting enzyme in cholesterol biosynthesis mainly in the liver. Statin-induced inhibition of HMG-CoA-R leads to a transient decrease in the cellular cholesterol content, which activates the transcription factor sterol regulatory element binding protein (SREBP), which results in upregulation of the expression of the LDL receptor gene. Enhanced LDL receptor expression results in a higher uptake of LDL particles from the plasma, leading to a reduction in plasma LDL-C concentrations.
Ezetimibe is an inhibitor of cholesterol absorption from the intestine and targets cholesterol uptake (via the cholesterol transport protein Nieman Pick C1 like protein) at the jejunal enterocyte brush border.
Alirocumab (Sanofi-Regeneron Pharmaceuticals, Inc., Paris, France and Tarrytown, New York) is a fully human monoclonal antibody currently approved in Europe for the treatment of adults with primary heterozygous familial and nonfamilial hypercholesterolemia or combined dyslipidemia. It can be used in addition to dietary measures and may be used in combination with a statin or in combination of a statin together with additional lipid-lowering therapies in patients who are not at LDL-C treatment goals with high-intensity statins (highest tolerated dose of a statin). Finally alirocumab has been approved alone or in combination with other lipid-lowering therapies in patients who cannot tolerate statins (statin intolerance).
In addition, evolocumab (the second fully human monoclonal antibody currently approved in Europe; Amgen, Thousand Oaks, CA, USA) is approved for adults and for adolescents over 12 years of age with homozygous familial hypercholesterolemia in addition to their lipid-lowering therapies.12
Alirocumab was investigated in three phase II trials that included patients with background statin therapy or the combination of statin plus ezetimibe [ClinicalTrials.gov identifiers: NCT01288443, NCT01266876, NCT01288469]. These studies revealed a reduction of LDL-C levels beyond 70% in alirocumab-treated patients. Moreover, in addition to LDL-C reduction, a decrease in the levels of apolipoprotein (ApoB), lipoprotein (a), and triglycerides (TGs) could be shown. Additionally, HDL-C and ApoA-1 levels significantly increased in alirocumab-treated patients.15–17
There are no signals of clinically relevant adverse events in patients undergoing treatment with alirocumab beyond mild reactions at the injection site.15–17 The phase III OPTIONS I and II studies investigated patients with LDL-C levels above the prespecified levels of more than 70 mg/dl or above 100 mg/dl and compared the lipid-lowering efficacy and safety of alirocumab with various therapeutic choices in patients at high cardiovascular (CV) risk on atorvastatin 20 mg or 40 mg daily or rosuvastatin 10 mg or 20 mg daily.
The phase III program with alirocumab includes a total of 14 studies (ODYSSEY alirocumab phase III program) with a total of more than 23,000 patients. The large CV outcomes trial (ODYSSEY OUTCOMES), investigating the long-term efficacy of alirocumab on the occurrence of CV events in more than 18,000 patients with a recent history of an acute coronary syndrome, is part of this program and the results are expected in 2018. This paper summarizes current knowledge about antibody-mediated proprotein convertase subtilisin/kexin type 9 (PCSK9) inhibition as add-on therapy to a statin.
PCSK-9 inhibition and the regulation of LDL-C metabolism
PCSK9 is secreted protease that is a 692-amino-acid mature protein. It consists of three domains (prodomain, catalytic, and C-terminal domain). It belongs to the family of serine proteases and plays a major role in the regulation of the activity of the hepatic LDL receptor.18 It is primarily expressed in liver, intestine and kidney, and has a rapid turnover in plasma (below 10 min). Removal from plasma occurs principally via the LDL receptor and its degradation in hepatic lysosomes.
PCSK9 regulates the surface expression of LDL receptors by targeting the LDL receptor–PCSK9 complex for lysosomal degradation in hepatocytes. In the presence of a high concentration of PCSK9, the number of LDL receptors on the hepatocyte cell surface decreases because of enhanced intracellular LDL-receptor (LDL-R) degradation. A low number of surface-bound LDL receptors results in decreased removal of LDL-C particles from plasma and rising LDL-C level. PCSK9 enhances the intracellular degradation of the PCSK9 target proteins in a nonenzymatic fashion.18
Genetics: the role of sequence variants in the gene coding for PCSK9
Variants in the gene coding for PCSK9 result in altered PCSK9 secretion or function through several heterogeneous mechanisms.
PCSK9-gain-of-function (GoF) mutations were first found in 2003 through genotype sequencing of two French families with familiar hypercholesterolemia.19 GoF mutations of the PCSK9 gene affect all domains of the protein and may reduce LDL receptor expression by several mechanisms. Patients with PCSK9 GoF mutations are characterized by high levels of LDL-C, premature atherosclerosis, ASCVD, and other CV complications.20–22 In contrast, subjects with loss-of-function (LoF) mutations in PCSK9 naturally have low levels of LDL-C and a reduced prevalence of coronary artery disease (CAD) relative to the general population. LoF variants may occur either as relatively rare mutations or as relatively more common genetic polymorphisms. They can affect all domains of the protein and may increase LDL-R expression by different mechanisms. The LDL-C-lowering effect of LoF mutations is usually variable and has been shown to be associated with decreased cardiovascular disease (CVD) risk. Such lower CAD risk has been found in Mendelian randomization studies in defined populations and particular cohorts.23 LoF mutations have not been associated with any detectable clinical abnormalities.24,25
These genetic findings have been a cornerstone to assess PCSK9 inhibition as a novel therapeutic approach to manage increased LDL-C levels.
Statins and the regulation of PCSK9
Cholesterol homeostasis is maintained by several mechanisms, including cholesterol uptake as a LDL-R-mediated process, cholesterol biosynthesis via HMG-CoA-R, conversion into bile acids, lipoprotein release from hepatocytes in the blood, and storage as cholesterol ester.
LDL-R expression and HMG-CoA-R activity are regulated according to the cellular sterol concentration by the transcription factors sterol regulatory element binding proteins (SREBP) that are responsible for the maintenance of cholesterol homeostasis.
A state of intracellular cholesterol depletion in hepatocytes as a result of statin therapy enhances the activity of SREBP-2. This SREBP activation may result in coexpression of both LDL-C receptors and PCSK9 (Figure 1).26,27 This autoregulatory mechanism is an important player in the field of cholesterol homeostasis.
Figure 1.
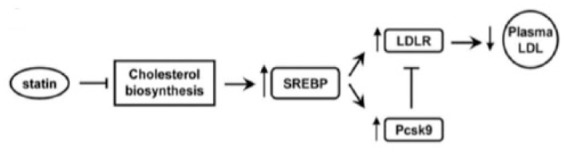
Intracellular cholesterol depletion in hepatocytes as an effect of statin therapy. Activation of the sterol regulatory element binding protein 2 (SREBP-2) may result in an enhanced expression of both low-density lipoprotein cholesterol receptor (LDLR) and proprotein convertase subtilisin/kexin type 9 (PCSK9).
However, by avoiding excessive cholesterol uptake in the liver cell, this SREBP-2-regulated mechanism may limit the efficacy of statin therapy. A large number of clinical trials, mainly with monoclonal antibody technology, have shown that PCSK9 inhibition either alone or in combination with statins can reduce serum LDL-C levels by a large amount. Furthermore, inhibition of PCSK9 used as add-on to a background statin therapy has additive LDL-C-lowering effects.
Moreover, a number of experimental data show that PCSK9 may induce endothelial dysfunction, inflammation, and hypertension by LDL-R-independent mechanisms. Such an effect may promote progression of atherosclerosis. Further research is needed to definitely rule out possible adverse effects and potential therapeutic effects of PCSK9 inhibition that may occur as target effects and could be currently unknown.28
Effects on atherosclerotic plaques when adding PCSK9 inhibitors to statin therapy
Several studies could demonstrate that PCSK9 is present in atherosclerotic plaques and that LDL-R expression in macrophages is influenced by PCSK9. Additionally, previous studies could demonstrate that PCSK9 is involved in atherosclerotic inflammation partially involving the toll-like receptor 4 (TLR4)/nuclear factor κB (NF-κB) pathway. In animal models (high-fat diet-fed ApoE knockout mice), PCSK9 gene interference reduced atherogenesis by decreasing vascular inflammation (e.g. via inhibition of the TLR4/NF-κB signaling pathway) not mediated by plasma cholesterol levels.
In a mouse model alirocumab dose-dependently decreased atherosclerotic lesion size and severity. These effects were enhanced when atorvastatin was added. Additionally, alirocumab reduced monocyte recruitment and modified the lesion composition towards a more stable morphology by increasing the amount of smooth muscle cell and collagen, and by decreasing the necrotic core size and its macrophage content.29 Moreover, alirocumab dose-dependently decreased atherosclerotic lesion size and enhanced the effects of atorvastatin compared with controls. Mice treated with alirocumab either alone or in combination with a high-potency statin (atorvastatin) had more lesion-free areas in their vasculature and fewer severe lesions compared with the controls. Plaque composition definition consisted of macrophage area and necrotic core area. Both were quantitatively assessed as inflammatory factors within atherosclerotic plaques. In contrast, the amount of smooth muscle cells in the fibrotic cap of the plaque and the collagen content were quantitatively assessed as protective factors.30 Alirocumab, either alone or when combined with atorvastatin, reduced the inflammatory factors compared with controls and atorvastatin-treated mice. Additionally, stabilizing protective factors increased in the alirocumab plus atorvastatin treated group compared with controls or with atorvastatin-treated animals.29
Alirocumab plus atorvastatin decreased circulating neutrophil granulocytes and blood monocytes. In particular, the proinflammatory Ly6Chi monocytes decreased excessively.31
When administered alone and together with atorvastatin, alirocumab decreased the adhering monocytes and the abundance of T cells. Alirocumab reduced monocyte adherence by a reduction in endothelial Intercellular Adhesion Molecule 1 (ICAM-1) expression.29
The ODYSSEY alirocumab phase III program
Alirocumab was intensively investigated in the ODYSSEY alirocumab global phase III clinical trial program representing a number of clinical studies in various patient populations. Individual studies from this large program32–39 and two pooled analyses of 8 and 10 (placebo- or ezetimibe-controlled) studies in patients with a background therapy with statins, respectively,40,41 suggest that alirocumab therapy results in a profound and sustained decrease in LDL-C levels. This treatment effect allows many more high CVD risk patients being treated at lipid goals compared with placebo or ezetimibe (Figures 2 and 3).
Figure 2.

Change in non-HDL-C levels over time [on-treatment (mITT) population]. The percent values represent the percent change from baseline at each time point. Pools 1–3 are on background statin therapy, pool 4 reveals no statin treatment in the control group. *p < 0.0001 versus control group. ApoB, apolipoprotein B; LS, least squares; mITT, modified intention to treat; non-HDL-C, non-high-density-lipoprotein cholesterol; Q2W, every 2 weeks; SE, standard error. Modified from Bays et al.41
Figure 3.

Change in ApoB levels over time [on-treatment (mITT) population]. The percent values represent the percent change from baseline at each time point. Pools 1–3 are on background statin therapy, pool 4 reveals no statin treatment in the control group. *p < 0.0001 versus control group. ApoB, apolipoprotein B; LS, least squares; mITT, modified intention to treat; non-HDL-C, non-high-density-lipoprotein cholesterol; Q2W, every 2 weeks; SE, standard error. Modified from Bays et al.41
In a pooled analysis, secondary endpoints included the rate of patients who reach guideline-directed treatment goals [National Lipid Association guidelines: non-high-density-lipoprotein cholesterol (non-HDL-C) <100 or <130 mg/dl for patients at very high CV risk and high CV risk, respectively; European Society of Cardiology/European Atherosclerosis Society guidelines: ApoB <80 mg/dl for patients at very high CV risk; Figures 4–6].
Figure 4.

Percent of all patients (regardless of cardiovascular risk) achieving non-HDL-C levels of <100 mg/dl during the studies, overall and by cardiovascular risk [on-treatment (mITT) population]. Pools 1–3 are on background statin therapy, pool 4 reveals no statin treatment in the control group. *p < 0.0001 versus control group at all time points in all study pools and patient categories. mITT, modified intention to treat; non-HDL-C, non-high-density-lipoprotein cholesterol; Q2W, every 2 weeks. Modified from Bays et al.41
Figure 5.

Percent of patients with very-high-cardiovascular risk achieving non-HDL-C levels of <100 mg/dl during the studies, overall and by cardiovascular risk [on treatment (mITT) population]. Pools 1–3 are on background statin therapy, pool 4 reveals no statin treatment in the control group. *p < 0.0001 versus control group at all time points in all study pools and patient categories. mITT, modified intention to treat; non-HDL-C, non-high-density-lipoprotein cholesterol; Q2W, every 2 weeks. Modified from Bays et al.41
Figure 6.

Percent of patients with high cardiovascular risk achieving non-HDL-C levels of <100 mg/dl during the studies, overall and by cardiovascular risk [on-treatment (mITT) population]. Pools 1–3 are on background statin therapy, pool 4 reveals no statin treatment in the control group. *p < 0.0001 versus control group at all time points in all study pools and patient categories, except for pool 3 and pool 4 of the ‘high cardiovascular risk’ category where †p = 0.0030, ‡p = 0.0008, §p = 0.0159, and ‖p = 0.0220. mITT, modified intention to treat; non-HDL-C, non-high-density-lipoprotein cholesterol; Q2W, every 2 weeks. Modified from Bays et al.41
ApoB plasma levels reflect the concentration of the proatherogenic lipoproteins very-low-density lipoprotein (VLDL) and LDL. Non-high-density-lipoprotein cholesterol (non-HDL-C) levels reflect the concentration of cholesterol transported by VLDL and LDL particles. Non-HDL-C is calculated as total cholesterol minus high-density-lipoprotein cholesterol (HDL-C).
Alirocumab therapy over approximately half a year enabled more than two thirds of alirocumab-treated patients on a background statin to achieve non-HDL-C less than 100 mg/dl or less than 130 mg/dl, and ApoB less than 80 mg/dl.41 Treatment with alirocumab was safe across all groups and there were no signals of adverse events beyond injection site reactions in line with previous reports. In general, alirocumab was well tolerated across all phase III clinical trials. There was no sign of an increase in muscle-related adverse events compared with placebo. A recently published meta-analysis has suggested a possible increase in the frequency of neurocognitive events,42 However, reporting of neurocognitive events was not systematically predefined in these trials. Additionally, the absolute numbers of such neurocognitive events were low. In a subanalysis of the Evolocumab and Clinical Outcomes in Patients with Cardiovascular Disease (FOURIER study),43 cognitive function using the Cambridge Neuropsychological Test Automated Battery was prospectively assessed.44 The results did not reveal any significant between-group difference in cognitive function over a median of 19 months.
Another recently published meta-analysis that included 21 phase III studies (12 studies of alirocumab and 9 of evolocumab) with a total of more than 10,000 patients revealed that PCSK9 inhibition with monoclonal antibody technology used in addition to other lipid-lowering treatments or as lipid-lowering monotherapy in patients who are statin intolerant may facilitate reaching LDL-C goals in patients with high or very high CV risk.45
The ODYSSEY OUTCOMES trial
The recently published FOURIER study43 has shown that inhibition of PCSK9 with evolocumab on a background of statin therapy lowered LDL-C levels to a median of 30 mg/dl and reduced the risk of cardiovascular events. These findings show that patients with ASCVD benefit from lowering of LDL-C levels far below current targets. The ongoing ODYSSEY OUTCOMES trial [ClinicalTrials.gov identifier: NCT01663402] is investigating the potential benefits of alirocumab in reducing MACEs. This study includes patients with the index event of an acute coronary syndrome (ACS) within 1 year prior to randomization. To become included in this study, patients had not achieved lipid management goals with intense statin therapy.46 Even in patients undergoing intensive statin treatment after ACS, the risk for future CV events remains elevated and is related to levels of LDL-C. The ODYSSEY OUTCOMES trial has randomized about 18,600 patients to receive biweekly injections of alirocumab (75–150 mg) or matching placebo. Treatment was started 4–52 weeks after hospitalization for acute MI or unstable angina. Patients who were included in this study were treated with background evidence-based medical and dietary management of dyslipidemia (atorvastatin 40–80 mg daily, rosuvastatin 20–40 mg daily) and had to have one of the following lipid criteria: LDL-C at least 70 mg/dl, or non-HDL-C at least 100 mg/dl, or ApoB at least 80 mg/dl. The primary efficacy outcome measurement is time to first occurrence of coronary heart disease death, acute MI, hospitalization for unstable angina, or ischemic stroke. The trial is an event-driven study and is expected to continue until sufficient primary endpoint events have occurred with minimum follow up of at least 2 years. This should result in a 90% power to significantly detect a 15% hazard reduction.
Secondary outcome events include any coronary heart disease events, major coronary heart disease events, any CV events, and a composite of all-cause mortality/nonfatal MI/non-fatal ischemic stroke, all-cause mortality. Additionally, this study is looking for safety and tolerability with a focus on adverse events, including allergic events and injection site reactions. Finally, changes from baseline in blood lipids and lipoprotein levels will be assessed. In summary, ODYSSEY Outcomes will reveal whether treatment with the PCSK9 antibody alirocumab in addition to high-dose intensive statin therapy reduces events including mortality after ACS and data will be presented in 2018.46
ODYSSEY Outcomes and FOURIER: similarities and differences
ODYSSEY Outcomes included about 18,600 patients and is smaller compared with FOURIER (27,564 patients). However, ODYSSEY Outcomes has a longer follow-up period of up to 5 years, and a different composite endpoint, which may influence the results and interpretation of data. The composite endpoint of ODYSSEY Outcomes includes MI, stroke, death from CV causes, and hospitalization for angina. FOURIER added coronary revascularization to the composite outcome and ODYSSEY Outcomes did not include coronary heart disease death to its endpoint.
PCSK9 inhibition using siRNA technology
Currently available preliminary phase II data for inclisiran look promising. ORION-3 [ClinicalTrials.gov identifier: NCT03060577] will compare 300 mg inclisiran every 180 days with 140 mg evolocumab every 14 days. Results of this head-to-head comparison of RNAi versus monoclonal antibody technology may be available in 2018.
Potential advantages of the siRNA technology may include a reduction in the number of injections (2 per year with inclisiran and 26 per year for evolocumab), a reduction in the amount of the drug (600 mg of siRNA versus 3640 mg of evolocumab), and the stability of siRNA versions under a wide variety of thermal conditions.
The clinical potential of antibody-mediated PCSK9 inhibition
Statin therapy has been shown to be highly effective in reducing LDL-C and future CV events, including CV deaths in patients with ASCVD. However, patients treated with high-intensity statins face substantial residual CV risk that is associated with achieved levels of LDL-C.47,48 Thus, a potential benefit of additional strategies to promote further LDL-C reduction has been suggested. Among statin-treated patients, PCSK9 inhibition using monoclonal antibody technology produces additional LDL-C lowering up to 60%. Both evolocumab and alirocumab were safe and well tolerated in clinical studies and meta-analyses.45,49 No significant differences in rates of common adverse events have been shown compared with placebo or ezetimibe controls. We are eagerly awaiting the results of ongoing trials evaluating their effects on CVD events.50
During the revision process of this paper, the ODYSSEY OUTCOMES study has been presented. In summary, this study could demonstrate that compared with placebo in patients with recent ACS, alirocumab 75 or 150 mg targeting LDL-C levels 25–50 mg/dL, and allowing levels as low as 15 mg/dL was able to reduce major adverse cardiovascular events, myocardial infarction, and ischemic stroke and was associated with a lower rate of all-cause death. Alirocumab was safe and well-tolerated over the duration of the trial.
Footnotes
Funding: This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Conflict of interest statement: Within the last 36 months, J. Auer was advisor and member of the speakers bureau for Sanofi and Amgen.
ORCID iD: Johann Auer  https://orcid.org/0000-0001-6464-7661
https://orcid.org/0000-0001-6464-7661
Contributor Information
Johann Auer, Department of Cardiology and Intensive Care, St Josef Hospital Braunau, Austria.
Robert Berent, Center of Cardiovascular Rehabilitation, HerzReha Bad Ischl, Austria.
References
- 1. Stone NJ Robinson JG Lichtenstein AH et al.;. American College of Cardiology/American Heart Association Task Force on Practice Guidelines. 2013 ACC/AHA guideline on the treatment of blood cholesterol to reduce atherosclerotic cardiovascular risk in adults: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol 2014; 63(pt B): 2889–2934. [DOI] [PubMed] [Google Scholar]
- 2. Expert Dyslipidemia Panel of the International Atherosclerosis Society Panel Members. An International Atherosclerosis Society Position Paper: global recommendations for the management of dyslipidemia: full report. J Clin Lipidol 2014; 8: 29–60. [DOI] [PubMed] [Google Scholar]
- 3. Reiner Z, Catapano AL, De Backer G, et al. ESC/EAS guidelines for the management of dyslipidaemias: the Task Force for the management of dyslipidaemias of the European Society of Cardiology (ESC) and the European Atherosclerosis Society (EAS). Eur Heart J 2011; 32: 1769–1818. [DOI] [PubMed] [Google Scholar]
- 4. Piepoli MF, Hoes AW, Agewall S, et al. 2016 European Guidelines on cardiovascular disease prevention in clinical practice: the Sixth Joint Task Force of the European Society of Cardiology and Other Societies on Cardiovascular Disease Prevention in Clinical Practice (constituted by representatives of 10 societies and by invited experts): developed with the special contribution of the European Association for Cardiovascular Prevention & Rehabilitation (EACPR). Eur Heart J 2016; 37: 2315–2381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Jones PH, Nair R, Thakker KM. Prevalence of dyslipidemia and lipid goal attainment in statin-treated subjects from 3 data sources: a retrospective analysis. J Am Heart Assoc 2012; 1: e001800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Banegas JR, López-García E, Dallongeville J, et al. Achievement of treatment goals for primary prevention of cardiovascular disease in clinical practice across Europe: the EURIKA study. Eur Heart J 2011; 32: 2143–2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kotseva K, Wood D, De Backer G, et al. Cardiovascular prevention guidelines in daily practice: a comparison of EUROASPIRE I, II, and III surveys in eight European countries. Lancet 2009; 373: 929–940. [DOI] [PubMed] [Google Scholar]
- 8. De Backer G, Besseling J, Chapman J, et al. ; EUROASPIRE Investigators. Prevalence and management of familial hypercholesterolaemia in coronary patients: an analysis of EUROASPIRE IV, a study of the European Society of Cardiology. Atherosclerosis 2015; 241: 169–175. [DOI] [PubMed] [Google Scholar]
- 9. Stein EA, Strutt K, Southworth H, et al. Comparison of rosuvastatin versus atorvastatin in patients with heterozygous familial hypercholesterolemia. Am J Cardiol 2003; 92: 1287–1293. [DOI] [PubMed] [Google Scholar]
- 10. Pijlman AH, Huijgen R, Verhagen SN, et al. Evaluation of cholesterol lowering treatment of patients with familial hypercholesterolemia: a large cross-sectional study in The Netherlands. Atherosclerosis 2010; 209: 189–194. [DOI] [PubMed] [Google Scholar]
- 11. Grundy SM, Cleeman JI, Merz CN, et al. Implications of recent clinical trials for the National Cholesterol Education Program Adult Treatment Panel III guidelines. Circulation 2004; 110: 227–239. [DOI] [PubMed] [Google Scholar]
- 12. Landmesser U Chapman MJ Farnier M et al.;. European Society of Cardiology (ESC); European Atherosclerosis Society (EAS). European Society of Cardiology/European Atherosclerosis Society Task Force consensus statement on proprotein convertase subtilisin/kexin type 9 inhibitors: practical guidance for use in patients at very high cardiovascular risk. Eur Heart J 2017; 38: 2245–2255. [DOI] [PubMed] [Google Scholar]
- 13. Catapano AL, Graham I, De Backer G, et al. 2016 ESC/EAS Guidelines for the management of dyslipidaemias: the task force for the management of dyslipidaemias of the European Society of Cardiology (ESC) and European Atherosclerosis Society (EAS). Eur Heart J 2016; 37: 2999–3058. [DOI] [PubMed] [Google Scholar]
- 14. Waters DD, Brotons C, Chiang CW, et al. Lipid treatment assessment project 2: a multinational survey to evaluate the proportion of patients achieving low-density lipoprotein cholesterol goals. Circulation 2009; 120: 28–34. [DOI] [PubMed] [Google Scholar]
- 15. Stein EA, Gipe D, Bergeron J, et al. Effect of a monoclonal antibody to PCSK9, REGN727/SAR236553, to reduce low-density lipoprotein cholesterol in patients with heterozygous familial hypercholesterolaemia on stable statin dose with or without ezetimibe therapy: a phase 2 randomised controlled trial. Lancet 2012; 380: 29–36. [DOI] [PubMed] [Google Scholar]
- 16. Roth EM, McKenney JM, Hanotin C, et al. Atorvastatin with or without an antibody to PCSK9 in primary hypercholesterolemia. N Engl J Med 2012; 367: 1891–1900. [DOI] [PubMed] [Google Scholar]
- 17. McKenney JM, Koren MJ, Kereiakes DJ, et al. Safety and efficacy of a monoclonal antibody to proprotein convertase subtilisin/kexin type 9 serine protease, SAR236553/REGN727, in patients with primary hypercholesterolemia receiving ongoing stable atorvastatin therapy. J Am Coll Cardiol 2012; 59: 2344–2353. [DOI] [PubMed] [Google Scholar]
- 18. Seidah NG, Awan Z, Chretien M, et al. PCSK9: a key modulator of cardiovascular health. Circ Res 2014; 14: 1022–1036. [DOI] [PubMed] [Google Scholar]
- 19. Abifadel M, Varret M, Rabès JP, et al. Mutations in PCSK9 cause autosomal dominant hypercholesterolemia. Nat Genet 2003; 34: 154–156. [DOI] [PubMed] [Google Scholar]
- 20. Abifadel M, Guerin M, Benjannet S, et al. Identification and characterization of new gain-of-function mutations in the PCSK9 gene responsible for autosomal dominant hypercholesterolemia. Atherosclerosis 2012; 223: 394–400. [DOI] [PubMed] [Google Scholar]
- 21. Tibolla G, Norata GD, Artali R, et al. Proprotein convertase subtilisin/kexin type 9 (PCSK9): from structure-function relation to therapeutic inhibition. Nutr Metab Cardiovasc Dis 2011; 21: 835–843 [DOI] [PubMed] [Google Scholar]
- 22. Norata GD, Garlaschelli K, Grigore L, et al. Effects of PCSK9 variants on common carotid artery intima media thickness and relation to ApoE alleles. Atherosclerosis 2010; 208: 177–182. [DOI] [PubMed] [Google Scholar]
- 23. Dron JS, Hegele RA. Complexity of mechanisms among human proprotein convertase subtilisin-kexin type 9 variants. Curr Opin Lipidol 2017; 28: 161–169. [DOI] [PubMed] [Google Scholar]
- 24. Benn M1, Nordestgaard BG, Grande P, et al. PCSK9 R46L, low-density lipoprotein cholesterol levels, and risk of ischemic heart disease: 3 independent studies and meta-analyses. J Am Coll Cardiol 2010; 55: 2833–2842. [DOI] [PubMed] [Google Scholar]
- 25. Cohen JC, Boerwinkle E, Mosley TH, Jr, et al. Sequence variations in PCSK9, low LDL, and protection against coronary heart disease. N Engl J Med 2006; 354: 1264–1272. [DOI] [PubMed] [Google Scholar]
- 26. Goldstein JL, Brown MS. The LDL receptor. Arterioscler Thromb Vasc Biol 2009; 29: 431–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Dubuc G, Chamberland A, Wassef H, et al. Statins upregulate PCSK9, the gene encoding the proprotein convertase neural apoptosis-regulated convertase-1 implicated in familial hypercholesterolemia. Arterioscler Thromb Vasc Biol 2004; 24: 1454–1459. [DOI] [PubMed] [Google Scholar]
- 28. Urban D, Pöss J, Böhm M, et al. Targeting the proprotein convertase subtilisin/kexin type 9 for the treatment of dyslipidemia and atherosclerosis. J Am Coll Cardiol 2013; 62: 1401–1408. [DOI] [PubMed] [Google Scholar]
- 29. Kühnast S, van der Hoorn JW, Pieterman EJ, et al. Alirocumab inhibits atherosclerosis, improves the plaque morphology, and enhances the effects of a statin. J Lipid Res 2014; 55: 2103–2112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Libby P, Sasiela W. Plaque stabilization: can we turn theory into evidence? Am J Cardiol 2006; 98: 26P–33P. [DOI] [PubMed] [Google Scholar]
- 31. Moore KJ, Sheedy FJ, Fisher EA. Macrophages in atherosclerosis: a dynamic balance. Nat Rev Immunol 2013; 13: 709–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Bays H, Gaudet D, Weiss R, et al. Alirocumab as add-on to atorvastatin versus other lipid treatment strategies: ODYSSEY OPTIONS I randomized trial. J Clin Endocrinol Metab 2015; 100: 3140–3148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kereiakes DJ, Robinson JG, Cannon CP, et al. Efficacy and safety of the proprotein convertase subtilisin/kexin type 9 inhibitor alirocumab among high cardiovascular risk patients on maximally tolerated statin therapy: the ODYSSEY COMBO I study. Am Heart J 2015; 169: 906–915e913. [DOI] [PubMed] [Google Scholar]
- 34. Farnier M, Jones P, Severance R, et al. Efficacy and safety of adding alirocumab to rosuvastatin versus adding ezetimibe or doubling the rosuvastatin dose in high cardiovascular-risk patients: the ODYSSEY OPTIONS II randomized trial. Atherosclerosis 2016; 244: 138–146. [DOI] [PubMed] [Google Scholar]
- 35. Kastelein JJ, Ginsberg HN, Langslet G, et al. ODYSSEY FH I and FH II: 78 week results with alirocumab treatment in 735 patients with heterozygous familial hypercholesterolaemia. Eur Heart J 2015; 36: 2996–3003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Moriarty PM, Thompson PD, Cannon CP, et al. Efficacy and safety of alirocumab vs ezetimibe in statin-intolerant patients, with a statin rechallenge arm: the ODYSSEY ALTERNATIVE randomized trial. J Clin Lipidol 2015; 9: 758–769. [DOI] [PubMed] [Google Scholar]
- 37. Roth EM, Taskinen MR, Ginsberg HN, et al. Monotherapy with the PCSK9 inhibitor alirocumab versus ezetimibe in patients with hypercholesterolemia: results of a 24 week, double-blind, randomized Phase 3 trial. Int J Cardiol 2014; 176: 55–61. [DOI] [PubMed] [Google Scholar]
- 38. Ginsberg HN, Rader DJ, Raal FJ, et al. Efficacy and safety of alirocumab in patients with heterozygous familial hypercholesterolemia and LDL-C of 160 mg/dl or higher. Cardiovasc Drugs Ther 2016; 30: 473–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Robinson JG, Farnier M, Krempf M, et al. Efficacy and safety of alirocumab in reducing lipids and cardiovascular events. N Engl J Med 2015; 372: 1489–1499. [DOI] [PubMed] [Google Scholar]
- 40. Farnier M, Gaudet D, Valcheva V, et al. Efficacy of alirocumab in high cardiovascular risk populations with or without familial hypercholesterolemia: pooled analysis of eight ODYSSEY phase 3 clinical program trials. Int J Cardiol 2016; 223: 750–757. [DOI] [PubMed] [Google Scholar]
- 41. Bays HE, Leiter LA, Colhoun HM, et al. Alirocumab treatment and achievement of non-high-density lipoprotein cholesterol and apolipoprotein b goals in patients with hypercholesterolemia: pooled results from 10 phase 3 ODYSSEY trials. J Am Heart Assoc 2017; 6: e005639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Lipinski MJ, Benedetto U, Escarcega RO, et al. The impact of proprotein convertase subtilisin-kexin type 9 serine protease inhibitors on lipid levels and outcomes in patients with primary hypercholesterolaemia: a network meta-analysis. Eur Heart J 2016; 37: 536–545. [DOI] [PubMed] [Google Scholar]
- 43. Giugliano RP Mach F Zavitz K et al.;. EBBINGHAUS Investigators. Cognitive function in a randomized trial of evolocumab. N Engl J Med 2017; 377: 633–643. [DOI] [PubMed] [Google Scholar]
- 44. Sabatine MS Giugliano RP Keech AC et al.;. FOURIER Steering Committee and Investigators. Evolocumab and clinical outcomes in patients with cardiovascular disease. N Engl J Med 2017; 376: 1713–1722. [DOI] [PubMed] [Google Scholar]
- 45. Gouni-Berthold I, Descamps OS, Fraass U, et al. Systematic review of published Phase 3 data on anti-PCSK9 monoclonal antibodies in patients with hypercholesterolaemia. Br J Clin Pharmacol 2016; 82: 1412–1443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Schwartz GG, Bessac L, Berdan LG, et al. Effect of alirocumab, a monoclonal antibody to PCSK9, on long-term cardiovascular outcomes following acute coronary syndromes: rationale and design of the ODYSSEY outcomes trial. Am Heart J 2014; 168: 682–689. [DOI] [PubMed] [Google Scholar]
- 47. Auer J, Berent R, Primus C. PCSK9 inhibitors and cardiovascular events. N Engl J Med 2015; 373: 773. [DOI] [PubMed] [Google Scholar]
- 48. Auer J, Sinzinger H, Franklin B, et al. Muscle- and skeletal-related side-effects of statins: tip of the iceberg? Eur J Prev Cardiol 2016; 23: 88–110. [DOI] [PubMed] [Google Scholar]
- 49. Zhang XL, Zhu QQ, Zhu L, et al. Safety and efficacy of anti-PCSK9 antibodies: a meta-analysis of 25 randomized, controlled trials. BMC Med 2015; 13: 123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Landmesser U, Chapman MJ, Stock JK, et al. 2017 Update of ESC/EAS Task Force on practical clinical guidance for proprotein convertase subtilisin/kexin type 9 inhibition in patients with atherosclerotic cardiovascular disease or in familial hypercholesterolaemia. Eur Heart J. Epub ahead of print 16 October 2017. DOI: 10.1093/eurheartj/ehx549. [DOI] [PubMed] [Google Scholar]