

Abstract
Proinflammatory cytokine activation of the Janus kinase/signal transducers and activators of transcription (JAK/STAT) signal transduction pathway is a critical event in the pathogenesis and progression of rheumatoid arthritis. Under normal conditions, JAK/STAT signaling reflects the influence of negative regulators of JAK/STAT, exemplified by the suppressor of cytokine signaling and protein inhibitor of activated STAT. However, in rheumatoid arthritis (RA) both of these regulators are dysfunctional. Thus, continuous activation of JAK/STAT signaling in RA synovial joints results in the elevated level of matrix metalloproteinase gene expression, increased frequency of apoptotic chondrocytes and most prominently ‘apoptosis resistance’ in the inflamed synovial tissue. Tofacitinib, a JAK small molecule inhibitor, with selectivity for JAK2/JAK3 was approved by the United States Food and Drug Administration (US FDA) for the therapy of RA. Importantly, tofacitinib has demonstrated significant clinical efficacy for RA in the post-US FDA-approval surveillance period. Of note, the success of tofacitinib has spurred the development of JAK1, JAK2 and other JAK3-selective small molecule inhibitors, some of which have also entered the clinical setting, whereas other JAK inhibitors are currently being evaluated in RA clinical trials.
Keywords: cytokine, Janus kinase, rheumatoid arthritis, signal transducers and activators of transcription, signal transduction
Introduction
Rheumatoid arthritis (RA) is a systemic, polyarticular, chronic, progressive, inflammatory musculoskeletal disorder of synovial joints.1 In addition, considerable tissue damage associated with RA can occur in the heart,2 as well as the lung, skin, eye, kidney, and blood vessels. RA is characterized at the molecular and pathophysiological level by abnormal innate, cellular and humoral immunity.1,3–5 Thus, abnormal proliferation kinetics results in an aberrant survival of activated T-lymphocytes, B-lymphocytes, mast cells, neutrophils, macrophages and accessory-antigen presenting cells (i.e. dendritic cells; DCs)6 as well as synovial tissue fibroblasts7 (e.g. fibroblast-like synoviocytes) which are the cardinal cellular hallmarks of the RA disease process.
In RA synovial joints, the normal single membrane synovium becomes hyperplastic. This change results from the stimulated migration and adhesion of activated immune and nonimmune cells under the direction of elevated levels of various chemokines and adhesion proteins.8–10 In addition, the significantly elevated levels of proinflammatory cytokines, exemplified, by tumor necrosis factor-α (TNF-α), interleukin (IL)-1ß, IL-6, IL-7, IL-8, IL-12/IL-23, IL-15, IL-17, IL-18, IL-32, and, interferon-γ (IFN-γ) produced by various cells, together with growth factors, such as fibroblast growth factor-2 (FGF-2) and vascular endothelial growth factor, the latter produced mainly by synovial-like fibroblasts and macrophages, have been shown to be crucial for RA to clinically progress whereby the destruction of articular cartilage and erosion of subchondral bone are the principal events that result in synovial joint failure.9,11–13 Thus, the overall changes occurring in RA synovial joints in response to these various factors, including suppression of cartilage-derived extracellular matrix production,14 an elevated frequency of apoptotic chondrocytes,15 synovial tissue ‘apoptosis resistance’,16 and an increased level of matrix metalloproteinase (MMP) gene expression17 as well as that class of enzymes, termed, a disintegrin and metalloproteinase (ADAM)18 gene expression are all critical components of the RA process.
Several signal transduction pathways have been implicated in RA progression. For example, although IL-1ß is noted to predominantly activate the stress-activated, mitogen-activated protein kinase (SAPK/MAPK) pathway19 and IL-6 and IFN-γ predominantly activate the Janus kinase/signal transducers and activators of transcription (JAK/STAT) pathway,20,21 compelling evidence points to activation of MAPK signaling by IL-622 and IFN-γ as well. Importantly, although TNF-α was reported to primarily activate the SAPK/MAPK pathway,23–26 TNF-α can also activate JAK/STAT as evidenced by results from our research group which showed that recombinant human (rh)-TNF-α caused the phosphorylation of the STAT3 protein (i.e. p-STAT3) by human chondrocytes in vitro without changing the content of STAT3.27 Of note, activation of JAKs by IL-6 was also reported to result in the activation of the SAPK/MAPK pathway and PI3K/Akt/mTOR signaling via ‘cross-talk’,22,28 whereby the PI3K/Akt/mTOR pathway, in particular, has been associated with aberrant survival of nonimmune and immune cells in RA.28
The crucial role played by JAK/STAT pathway activation in RA was further established following the US FDA approval of the JAK3-selective small molecule inhibitor (SMI), tofacitinib, for the medical therapy of RA.29 Indeed, the successful incorporation of tofacitinib into the armamentarium of RA therapies has resulted in the further development of JAK1-selective, JAK2-selective, TYK2-selective and pan-JAK SMIs for RA.
In that regard, we have evaluated the peer-reviewed published literature primarily employing the PubMed database (https://www.ncbi.nlm.nih.gov/pubmed). This literature search focused on the role of JAK/STAT signaling in RA. In particular we have discussed the molecular mechanisms underpinning the activity of the JAK-selective SMIs. We also point out some important gaps in our knowledge relative to how these JAK-selective SMIs actually regulate the changes consonant with RA progression at the level of synovial joints.
An overview of JAK/STAT signaling
The Janus family of kinases (JAKs), namely, JAK1, JAK2, JAK3 and TYK2, are nonreceptor protein tyrosine kinases.30 Abnormal activation of JAK/STAT signaling via JAK mutations or constitutive TYK2 signaling was shown to be critical for the induction of aberrant hematopoietic stem cell development, hematological malignancies, autoimmunity and certain immunodeficiency syndromes.31 In that regard, inhibitors of JAK activation altered T-cell, natural killer cell and DC activity, all of which are pertinent to the pathogenesis and progression of autoimmune disorders.32 Of note, pharmacological inhibition of JAKs was shown to efficiently block the downstream events associated with type I/II cytokines33 and JAK SMIs (now often referred to as Jakinibs)34 have become useful as potent and efficacious medical therapies for a host of autoimmune diseases, such as RA, psoriasis and inflammatory bowel diseases.35
All JAKs share a common structural region referred to as the JAK homology (JH) region22 (Figure 1). In this schematic, the JH domains are structurally numbered, JH1 to JH7 based on their consecutive structural domains beginning at the carboxy-terminal and continuing through to the amino-terminus.36 Of note, structural analysis also proved that the JH2 region once thought to be the catalytic domain was not fully functional and therefore was redefined as a pseudo-kinase.37 Importantly, the JH4-JH7 regions were shown to be critical for regulating the interactions between JAKs and other protein kinases as well as for receptor binding, catalytic activity, JAK autophosphorylation, and in some cases, for even suppressing JAK activity.38
Figure 1.
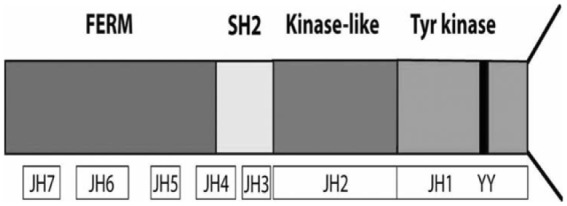
JH domains and JAK3 phosphorylation sites. (Figure was originally published in Malemud and Pearlman22).
FERM, four-point.1-ezrin-radaxin-moesin domain; JAK, Janus kinase; JH, JAK homology; kinase-like, pseudokinase domain; SH2, Src homology domain; Tyr kinase, tyrosine kinase domain.
Normally, STAT proteins are inactive cytoplasmic proteins. However, after cytokine activation, perhaps best illustrated by the binding of IL-6 to the IL-6Rα/gp130 complex, STAT proteins are recruited to the cytokine/receptor complex via the SH2 domain where they become phosphorylated. This recruitment promotes the formation of p-STAT dimers22 (Figure 2). In fact, it is the p-STAT homodimers or heterodimers that provide a primary mechanism for STAT proteins to be efficiently translocated to the nucleus where they bind to STAT-response DNA motifs and, in that manner, act as transcription factors.39,40
Figure 2.

The interaction of IL-6 with the IL-6Rα/gp130 complex activates JAK3 resulting in the phosphorylation of STAT3 (p-STAT3) (ON). SHP-1 is a phosphatase which regulates STAT phosphorylation by de-phosphorylating p-STAT3 (OFF). (Figure was originally published in Malemud and Pearlman22).
IL, interleukin; JAK, Janus kinase.
Suppressor of cytokine signaling (SOCS): a primary negative regulator of JAK/STAT activation
The constitutive activation of JAK/STAT signaling is characteristic of various types of cancers, including, lymphoma, leukemia and myeloproliferative diseases,41,42 as well as solid tumors43 and certain immunodeficiency syndromes.44 The finding that a class of proteins, termed, suppressor of cytokine signaling (SOCS) were upregulated under those conditions where STAT proteins were also activated indicated that the presence of SOCS most likely constituted the primary mechanism for the negative regulation of JAK/STAT signaling.45–48 In fact, it is important to note that in RA, negative regulation of STAT protein activation via SOCS was found to be seriously deficient.47 Moreover, it was speculated that experimental over-expression of SOCS3 in RA synovial tissue might provide a mechanism for dampening the inflammatory milieu associated with RA.49,50 In addition to SOCS proteins, ongoing studies of protein inhibitor of activated STAT (PIAS) have shed light on several additional targets for regulating JAK/STAT activation.51,52
Development of JAK-selective SMIs for RA
Tofacitinib
Borie and colleagues53 proposed tofacitinib as a JAK inhibitor in the context of preventing transplant rejection following the results of several studies in rodents which validated the effectiveness of tofacitinib as an immune suppression drug. Thus, tofacitinib significantly improved allograft survival in a series of primate studies while also exhibiting an acceptable safety profile in nonhuman primates. As previously indicated, tofacitinib, was the first Jakinib to be approved by the US FDA in 2012 for the therapy of moderate-to-severe active RA54 in patients whose response to methotrexate was deemed to be inadequate. Tofacitinib was reported to inhibit JAK3, JAK2, JAK1 with IC50 values of 1 nM, 20 nM and 112 nM, respectively.55 Tofacitinib was developed from the sequential optimization of pyrrolopyrimidine-based JAK3 inhibitors.56 The effectiveness of tofacitinib was achieved in numerous randomized clinical trials involving RA patients57,58 where the majority of those enrolled RA patients achieved an American College of Rheumatology-20 (ACR20) response criteria59 as early as week 2 which was sustained by week 24.60 In addition, long-term efficacy with tofacitinib in for RA patients of up to 48 months was reported.61
At the pathophysiological level, tofacitinib was reported to modulate the activity of proinflammatory cytokines that appear to be critical for the progression of RA.62 Although tofacitinib is reported to be JAK3-selective, Yamaoka63 contended that tofacitinib actually targeted multiple JAKs, whereas other recently developed ‘Jakinibs’ have been developed to target a single JAK. Of note, Fleischmann64 reported on whether tofacitinib monotherapy in RA subjects had similar efficacy to dual therapy with methotrexate. Therefore, the cumulative data from these human clinical trial studies indicated that although tofacitinib in combination with methotrexate was statistically more effective in RA, tofacitinib alone was also clinically effective.
Importantly, JAK inhibition can result in serious and opportunistic infections where viral infections (including herpes zoster) are reported to be particularly worrisome.65 However, the incidence of malignancy, with the exclusion of nonmelanoma skin cancers, was found to be similar in patients treated with tofacitinib, compared with those in the general population.66
Ruxolitinib
Ruxolitinib, formally termed ruxolitinib/INCB01824, is a JAK1/JAK2-selective Jakinib67 which was approved for treating myeloproliferative diseases and psoriasis. As indicated by Gadina, developing JAK SMIs that target more than one JAK does not appear on the face of it to be ‘problematic’.68 Considered to be generally safe and well tolerated in normal volunteers and RA patients, study results with ruxolitinib showed that the level of p-STAT3 inhibition in whole blood correlated with the plasma levels of the drug.69 In a randomized clinical trial conducted by Williams and colleagues,70 with active RA patients an ACR70 response criteria was achieved in 33% of those patients receiving ruxolitinib compared with 0% in the placebo arm.
At the pathophysiological level, ruxolitinib-mediated inhibition of JAK1/JAK2 reduced the plasma levels of IL-6 and CD-40, the latter of considerable importance as a co-stimulatory biomarker protein on antigen-producing cells. Ruxolitinib also inhibited p-STAT3 in an ex vivo analysis conducted on blood cells from RA patients. In that regard, Menet and colleagues,71 confirmed that JAK1 played a crucial role not only in the transduction of the common γ chain cytokines, but also in IL-6 signaling. However, despite the high level of structural homology between JAK1 and JAK2 including similar binding profiles at the adenosine triphosphate (ATP)-binding site, according to Vrontaki and colleagues,72 there continues to be a persuasive rationale for the development of JAK1 and JAK2-specific SMIs
Baricitinib, decernotinib and filgotinib
Baricitinib is one of several JAK-selective SMIs59,73 approved by the European Medicines Agency and the US FDA for the treatment of RA. In that regard, baricitinib is an orally-administered Jakinib with selectivity towards JAK-1/JAK-240,74 with an IC50 of 5.9 nM and 5.7 nM, respectively, in cell-free assays and a ~70 and ~10-fold selective versus JAK3 and Tyk2 with no inhibition of tyrosine-protein kinase Met and checkpoint kinase-2. Baricitinib was also shown to inhibit IL-6–stimulated phosphorylation of STAT3 (pSTAT3) as well as the downstream synthesis of the chemokine, monocyte chemoattractant protein-1, with IC50 values of 44 nM and 40 nM, respectively, in PBMCs. Baricitinib also inhibited pSTAT3 stimulated by IL-23 with IC50 of 20 nM in isolated naïve T-cells.75 In fact, a clinical trial was conducted on normal volunteers which indicated that baricitinib exhibited dose–linear and time-dependent pharmacokinetics with low oral-dose clearance of around 17 l/h and minimal accumulation in tissues and organs.76 Thus, in a manner similar to tofacitinib, baricitinib inhibited p-STAT3 in whole blood ex vivo which correlated with the level of baricitinib in plasma. Although baricitinib exhibited negligible side effects in normal volunteers, its use was associated with reduced neutrophil counts.72 Recently, Richez and colleagues,77 reported that treatment of RA patients with baricitinib monotherapy, or when baricitinib was combined with conventional synthetic disease-modifying antirheumatic drugs (csDMARDs) showed efficacy and an acceptable safety profile in early active naïve csDMARD-treated RA patients who had exhibited an inadequate response to csDMARDs or biologic DMARDs. Their review also pointed out that baricitinib offered several advantages over other DMARDs in terms of oral administration, onset of response, and clinical efficacy as a monotherapy compared with the TNF blockade biologic, adalimumab. In a large phase III randomized clinical trial of 527 RA patients who had shown an inadequate response to TNF blockade or other biologic DMARDs, 55% [versus 27% in the placebo arm (p < 0.001)] treated with 2 or 4 mg/daily for 24 weeks exhibited an ACR20 response as well as reductions in the Health Assessment Questionnaire-Disability Index and 28 Joint Disease Activity Score based on c-reactive protein (DAS28-CRP), but not in the Simplified Disease Activity Index of 3.3 or less. Overall, two nonmelanoma skin cancers and two major cardiovascular events were reported, including a fatal stroke, which was associated with the higher dose (4 mg) of baricitinib. Treatment with the drug was also associated with reduced neutrophil counts as well as increased serum creatinine and low-density cholesterol,78,79 the latter a potential contributor to atherosclerosis which has been shown to be a major comorbidity in RA.80 Of note, treatment of RA patients with baricitinib was associated not only with clinical improvement, but also with inhibition of radiographic joint damage.
Decernotinib
Decernotinib is an orally-administered JAK3-selective reversible SMI59 which was shown to possess clinical efficacy for the treatment of RA.81–83 The clinical efficacy of decernotinib was demonstrated by improvement in the ACR criteria and the DAS28-CRP compared with placebo.81 In addition to assessing the effect of decernotinib on RA progression, this drug was also evaluated for its effects on JAK/STAT-mediated signaling. Thus, it was reported that when decernotinib and other JAK3 and JAK1/JAK2-selective SMIs were compared with one another, a common component in the response was identified for the IFN-α and IFN-γ signaling pathways, although IL-15, IL-21, IL-6 and IL-27-mediated signaling was more effectively blocked by tofacitinib and baricitinib than by either decernotinib or filgotinib (see below). However, these JAK SMIs had less of an effect on IL-10, IL-12, IL-23 or erythropoietin which under certain conditions are also capable of inducing JAK/STAT activation.40 The results of two additional experimental studies with decernotinib are also worthy of comment here. Thus, Mahajan and colleagues,84 showed that decernotinib effectively inhibited JAK3 activity in vitro and in vivo which was characterized by the lack of potency on JAK1/JAK2 activity in vitro. Decernotinib also had the capacity to reduce paw swelling, and paw weight while improving the histopathological score in rat collagen-induced arthritis (CIA). Moreover, in the mouse model of oxazolone-induced delayed-type hypersensitivity, decernotinib reduced T-cell mediated skin inflammation.84
Irreversible JAK3-selective SMIs
Decernotinib and tofacitinib are both reversible SMIs. This being the case, Elwood and colleagues,85 contended that the development of irreversible JAK3 SMIs were likely to be useful and highly effective for altering JAK-directed STAT activation. Thus, a newly synthesized irreversible JAK-3 inhibitor, called Compound 2, was shown to be 4300-fold selective towards JAK3 versus JAK1 in enzyme assays and >35-fold selective in human peripheral blood mononuclear cell assays in assessing JAK/STAT activation by IL-7 versus IL-6 or granulocyte/macrophage colony stimulating factor. Importantly, the irreversible JAK3 SMI blocked inflammation in a rat model of arthritis without affecting hematopoiesis which is considered an important step forward in the use of such as drug for the treatment of chronic diseases such as RA.
Filgotinib
Filgotinib is an investigational selective JAK1 inhibitor.86 The preclinical data for filgotinib were impressive in that they revealed selectivity for JAK1 versus JAK2 of nearly 30-fold87 as well as the capacity of filgotinib to inhibit Th1/Th2 differentiation and to a lesser extent Th17 differentiation. Filgotinib also attenuated the progression of arthritis in rodent CIA as evidenced by reduced paw swelling, reduced cartilage and bone degradation as well as through a lowering of the level of proinflammatory cytokines. Of note, the efficacy of filgotinib was comparable in CIA with that obtained with the TNF biologic, etanercept.
In a phase IIb clinical trial in 283 RA patients, filgotinib employed as a monotherapy was effective in treating the signs and symptoms of RA with a rapid onset of activity.88 In that study no opportunistic infections or tuberculosis was reported. In another clinical trial, Westhovens and colleagues,89 reported that filgotinib added to methotrexate also demonstrated a rapid onset of activity, was well tolerated and improved the clinical picture of RA. In a dosage study, Vanhoutte and colleagues,90 showed that filgotinib employed at 75–300 mg daily gradually improved the ACR20 response and the DAS28-CRP score, without causing anemia, or altering the activity of liver transaminases. Importantly, there was no increase in the level of low-density lipoprotein or total cholesterol, although a small decrease in neutrophils was reported which was attributed to the effect of specifically inhibiting JAK1. There were no reported infections and treatment was well tolerated with the most common adverse event reported as nausea. Filgotinib remains under clinical investigation where a phase III clinical trial evaluation is being conducted using filgotinib and ABT-494 (another JAK1 inhibitor).91
Additional JAK SMIs in development
Upadacitinib and peficitinib
Upadacitinib92 and peficitinib93 are two JAK SMIs currently undergoing evaluation as potential therapies for RA. Upadacitinib was shown to be JAK1-selective,92 whereas peficitinib was shown to inhibit JAK1 and JAK3 with 50% inhibitory concentrations of 3.9 and 0.7 nM, respectively, indicating a relatively selective effect of peficitinib for JAK3.92
Klünder and colleagues,94 reported in a study of 107 healthy volunteer subjects and 466 RA patients in three phase I and two phase IIb clinical trials that upadacitinib, had an acceptable safety profile and followed dose-proportional, bi-exponential disposition. However, a somewhat lower clearance of the drug was also reported in RA patients compared with healthy patients. Other potential side effects possibly attributed to upadacitinib such as changes in weight, sex drive, or mild or moderate renal impairment were unchanged. Peficitinib has been evaluated in several RA clinical trials. In one of these trials, Genovese and colleagues,95 orally-administered peficitinib at varying doses (25–150 mg) for 12 weeks to RA patients with moderate-to-severe disease. A positive ACR20 response was obtained at the 100 mg and 150 mg doses. Adverse events were similar in the RA and the placebo arm of the trial with satisfactory tolerability. In another clinical trial, Kivitz and colleagues,96 reported that peficitinib (50 mg) employed in combination with methotrexate accelerated the ACR20 response in 378 RA patients compared with those patients in the methotrexate arm (i.e. the placebo arm) of the trial where, as would be expected, the placebo ACR20 response was high. They concluded from these results that peficitinib was effective in RA and well tolerated with limited safety concerns.
A consequence of the emerging comorbidity of atherosclerosis with RA,80 Zhu and colleagues,97 conducted an open-label clinical trial on 24 healthy adults treated with peficitinib added to rosuvastatin. The overall conclusion from that study was that peficitinib, through its major metabolite H2, did not significantly alter the pharmacokinetics of rosuvastatin as determined from measurements of hepatic uptake transporter anion transporting polypeptide 1B1. Finally, the potential underlying mechanism for the effectiveness of peficitinib in RA was studied by Ito and colleagues,93 who reported a dose-dependent suppression of bone destruction and paw swelling in a rat antigen-arthritis model where the drug was administered either via prophylactic or therapeutic dosing or by continuous intraperitoneal infusion. Peficitinib also inhibited IL-2-dependent T-cell proliferation in vitro and STAT5 activation in vitro and in vivo.
Conclusions and future perspectives
We conclude from the preceding analysis that constitutive activation or perturbations in JAK/STAT signaling produces changes crucial to many of the clinical aspects of RA associated with its pathogenesis and progression. In fact, more than a decade ago, Sweeney and Firestein98 implicated JAK/STAT signaling (as well as p38 kinase MAPK) as one of the critical regulators of matrix metalloproteinase (MMP) gene expression.17 Therefore, it will be important to determine the extent to which JAK SMIs alter MMP gene expression by chondrocytes, a major producer of MMPs in RA synovial joints.
JAKs and TYK2 have also been implicated in several aspects of innate and adaptive immunity.30,53 In addition to the effect of Jakinibs on T-cell and B-cells, several JAK inhibitors have also been shown to alter the activity of osteoclasts and DC both of which are crucial to mediating bone erosions and antigen-presentation, respectively.53
Mutations that inactivate JAK3 are responsible for severe combined immunodeficiency syndromes, TYK2 mutations were associated with autosomal recessive hyperimmunoglobulin E syndrome and a JAK2 ‘gain-of-function’ mutation causes polycythemia vera and other myeloproliferative diseases. In addition, several other molecules pertinent to RA pathology are also regulated by JAK/STAT signaling. These include, IP-10,99 TNFRSF12, a mediator of apoptosis100 and IL-15.101 In fact, Shenoy and colleagues,102 showed that IL-15 regulated the Bcl-2 family proteins, Bim and Mcl-1 in T-cells. The results of that study also suggested that down-regulating short-lived Mcl-1 could induce Bim-dependent apoptosis which might be useful in promoting apoptosis in the perpetually activated T-cells. IL-2, a cytokine responsible, in part, for T-cell activation, also was shown to enhance IL-10 production through activation of STAT5103 in the Treg cell line, HOZOT. Thus, if IL-10 production could be increased in this manner and if IL-10 was properly regulated under those conditions then this strategy could potentially restore one of the functions of IL-10 believed to be compromised in RA.
Other molecules, including, programmed cell death protein-1 (PD-1) and its ligand PD-L1, cytotoxic T-lymphocyte-associated protein-4 (CTLA-4), lymphocyte activation gene-3 (LAG-3), T-cell immunoglobulin and mucin domain-1 (TIM-1) and various interferons are critical for maintaining immune balance.104,105 For example, it may be informative to connect JAK/STAT signaling with PD-L1 since Doi and colleagues106 recently demonstrated that the JAK/STAT pathway regulated PD-L1 gene expression in pancreatic cancer cells which was suppressed by a JAK1 inhibitor that also reduced the activation of STAT1. However, the results of this study106 also points to a potential flaw in reasoning that merely associating JAK/STAT signaling with a particular regulatory mechanism involving STAT-responsive genes means that inhibiting the latter will alter the course of disease. In that regard, inhibitors of PD-1/PD-L1 activate T-cells. This establishes an immunotherapeutic paradigm for suppressing cancer cell proliferation. However, the extent to which checkpoint inhibition would result in suppression of the dysregulated proliferation of RA synovial fibroblasts remains to be determined.
The results of two clinical trials which assessed the JAK inhibitor, tofacitinib, against adalimumab or placebo107 or tofacitinib monotherapy108 in RA patients showed that tofacitinib was clinically efficacious yet was also associated with increased levels of low-density and high-density lipoprotein cholesterol as well as with reduced neutrophil counts. Thus, it remains to be determined as to the extent to which ‘Jakinibs’ will either replace or supplement conventional synthetic csDMARDs or biologic drugs as first-line therapies for RA. Presently, moderate-to-severe RA continues to be treated with methotrexate plus/minus biological drugs; the latter targeting either proinflammatory cytokines, TNF-α, IL-6R or T-cell/B-cell proliferation, survival or biological activity. In addition, future studies should be conducted to ask whether or not certain RA subgroups (e.g. rheumatoid factor positive RA versus rheumatoid factor negative RA; high titer versus low titer anti-cyclic citrullinated peptide antibody) will derive greater benefit from ‘Jakinibs’ compared with conventional DMARDs or biologic drugs.
Acknowledgments
The author wishes to thank Evan Meszaros, and Sam Mesiano for helpful discussions.
Footnotes
Funding: The experimental results reported from the Malemud research group at CWRU18,23,27 were supported, in part, by contracts from Takeda Pharmaceuticals of North America, Genentech/Roche Group and NICHD (R01-HD-061819; PI: Sam Mesiano), and the CWRU Visual Sciences Research Center (P30-EY-11373).
Conflict of interest statement: The authors declare that there is no conflict of interest.
References
- 1. Firestein GS. Immunologic mechanisms in the pathogenesis of rheumatoid arthritis. J Clin Rheumatol 2005; 11: S39–S44. [DOI] [PubMed] [Google Scholar]
- 2. Hollan I, Meroni PL, Ahearn JM, et al. Cardiovascular disease in autoimmune rheumatic diseases. Autoimmune Rev 2013; 12: 1004–1015. [DOI] [PubMed] [Google Scholar]
- 3. Mankan AK, Lawless MW, Gray SG, et al. NF-κB regulation: the nuclear response. J Cell Mol Med 2009; 13: 631–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Toh ML, Moissec P. The role of T cells in rheumatoid arthritis: new subsets and new targets. Curr Opin Rheumatol 2007; 19: 284–288. [DOI] [PubMed] [Google Scholar]
- 5. Bugatti S, Codullo V, Caporali R, et al. B cells in rheumatoid arthritis. Autoimmun Rev 2007; 6: 482–487. [DOI] [PubMed] [Google Scholar]
- 6. Karouzakis E, Neidhart M, Gay RE, et al. Molecular and cellular basis of rheumatoid arthritis destruction. Immunol Lett 2006; 106: 8–13. [DOI] [PubMed] [Google Scholar]
- 7. Huber LC, Distler O, Tamer I, et al. Synovial fibroblasts: key player in rheumatoid arthritis. Rheumatology (Oxford) 2006; 45: 669–675. [DOI] [PubMed] [Google Scholar]
- 8. Malemud CJ, Reddy SK. Targeting cytokines, chemokines and adhesion molecules in rheumatoid arthritis. Curr Rheum Rev 2008; 4: 219–234. [Google Scholar]
- 9. Fox DA, Gizinski A, Morgan B, et al. Cell-cell interactions in rheumatoid arthritis synovium. Rheum Dis Clin North Am 2010; 36: 311–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bartok B, Firestein GS. Fibroblast-like synoviocytes: key effector cells in rheumatoid arthritis. Immunol Rev 2010; 233: 233–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Asquith DL, McInnes IB. Emerging cytokine targets in rheumatoid arthritis. Curr Opin Rheumatol 2007; 19: 246–251. [DOI] [PubMed] [Google Scholar]
- 12. Brennan F, Beech J. Update on cytokines in rheumatoid arthritis. Curr Opin Rheumatol 2007; 16: 218–222. [DOI] [PubMed] [Google Scholar]
- 13. Imboden JB. The immunopathogenesis of rheumatoid arthritis. Ann Rev Path 2009; 4: 417–434. [DOI] [PubMed] [Google Scholar]
- 14. Malemud CJ. Defining novel targets for intervention in rheumatoid arthritis: an overview. Curr Rheum Rev 2008; 4: 214–218. [Google Scholar]
- 15. Malemud CJ, Gillespie HJ. The role of apoptosis in arthritis. Curr Rheum Rev 2005; 1: 131–142. [Google Scholar]
- 16. Malemud CJ. Apoptosis resistance in rheumatoid arthritis synovial tissue. J Clin Cell Immunol 2012; S3: 006. [Google Scholar]
- 17. Malemud CJ. Regulation of chondrocyte matrix metalloproteinase gene expression. In: Dhalla NS, Chakraborti S. (Eds) Role of Proteases in Cellular Dysfunction. UK: Springer, 2013, pp.63–77. [Google Scholar]
- 18. Akeson G, Malemud CJ. A role for soluble IL-6 receptor in osteoarthritis. J Funct Morphol Kinesiol 2017; 2: 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ohori M. Erk inhibitors as a potential new therapy for rheumatoid arthritis. Drug News Perspect 2008; 21: 245–250. [DOI] [PubMed] [Google Scholar]
- 20. Heim MH. The JAK-STAT pathway: signaling from the receptor to nucleus. J Recept Signal Transduction Res 1999; 19: 75–120. [DOI] [PubMed] [Google Scholar]
- 21. Malemud CJ. Recent advances in neutralizing the IL-6 pathway in arthritis. Open Access Rheumatol 2009; 1: 133–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Malemud CJ, Pearlman E. Targeting JAK/STAT signaling pathway in inflammatory diseases. Curr Signal Transduct Ther 2009; 4: 201–221. [Google Scholar]
- 23. Wylie MA, Meszaros EC, Malemud CJ. The effect of interleukin-6-type cytokines and adiponectin on MAPK activation in the immortalized human chondrocyte C28/I2 line and normal human chondrocytes. J Cell Biol Histol 2015; 1: 101. [Google Scholar]
- 24. Wajant H, Pfizernmaier K, Scheurich P. Tumor necrosis factor signaling. Cell Death Differ 2003; 10: 45–65. [DOI] [PubMed] [Google Scholar]
- 25. Meyer LH, Pap T. MAPK signalling in rheumatoid arthritis destruction: can we unravel the puzzle? Arthritis Res Ther 2005; 7: 177–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Malemud CJ. Small molecular weight inhibitors of stress-activated and mitogen-activated protein kinases. Mini Rev Med Chem 2006; 6: 689–698. [DOI] [PubMed] [Google Scholar]
- 27. Malemud CJ, Sun Y, Pearlman E, et al. Monosodium urate and tumor necrosis factor-α increase apoptosis in human chondrocyte cultures. Rheumatolology (Sunnyvale) 2012; 2: 113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Benham-Hall E, Clatworthy MR, Okkenhaug K. The therapeutic potential for PI3K inhibitors in autoimmune rheumatic diseases. Open Rheumatol J 2012; 6: 245–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kaur S, Kaira S, Kaushal S. Systematic review of tofacitinib: a new drug for the management of rheumatoid arthritis. Clin Ther 2014; 36: 1074–1086. [DOI] [PubMed] [Google Scholar]
- 30. Ghoreschi K, Lawrence A, O’Shea JJ. Janus kinases in immune cell signaling. Immunol Rev 2009; 228: 273–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Meyer SC, Levine RL. Molecular pathways: molecular basis for sensitivity and resistance to JAK kinase inhibitors. Clin Cancer Res 2014; 20: 2051–2059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. McLornan DP, Khan AA, Harrison CN. Immunological consequences of JAK inhibition: friend or foe? Curr Hematol Malig Rep 2015; 10: 370–379. [DOI] [PubMed] [Google Scholar]
- 33. Schwartz DM, Bonelli M, Gadina M, et al. Type I/II cytokines, JAKs, and new strategies for treating autoimmune diseases. Nat Rev Rheumatol 2016; 12: 25–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kontzias A, Kotlyar A, Laurence A, et al. Jakinibs: a new class of kinase inhibitors in cancer and autoimmune diseases. Curr Opin Pharmacol 2012; 12: 464–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Schwartz DM, Kanno Y, Villarino A, et al. JAK inhibition as a therapeutic strategy for immune and inflammatory diseases. Nat Rev Drug Discov 2017; 16: 843–862. [DOI] [PubMed] [Google Scholar]
- 36. Leonard WJ, O’Shea JJ. JAKs and STATs: biological implications. Annu Rev Immunol 1998; 16: 293–322. [DOI] [PubMed] [Google Scholar]
- 37. Giordanetto P, Kroemer RT. Prediction of the structure of human Janus kinase (JAK2) comprising JAK homology domains 1 through 7. Protein Eng 2002; 15: 727–737. [DOI] [PubMed] [Google Scholar]
- 38. Haan S, Margue C, Engrand A, et al. Dual role of the JAK1 FERM and kinase domains in cytokine receptor binding and stimulation-dependent JAK activation. J Immunol 2008; 180: 998–1007. [DOI] [PubMed] [Google Scholar]
- 39. Darnell JR., Jr. STATS and gene regulation. Science 1997; 277: 1630–1635. [DOI] [PubMed] [Google Scholar]
- 40. Malemud CJ. Suppression of pro-inflammatory cytokines via targeting of STAT-responsive genes. In: El-Shemy H. (ed) Drug Discovery. Rijeka, Croatia: InTech Publishing, 2013, pp. 373–411. [Google Scholar]
- 41. Munoz J, Dhillon N, Janku F, et al. STAT3 inhibitors: finding a home in lymphoma and leukemia. Oncologist 2014; 19: 536–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Derenzini E, Younes A. Targeting the JAK-STAT pathway in lymphoma: a focus on pericitinib. Expert Opin Investig Drugs 2013; 22: 775–785. [DOI] [PubMed] [Google Scholar]
- 43. Meissl K, Macho-Maschler S, Miller A, et al. The good and bad faces of STAT1 in solid tumors. Cytokine 2017; 89: 12–20. [DOI] [PubMed] [Google Scholar]
- 44. Bovolenta C, Camorali L, Lorini AL, et al. Constitutive activation of STATs upon in vivo human immunodeficiency virus infection. Blood 1999; 94: 4202–4209. [PubMed] [Google Scholar]
- 45. Greenlaigh CJ, Hilton DJ. Negative regulation of cytokine signaling. J Leukoc Biol 2001; 70: 348–356. [PubMed] [Google Scholar]
- 46. Krebs DL, Hilton DJ. SOCS proteins: negative regulators of cytokine signaling. Stem Cells 2001; 19: 378–387. [DOI] [PubMed] [Google Scholar]
- 47. Malemud CJ. Suppressor of cytokine signaling and rheumatoid arthritis. Integr Mol Med 2016; 3: 17–20. [Google Scholar]
- 48. Malemud CJ. Negative regulators of JAK/STAT signaling in rheumatoid arthritis and osteoarthritis. Int J Mol Sci 2017; 18: 484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Shouda T, Yoshida T, Hanada T, et al. Induction of the cytokine signal regulator SOCS3/CIS3 as a therapeutic strategy for treating inflammatory arthritis. J Clin Invest 2001; 108: 1781–1788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Mahony R, Ahmed S, Diskin C, et al. SOCS revisited: a broad regulator of disease, now ready for therapeutic use? Cell Mol Life Sci 2016; 73: 1745–1747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Lao M, Shi M, Zou Y, et al. Protein inhibitor of activated STAT3 regulates migration, invasion, and activation of fibroblast-like synoviocytes in rheumatoid arthritis. J Immunol 2016; 196: 596–606. [DOI] [PubMed] [Google Scholar]
- 52. Gadina M. Advances in kinase inhibition: treating rheumatic diseases and beyond. Curr Opin Rheumatol 2014; 26: 237–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Borie DC, Si MS, Morris RE, et al. JAK3 inhibition as a new concept for immune suppression. Curr Opin Investig Drugs 2003; 4: 1297–1303. [PubMed] [Google Scholar]
- 54. Kawalec P, Mikrut A, Wiśniewska N, et al. The effectiveness of tofacitinib: a novel Janus kinase inhibitor, in the treatment of rheumatoid arthritis: a systematic review and meta-analysis. Clin Rheumatol 2013; 32: 1415–1424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Jiang JK, Ghoreschi K, Deflorian F, et al. Examining the chirality, conformation and selective kinase inhibition of 3-((3R,4R)-4-methyl-3-(methyl(7H-pyrrolo2,3-d.pyrimidin-4-yl)amino)piperidin-1-yl)-3-oxopropanenitrile (CP-690,550). J Med Chem 2008; 51: 8012–8018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Malemud CJ, Blumenthal DE. Protein kinase small molecule inhibitors for rheumatoid arthritis: medicinal chemistry/clinical perspectives. World J Orthopedics 2014; 5: 496–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Lee FB, Fleischmann R, Hall S, et al. Tofacitinib versus methotrexate in rheumatoid arthritis. N Engl J Med 2014; 370: 2377–2386. [DOI] [PubMed] [Google Scholar]
- 58. Vyas D, O’Dell KM, Bandy JL, et al. Tofacitinib: the first Janus kinase (JAK) inhibitor for the treatment of rheumatoid arthritis. Ann Pharmacother 2013; 47: 1524–1531. [DOI] [PubMed] [Google Scholar]
- 59. Norman P. Selective JAK inhibitors in development for rheumatoid arthritis. Expert Opin Investig Drugs 2014; 23: 1067–1077. [DOI] [PubMed] [Google Scholar]
- 60. Zerbini CA, Lomonte AB. Tofacitinib for the treatment of rheumatoid arthritis. Expert Rev Clin Immunol 2012; 8: 319–331. [DOI] [PubMed] [Google Scholar]
- 61. Wollenhaupt J, Silverfield J, Lee EB, et al. Safety and efficacy of tofacitinib, an oral janus kinase inhibitor, for the treatment of rheumatoid arthritis in open-label, long-term extension studies. J Rheumatol 2014; 41: 837–852. [DOI] [PubMed] [Google Scholar]
- 62. Hodge JA, Kawabata TT, Krishnaswami S, et al. The mechanism of action of tofacitinib – an oral Janus kinase inhibitor for the treatment of rheumatoid arthritis. Clin Exp Rheumatol 2016; 34: 318–328. [PubMed] [Google Scholar]
- 63. Yamaoka K. Janus kinase inhibitors for rheumatoid arthritis. Curr Opin Chem Biol 2016; 32: 29–33. [DOI] [PubMed] [Google Scholar]
- 64. Fleischmann R. A review of tofacitinib efficacy in rheumatoid arthritis patients who have had an inadequate response or intolerance to methotrexate. Expert Opin Pharmacother 2017; 18: 1525–1533. [DOI] [PubMed] [Google Scholar]
- 65. Winthrop KL. The emerging safety profile of JAK inhibitors in rheumatic diseases. Nat Rev Rheumatol 2017; 13: 234–243. [DOI] [PubMed] [Google Scholar]
- 66. Sivaraman P, Cohen SB. Malignancy and Janus kinase inhibition. Rheum Dis Clin North Am 2017; 43: 79–93. [DOI] [PubMed] [Google Scholar]
- 67. Mesa RA. Ruxolitinib, a selective JAK1 and JAK2 inhibitor for the treatment of myeloproliferative neoplasms and psoriasis. Drugs 2010; 13: 394–403. [PubMed] [Google Scholar]
- 68. Gadina M. Janus kinases: an ideal target for the treatment of autoimmune diseases. J Investig Dermatol Symp Proc 2013; 16: S70–S72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Shi JG, Chen X, McGee RF, et al. The pharmacokinetics, pharmacodynamics, and safety of orally dosed INCB018424 phosphate in healthy volunteers. J Clin Pharmacol 2011; 51: 1644–1654. [DOI] [PubMed] [Google Scholar]
- 70. Williams W, Scherle P, Shi P, et al. A randomized placebo-controlled study of INCB018424, a selective Janus kinase 1&2 (JAK1&2) inhibitor in rheumatoid arthritis. Arthritis Rheum 2008; 58: S431. [Google Scholar]
- 71. Menet CJ, Mammoliti O, López-Ramos M. Progress towards JAK1-selective inhibitors. Future Med Chem 2015; 7: 203–235. [DOI] [PubMed] [Google Scholar]
- 72. Vrontaki E, Melagraki G, Afantitis A, et al. Searching for novel Janus kinase-2 inhibitors using a combination of pharmacophore modeling, 3D-QSAR studies and virtual screening. Mini Rev Med Chem 2017; 17: 268–294. [DOI] [PubMed] [Google Scholar]
- 73. Kremer JM, Schiff M, Muram M, et al. Response to baricitinib therapy in patients with rheumatoid arthritis with inadequate response to csDMARDs as a function of baseline characteristics. RMD Open 2018; 4: e000581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. van Vollenhoven RF. Small molecular compounds in development for rheumatoid arthritis. Curr Opin Rheumatol 2013; 25: 391–397. [DOI] [PubMed] [Google Scholar]
- 75. Fridman JS, Scherle PA, Collins R, et al. Selective inhibition of JAK1 and JAK2 is efficacious in rodent models of arthritis: preclinical characterization of INCB028050. J Immunol 2010; 184: 5298–5307. [DOI] [PubMed] [Google Scholar]
- 76. Shi JG, Chen X, Lee F, et al. The pharmacokinetics, pharmacodynamics and safety of baracitinib, an oral JAK1/2 inhibitor, in healthy volunteers. J Clin Pharmacol 2014; 54: 1354–1361. [DOI] [PubMed] [Google Scholar]
- 77. Richez C, Truchetet ME, Kostine M, et al. Efficacy of baracitinib in the treatment of rheumatoid arthritis. Expert Opin Pharmacother 2017; 18: 1399–1407. [DOI] [PubMed] [Google Scholar]
- 78. Genovese MC, Kremer J, Zamani O, et al. Baricitinib in patients with refractory rheumatoid arthritis. N Engl J Med 2016; 374: 1243–1252. [DOI] [PubMed] [Google Scholar]
- 79. Dougados M, van der Heijde D, Chen YC, et al. Baricitinib in patients with inadequate response or intolerance to conventional synthetic DMARDs: results from the RA-BUILD study. Ann Rheum Dis 2017; 76: 88–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Mattar M, Jandali B, Malemud CJ, et al. Atherosclerosis and rheumatic diseases. Rheumatology (Sunnyvale) 2015; 5: 147. [Google Scholar]
- 81. Genovese MC, van Vollenhoven RE, Pacheco-Tena C, et al. VX-509 (Decernotinib), an oral selective JAK-3 inhibitor, in combination with methotrexate in patients with rheumatoid arthritis. Arthritis Rheum 2016; 68: 46–55. [DOI] [PubMed] [Google Scholar]
- 82. Genovese MC, Yang F, Østergaard M, et al. Efficacy of VX-509 (decernotinib) in combination with a disease-modifying antirheumatic drug in patents with rheumatoid arthritis: clinical and MRI findings. Ann Rheum Dis 2016; 75: 1979–1983. [DOI] [PubMed] [Google Scholar]
- 83. Gadina M, Schwartz DM, O’Shea JJ. Decernotinib: a next-generation Jakinib. Arthritis Rheumatol 2016; 68: 31–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Mahajan S, Hogan JK, Shlyakhter D, et al. VX-509 (decernotinib) is a potent and selective Janus kinase inhibitor that attenuates inflammation in animal models of autoimmune disease. J Pharmacol Exp Ther 2015; 353: 405–414. [DOI] [PubMed] [Google Scholar]
- 85. Elwood F, Witter DJ, Piesvaux J, et al. Evaluation of JAK3 biology in autoimmune disease using a highly selective, irreversible JAK3 inhibitor. J Pharmacol Exp Ther 2017; 361: 229–244. [DOI] [PubMed] [Google Scholar]
- 86. Taylor PC, Abdul Azeez M, Kiriakidis S. Filgotinib for the treatment of rheumatoid arthritis. Expert Opin Investig Drugs 2017; 26: 1181–1187. [DOI] [PubMed] [Google Scholar]
- 87. Van Rompaey L, Galien R, van der Aar EM, et al. Preclinical characterization of GLPG0634, a selective inhibitor of JAK1, for the treatment of inflammatory diseases. J Immunol 2013; 191: 3568–3577. [DOI] [PubMed] [Google Scholar]
- 88. Kavanaugh A, Kremer J, Ponce CL, et al. Filgotinib (GLPG0634/GS-6034), an oral, selective JAK1 inhibitor, is effective as monotherapy in patients with active rheumatoid arthritis: results from a randomised, dose-finding study (DARWIN 2). Ann Rheum Dis 2017; 76: 1009–1019. [DOI] [PubMed] [Google Scholar]
- 89. Westhovens R, Taylor PC, Alten R, et al. Filgotinib (GLPG0634/GS-6034), an oral JAK1 selective inhibitor, is effective in combination with methotrexate (MTX) in patients with active rheumatoid arthritis and insufficient response to MTX: results from a randomised, dose-finding study (DARWIN 1). Ann Rheum Dis 2017; 76: 998–1008. [DOI] [PubMed] [Google Scholar]
- 90. Vanhoutte F, Mazur M, Voloshyn O, et al. Efficacy, safety, pharmacokinetics, and pharmacodynamics of filgotinib, a selective JAK-1 inhibitor, after short-term treatment of rheumatoid arthritis: results of two randomized phase IIa trials. Arthritis Rheumatol 2017; 69: 1949–1959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Nakayamada S, Kubo S, Iwata S, et al. Recent progress in JAK inhibitors for the treatment of rheumatoid arthritis. BioDrugs 2016; 30: 407–419. [DOI] [PubMed] [Google Scholar]
- 92. Baker KF, Isaacs JD. Novel therapies for immune-mediated inflammatory diseases: what can we learn for their use in rheumatoid arthritis, spondyloarthritis, systemic lupus erythematosus, psoriasis, Crohn’s disease and ulcerative colitis? Ann Rheum Dis 2018; 77: 175–187. [DOI] [PubMed] [Google Scholar]
- 93. Ito M, Yamazaki S, Yamagami K, et al. A novel JAK inhibitor, peficitinib, demonstrates potent efficacy in a rat adjuvant-induced arthritis model. J Pharmacol Sci 2017; 133: 25–33. [DOI] [PubMed] [Google Scholar]
- 94. Klünder B, Mohamed MF, Othman AA. Population pharmacokinetics of upadacitinib in healthy subjects and subjects with rheumatoid arthritis: analysis of phase I and II clinical trials. Clin Pharmacokinet. Epub ahead of print 26 October 2017. DOI: 1007/s40262-017-0605-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Genovese MC, Greenwald M, Codding C, et al. Peficitinib, a JAK inhibitor, in combination with limited conventional synthetic disease-modifying antirheumatic drugs in the treatment of moderate-severe rheumatoid arthritis. Arthritis Rheumatol 2017; 69: 932–942. [DOI] [PubMed] [Google Scholar]
- 96. Kivitz AJ, Gutierrez-Ureῆa SR, Poiley J, et al. Peficitinib, a JAK inhibitor, in the treatment of moderate-severe rheumatoid arthritis in patients with an inadequate response to methotrexate. Arthritis Rheumatol 2017; 69: 709–719. [DOI] [PubMed] [Google Scholar]
- 97. Zhu T, Parker B, Wojtkowski T, et al. Drug interactions between peficitinib, an orally administered, once-daily Janus kinase inhibitor, and rosuvastatin in healthy subjects. Clin Pharmacokinet 2017; 56: 747–757. [DOI] [PubMed] [Google Scholar]
- 98. Sweeney SE, Firestein GS. Signal transduction in rheumatoid arthritis. Curr Opin Rheumatol 2004; 16: 231–237. [DOI] [PubMed] [Google Scholar]
- 99. Wang W, Tan J, Xing Y, et al. p43 induces IL-10 expression through the JAK-STAT signaling pathway in HMEC-1 cells. Int J Mol Med 2016; 38: 1217–1224. [DOI] [PubMed] [Google Scholar]
- 100. Chapman MS, Wu L, Amatucci A, et al. TWEAK signals through JAK-STAT to induce tumor cell apoptosis. Cytokine 2013; 61: 210–217. [DOI] [PubMed] [Google Scholar]
- 101. Krolopp JE, Thornton SM, Abbott MJ. IL-15 activates the Jak3/STAT3 signaling pathway to mediate glucose uptake in skeletal muscle cells. Front Physiol 2016; 7: 626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Shenoy AR, Kirschnek S, Häcker G. IL-15 regulates Bcl-2 family members Bim and Mcl-1 through JAK/STAT and PI3K/AKT pathways in T-cells. Eur J Immunol 2014; 44: 2500–2507. [DOI] [PubMed] [Google Scholar]
- 103. Tsuji-Takayama K, Suzuki M, Yamamoto M, et al. IL-2 activation of STAT5 enhances production of IL-10 from human cytotoxic regulatory T-cells, HOZOT. Exp Hematol 2008; 36: 181–192. [DOI] [PubMed] [Google Scholar]
- 104. Ok CY, Young KH. Checkpoint inhibitors in hematological malignancies. J Hematol Oncol 2017; 10: 103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. McInnes IB, Schett G. Pathogenetic insights from the treatment of rheumatoid arthritis. Lancet 2017; 389: 2328–2337. [DOI] [PubMed] [Google Scholar]
- 106. Doi T, Ishikawa T, Okayama T, et al. The JAK/STAT pathway is involved in the upregulation of PD-L1 expression in pancreatic cancer cell lines. Oncol Rep 2017; 37: 1545–1554. [DOI] [PubMed] [Google Scholar]
- 107. Van Vollenhoven RF, Fleischmann R, Cohen S, et al. Tofacitinib or adalimumab versus placebo in rheumatoid arthritis. New Engl J Med 2012; 367: 509–519. [DOI] [PubMed] [Google Scholar]
- 108. Fleischmann R, Kremer J, Cush J, et al. Placebo-controlled trial of tofacitinib monotherapy in rheumatoid arthritis. N Engl J Med 2012; 367: 495–507. [DOI] [PubMed] [Google Scholar]