

Abstract
The homeostasis of intracellular cholesterol in animal cells is highly regulated by a complex system in which the microsomal rate-limiting enzyme 3-hydroxy-3-methylglutaryl CoA (HMG-CoA) reductase plays a key role in cholesterol synthesis. Substantial evidence has demonstrated that the cytosolic antioxidant enzyme CuZn superoxide dismutase (SOD1) inhibits the HMG-CoA reductase activity in rat hepatocytes and in human fibroblasts by decreasing cholesterol synthesis. Although these data suggest that SOD1 exerts a physiological role in cholesterol metabolism, it is still unclear whether the decrease of HMG-CoA reductase activity is mediated by transcriptional or by posttranscriptional events. The results of the present study, obtained by one-step RT-PCR assay, demonstrated that both SOD1 and the metal-free form of enzyme (Apo SOD1) inhibit HMG-CoA reductase gene expression in hepatocarcinoma HepG2 cells, in normal human fibroblasts, and in fibroblasts of subjects affected by familiar hypercholesterolemia. Accordingly, SOD1 could be used as a potential agent in the treatment of hypercholesterolemia, even in subjects lacking a functional LDL receptor pathway.
Key words: Cholesterol, CuZn superoxide dismutase, Familial hypercholesterolemia, 3-Hydroxy 3-methylglutaryl-CoA reductase, Human fibroblasts, HepG2 cells
THE intracellular cholesterol content is tightly controlled. Brown and Goldstein’s classical experiments (4) have, in fact, demonstrated that when intracellular cholesterol is too high, cells downregulate cholesterol synthesis and LDL cholesterol uptake. By contrast, when the intracellular cholesterol is insufficient, cholesterol synthesis and LDL cholesterol uptake increase.
In mammalian cells, cholesterol synthesis is mainly regulated by the microsomal rate-limiting enzyme 3-hydroxy-3-methylglutaryl-CoA (HMG-CoA) reductase, which constitutes the limiting step in cholesterol biosynthesis, and by the LDL receptor pathway, which is involved in the uptake of exogenous cholesterol (10). HMG-CoA reductase is an enzyme regulated by a complex system in which both cholesterol and nonsterol mevalonate metabolites carry out a feedback suppression (5).
Current studies suggest that the suppression of cholesterol is mainly mediated by oxysterols produced within the cell by oxidation of intracellular cholesterol; however, many other physiological mechanisms, as enzyme oxidation or phosphorylation caused by many types of kinases, are capable of inactivating HMG-CoA reductase and of increasing its catabolism (24).
Consistent with these findings, we have found that CuZn superoxide dismutase (SOD1) inhibits HMG-CoA reductase activity in rat hepatocyte cells (BRL-3A) and in human fibroblasts. Furthermore, we previously showed that such inhibitory effect on HMG-CoA reductase activity is also paralleled by a decrease in HMG-CoA reductase protein levels (17,22) and that the circulating SOD1 binds to lipoproteins, mainly to LDL and HDL (19).
Furthermore, we have also recently reported that SOD1 affects cholesterol metabolism by decreasing cholesterol synthesis and LDL binding to human hepatocarcinoma HepG2 cells. Interestingly, this effect was shown to be independent of the dismutase activity of the enzyme and was mediated by PKC activation (22).
Although these data further confirm the relevant role played by SOD1 in cholesterol metabolism, it is still not fully understood whether the decrease of HMG-CoA reductase activity is mediated by transcriptional or posttranscriptional events.
Therefore, to have a better understanding of the mechanisms involved in cholesterol synthesis suppression, we evaluated the effect of SOD1 on HMG-CoA reductase gene expression in HepG2 cells and in human fibroblasts, deriving either from normocholesterolemic subjects or from subjects affected by familial heterozygotic hypercholesterolemia.
MATERIALS AND METHODS
Cells
Human hepatocarcinoma cells (HepG2 cells) and human fibroblasts of normal and hypercholesterolemic subjects (obtained from the “Cell Line and DNA Bank of patients affected by Genetic Diseases”) were grown in Dulbecco’s modified Eagle’s medium (DMEM), containing 10% fetal bovine serum (FBS), 2 mM l-glutamine, 50 μg/ml streptomycin, and 50 IU/ml penicillin (all purchased from Life Technologies, Italy); the cells were kept in 5% CO2 at 37°C.
To measure the mRNA of HMG-CoA reductase, human fibroblasts and HepG2 cells were starved in DMEM without serum for 18 h. Next, the cells were incubated with 150 ng/ml SOD1 at different time intervals, as reported in the Results section. In one series of experiments cells were incubated with SOD1 in the presence of 10% FBS.
RNA Preparation
Total RNAs of cell lines were extracted with High Pure RNA isolation kit (Roche), according to the manufacturer’s instructions, using 1 × 106 cells. Traces of contaminated DNA were removed with DNAse I treatment.
[Ca2+]i Measurements
To evaluate whether the modulation of SOD1 on HMG-CoA reducatese gene expression involved an increase of intracellular calcium, via PLC-PKC pathway activation, [Ca2+]i measurements were performed using a microfluorimetric technique as previously reported (9). Briefly, cells grown on glass coverslips were loaded with 5 μM fura-2 AM in Krebs-Ringer saline solution for 1 h at 22°C. At the end of fura-2 AM loading, the coverslips were introduced into a microscope chamber (Medical System Co., Greenvale, NY) mounted on an inverted Nikon Diaphot fluorescence microscope. Cells were kept in Krebs-Ringer saline solution throughout the experiment. All the solutions were prepared as previously reported (26). The substances tested were introduced into the microscope chamber by fast injection. A 100-W Xenon Lamp (Osram, Frankfurt, Germany) with a computer-operated filter wheel, bearing two different interference filters (340 and 380 nm), illuminated the microscopic field with UV light, alternating the wavelength at 500-ms intervals. The interval between each pair of illuminations was 2 s, and the interval between filter movements was 1 s. Consequently, [Ca2+]i was measured every 3 s. Emitted light was passed through a 400-nm dichroic mirror, was filtered at 510 nm, and was collected by a CCD camera (Photonic Science, Robertsbridge, UK) connected to a light amplifier (Applied Imaging Ltd, Dukesway Gateshead, UK). Images were digitized and analyzed with a Magiscan image processor (Applied Imaging Ltd, Dukesway Gateshead, UK). Using a calibration curve, the AUTOLAB software (RBR Altair, Florence, Italy) calculated the [Ca2+]i corresponding to each pair of images from the ratio between the intensity of the light emitted when cells were illuminated at both 340 and 380 nm. At the end of each experimental session, the calibration was performed according to the procedure described by Grynkievicz et al. (11). In particular, cells were lyzed with ionomycin (2–10 μM) in the presence of 1.5 mM extracellular Ca2+. The addition of ionomycin produced a rapid increase in fluorescence intensity, allowing us to calculate the R max value. To determine the R min value, cells were subsequently exposed to a Ca2+-free solution containing 1–20 mM EGTA. Given that K d for Ca2+ of fura-2 AM is 224 nM at 37°C, the R min and R max values were introduced into the Grynkievicz formula to convert the values of the fluorescence ratio, ranging between 340 and 380 nm, to [Ca2+]i. The values were subtracted for background fluorescence obtained from images taken from a region of the coverslip devoid of cells. No interference was detected between any of the compounds utilized in the present study and the excitation or the emission spectra of fura-2 AM.
Semiquantitative RT-PCR
To improve the accuracy and sensitivity of the procedure, we performed 1) a one-step quantitative reverse transcription polymerase chain reaction, in which the reverse transcriptase enzyme and Pwo and Taq DNA polymerase were combined in the one tube, and 2) a single, noninterrupted thermal cycling program. Quantitation was achieved in a single reaction by using the housekeeping β-actin gene as internal standard.
RNA template (100 ng) was reverse-transcribed in 50 μl reaction mix containing 200 μM dNTPs, 100 mM DTT, 0.25 μl RNAse inhibitor, 1.5 mM MgCl2, and 1 μl enzyme mix (Titan One-tube RT-PCR kit, Roche).
The solution was incubated for 30 min at 50°C in an automated DNA thermal cycler (GeneAmp 2400 Perkin Elmer). To perform RT-PCR at optimal conditions and to stay within the logarithmically linear product formation, 40 cycles were chosen (30 s at 94°C, 30 s at 59°C, 60 s at 68°C) and were followed by a final extension for 7 min at 68°C.
β-Actin and Hmg primer pairs were designed to yield PCR products of different sizes (587 bp for β-actin and 391 bp for Hmg). The forward and reverse β-actin primers were 5′-CCAAGGCCAACCGCGAGAAGATGAC-3′ and 5′-AGGGTACATGGTGGTGCCGCCAGAC-3′, respectively; the forward and reverse HMG-CoA reductase primers were 5′-AGCTTTGCCCTTTTTCCTACTTTT-3′ and 5′-AGGGTACATGGTGGTGCCGCCAGAC-3′, respectively.
SREBP-2, squalene synthase, and LDL receptor gene primer pairs were designed to yield PCR products of different sizes, such as 401, 319, and 258 bp, respectively. The forward and reverse SREBP-2 primers were 5′-CCCTTCAGTGCAACGGTCATTCAC-3′ and 5′-TGCCATTGGCCGTTTGTGTC-3′, respectively; the forward and reverse squalene synthase primers were 5′-TTCTACAACCTGGTGCGCTT-3′ and 5′-CGATCCTTCTCCTTGCTCTC-3′, respectively; the forward and reverse LDL receptor primers were 5′-CAATGTCTCACCAAGCTCTG-3′ and 5′-TCTGTCTCGAGGGGTAGCTG-3′, respectively. To perform RT-PCR at optimal conditions and to stay within the logarithmically linear product formation, 40 cycles were chosen (30 s at 94°C, 30 s at 60°C, 60 s at 68°C) for SREBP-2 and 40 cycles (30 s at 94°C, 30 s at 57°C, 60 s at 68°C) for squalene synthase and LDL receptor, followed by a final extension for 7 min at 68°C.
To rule out genomic DNA contamination we performed a negative control that contained RNA instead of cDNA.
The signal intensities of PCR products were separated on a 1.2% agarose gel and were visualized by ethidium bromide staining. The products’ signal intensities were determined by computerized densitometric analysis using Fotoplot software. The expression of HMG-CoA reductase, SREBP-2, squalene synthase, and LDL receptor mRNAs were normalized to β-actin mRNA levels.
Statistical Analysis
All results shown are mean ± SD of at least three separate experiments; each point represents a triplicate of each experiment. Statistically significant differences were tested by one-way analysis of variance (ANOVA).
DNA Sequencing
To check the specificity of the amplified products, DNA bands were eluted from the gel and purified; sequence analysis was determined by the Big Dye Terminator Cycle Sequencing method (ABI-PRISM Sequencer 310 Perkin-Elmer).
RESULTS
Commonly, gene expression has been determined by the one-step RT-PCR technique (14,23), because it is considered a useful method for investigating transcriptional gene expression. However, because the relevance of assay conditions should be reassessed whenever applied to mRNA levels of different types of cells and tissues (2,6,27), the following experimental procedure was carried out. Firstly, we subjected HepG2 cells to the one-step RT-PCR assay; secondly, we examined the correlation between the amounts of RNA used and those of the PCR products, deriving from HMG-CoA reductase gene (the target) and β-actin (the internal standard) mRNAs. The bands corresponding to the expected size of PCR products for HMG-CoA reductase mRNA (587 bp) and β-actin mRNA (391 bp) were observed on the agarose gel. The intensity of these bands increased in proportion to the increasing total RNA from 0.1 to 2 μg/tube. Moreover, during the PCR cycles the cell lines showed a linear increment in both PCR products, ranging from 25 to 40 cycles (data not shown).
The purification and sequencing of these bands confirmed the HMG-CoA and the β-actin transcripts. The same procedure was used to assay SREBP-2, squalene synthase, and LDL receptor gene expression. , 25 Apr 2018 06:47:21
Similar results were also obtained using total RNA deriving from human fibroblasts of both normal and hypercholesterolemic subjects (data not shown).
In effect, in Hep-G2 cells, we observed a significant reduction in HMG-CoA reductase expression after 1 h of SOD1 incubation (Fig. 1A and B) and a linear decrease in HMG-CoA reductase expression up to 4 h of SOD1 incubation. Four hours of Apo SOD incubation produced similar results to the ones obtained during 4 h of SOD1 incubation.
Figure 1.
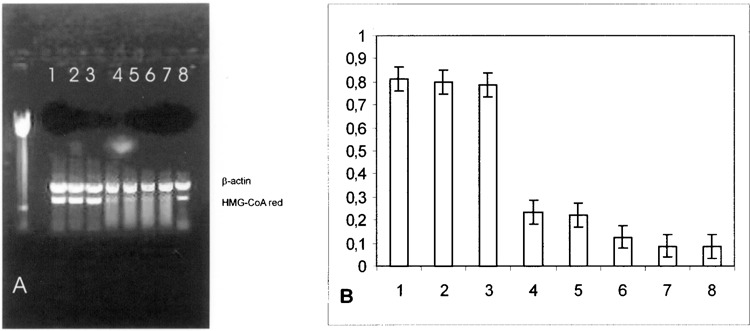
(A) Effect of SOD1 on HMG-CoA reductase mRNA in HepG2 cells. Quantitation was achieved in a single reaction in all the cell lines by the use of β-actin gene as internal standard. Left lane: marker 100 bp. Lane 1: untreated HepG2 cells; lane 2: 10-min SOD1 incubation; lane 3: 30-min SOD1 incubation; lane 4: 1-h SOD1 incubation; lane 5: 1-h Apo SOD1 incubation; lane 6: 2-h SOD1 incubation; lane 7: 4-h SOD1 incubation; lane 8: 4-h Apo SOD incubation. (B) Quantitation of the effect of SOD1 on HMG-CoA reductase mRNA expression in HepG2 cells. Each bar represents the mean of three independent experiments in which the ratio of target PCR product/internal standard PCR product was plotted. 1: Untreated HepG2 cells; 2: 10-min SOD1 incubation; 3: 30-min SOD1 incubation; 4: 1-h SOD1 incubation; 5: 1-h Apo SOD1 incubation; 6: 2-h SOD1 incubation; 7: 4-h SOD1 incubation; 8: 4-h Apo SOD incubation.
Data from the same series of experiments was performed in normal fibroblasts, as shown in Figure 2A and B. Similarly, in normal fibroblasts, a linear HMG-CoA reductase expression reduction followed SOD1 incubation. SOD1 incubation of human fibroblasts, deriving from subjects affected by familial heterozygotic hypercholesterolemia, led to a reduction of HMG-CoA expression for up to 4 h (Fig. 3A, lanes 3–5, and B). Interestingly, because similar results were observed after 4 h of Apo SOD and of SOD1 incubations in hypercholesterolemic fibroblasts, as well as in HepG2 cells and in normal fibroblasts, we concluded that the HMG-CoA reductase expression reduction is independent of the dismutase activity of SOD1.
Figure 2.
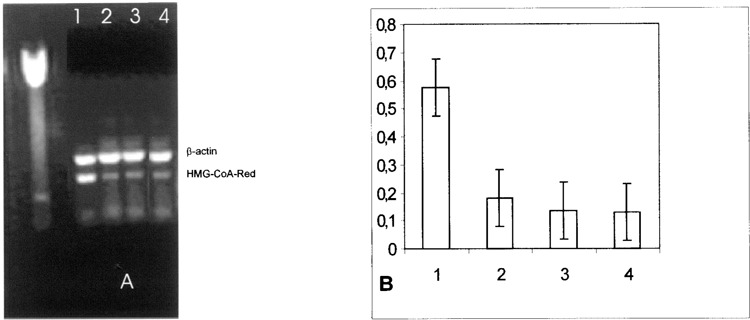
(A) Effect of SOD1 on HMG-CoA reductase mRNA in fibroblast cells from normal subjects. Quantitation was achieved in a single reaction in all the cell lines by the use of β-actin gene as internal standard. Left lane: marker 100 bp. Lane 1: untreated fibroblasts cells from normal subjects; lane 2: 1-h SOD1 incubation; lane 3: 2-h SOD1 incubation; lane 4: 2-h Apo SOD incubation. (B) Quantitation of the effect of SOD1 on HMG-CoA reductase mRNA expression in normal subjects. Each bar represents the mean of three independent experiments in which the ratio of target PCR product/internal standard PCR product was plotted. 1: Untreated fibroblast cells from normal subjects; 2: 1-h SOD1 incubation; 3: 2-h SOD1 incubation; 4: 2-h Apo SOD incubation.
Figure 3.
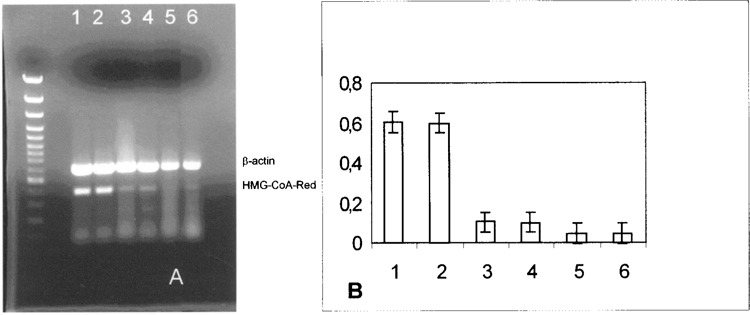
(A) Effect of SOD1 on HMG-CoA reductase mRNA in fibroblasts from hypercholesterolemic subjects. Quantitation was achieved in a single reaction in all the cell lines by the use of β-actin gene as internal standard. Left lane: marker 100 bp. Lane 1: untreated fibroblasts cells from hypercholesterolemic subjects; lane 2: 30-min SOD incubation; lane 3: 1-h SOD incubation; lane 4: 2-h SOD incubation; lane 5: 4-h SOD incubation; lane 6: 4-h Apo SOD incubation. (B) Quantification of the effect of SOD1 on HMG-CoA reductase mRNA expression in fibroblasts from hypercholesterolemic subjects. Each bar represents the mean of three independent experiments in which the ratio of target PCR product/internal standard PCR product was plotted. 1: Untreated fibroblast cells from hypercholesterolemic subjects; 2: 30-min SOD incubation; 3: 1-h SOD incubation; 4: 2-h SOD incubation; 5: 4-h SOD incubation; 6: 4-h Apo SOD incubation.
The microfluorimetric determination of intracellular calcium concentration in HepG2 cells, starved overnight in a medium without serum and then incubated with 150 ng/ml of SOD1, was assessed (Fig. 4). After about 5 min, the cytosolic calcium started to increase, reaching an increment of 48%.
Figure 4.
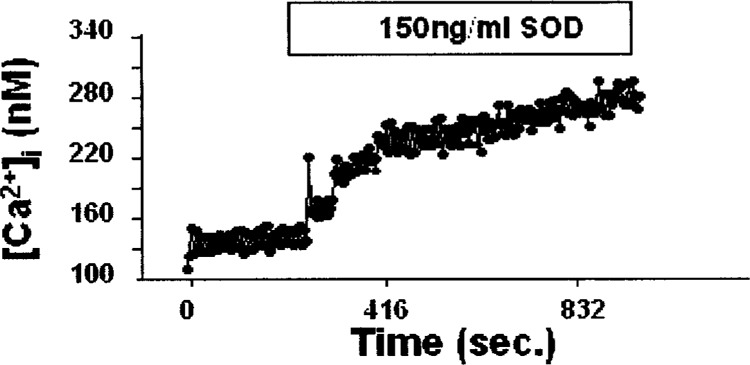
Microfluorimetric intracellular calcium concentration in HepG2 cells incubated with 150 ng/ml of SOD1.
To evaluate whether the cytosolic calcium increase depended on the activation of the phospholipase C(PLC)-protein kinase C (PKC) pathway, we determined the cytosolic calcium concentration in HepG2 cells. The cells were starved and were incubated with 150 ng/ml of SOD1 in the presence and in the absence of 10 μM of U73122 (PLC inhibitor) and 1 μM of BDM (PKC inhibitor). The PLC inhibitor completely reversed the calcium increase determined by SOD1 (Fig. 5A), whereas the PKC inhibitor only partially reduced the intracellular calcium increase induced by SOD1 treatment (Fig. 5B and C).
Figure 5.
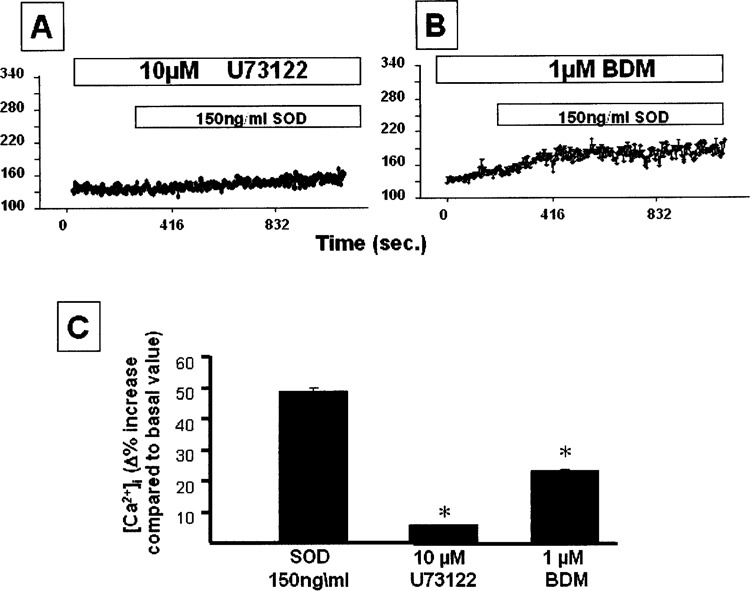
Microfluorimetric calcium concentration in HepG2 cells preincubated for 15 min with 10 μM U73122 (A) and 1 μM BDM (B), respectively, and then treated with 150 ng/ml of SOD1. (C) The percentage increase of intracellular calcium in SOD1-treated cells and in cells treated with PLC and PKC inhibitors.
To verify whether the inhibition of PLC–PKC pathway induced by U73122 and by BDM, respectively, also contributed to SOD1 downregulation of HMG-CoA reductase gene expression, the effect of SOD1 on HMG-CoA reductase mRNA was evaluated (Fig. 6). In effect, both substances reversed the inhibitory effect of SOD1 on HMG-CoA reductase gene expression.
Figure 6.
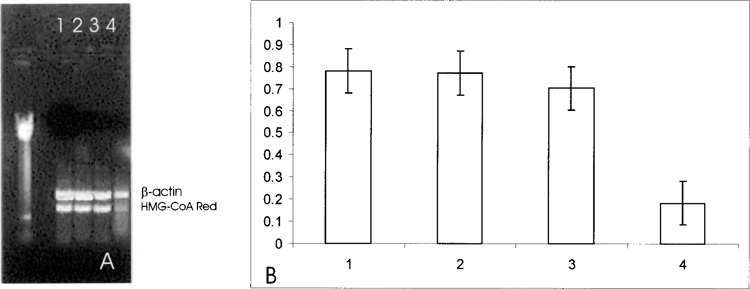
Effect of SOD1 on HMG-CoA reductase mRNA in HepG2 cells. Quantitation was achieved in a single reaction in all the cell lines by the use of β-actin gene as internal standard. Left lane: marker 100 bp. Lane 1: untreated HepG2 cells; lane 2: 20-min preincubation with BDM and 1-h incubation with 150 ng/ml SOD1; lane 3: 20-min preincubation with U73122 and 1-h incubation with 150 ng/ml SOD1; lane 4: HepG2 incubated for 1 h with 150 ng/ml SOD 1 alone.
Furthermore, to assess whether the inhibitory effect of SOD1 on the reductase gene expression occurred also in the presence of serum, we performed a further experiment in the presence of 10% FBS. The incubation of HepG2 cells in the presence of the serum led to a downregulation of HMG-CoA reductase gene expression, as expected from the presence of serum cholesterol; instead, the concomitant incubation of HepG2 cells in the presence of both serum and SOD1 caused only a slighter decrease in HMG-CoA gene expression (Fig. 7). To evaluate whether SOD1 affected other sterol-responsive genes involved in cholesterol metabolism, we evaluated squalene synthase and LDL receptor gene expression. The same downregulation was observed for the expression of squalene synthase (Fig. 8) and for LDL receptor (Fig. 9). Moreover, to investigate whether SOD1 regulated the HMG-CoA reductase, squalene synthase, and LDL receptor at a transcriptional level, we studied the effect of SOD1 on the gene expression of sterol regulatory element-binding proteins (SREBP-2) belonging to a family of membrane-bound transcriptional factors that regulate many enzymes of cholesterol metabolism (12). As shown in Figure 10, the incubation of both SOD1 and Apo SOD (150 ng/ml) in HepG-2 strongly decreased SREBP-2 gene expression.
Figure 7.
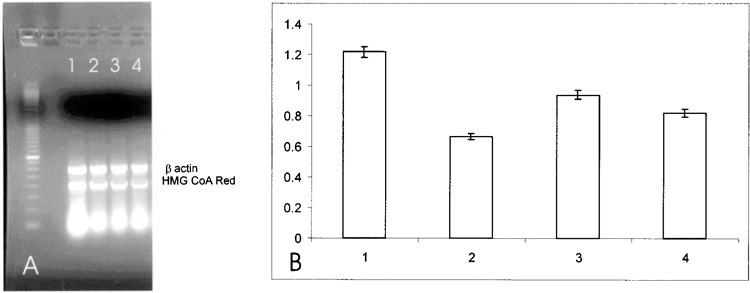
(A) Effect of SOD1 on HMG-CoA reductase mRNA in HepG2 cells. Quantitation was achieved in a single reaction in all the cell lines by the use of β-actin gene as internal standard. Left lane: marker 100 bp. Lane 1: untreated HepG2 cells without serum; lane 2: HepG2 cells without serum + 1-h SOD1 incubation; lane 3: untreated HepG2 cells with serum; lane 4: HepG2 cells with serum + 1-h SOD1 incubation. (B) Quantification of the effect of SOD1 on HMG-CoA reductase mRNA expression in HepG2 cells. Each bar represents the mean of three independent experiments in which the ratio of target PCR product/internal standard PCR product was plotted. 1: Untreated HepG2 cells without serum; 2: untreated HepG2 cells without serum + 1-h SOD1 incubation; 3: untreated HepG2 cells with serum; 4: untreated HepG2 cells with serum + 1-h SOD1 incubation.
Figure 8.
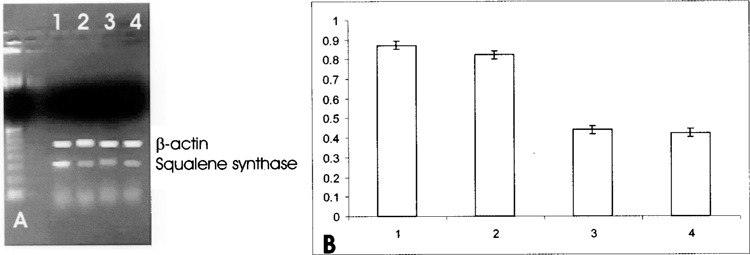
(A) Effect of SOD1 on squalene synthase mRNA in HepG2 cells. Quantitation was achieved in a single reaction in all the cell lines by the use of β-actin gene as internal standard. Left lane: marker 100 bp. Lane 1: untreated HepG2 cells; lane 2: 30-min SOD1 incubation; lane 3: 2-h SOD1 incubation; lane 4: 2-h Apo SOD incubation. (B) Quantitation of the effect of SOD1 on squalene synthase mRNA expression in HepG2 cells. Each bar represents the mean of three independent experiments in which the ratio of target PCR product/ internal standard PCR product was plotted. 1: Untreated HepG2 cells; 2: 30-min SOD1 incubation; 3: 2-h SOD1 incubation; 4: 2-h Apo SOD1 incubation.
Figure 9.
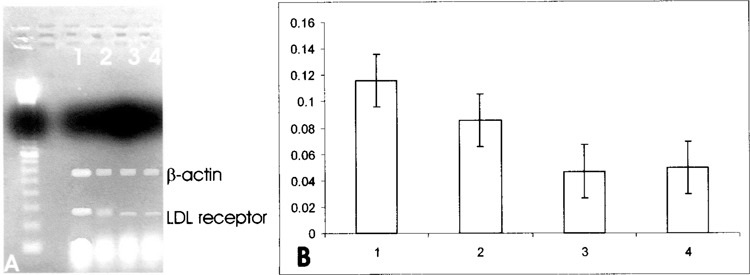
(A) Effect of SOD1 on LDL receptor mRNA in HepG2 cells. Quantitation was achieved in a single reaction in all the cell lines by the use of β-actin gene as internal standard. Left lane: marker 100 bp. Lane 1: untreated HepG2 cells; lane 2: 30-min SOD1 incubation; lane 3: 2-h SOD1 incubation; lane 4: 2-h Apo SOD incubation. (B) Quantitation of the effect of SOD1 on LDL receptor mRNA expression in HepG2 cells. Each bar represents the mean of three independent experiments in which the ratio of target PCR product/internal standard PCR product was plotted. 1: Untreated HepG2 cells; 2: 30-min SOD1 incubation; 3: 2-h SOD1 incubation, 4: 2-hours Apo SOD1 incubation.
Figure 10.
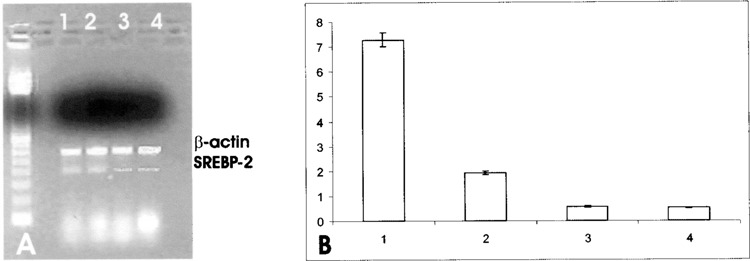
(A) Effect of SOD1 on SREBP-2 mRNA in HepG2 cells. Quantitation was achieved in a single reaction in all the cell lines by the use of β-actin gene as internal standard. Left lane: marker 100 bp. Lane 1: untreated HepG2 cells; lane 2: 30-min SOD1 incubation; lane 3: 2-h SOD1 incubation; lane 4: 2-h ApoSOD incubation. (B) Quantitation of the effect of SOD1 on SREBP-2 mRNA expression in HepG2 cells. Each bar represents the mean of three independent experiments in which the ratio of target PCR product/internal standard PCR product was plotted. 1: Untreated HepG2 cells; 2: 30-min SOD1 incubation; 3: 2-h SOD1 incubation; 4: 2-h Apo SOD1 incubation.
DISCUSSION
In this study, we found that the SOD1 is able to inhibit HMG-CoA reductase gene expression in HepG2 cells and other genes correlated with cholesterol metabolism, such as squalene synthase and LDL receptor. Furthermore, SOD1 strongly downregulated the membrane-bound transcriptional factor (SREBP-2) that controls many enzymes involved in the cholesterol biosynthetic pathway. As already documented on cholesterol synthesis (22), the effect is independent of SOD1 activity, because the metal-free enzyme (Apo SOD) yields the same inhibitory effect on HMG-CoA reductase gene expression. Similar inhibitory effects are also observed in HepG2 cells incubated in 10% FBS in the presence of SOD1. In fact, a synergic effect of both serum cholesterol and SOD1 led to a decrease in HMG-CoA gene expression. This effect was less evident when HepG2 cells were incubated only with FBS. CuZn SOD is secreted by many cellular lines (18,20) and is transported in the blood by binding to lipoproteins, mainly LDL and HDL (19) and to albumin (13).
Our data demonstrated that in the cell lines tested, the SOD1 strongly inhibits the HMG-CoA reductase gene expression affecting cholesterol metabolism, both in wild-type fibroblasts and in fibroblasts of patients affected by familial hypercolesterolemia. This effect is in agreement with our previous research in which a strong decrease in HMG-CoA reductase protein amount was observed in cells treated with SOD1 (22). The effect carried out by SOD1, as well as by Apo SOD, on enzymes involved in cholesterol synthesis is exerted at the transcriptional level, as shown by the strong downregulation of SREBP-2. It is in this respect that SOD1 works differently from statins, such as compactin and lovastatin, potent inhibitors of HMG-CoA reductase (1,9). In fact, when native cells are exposed to statins in their growth medium, such inhibitors induce an increase in the rate of HMG-CoA reductase gene expression (7,16). Moreover, the mechanism involved in the downregulation of HMG-CoA reductase gene expression, induced by SOD1, seems to be correlated with the activation of PKC activity (data not shown) and with an increase in intracellular calcium concentration.
Evidence that the activation of PLC–PKC pathway elicits a strong increase in intracellular calcium is supported by the finding that in HepG2 cells U73122 (PLC inhibitor) and BDM (PKC inhibitor) can reverse either the increased intracellular calcium concentration or the downregulation of HMG-CoA reductase gene expression induced by SOD1.
Although our present results do not reveal how SOD1 is transported into the cells, our unpublished data do indicate that SOD1 can be internalized in HepG2 cells by endocytosis, occurring 15 min after incubation.
More importantly, the experimental outcomes of our study reflect the hypothesis that the inhibitory effect of SOD1 on cholesterol metabolism can be ascribed to a signal transduction mechanism that involves the activation of PLC–PKC–intracellular calcium concentration.
These data, together with previous studies (8,15,20,21), which demonstrate that free SOD1 is bound to plasma lipoproteins (19) and is internalized in rat hepatocytes, indicate that CuZn superoxide dismutase plays a key role in affecting the cellular cholesterol synthesis via HMG-CoA reductase gene expression regulation.
Theoretically, the experimental and therapeutic potentials of SOD1 could be limited, because the protein is rapidly cleared by the kidneys (25,28). However, it has been reported that when SOD1 is combined with polyethylene glycol (PEG) its half-life is increased up to 40 h, its immunogenicity is reduced, and its sensitivity to proteolysis diminished (3).
Finally, we conclude that the inhibitory role of SOD1 on cholesterol synthesis could prospectively be of particular interest also in relation to the treatment of familial hypercholesterolemia, thus suggesting a potential therapeutic use of this antioxidant enzyme.
ACKNOWLEDGMENTS
The fibroblasts of subjects affected by familial hypercholesterolemia were obtained from the “Cell Line and DNA Bank from Patients affected by Genetic Diseases” collection for the kind concern of Prof. A. Daniele. The research was supported by Italian Telethon grants (project No. GTF02012). The Apo SOD was kindly supplied by Prof. Maria Rosa Ciriolo, Department of Biology, Università di Roma “Tor Vergata.” We would also like to thank Dr. Paola Merolla for her assistance in the English editorial revision and Dr. Agnese Secondo (Division of Pharmacology) for the intracellular calcium determination.
REFERENCES
- 1. Alberts A. W.; Chen J.; Kuron G.; Hunt V.; Huff J.; Hoffman C.; Rothrock J.; Lopez M.; Joshua H.; Harris E.; Patchett A.; Monaghan R.; Currie S.; Stapley E.; Albers-Schonberg G.; Hensens O.; Hirshfield J.; Hoogsteen K.; Liesch J.; Springer J. Mevinolin: A highly potent competitive inhibitor of hydroxymethylglutarylco-enzyme A reductase and a cholesterol-lowering agent. Proc. Natl. Acad. Sci. USA 77:3957–3961; 1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bachelor A. M.; Silvers A. L.; Bowden G. T. The role of p38 in UVA-induced cyclooxygenase-2 expression in the human keratinocyte cell line, HaCaT. Oncogene 21:7092–7099; 2002. [DOI] [PubMed] [Google Scholar]
- 3. Beckman J. S.; Minor R. L. Jr.; White C. W.; Repine J. E.; Rosen G. M.; Freeman B. A. Superoxide dismutase and catalase conjugated to polyethylene glycol increases endothelial enzyme activity and oxidant resistance. J. Biol. Chem. 263:6884–6892; 1988. [PubMed] [Google Scholar]
- 4. Brown M. S.; Goldstein J. L. A receptor-mediated pathway for cholesterol homeostasis. Science 232:34–47; 1986. [DOI] [PubMed] [Google Scholar]
- 5. Brown M. S.; Goldstein J. L. Multivalent feedback regulation of HMG CoA reductase, a control mechanism coordinating isoprenoid synthesis and cell growth. J. Lipid Res. 21:505–517; 1980. [PubMed] [Google Scholar]
- 6. Chen L.; Segal D. M.; Mash D. C. Semi-quantitative reverse-transcriptase polymerase chain reaction: An approach for the measurement of target gene expression in human brain. Brain Res. Brain Res. Protoc. 4: 132–139; 1999. [DOI] [PubMed] [Google Scholar]
- 7. Clarke C. F.; Fogelman A. M.; Edwards P. A. Transcriptional regulation of the 3-hydroxy-3-methylglutaryl coenzyme A reductase gene in rat liver. J. Biol. Chem. 260:14363–14367; 1985. [PubMed] [Google Scholar]
- 8. Dini L.; Rotilio G. Electron microscopic evidence for endocytosis of superoxide dismutase by hepatocytes using protein-gold adducts. Biochem. Biophys. Res. Commun. 162:940–944; 1989. [DOI] [PubMed] [Google Scholar]
- 9. Endo A. The discovery and development of HMG-CoA reductase inhibitors. J. Lipid Res. 33:1569–1582; 1992. [PubMed] [Google Scholar]
- 10. Goldstein J. L.; Brown M. S. Progress in understanding the LDL receptor and HMG-CoA reductase, two membrane proteins that regulate the plasma cholesterol. J. Lipid Res. 25:1450–1461; 1984. [PubMed] [Google Scholar]
- 11. Grynkiewicz G.; Poenie M.; Tsien R. Y. A new generation of Ca2+ indicators with greatly improved fluorescence properties. J. Biol. Chem. 264:3440–3450; 1985. [PubMed] [Google Scholar]
- 12. Horton J. L.; Goldstein J. D.; Brown M. S. SREBPs: Activators of the complete program of cholesterol and fatty acid synthesis in the liver. J. Clin. Invest. 109: 1125–1131; 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Inoue M.; Ebashi I.; Watanabe N.; Morino Y. Synthesis of a superoxide dismutase derivative that circulates bound to albumin and accumulates in tissues whose pH is decreased. Biochemistry 28:6619–6624; 1989. [DOI] [PubMed] [Google Scholar]
- 14. Kelleher K. L.; Leck K. J.; Hendry I. A.; Matthaei K. I. A one-step quantitative reverse transcription polymerase chain reaction procedure. Brain Res. Brain Res. Protoc. 6:100–107; 2001. [DOI] [PubMed] [Google Scholar]
- 15. Li L.; Wattiaux De-Conincks S.; Wattiaux R. Endocytosis of superoxide dismutase by rat liver. Biochem. Biophys. Res. Commun. 184:727–732; 1992. [DOI] [PubMed] [Google Scholar]
- 16. Liscum L.; Luskey K. L.; Chin D. J.; Ho Y. K.; Goldstein J. L.; Brown M. S. Regulation of 3-hydroxy-3-methylglutaryl coenzyme A reductase and its mRNA in rat liver as studied with a monoclonal antibody and a cDNA probe. J. Biol. Chem. 258:8450–8455; 1983. [PubMed] [Google Scholar]
- 17. Mondola P. The calf thymus superoxide dismutase: A protein active on cholesterol metabolism. Comp. Biochem. Physiol. B 105:457–464; 1993. [DOI] [PubMed] [Google Scholar]
- 18. Mondola P.; Annella T.; Santillo M.; Santangelo F. Evidence for secretion of cytosolic CuZn superoxide dismutase by Hep G2 cells and human fibroblasts. Int. J. Biochem. Cell Biol. 28:677–681; 1996. [DOI] [PubMed] [Google Scholar]
- 19. Mondola P.; Bifulco M.; Seru R.; Annella T.; Ciri-olo M.; Santillo M. Presence of CuZn superoxide dismutase in human serum lipoproteins. FEBS Lett. 467: 57–60; 2000. [DOI] [PubMed] [Google Scholar]
- 20. Mondola P.; Ruggiero G.; Serù R.; Damiano S.; Grimaldi S.; Garbi C.; Monda M.; Greco D.; San-tillo M. The Cu,Zn superoxide dismutase in neuroblastoma SK-N-BE cells is exported by a microvesicles dependent pathway. Mol. Brain Res. 110:45–51; 2003. [DOI] [PubMed] [Google Scholar]
- 21. Mondola P.; Santillo M.; Santangelo F.; Garbi C.; Daniele A. The calf superoxide dismutase receptor of rat hepatocytes. Comp. Biochem. Physiol. Biochem. Mol. Biol. 108:309–313; 1994. [DOI] [PubMed] [Google Scholar]
- 22. Mondola P.; Serù R.; Santillo M.; Damiano S.; Bifulco M.; Laezza C.; Formisano P.; Rotilio G.; Ciriolo M. Effect of Cu,Zn superoxide dismutase on cholesterol metabolism in human hepatocarcinoma (HepG2) cells. Biochem. Biophys. Res. Commun. 295: 603–609; 2002. [DOI] [PubMed] [Google Scholar]
- 23. Morita K.; Kuwada A.; Fujihara H.; Morita Y.; Sei H. Influence of sleep disturbance on steroid 5alpha-reductase mRNA levels in rat brain. Neuroscience 115: 341–348; 2002. [DOI] [PubMed] [Google Scholar]
- 24. Myant N. B. LDL and LDL receptor. In: Cholesterol metabolism. New York: Academic Press, Inc.; 1990. [Google Scholar]
- 25. Petkau A.; Chelack W. S.; Kelly K.; Barefoot C.; Monasterski L. Tissue distribution of bovine 125I-superoxide dismutase in mice. Res. Commun. Chem. Pathol. Pharmacol. 15:641–654; 1976. [PubMed] [Google Scholar]
- 26. Secondo A.; Taglialatela M.; Cataldi M.; Giorgio G.; Valore M.; Di Renzo G. F.; Annunziato L. Pharmacological blockade of ERG K(+) channels and Ca(2+) influx through store-operated channels exerts opposite effects on intracellular Ca(2+) oscillations in pituitary GH(3) cells. Mol. Pharmacol. 58:1115–1128; 2000. [DOI] [PubMed] [Google Scholar]
- 27. Semprini S.; Capon F.; Tacconelli A.; Giardina E.; Orecchia A.; Mingarelli R.; Gobello T.; Zambruno G.; Botta A.; Fabrizi G.; Novelli G. Evidence for differential S100 gene over-expression in psoriatic patients from genetically heterogeneous pedigrees. Hum. Genet. 111:310–313; 2002. [DOI] [PubMed] [Google Scholar]
- 28. Turrens J. F.; Crapo J. D.; Freeman B. A. Protection against oxygen toxicity by intravenous injection of liposome-entrapped catalase and superoxide dismutase. J. Clin. Invest. 73:87–95; 1984. [DOI] [PMC free article] [PubMed] [Google Scholar]