

Abstract
NF-Y is a bifunctional transcription factor capable of activating or repressing transcription. NF-Y specifically recognizes CCAAT box motifs present in many eukaryotic promoters. The mechanisms involved in regulating its activity are poorly understood. Previous studies have shown that the FGF-4 promoter is regulated positively by its CCAAT box and NF-Y in embryonal carcinoma (EC) cells where the distal enhancer of the FGF-4 gene is active. Here, we demonstrate that the CCAAT box functions as a negative cis-regulatory element when cis-regulatory elements of the FGF-4 enhancer are disrupted, or after EC cells differentiate and the FGF-4 enhancer is inactivated. We also demonstrate that NF-Y mediates the repression of the CCAAT box and that NF-Y associates with the endogenous FGF-4 gene in both EC cells and EC-differentiated cells. Importantly, we also determined that the orientation and the position of the CCAAT box are critical for its role in regulating the FGF-4 promoter. Together, these studies demonstrate that the distal enhancer of the FGF-4 gene determines whether the CCAAT box of the FGF-4 promoter functions as a positive or a negative cis-regulatory element. In addition, these studies are consistent with NF-Y playing an architectural role in its regulation of the FGF-4 promoter.
Key words: NF-Y, F9-differentiated cells, EC cells, Transcription, Differentiation, Enhancer, Immunoprecipitation
TRANSCRIPTIONAL regulation of gene expression involves the proper activation and repression of specific genes. This process is controlled by sequence-specific transcription factors binding to promoters and enhancers and interacting with co-regulators, chromatin remodeling complexes, and/or components of the basal transcriptional machinery (34,38,65). Many transcription factors activate transcription, whereas others repress it. However, several transcription factors are bifunctional and act either as activators or as repressors. Thyroid hormone receptors and retinoic acid receptors interact with a co-repressor complex to confertranscriptional repression in the absence of hormone (65). Upon hormone binding, the receptor undergoes a conformational change resulting in the dissociation of co-repressors and the recruitment of coactivators to activate transcription. Other bifunctional transcription factors include YY1 (43,81), NF-κB (90) and NF-Y (51,62,75,89).
NF-Y is a ubiquitously expressed transcription factor that is composed of three subunits: NF-YA, NF-YB, and NF-YC (also known as CBF-B, CBF-A, and CBF-C, respectively). Each subunit contains an evolutionarily conserved HAP domain that is critical for subunit interactions and DNA binding (30,45,77). NF-YA and NF-YC each possess large glutamine-rich activation domains outside of the HAP domain (14,15,19). Located within the HAP domains of NF-YB and NF-YC is a histone-fold motif that is found in all core histones and that mediates dimerization and contributes to DNA binding (3,4,47). NF-YB and NF-YC subunits dimerize to form a complex that associates with NF-YA (77). Once the trimer forms, it can bind specifically to CCAAT boxes, which are cis-regulatory elements found in either orientation in 25–30% of eukaryotic promoters, and typically located between 60 and 100 bp upstream of the transcriptional start site (11,52). Analysis of 178 NF-Y-binding CCAAT boxes demonstrated that their orientation and position relative to neighboring TATA boxes are not distributed randomly (52). This suggests that CCAAT boxes and NF-Y may serve an important architectural role in the organization of promoters. In this regard, NF-Y is capable of bending DNA 60–80° (46,72), and it has been shown to interact with components of the basal transcriptional machinery (5,16,26). Thus far, the role of NF-Y as an architectural factor when bound to CCAAT boxes has not been systematically tested by altering its position or orientation.
The binding of NF-Y to gene promoters appears to be regulated carefully during cell growth and differentiation. Although NF-Y is a ubiquitous transcription factor, it may not bind to CCAAT boxes constitutively. The binding of NF-Y to the promoter of several cell cycle genes fluctuates during the cell cycle (12). In other cases, loss of NF-YA expression in terminally differentiated muscle cells results in the loss of a functional NF-Y complex (24,28). Moreover, decreased NF-YA expression has been reported after serum deprivation of fibroblasts (13), whereas an increase in NF-YA expression was observed during monocyte maturation (55). Although CCAAT boxes function primarily as positive cis-regulatory elements, CCAAT boxes negatively regulate transcription in several promoters, and both the positive and the negative effects of these CCAAT boxes are likely to be mediated by NF-Y (9,27,29,31,39,51,75,85,89). Importantly, NF-Y has been shown to interact with coactivators (18,23,35,44) or co-repressors in different promoters (27,63). It remains unclear what factors or other variables determine whether a CCAAT box and NF-Y will activate or repress transcription of a given gene. However, adjacent cis-regulatory elements have been shown to be required for the negative effect of CCAAT boxes (27,29).
Transcription of the fibroblast growth factor-4 (FGF-4) gene has been shown to be activated by a CCAAT box located in its promoter approximately 100 bp upstream of the transcription start site (41). The FGF-4 gene is differentially expressed during early embryogenesis (57,66), and gene targeting studies have shown that FGF-4 is essential for early development and the survival of extraembryonic tissues (25,79,83). FGF-4 expression is restricted to cells of the inner cell mass at the late blastocyst stage, whereas cells of the extraembryonic endoderm, which are derived from the inner cell mass, do not express FGF-4 (57,66). Expression of FGF-4 is dependent on its promoter and a powerful downstream distal enhancer located within the untranslated region of the third and last exon (17,50). Binding motifs for known transcription factors have been identified in both the promoter and the enhancer. The cis-regulatory elements of the promoter include a TATA box, two GC boxes that bind Sp1 and Sp3 in vitro (40,48), and an inverted CCAAT box that binds NF-Y in vitro (41). The cis-regulatory elements identified within the enhancer include: two high mobility group (HMG) motifs, referred to as upstream HMG (USHMG) and downstream HMG (DSHMG), which bind Sox2 in vitro and in vivo (49,58,87); a POU motif that binds Oct-3 in vitro (50); and a GT box that binds Sp1 and Sp3 in vitro (48). Sox2 and Oct-3 bind cooperatively to the DSHMG and POU motifs, respectively, to synergistically activate transcription of promoter/reporter gene constructs (1,2,58,59,87). In addition, the coactivator p300 appears to mediate the effect of Sox2/Oct-3 synergism (59,58), and p300 has been shown to associate with the endogenous FGF-4 enhancer in embryonal carcinoma (EC) cells (58).
EC cells serve as an excellent model for studying the expression of FGF-4 during early development. These cells are similar both biochemically and morphologically to cells of the inner cell mass (54). Moreover, transcription of the FGF-4 gene decreases substantially after EC cells undergo differentiation (64,69,82,86). The downregulation of the FGF-4 gene is thought to be due, at least in part, to the decrease in Sox2 and Oct-3 that accompanies differentiation (37,42,56,60,87). To better understand the regulation of the FGF-4 gene in this model system, we addressed four questions in this study. First, does NF-Y bind to the FGF-4 promoter in EC cells? Second, if it does, does it remain associated with the FGF-4 promoter in the differentiated cells, and does it influence the FGF-4 promoter when EC cells differentiate and transcription of the FGF-4 gene is downregulated? Third, is the function of the CCAAT box dependent on adjacent cis-regulatory elements in the FGF-4 promoter, in particular the two upstream GC boxes? Fourth, does the CCAAT box play an architectural role in the regulation of the FGF-4 promoter? Our results demonstrate that NF-Y binds to the endogenous FGF-4 promoter in both F9 EC cells and F9-differentiated cells. In addition, we show that, upon differentiation, the FGF-4 enhancer becomes inactive, the CCAAT box becomes a negative cis-regulatory element, and NF-Y mediates the repression of the CCAAT box. Therefore, NF-Y can both activate and repress the FGF-4 promoter, and the role of NF-Y is dependent upon the FGF-4 enhancer. Moreover, we show that the CCAAT box can negatively regulate transcription independent of the two upstream GC boxes. Finally, we demonstrate that the orientation and the position of the CCAAT box are critical for its regulatory role in FGF-4 promoter activity. Collectively, our results address two important issues concerning NF-Y-mediated gene regulation. First, we demonstrate that the behavior of NF-Y and the CCAAT box of the FGF-4 promoter is regulated by an unexpected mechanism, specifically, by the downstream distal enhancer of the FGF-4 gene. Second, we provide direct evidence that the position and the orientation of the CCAAT box critically affect its function. Thus, our data argue strongly that NF-Y can function in an architectural manner.
MATERIALS AND METHODS
Cell Culture and Conditions
Mouse F9 EC cells were cultured on gelatinized dishes in Dulbecco’s modified Eagle’s medium (DMEM, Invitrogen, Carlsbad, CA) supplemented with 10% fetal bovine serum (FBS, HyClone, Logan, UT) and antibiotics, as described previously (68,70). Differentiation of F9 EC cells was induced by treatment with 5 μM retinoic acid (RA). Cells were cultured at 37°C in a humidified atmosphere of 5% CO2 in air.
Transient Transfection Assay
F9 EC cells were seeded at 5 × 105 cells per 100-mm dish. After 24 h, the cells were transfected with promoter/reporter gene constructs and expression vectors by the calcium phosphate precipitation method (50). Following a 24-h incubation with the precipitate, the cells were refed with DMEM + 10% FBS. Chloramphenicol acetyltransferase (CAT) and β-galactosidase activities were determined 48 h after transfection, as described previously (50). For experiments involving F9-differentiated cells, F9 EC cells were seeded at 1 × 105 cells per 100-mm dish in DMEM + 10% FBS and 5 μM RA and transfected with promoter/reporter gene constructs and expression vectors 72 h later. Following a 24-h incubation with the precipitate, the cells were refed with DMEM + 10% FBS and 5 μM RA. CAT and β-galactosidase activities were determined 72 h after transfection. Molar amounts of DNA were kept constant with the addition of null plasmid DNA, pCMV5. All transfections were normalized by addition of 1 μg of CMVβ-galactosidase (Clontech, Palo Alto, CA). All transfections were performed in duplicate. Error bars indicate the SD of either the fold induction or the relative CAT activity obtained by the compilation of multiple experiments as indicated in the figure legends. Data were evaluated for statistical significance using the Student’s t-test.
Plamid Constructs
The following promoter/reporter constructs have been described elsewhere: 427T+E (also known as pKFGF427T+E) (48,73), 427T, CCAATmut+E (also known as −427T+E CCAATmut), CCAATmut (also known as −427T CCAATmut) (36), TKCat, TKCat+E, 427T+EnGSp, GCmut (also known as 427T Sp1DP-E), CCAATmut+EnGSp (also known as 427TCmut+EnGSp) (58), and GCmut+E (also known as Sp1DP) (48). GCmut/CCAATmut contains mutations in both the GC boxes and the CCAAT box of the FGF-4 promoter but lacks the enhancer and was constructed by removing the enhancer from 427TGC/Cmut+EnGSp (58) by digestion and religation of the SacI sites flanking the enhancer. CCAATmut+EGTmut and CCAATmut+EHMGmut were constructed by isolating the enhancers from Sp1E (48) and 427T+DSHMGmut (49), respectively, by digestion with SacI and ligating them into the SacI site of CCAATmut. 427TG4/CCAAT was constructed using PCR with 427T as the template and the following primers: upper primer, 5′-CCTCggatccGGAGGACAGTCCACCGACCTGCGCCCCGGCTCCCAGG-3′; lower primer, 5′-TCACCGggatccGAGGAGGCGCGGGCCGGGCGGTGGC-3′. Lower case sequence denotes BamHI sites, sequence in bold denotes the Gal4 binding site, and the underlined sequence is wild-type promoter sequence. This PCR amplified the entire plasmid except for the CCAAT box, which was replaced by the Gal4 binding site. The BamHI sites were then used to circularize the plasmid. Upstream CCAAT+E, reversed CCAAT+E, upstream CCAAT, and reversed CCAAT were generated by the Quick-Change method of mutagenesis (Stratagene, La Jolla, CA) using the promoter/reporter constructs CCAATmut+E and CCAATmut, respectively. The following primer and its complement were used to replace sequence upstream of the two GC boxes with a CCAAT box in the constructs upstream CCAAT+E and upstream CCAAT: 5′-CAGCTCAAGTCAGTGATTGGCAGAGCAGAAAGGTTCTGGCGG-3′. The sequence in bold denotes the CCAAT box. The following primer and its complement were used to create a CCAAT box in the opposite orientation of the original CCAAT box in the constructs reversed CCAAT+E and reversed CCAAT: 5′-GCGCCTCCTCCCCCGGTGCTGCCAATCACGCGACCTGC-3′. The sequence in bold denotes the inverted CCAAT box.
The following expression plasmids were utilized: CMVGal4 and Gal4/Sox2 121–319 (59); the SV40-Gal4 fusion vector, pM3 (74); Gal4/p300 1–2414 (full-length p300 fused to the DNA binding domain of the yeast transcription factor Gal4) and Gal4/p300 964–1922 (p300 amino acids 964–1922 fused to the DNA binding domain of Gal4) (88); pNF-YA29 (53); and pSG424-NF-YC Δ1, which was received from Keiko Funa (Göteborg University) (32). CMVGal4/NF-YA 1-261 was generated by amplifying amino acids 1–261 of NF-YA and inserting the amplified fragment into the HindIII site of CMVGal4 using the following primers: upper primer, 5′-AGCTaagcttATGGAGCAGTATACGACAAACAGC-3′; lower primer, 5′-AGCTaagcttCTCTTCTTCCAGCATCTCTGCTCCAG-3′. The lowercase sequences denote HindIII sites. CMVGal4/NF-YB Δ50–139 was made in two steps. The first step involved amplifying the entire NF-YB sequence and inserting it into the BamHI site of CMVGal4 to create CMVGal4/NF-YB using the following primers: upper primer, 5′-CTACGTggatccTATGACAATGGACGGCGACAG-3′; lower primer, 5′-CGAACTggatccTCATGAAAACTGAATTTGCTG-3′. The lowercase sequences denote BamHI sites. The second step involved amplifying the entire CMV-Gal4/NF-YB sequence except for the sequence that encodes amino acids 50 to 139 of NF-YB. The following primers used for this step inserted a KpnI site to allow the amplified plasmid to be religated: upper primer, 5′-CCTCggtaccAAGTTCAGAGAGGCCATGAAAG-3′; lower primer, 5′-CCTCggtaccACTTTCTTTTGAACCATTTG-3′. The lowercase sequences denote KpnI sites.
Chromatin Immunoprecipitation (ChIP)
ChIP was performed as follows. On day 1, F9 EC cells were seeded at a density of 2 × 105 on 100-mm dishes and induced to differentiate with 5 μM RA. On day 4, F9 EC cells were seeded at 2 × 106 cells per 100-mm dish. On day 5, cross-linking was performed by addition of formaldehyde to the culture medium at a final concentration of 1% for 10 min at room temperature. Cells were then washed and scraped into PBS containing 1 mM PMSF, 1 μg/ml aprotinin, and 1 μg/ml pepstatin A. Cells were lysed in SDS lysis buffer (1% SDS, 10 mM EDTA, 50 mM Tris, pH 8.1) containing 1 mM PMSF, 1 μg/ml aprotinin, and 1 μg/ml pepstatin A by adding 200 μl of lysis buffer per 1 × 106 cells. Following incubation on ice for 10 min, the DNA was sheared to lengths between 200 and 1000 bp by sonication, and cell debris was pelleted. Aliquots (200 μl) of the sonicated cell supernatant were diluted 10-fold by the addition of ChIP dilution buffer (0.01% SDS, 1.1% Triton X-100, 1.2 mM EDTA, 16.7 mM Tris-HCl, pH 8.1, 167 mM NaCl) plus 1 mM PMSF, 1 μg/ml aprotinin, and 1 μg/ml pepstatin A. To reduce nonspecific background, the samples were precleared with 80 μl of salmon sperm DNA/protein A agarose (50% slurry) (Upstate Biotechnology, Lake Placid, NY) for 30 min at 4°C with rotation. The precleared samples were then incubated with 15 μg NF-YA antibody (H-209; Santa Cruz Biotechnology, Inc., Santa Cruz, CA), 15 μg NF-YB antibody (FL-207; Santa Cruz Biotechnology, Inc.), or 15 μg preimmune normal rabbit IgG (Santa Cruz Biotechnology, Inc.) at 4°C with rotation overnight. Immune complexes were collected with 60 μl of salmon sperm DNA/protein A agarose (50% slurry) for 1 h at 4°C with rotation. The samples were washed once with low-salt wash buffer (0.1% SDS, 1% Triton X-100, 2 mM EDTA, 20 mM Tris-HCl, pH 8.1, 150 mM NaCl), high-salt buffer (0.1% SDS, 1% Triton X-100, 2 mM EDTA, 20 mM Tris-HCl, pH 8.1, 500 mM NaCl), and LiCl wash buffer (2.25 M LiCl, 1% IGEPAL-CA630, 1% deoxycholic acid, 1 mM EDTA, 10 mM Tris, pH 8.1) and twice with TE buffer (10 mM Tris-HCl, 1 mM EDTA, pH 8.0). Complexes were eluted by two incubations with 250 μl of elution buffer (1% SDS, 0.1 M NaHCO3) at room temperature for 10 min with rotation. NaCl was added to a final concentration of 0.2 M, and the samples were incubated at 65°C for 4 h to reverse the cross-links. The samples were treated with 20 μg of Proteinase K for 1 h at 45°C after the addition of 10 μl 0.5 M EDTA and 20 μl 1 M Tris-HCl, pH 6.5. DNA was purified using the GeneClean turbo kit (Q Biogene, Carlsbad, CA) following the manufacturer’s directions. In studies not shown, ChIP analysis was performed with HDAC1 antibodies (H-51, Santa Cruz Biotechnology).
The following primer pairs were used to detect the presence of DNA fragments containing the indicated regions. FGF-4 promoter: FGFproU2, 5′-GTAAGG AAGGAGCACAGGAGAT-3′ and FGFproL3, 5′-CAGACCTAAGTAGCGAGAGCAA. TβR-II promoter: TβRIIproU, 5′-ACACCACTTTCGAGCTTGTTTTGC and TβRIIproL, 5′-CGCGAGTGACTCACTCATCAAC. FGF-4 5′ region: FGF5′U, 5′-ACCTCACCTGTGCCCTGTTGAAT-3′ and FGF5′L, 5′-GTGCTCATCTCCCCAGACTGTTTCT-3′. PCR was performed on equivalent volumes of each sample and analyzed at multiple cycles. Results are presented for one cycle.
RESULTS
The FGF-4 Enhancer Is Inactive and the FGF-4 Promoter Can Be Activated in F9-Differentiated Cells
The transcription of the FGF-4 gene depends on a powerful distal enhancer located 3 kb downstream of the transcription start site. In F9 EC cells, the FGF-4 enhancer increases the activity of FGF-4 promoter/reporter gene constructs over 50-fold (17,50). Previous studies demonstrated that the activity of FGF-4 promoter/reporter gene constructs is greatly reduced after differentiation. Although the FGF-4 enhancer appears to be inactivated after EC cells differentiate (17,50), it was unclear whether the enhancer is completely inactivated after differentiation and whether differentiation influences the function of the FGF-4 promoter. To address the first issue, we tested the effect of the 316 bp FGF-4 enhancer on the heterologous herpes simplex virus thymidine kinase (TK) promoter. We chose the TK promoter because it is active in F9-differentiated cells, and because this promoter is stimulated five- to sixfold by the FGF-4 enhancer in F9 EC cells (58). Here we show that the FGF-4 enhancer hardly affected the TK promoter in F9-differentiated cells (Fig. 1A). This argues that the FGF-4 enhancer exhibits little or no activity after EC cells differentiate. To determine whether differentiation influences the function of the FGF-4 promoter, we initially tested whether the FGF-4 promoter can be activated in F9-differentiated cells. For this purpose, we examined whether the activation domains of Sox2 and p300 could stimulate the FGF-4 promoter in F9-differentiated cells, because both factors have been shown to activate the FGF-4 promoter (37,58,87). For these studies, we utilized the promoter/reporter gene construct 427T+EnGSp, in which the CAT reporter gene is under the control of the FGF-4 promoter and a modified FGF-4 enhancer (58). More specifically, in this construct the DSHMG and POU motifs of the enhancer have been replaced by a Gal4 DNA binding site, allowing Gal4-chimeric fusion proteins to bind to the FGF-4 enhancer. Cotransfection of 427T+EnGSp with an expression plasmid for Gal4/Sox2 or Gal4/p300 resulted in over a 150-fold induction in reporter gene activity in F9-differentatiated cells (Fig. 1B and C, respectively). This is similar to the 150-fold induction that is observed with Gal4/Sox2 in F9 EC cells (Fig. 1D). These results demonstrate that the FGF-4 promoter can be activated to comparable levels in both F9 EC and F9-differentiated cells. Therefore, inactivation of the FGF-4 gene in F9-differentiated cells appears to be due primarily to loss of enhancer function and does not appear to be due to an FGF-4 promoter that is irreversibly repressed.
Figure 1.
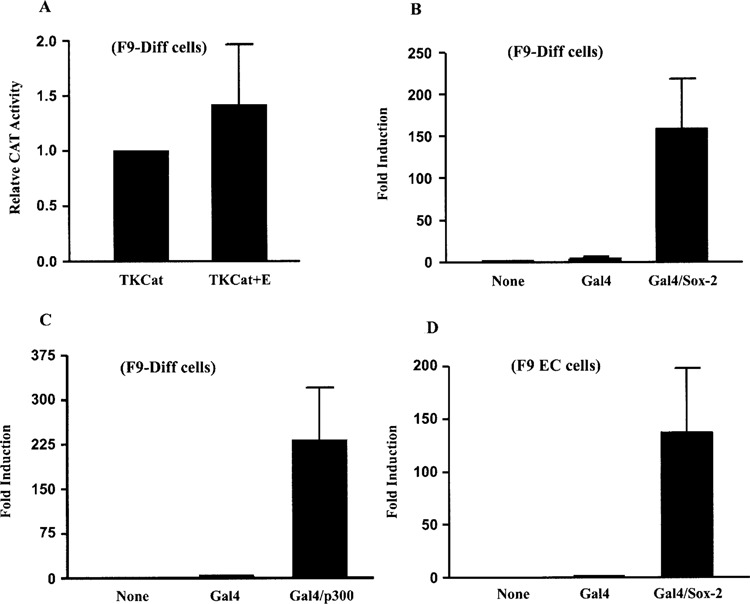
The FGF-4 enhancer is inactive and the FGF-4 promoter can be activated in F9 differentiated cells. (A) Transfection of F9-differentiated cells with 15 μg of TKCat or TKCat+E. Results are presented as CAT expression relative to the activity of TKCat, which was set to 1. This experiment was performed three times. The data shown are the compilation of the three experiments, and the standard deviation for each condition is represented by error bars. Analysis of the data by the Student’s t-test indicated that the activity of TKCat+E was not significantly different than the activity of TKCat. (B) Transfection of F9-differentiated cells with 15 μg of 427T+EnGSp alone or with 1 μg of a vector expressing Gal4 or Gal4/Sox2. Results are presented as fold induction over the activity of 427T+EnGSp alone, which was set to 1. This experiment was performed two times. The data shown are the compilation of the two experiments, and the standard deviation for each condition is represented by error bars. Analysis of the data by the Student’s t-test indicated that the activation by Gal4/Sox2 was significant with a p-value of less than 0.05. (C) Transfection of F9-differentiated cells with 15 μg of 427T+EnGSp alone or with 5 μg of a vector expressing Gal4 or Gal4/p300. Results are presented as fold induction over the activity of 427T+EnGSp alone, which was set to 1. This experiment was performed three times. The data shown are the compilation of the three experiments, and the standard deviation for each condition is represented by error bars. Analysis of the data by the Student’s t-test indicated that the activation by Gal4/p300 was significant with a p-value of less than 0.05. (D) Transfection of F9 EC cells with 15 μg of 427T+EnGSp alone or with 5 μg of a vector expressing Gal4 or Gal4/Sox2. Results are presented as fold induction over the activity of 427T+EnGSp alone, which was set to 1. This experiment was performed five times. The data shown are the compilation of the five experiments, and the standard deviation for each condition is represented by error bars. Analysis of the data by the Student’s t-test indicated that the activation by Gal4/Sox2 was significant with a p-value of less than 0.05.
The CCAAT Box of the FGF-4 Promoter Can Function as a Positive or a Negative cis-Regulatory Element
Several cis-regulatory elements have been identified in the FGF-4 promoter, including an inverted CCAAT box and a pair of GC boxes (41,48). Previous studies have shown that disruption of the CCAAT box and the GC boxes in FGF-4 promoter/reporter gene constructs lowers reporter gene activity when these constructs and their wild-type counterparts are compared in F9 EC cells. In the case of the CCAAT box, its disruption can reduce FGF-4 promoter activity as much as 70% (41,48). Surprisingly, in F9-differentiated cells, disruption of the CCAAT box resulted in a twofold increase in reporter gene activity relative to a construct with a wild-type promoter (Fig. 2A). This finding suggests that the CCAAT box functions as a negative cis-regulatory element in F9-differentiated cells. Because the FGF-4 enhancer is inactivated in F9-differentiated cells, we examined the effect of disrupting the CCAAT box in F9 EC cells utilizing promoter/reporter gene constructs that do not contain the FGF-4 enhancer. In this context, disruption of the CCAAT box resulted in a four- to fivefold increase in reporter gene activity (Fig. 2B). This observation argues that the CCAAT box functions as a negative regulatory element in the absence of the FGF-4 enhancer. This property is unique to the CCAAT box, because disruption of the two GC boxes (GCmut) resulted in a decrease in promoter activity when the FGF-4 enhancer was not present (Fig. 2B). Moreover, the ability of the CCAAT box to function negatively in the absence of the enhancer was not impaired when the two GC boxes were disrupted. This was illustrated by the increase in activity of the construct in which both the CCAAT box and the two GC boxes were disrupted (GCmut/CCAATmut) relative to the construct in which only the two GC boxes had been disrupted (GCmut) (Fig. 2B). Importantly, our studies indicate that disruption of the CCAAT box did not create a sequence for a positive acting factor. In this regard, similar results were obtained when a different DNA sequence was used to disrupt the CCAAT box (data not shown). Moreover, although the CCAAT box motif of the FGF-4 promoter has been shown to bind NF-Y in vitro (41), no DNA/protein complexes formed in a gel electrophoretic mobility shift assay in which the DNA probe contained the mutant CCAAT box sequence (data not shown). Collectively, these results argue that the CCAAT box of the FGF-4 promoter can function both as a positive and a negative cis-regulatory element, and that the function of the CCAAT box is dependent upon the activity of the FGF-4 enhancer.
Figure 2.
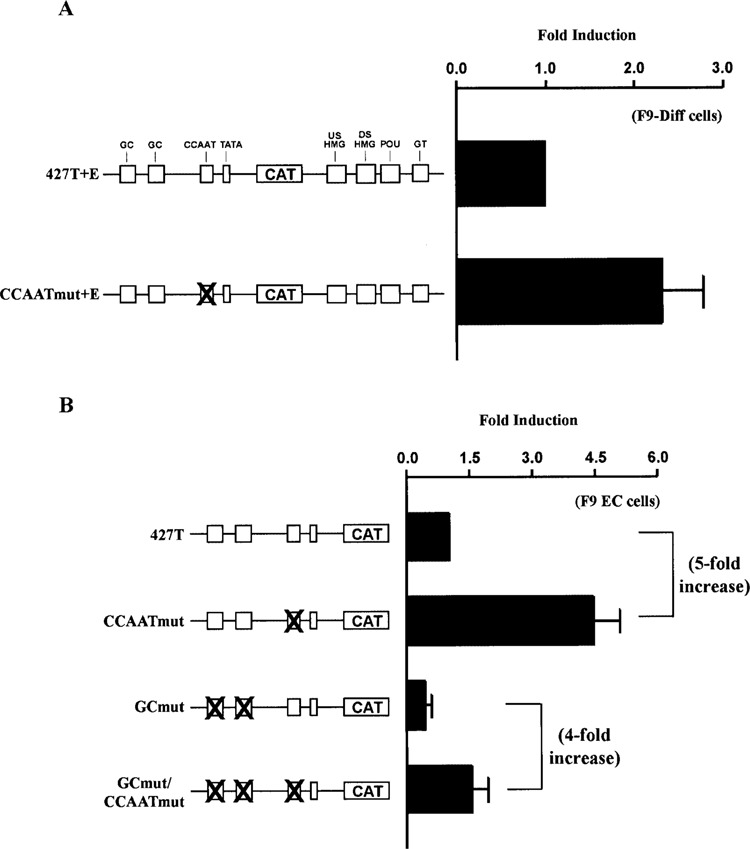
The FGF-4 CCAAT box functions as a negative cis-regulatory element in the absence of a functional enhancer. (A) Transfection of F9-differentiated cells with 15 μg of 427T+E or CCAATmut+E. Results are presented as fold induction over the activity of 427T+E, which was set to 1. This experiment was performed four times. The data shown are the compilation of the four experiments, and the standard deviation for each condition is represented by error bars. Analysis of the data by the Student’s t-test indicated that the activity of CCAATmut+E was significantly greater than the activity of 427T+E with a p-value of less than 0.05. (A) Transfection of F9 EC cells with 15 μg of 427T, CCAATmut, GCmut, or GCmut/CCAATmut. Results are presented as fold induction over the activity of 427T, which was set to 1. The fivefold increase indicates fold induction of CCAATmut relative to 427T, and the fourfold increase indicates fold induction of GC/CCAATmut relative to GCmut. This experiment was performed three times. The data shown are the compilation of the three experiments, and the standard deviation for each condition is represented by error bars. Analysis of the data by the Student’s t-test indicated that the activity of CCAATmut was significantly greater than the activity of 427T with a p-value of less than 0.05, and the activity of GCmut/CCAATmut was significantly greater than the activity of GCmut with a p-value of less than 0.05.
Several important cis-regulatory elements have been identified in the FGF-4 enhancer, including two HMG motifs that bind Sox2, a POU motif that binds Oct-3, and a GT box that binds Sp1 and Sp3 (40,48–50,87). Moreover, several studies have shown that at least two of the cis-regulatory elements of the FGF-4 enhancer work together to cooperatively activate the FGF-4 promoter (1,2,49,59,87). To determine which of these cis-regulatory elements influence the function of the CCAAT box in F9 EC cells, we utilized promoter/reporter gene constructs in which the different cis-regulatory elements of the enhancer were disrupted along with the CCAAT box. The activities of these constructs were compared to constructs that contain a wild-type CCAAT box. Disruption of the CCAAT box in conjunction with the GT box, the DSHMG motif, or both the DSHMG and POU motifs each resulted in a two- to threefold increase in reporter gene activity in F9 EC cells (Fig. 3). Although this is less than the four- to fivefold increase that is observed when the entire enhancer is absent, these findings argue that the GT box, the DSHMG motif, and the POU motif of the FGF-4 enhancer are required for the CCAAT box to function positively. Moreover, these findings argue that disruption of any of these cis-regulatory elements in the enhancer is sufficient to convert the CCAAT box of the FGF-4 promoter from a positive to a negative cis-regulatory element. Interestingly, disruption of the CCAAT box in conjunction with the USHMG motif did not lead to an increase in reporter gene activity (data not shown). Thus, although the USHMG site contributes to the function of the FGF-4 enhancer (49), the USHMG site does not appear to influence the inhibitory behavior of the CCAAT box.
Figure 3.
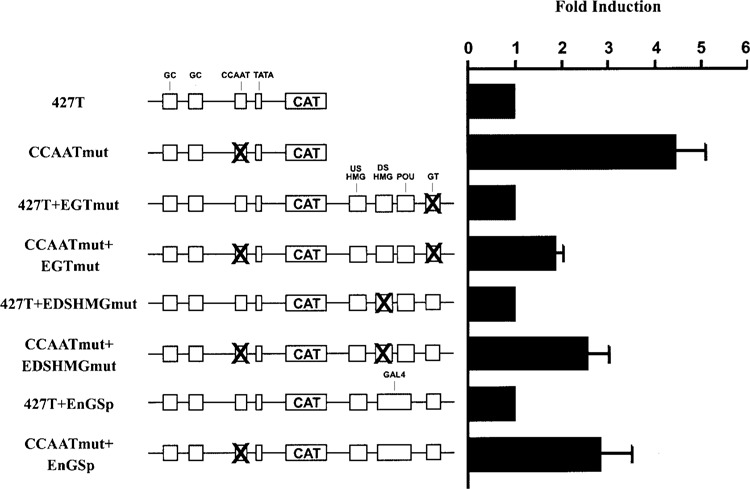
The CCAAT box functions as a negative cis-regulatory element when different cis-regulatory elements of the FGF-4 enhancer are disrupted. Transfection of F9 EC cells with 15 μg of the indicated plasmid. Results are presented as fold induction over the activity of the constructs that contain a wild-type CCAAT box. Each construct that contains a wild-type CCAAT box was set to 1. This experiment was performed five times. The data shown are the compilation of the five experiments, and the standard deviation for each condition is represented by error bars. Analysis of the data by the Student’s t-test indicated that the activity of CCAATmut+EGTmut was significantly greater than the activity of 427T+EGTmut with a p-value of less than 0.05, the activity of CCAATmut+EDSHMGmut was significantly greater than the activity of 427T+EDSHMGmut with a p-value of less than 0.05, and the activity of CCAATmut+EnGSp was significantly greater than the activity of 427T+EnGSp with a p-value of less than 0.05.
The Coactivator p300 Converts the FGF-4 CCAAT Box From a Negative to a Positive cis-Regulatory Element
The transcription factors Sox2 and Oct-3 bind cooperatively to the DSHMG and POU motifs of the FGF-4 enhancer to synergistically activate transcription (1,59,87), and p300 appears to mediate this synergism (58,59). In addition, p300 can physically interact with NF-Y in other promoters (23,44). These findings led us to test directly the effect of p300 on the function of the FGF-4 CCAAT box. For these studies, we employed the promoter/reporter gene construct 427T+EnGSp, which was described above, and the construct CCAATmut+EnGSp in which the CCAAT box of 427T+EnGSp has been disrupted. Cotransfection of F9 EC cells with CCAATmut+EnGSp plus an expression plasmid for Gal4/p300 resulted in 50% less reporter gene activity relative to the activation induced by Gal4/p300 with the construct that contains a wild-type CCAAT box (Fig. 4A). Importantly, previous studies demonstrated that Gal4/p300 activates the activity of 427T+EnGSp, but does not influence the activity of the parent construct, 427T+E, which lacks a binding site for the yeast transcription factor Gal4 (58). Therefore, the CCAAT box functions as a positive cis-regulatory element in response to p300, as well as the FGF-4 enhancer. It should be noted that the difference in the activity induced by Gal4/p300 in F9 EC cells (approximately 25-fold) (Fig. 4A) relative to the activity that is induced by Gal4/p300 in F9-differentiated cells (approximately 150-fold) (Fig. 1C) may be due to different expression levels of Gal4/p300 or to differential posttranslational modifications of p300. In this regard, the levels of p300 and its associated histone acetyltransferase activity have been reported to change after F9 EC cells undergo differentiation (10,61). To extend these findings, we examined whether the ability of the FGF-4 CCAAT box to function positively is specific for the FGF-4 enhancer. For this purpose, the FGF-4 enhancer of 427T+E was replaced with the heterologous SV40 enhancer to generate 427T+ SV40E. In F9 EC cells, the activities of 427T+E and 427T+SV40E were similar (data not shown). We chose the SV40 enhancer because it has been shown to mediate its effect, at least in part, through p300 (21). Moreover, unlike the FGF-4 enhancer, the activity of the SV40 enhancer increases with differentiation (78). In the presence of the SV40 enhancer, disruption of the CCAAT box results in a decrease in reporter gene activity in both F9 EC and F9-differentiated cells (Fig. 4B). Similar to the FGF-4 enhancer, the SV40 enhancer enables the CCAAT box to function as a positive cis-regulatory element. Together, these findings demonstrate that p300 is not only capable of activating the FGF-4 promoter, but it is also able to convert the FGF-4 CCAAT box from a negative to a positive cis-regulatory element.
Figure 4.
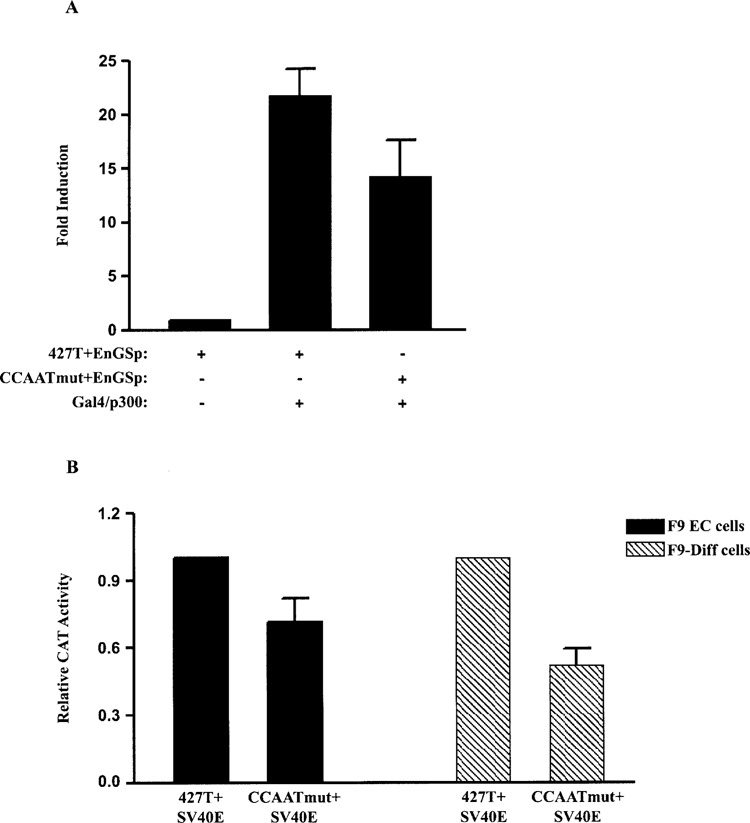
The CCAAT box functions as a positive cis-regulatory element when p300 is tethered to the FGF-4 enhancer or in the presence of the SV40 enhancer. (A) Transfection of F9 EC cells with 15 μg of 427T+EnGSp or CCAATmut+EnGSp alone or with 5 μg of Gal4/p300. Results are presented as fold induction over the activity of 427T+EnGSp, which was set to 1. This experiment was performed three times. The data shown are the compilation of the three experiments, and the standard deviation for each condition is represented by error bars. Analysis of the data by the Student’s t-test indicated that the activation of CCAATmut+EnGSp by Gal4/p300 was significantly less than the activation of 427T+EnGSp by Gal4/p300 with a p-value of less than 0.05. (B) Transfection of F9 EC and F9-differentiated cells with 15 μg of 427T+SV40E or CCAATmut+SV40E. Results are presented as CAT expression relative to the activity of 427T+SV40E, which was set to 1. This experiment was performed four times. The data shown are the compilation of the four experiments, and the standard deviation for each condition is represented by error bars. Analysis of the data by the Student’s t-test indicated that the activity of CCAATmut+SV40E was significantly less than the activity of 427T+SV40E in both F9 EC and F9-differentiated cells with a p-value of less than 0.05.
NF-Y Mediates the Repression of the FGF-4 CCAAT Box
Previous studies have shown NF-Y can bind to the FGF-4 CCAAT box sequence in vitro when nuclear extracts from F9 EC and F9-differentiated cells were examined by electrophoretic mobility shift assays (41). Moreover, functional studies utilizing a dominant negative form of NF-YA, NF-YA29, have shown NF-Y regulates FGF-4 promoter activity positively in F9 EC cells (41). NF-YA29 contains a mutation in the DNA binding domain of NF-YA that allows NF-YA29 to bind to the NF-YB–NF-YC dimer, but prohibits the trimeric complex from binding to DNA (53). Ectopic expression of NF-YA29 in F9 EC cells reduces the activity of the promoter/reporter gene construct 427T+E by as much as 70% (41). In the current study, we used NF-YA29 to examine whether NF-Y represses the FGF-4 promoter in the absence of the FGF-4 enhancer. Cotransfection of F9 EC cells with 427T and increasing amounts of NF-YA29 resulted in a dose-dependent increase in reporter gene activity (Fig. 5A). In addition, we tested the effect of NF-YA29 on 427T+E in F9-differentiated cells, where the FGF-4 enhancer is inactive. Again, increasing amounts of NF-YA29 led to a dose-dependent increase in reporter gene activity (Fig. 5B). Moreover, NF-YA29 had no effect on the FGF-4 promoter/reporter construct in which the CCAAT box was disrupted. Together, these findings argue NF-Y is functionally involved in the repression of the FGF-4 promoter in the absence of an active enhancer.
Figure 5.
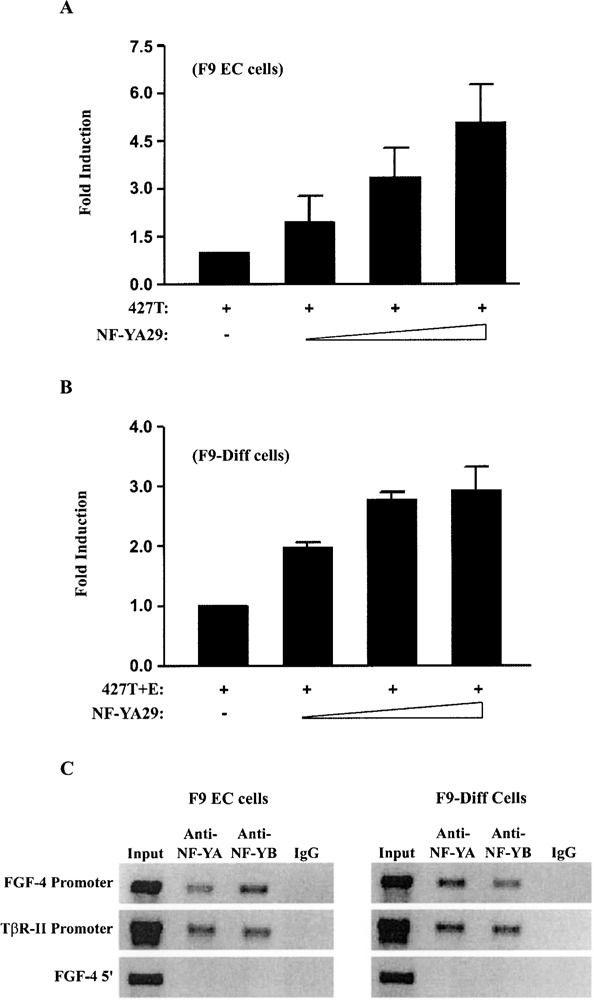
NF-Y is functionally involved in the repression of the FGF-4 promoter and binds to the endogenous FGF-4 promoter in F9 EC and F9-differentiated cells. (A) Transfection of F9 EC cells with 15 μg of 427T alone or with 0, 1, 2, or 4 μg of an expression plasmid for NF-YA29. Results are presented as fold induction over the activity of 427T alone, which was set to 1. This experiment was performed three times. The data shown are the compilation of the three experiments, and the standard deviation for each condition is represented by error bars. Analysis of the data by the Student’s t-test indicated that the activation observed by the transfection of 2 μg of NF-YA29 was significantly greater than the activity of 427T with a p-value of less than 0.05. (B) Transfection of F9-differentiated cells with 15 μg of 427T+E alone or with 0, 2, 4, or 6 μg of an expression plasmid for NF-YA29. Results are presented as fold induction over the activity of 427T+E alone, which was set to 1. This experiment was performed two times. The data shown are the compilation of the two experiments, and the standard deviation for each condition is represented by error bars. Analysis of the data by the Student’s t-test indicated that the activation observed by the transfection of 0.5 μg of NF-YA29 was significantly greater than the activity of 427T+E with a p-value of less than 0.05. (C) ChIP assays were performed on nontransfected F9 EC and F9-differentiated cells as described in Materials and Methods. NF-Y-associated DNA fragments were immunoprecipitated with an antibody to NF-YA and NF-YB. Preimmune normal rabbit IgG was used as a negative control. DNA fragments were amplified using primers for the FGF-4 promoter, the TβR-II promoter, or a region upstream of the FGF-4 promoter (FGF-4 5′). This experiment was performed three times with similar results.
To extend these studies, ChIP analyses were performed to determine whether NF-Y is bound to the endogenous FGF-4 promoter before and after F9 EC cells differentiate. For this purpose, antibodies specific for NF-YA and NF-YB were used. We determined that both NF-Y antibodies immunoprecipitated chromatin fragments containing the FGF-4 gene promoter from F9 EC cells, as well as from F9-differentiated cells (Fig. 5C). As a control, we demonstrated that NF-Y antibodies did not immunoprecipitate chromatin fragments from a region of the genome 3 kb upstream of the FGF-4 promoter. To help confirm these studies, we examined the in vivo binding of NF-Y to the promoter of the type II transforming growth factor-β receptor (TβR-II) gene in both F9 EC and F9-differentiated cells. The TβR-II promoter was used as a positive control, because NF-Y has been shown to positively regulate this promoter in F9-differentiated cells (6). Together, the functional studies with NF-YA29 and the ChIP analyses suggest that NF-Y binds to and activates the FGF-4 promoter in the presence of a functional enhancer, whereas in the absence of a functional enhancer, NF-Y binds to and represses the FGF-4 promoter. Currently, it is unclear how NF-Y represses the FGF-4 promoter. Recent studies argue that NF-Y can interact with the co-repressor, histone deacetylase 1 (HDAC1), and repress two other promoters (63,75). To examine whether this was the case for NF-Y and the FGF-4 promoter, we performed ChIP analysis with an HDAC1 antibody. However, in F9-differentiated cells, we did not observe enrichment of the FGF-4 promoter with antibodies that recognize HDAC1 (data not shown).
Although NF-Y has been reported to repress transcription from several promoters (27,51,63,75,89), no studies have mapped the region(s) of NF-Y responsible for repression. The transactivation domains of NF-Y have been mapped to glutamine-rich regions of NF-YA and NF-YC (14,15,19). When isolated and fused to heterologous DNA binding domains, such as LexA and Gal4, these domains can activate transcription. We used a similar approach to examine whether the individual subunits of NF-Y can mediate FGF-4 promoter repression. For these studies, the promoter/reporter gene construct 427T was modified to include a Gal4 DNA binding site in place of the CCAAT box in the FGF-4 promoter (427TG4/CCAAT). This modification allows Gal4/NF-Y fusion proteins to bind to the FGF-4 promoter. The HAP domain of each of the NF-Y subunits was removed from the Gal4/NF-Y expression plasmids used in this study (Gal4/NF-YA 1-261, Gal4/NF-YC Δ1 and Gal4/NF-YB Δ50–139) (Fig. 6A), because these domains are involved in subunit interactions and DNA binding (30,45,77). Cotransfection of F9 EC cells with 427TG4/CCAAT and increasing amounts of an expression plasmid for Gal4/NF-YA 1–261 or Gal4/NF-YC Δ1 generated a dose-dependent increase in reporter gene activity (Fig. 6B and C, respectively). In contrast, expression of Gal4/NF-YB Δ50–139 appears to inhibit reporter gene activity (Fig. 6D). However, the decrease in activity is similar to the decrease that is observed with an expression plasmid for the Gal4 DNA binding domain alone. This result is most likely a reflection of the very weak activation potential of Gal4/NF-YB (15,32). In this regard, when F9 EC cells were cotransfected with Gal4/NF-YB Δ50–139 and the promoter/reporter gene construct 427T+EnGSp, which contains a Gal4 DNA binding site inserted into the FGF-4 enhancer, Gal4/NF-YB Δ50–139 induced a small increase (approximately four- to fivefold) in reporter gene activity, whereas Gal4/NF-YA 1–261 and Gal4/NF-YC Δ1 induced approximately 15- and 50-fold increases in reporter gene activity, respectively (data not shown). Together, these results argue that the individual subunits of NF-Y are not sufficient to repress FGF-4 promoter activity.
Figure 6.
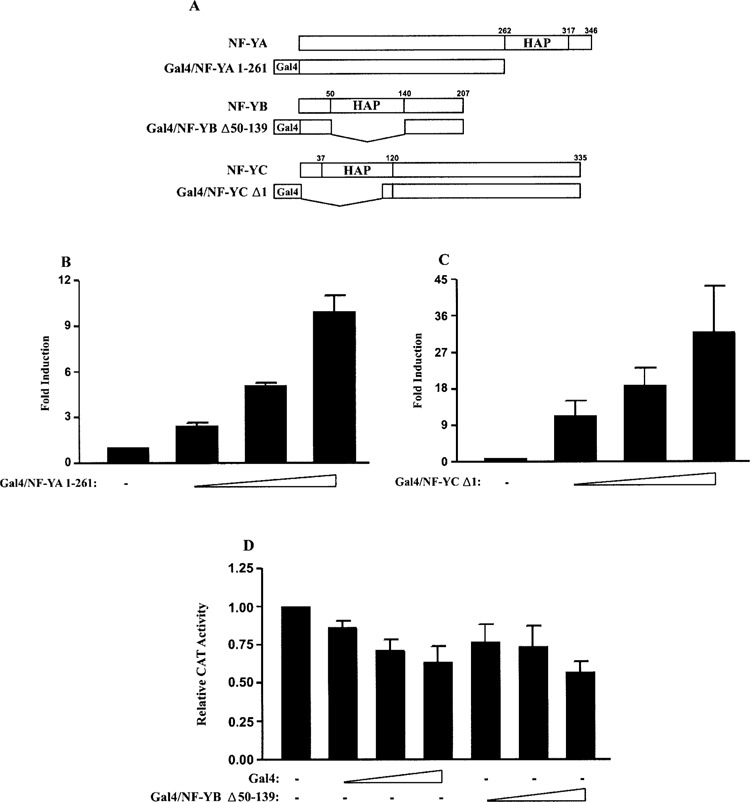
The individual subunits of NF-Y are not sufficient to repress the FGF-4 promoter. (A) Schematic representation of the Gal4/NF-Y fusion proteins compared to the wild-type protein. HAP, HAP domain. (B) Transfection of F9 EC cells with 15 μg of 427TG4/CCAAT alone or with 0, 1, 2.5, or 5 μg of an expression plasmid for Gal4/NF-YA 1–261. Results are presented as fold induction over the activity of 427TG4/CCAAT, which was set to 1. This experiment was performed three times. The data shown are the compilation of the three experiments, and the standard deviation for each condition is represented by error bars. Analysis of the data by the Student’s t-test indicated that the activation observed by the transfection of 1 μg of Gal4/NF-YA 1–261 was significantly greater than the activity of 427TG4/CCAAT with a p-value of less than 0.05. (C) Transfection of F9 EC cells with 15 μg of 427TG4/CCAAT alone or with 0, 1, 2, or 4 μg of an expression plasmid for Gal4/NF-YC Δ1. Results are presented as fold induction over the activity of 427TG4/CCAAT, which was set to 1. This experiment was performed four times. The data shown are the compilation of the four experiments, and the standard deviation for each condition is represented by error bars. Analysis of the data by the Student’s t-test indicated that the activation observed by the transfection of 1 μg of Gal4/NF-YC Δ1 was significantly greater than the activity of 427TG4/CCAAT with a p-value of less than 0.05. (D) Transfection of F9 EC cells with 15 μg of 427TG4/CCAAT alone or with 0, 0.5, 1, or 2 μg of an expression plasmid for Gal4 or Gal4/NF-YB Δ50–139. Results are presented as CAT expression relative to the activity of 427TG4/CCAAT, which was set to 1. This experiment was performed three times. The data shown are the compilation of the three experiments, and the standard deviation for each condition is represented by error bars. Analysis of the data by the Student’s t-test indicated that the decrease in CAT activity observed by the transfection of 2 μg of Gal4/NF-YB Δ50–139 was not significantly different than the decrease in CAT activity observed by the transfection of 2 μg of Gal4.
The Orientation and the Position Are Important for CCAAT Box Function
Because none of the three subunits of NF-Y appeared to repress the FGF-4 promoter on their own and all three subunits are required for binding to DNA, we considered the possibility that repression by NF-Y may be architectural in nature. In this regard, analysis of bona fide NF-Y binding CCAAT boxes demonstrated that CCAAT boxes of TATA-containing promoters are most often located between 80 and 100 bp upstream of the transcription start site (52). In addition, NF-Y has been shown to bend DNA 60–80° (46,72). To determine whether the orientation and/or the position of the CCAAT box in the FGF-4 promoter influence its function, we generated a set of promoter/reporter gene constructs in which the wild-type CCAAT box sequence was replaced with a CCAAT box of the opposite orientation (reversed CCAAT+E and reversed CCAAT) (Fig. 7A). In another set of promoter/reporter gene constructs, the CCAAT box sequence was moved from its wild-type position (−101 to −111) to an upstream position (−306 to −296) within the FGF-4 promoter (upstream CCAAT+E and upstream CCAAT). The sequence used to change the orientation and the position of the CCAAT box consisted of 11 nucleotides that included the CCAAT pentanucleotide plus three 5′- and three 3′-flanking bp. These 11 bp constitute an NF-Y consensus site that has been derived from both functional and statistical studies (7,52). These promoter/reporter gene constructs were tested in F9 EC cells to examine the effect of the orientation and the position on the function of the CCAAT box in both the presence and the absence of the FGF-4 enhancer. In the presence of the FGF-4 enhancer, reversing the orientation of the CCAAT box resulted in reporter gene activity similar to that of the wild-type promoter/reporter gene construct (Fig. 7B). In contrast, moving the CCAAT box to an upstream site led to a decrease in reporter gene activity, to levels similar to those with CCAATmut+E. Therefore, when the FGF-4 enhancer is present, the FGF-4 CCAAT box can function in either orientation to positively affect promoter activity. In direct contrast, when the FGF-4 enhancer is absent, either reversing the orientation of the CCAAT box or moving the CCAAT box upstream neutralized its ability to repress the FGF-4 promoter (Fig. 7C). Therefore, both the orientation and the position of the CCAAT box are important for the CCAAT box to function as a negative cis-regulatory element in the absence of the FGF-4 enhancer. Collectively these results demonstrate that both the orientation and the position of the CCAAT box influence its ability to properly regulate the FGF-4 promoter and argue for an architectural role by the CCAAT box in FGF-4 promoter repression.
Figure 7.
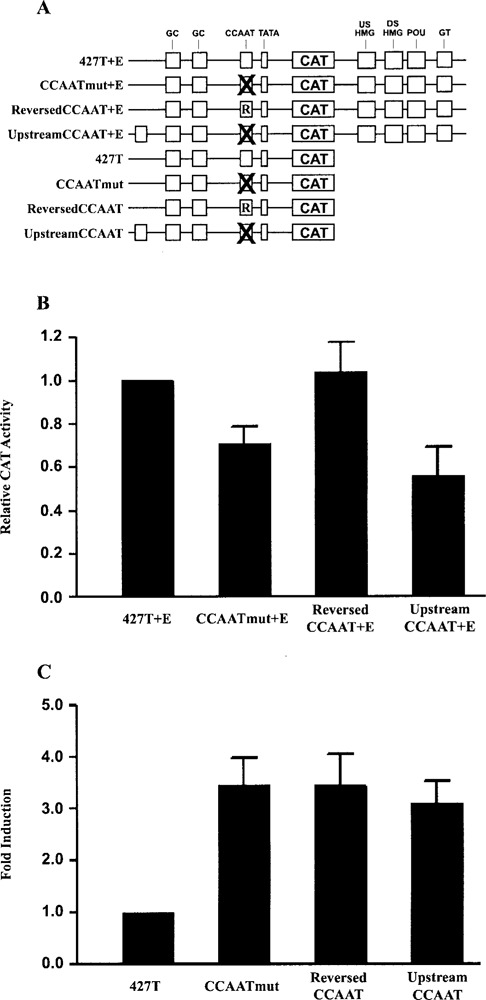
The orientation and the position of the CCAAT box is important for the regulation of the FGF-4 promoter. (A) Physical map of the promoter/reporter gene constructs that contain a reversed or upstream CCAAT box. (B) Transfection of F9 EC cells with 15 μg of 427T+E, CCAATmut+E, invert CCAAT+E, and upstream CCAAT+E. Results are presented as CAT expression relative to the activity of 427T+E, which was set to 1. This experiment was performed five times. The data shown are the compilation of the five experiments, and the standard deviation for each condition is represented by error bars. Analysis of the data by the Student’s t-test indicated that the activity of CCAATmut+E and upstream CCAATmut+E was significantly less than the activity of 427T+E with a p-value of less than 0.05, whereas the activity of reversed CCAAT+E was not significantly different than the activity of 427T+E. (C) Transfection of F9 EC cells with 15 μg of 427T, CCAATmut, invert CCAAT and upstream CCAAT. Results are presented as fold induction over the activity of 427T, which was set to 1. This experiment was performed four times. The data shown are the compilation of the four experiments, and the standard deviation for each condition is represented by error bars. Analysis of the data by the Student’s t-test indicated that the activity of CCAATmut, reversed CCAATmut, and upstream CCAATmut were significantly greater than the activity of 427T with a p-value of less than 0.05.
DISCUSSION
The work described in this study demonstrates that the CCAAT box of the FGF-4 promoter can function as a positive cis-regulatory element in the presence of an active FGF-4 enhancer or function as a negative cis-regulatory element when the FGF-4 enhancer is inactive. Previous studies have shown that NF-Y mediates the positive effect of the CCAAT box of the FGF-4 promoter in F9 EC cells (41). In this study, we demonstrate that NF-Y mediates the repressive effect of this CCAAT box and that the status of the FGF-4 enhancer determines whether NF-Y activates or represses the FGF-4 promoter. In addition, we show that NF-Y associates with the endogenous FGF-4 promoter in both F9 EC cells and F9-differentiated cells. Studies to determine how NF-Y mediates repression demonstrate that the individual subunits of NF-Y are not sufficient for repression, and that the orientation and the position of the CCAAT box are important for its ability to repress the FGF-4 promoter.
Differentiation of EC cells results in the downregulation of FGF-4 transcription (64,69,82,86). Although previous studies have suggested that the FGF-4 enhancer is inactivated after EC cells differentiate (17,50), it was unclear whether the enhancer is completely inactivated after differentiation or whether the function of the FGF-4 promoter is actively repressed after differentiation. We demonstrate here that the FGF-4 enhancer is incapable of activating the heterologous TK promoter in F9-differentiated cells. The most likely explanation for the loss of FGF-4 enhancer activity is the loss of Sox2 and Oct-3 expression that occurs with differentiation (56,87). In this regard, Sox2 and Oct-3 bind cooperatively to the DSHMG and POU motifs to synergistically activate transcription of the FGF-4 gene (1,2,59,87), and both Sox2 and Oct-3 mRNA are nearly undetectable 72 h after EC cells have been induced to differentiate (56,87). Unlike Sox2 and Oct-3, the transcription factors NF-Y, Sp1, and Sp3 that bind to the FGF-4 promoter are expressed before and after EC cells differentiate and differentiation does not significantly alter their binding to DNA in vitro (41,42).
Examination of the FGF-4 promoter led to the surprising finding that the promoter is subject to repression in the absence of a functional FGF-4 enhancer. More specifically, we show that disruption of the CCAAT box results in an increase in FGF-4 promoter activity in F9 EC cells in the absence of the FGF-4 enhancer and in F9-differentiated cells where the FGF-4 enhancer is inactive. The increased FGF-4 promoter activity (four- to fivefold) that is observed when the CCAAT box is disrupted in F9 EC cells, relative to the increase that is observed in differentiated cells (twofold) where the enhancer is inactive, is most likely the result of other changes that occur within the promoter after differentiation. Moreover, it appears that the GC boxes are not involved in the repression of the FGF-4 promoter, and the GC boxes are not required by the CCAAT box to mediate repression. These latter findings are of particular interest, because neighboring cis-regulatory elements are involved with CCAAT boxes in the repression of other promoters. Specifically, the CCAAT box of the RUSH/SMARCA3 promoter is only repressive in conjunction with two upstream Sp1 sites (29), while the CCAAT box of the growth hormone receptor promoter is involved in the recruitment of a “represso-some” complex that involves several neighboring cis-regulatory elements (27).
Like the CCAAT box of the FGF-4 promoter, the CCAAT boxes of several cell cycle-dependent promoters also function in a context-dependent manner. Cyclin B1, B2, cdc25, and cdc2 are all required for progression through the G2 phase of the cell cycle. The promoters of these genes contain CCAAT boxes that positively modulate their activity through the progression of the cell cycle (8,24,80,91). However, during p53-induced G2 arrest, the CCAAT boxes of these promoters negatively regulate the transcription of these genes (51,89). Therefore the regulation of the cyclin B1, B2, cdc25, and cdc2 promoters by the CCAAT box is dependent upon the cell cycle. However, the mechanism involved remains to be determined. Our studies indicate that the regulation of the FGF-4 promoter by the CCAAT box is dependent upon the FGF-4 enhancer, which is located more than 3 kb downstream from the FGF-4 promoter CCAAT box. Furthermore, our results demonstrate that disruption of the DSHMG motif, the DSHMG and POU motifs, or the GT box in the enhancer is sufficient to convert the CCAAT box from a positive to a negative cis-regulatory element. The effects of disrupting the DSHMG and POU motifs are particularly interesting, because Sox2 and Oct-3 synergistically activate transcription by binding to these cis-regulatory elements in the FGF-4 enhancer (1,59,87). Moreover, p300 has been shown to mediate the effects of Sox2 and Oct-3 on gene transcription (58,59), and p300 is associated with the endogenous FGF-4 enhancer in F9 EC cells (58). This led us to examine the effect of p300 on the function of the CCAAT box by physically tethering p300 to the disabled FGF-4 enhancer in 427T+EnGSp. Our results demonstrate that p300 can convert the CCAAT box in the disabled FGF-4 enhancer from a negative to a positive cis-regulatory element. Together, our findings argue that the FGF-4 enhancer strongly influences the behavior of the CCAAT box and further suggest that p300 is likely to mediate, at least in part, the positive influence of the FGF-4 enhancer on the promoter CCAAT box.
Previous work utilizing the dominant negative NF-YA29 has shown that NF-Y is functionally involved in the activation of the FGF-4 promoter (41). In this study, NF-YA29 was used to demonstrate that NF-Y can function to repress the FGF-4 promoter when the FGF-4 enhancer is either absent or inactive. NF-YA29 has been utilized by others to show that NF-Y mediates the repression of certain cell cycle-dependent promoters during p53-induced G2 arrest and the repression of the growth hormone receptor promoter (27,51,89). NF-Y is one of several bifunctional transcription factors that include the nonsteroidal nuclear receptors, YY1 and NF-κB. The context of bifunctional transcription factors dictates whether they activate or repress transcription. NF-Y functions as a bifunctional transcription factor in the regulation of several cell cycle-dependent promoters. In the case of the von Willebrand factor promoter, NF-Y binds to the canonical CCAAT box of the von Willebrand factor promoter in endothelial cells to activate transcription, whereas in nonendothelial cells, NF-Y binds to a noncanonical NF-Y binding site in this promoter to repress transcription (62).
NF-Y has been reported to mediate repression and several mechanisms appear to be utilized. In the case of the growth hormone receptor promoter, NF-Y has been shown to be part of a “repressosome” complex that interacts with the co-repressor mSin3b (27). In addition, NF-Y has been shown to interact with the co-repressor, HDAC1, in the regulation of at least two promoters (63,75). The interaction of NF-Y with either coactivators or co-repressors in response to different signals appears to parallel the bifunctional action of nuclear hormone receptors, which associate with coactivators in the presence of hormone and activate transcription, but associate with co-repressors in the absence of hormone and inhibit transcription (65). Consequently, this prompted us to examine by ChIP analyses whether HDAC1 is associated with the FGF-4 promoter in F9-differentiated cells. We were unable to show enrichment of the FGF-4 promoter with antibodies that recognize HDAC1. However, this negative result does not rule out the possibility that HDAC1 or another co-repressor associates with the FGF-4 promoter in differentiated EC cells. NF-Y may also function by influencing the binding of transcription factors to neighboring cis-regulatory elements. In this regard, the binding of NF-Y to DNA can induce bends of 60–80° in the DNA (46,72). Thus, NF-Y could act as an architectural factor to enhance transcription by increasing the affinity of factors that bind to adjacent cis-regulatory elements, as it does in the cases of Sp1, SREBP1, and RF-X binding to four different promoters. (33,67,71,84). Similarly, NF-Y could to repress transcription by sterically blocking the binding of activating transcription factors to adjacent regulatory regions as it does in the insulin gene and the renin gene (22,76).
Our studies demonstrate that the individual NF-Y subunits are not sufficient to mediate repression of the FGF-4 promoter, which suggests that multiple subunits of NF-Y are required for repression. In this regard, recruitment of a co-repressor may require a binding surface that is formed by two or more of the NF-Y subunits. For example, all three subunits of NF-Y are required to interact with the sterol regulatory element binding protein, a transcription factor involved in cholesterol and fatty acid metabolism (20). Alternatively, repression by NF-Y may be a result of NF-Y binding to and bending DNA, thereby altering the architecture of the promoter. For this reason, we examined the effect of altering the orientation and the position of the CCAAT box on its ability to regulate the FGF-4 promoter. We show that the CCAAT box can activate the FGF-4 promoter in either orientation in the presence of the FGF-4 enhancer, but it is unable to activate when it is moved almost 200 bp upstream of its wild-type position. The ability of the CCAAT box to activate transcription regardless of orientation is not surprising considering that CCAAT boxes are found throughout the genome in either orientation, with roughly 60% of CCAAT boxes being in the inverted ATTGG orientation (52). The loss of function when the CCAAT box is moved to an upstream position within the promoter may reflect the need for the CCAAT box to be within an optimal distance of the TATA box. In promoters that contain both a CCAAT box and a TATA box, the CCAAT box is found most often between 80 and 100 bp upstream of the transcription start site (52). In addition, NF-Y has been shown to interact with components of the basal transcriptional machinery (5,16,26). Although changing the orientation of the CCAAT box does not alter its function when the FGF-4 enhancer is active, this is not the case when the FGF-4 enhancer is absent. In the absence of the enhancer, the CCAAT box fails to inhibit the FGF-4 promoter if its orientation is reversed or if the CCAAT box is moved to an upstream position. These results suggest that the ability of NF-Y to repress the FGF-4 promoter is strictly dependent upon the orientation and the position of the CCAAT box. Previous studies have suggested that the CCAAT box and NF-Y may play a structural role in gene regulation (46,52,72); our findings provide direct evidence for an architectural role of NF-Y in the case of the FGF-4 promoter.
In conclusion, our studies provide evidence that the CCAAT box of the FGF-4 promoter can activate or repress transcription. We show that the FGF-4 enhancer, and more specifically p300, regulates the function of the CCAAT box. Moreover, our findings provide both functional and physical evidence that NF-Y mediates the effect of the CCAAT box in both F9 EC cells and F9-differentiated cells. Finally, we demonstrate that orientation and position of the CCAAT box are important and argue for an architectural role by the CCAAT box and NF-Y in the regulation of the FGF-4 promoter. Given these findings, the system described in this study appears to provide an excellent model for exploring the architectural role of CCAAT boxes and for investigating the bifunctional behavior of NF-Y and its regulation of genes that play key roles during mammalian development.
ACKNOWLEDGMENTS
Keiko Funa is thanked for his gift of the Gal4/NF-YC expression plasmid, GAL4/NF-YC Δ1. Heather Rizzino is thanked for editorial assistance. Dr. Phillip Wilder is thanked for reading this manuscript and for his helpful comments. This work was supported by the National Cancer Institute (CA74771). Core facilities of the UNMC Eppley Center were supported in part by a Cancer Center Support Grant (CA36727).
REFERENCES
- 1. Ambrosetti D. C.; Basilico C.; Dailey L. Synergistic activation of the fibroblast growth factor 4 enhancer by Sox2 and Oct-3 depends on protein–protein interactions facilitated by a specific spatial arrangement of factor binding sites. Mol. Cell. Biol. 17:6321–6329; 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ambrosetti D. C.; Scholer H. R.; Dailey L.; Basilico C. Modulation of the activity of multiple transcriptional activation domains by the DNA binding domains mediates the synergistic action of Sox2 and Oct-3 on the fibroblast growth factor-4 enhancer. J. Biol. Chem. 275:23387–23397; 2000. [DOI] [PubMed] [Google Scholar]
- 3. Arents G.; Moudrianakis E. N. The histone fold: A ubiquitous architectural motif utilized in DNA compaction and protein dimerization. Proc. Natl. Acad. Sci. USA 92:11170–11174; 1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Baxevanis A. D.; Arents G.; Moudrianakis E. N.; Landsman D. A variety of DNA-binding and multimeric proteins contain the histone fold motif. Nucleic Acids Res. 23:2685–2691; 1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bellorini M.; Lee D. K.; Dantonel J. C.; Zemzoumi K.; Roeder R. G.; Tora L.; Mantovani R. CCAAT binding NF-Y-TBP interactions: NF-YB and NF-YC require short domains adjacent to their histone fold motifs for association with TBP basic residues. Nucleic Acids Res. 25:2174–2181; 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bernadt C. T.; Rizzino A. Roles of the conserved CCAAT and GC boxes of the human and mouse type II transforming growth factor-beta receptor genes. Mol. Reprod. Dev. 65:353–365; 2003. [DOI] [PubMed] [Google Scholar]
- 7. Bi W.; Wu L.; Coustry F.; de Crombrugghe B.; Maity S. N. DNA binding specificity of the CCAAT-binding factor CBF/NF-Y. J. Biol. Chem. 272:26562–26572; 1997. [DOI] [PubMed] [Google Scholar]
- 8. Bolognese F.; Wasner M.; Dohna C. L.; Gurtner A.; Ronchi A.; Muller H.; Manni I.; Mossner J.; Piaggio G.; Mantovani R.; Engeland K. The cyclin B2 promoter depends on NF-Y, a trimer whose CCAAT-binding activity is cell-cycle regulated. Oncogene 18:1845–1853; 1999. [DOI] [PubMed] [Google Scholar]
- 9. Boucher P. D.; Piechocki M. P.; Hines R. N. Partial characterization of the human CYP1A1 negatively acting transcription factor and mutational analysis of its cognate DNA recognition sequence. Mol. Cell. Biol. 15:5144–5151; 1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Brouillard F.; Cremisi C. E. Concomitant increase of histone acetyltransferase activity and degradation of p300 during retinoic acid-induced differentiation of F9 cells. J. Biol. Chem. 278:39509–39516; 2003. [DOI] [PubMed] [Google Scholar]
- 11. Bucher P. Weight matrix descriptions of four eukaryotic RNA polymerase II promoter elements derived from 502 unrelated promoter sequences. J. Mol. Biol. 212:563–578; 1990. [DOI] [PubMed] [Google Scholar]
- 12. Caretti G.; Salsi V.; Vecchi C.; Imbriano C.; Mantovani R. Dynamic recruitment of NF-Y and histone acetyltransferases on cell-cycle promoters. J. Biol. Chem. 278:30435–30440; 2003. [DOI] [PubMed] [Google Scholar]
- 13. Chang Z. F.; Liu C. J. Human thymidine kinase CCAAT-binding protein is NF-Y, whose A subunit expression is serum-dependent in human IMR-90 diploid fibroblasts. J. Biol. Chem. 269:17893–17898; 1994. [PubMed] [Google Scholar]
- 14. Coustry F.; Maity S. N.; de Crombrugghe B. Studies on transcription activation by the multimeric CCAAT-binding factor CBF. J. Biol. Chem. 270:468–475; 1995. [DOI] [PubMed] [Google Scholar]
- 15. Coustry F.; Maity S. N.; Sinha S.; de Crombrugghe B. The transcriptional activity of the CCAAT-binding factor CBF is mediated by two distinct activation domains, one in the CBF-B subunit and the other in the CBF-C subunit. J. Biol. Chem. 271:14485–14491; 1996. [DOI] [PubMed] [Google Scholar]
- 16. Coustry F.; Sinha S.; Maity S. N.; Crombrugghe B. The two activation domains of the CCAAT-binding factor CBF interact with the dTAFII110 component of the Drosophila TFIID complex. Biochem. J. 331(Pt. 1):291–297; 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Curatola A. M.; Basilico C. Expression of the K-fgf proto-oncogene is controlled by 3′ regulatory elements which are specific for embryonal carcinoma cells. Mol. Cell. Biol. 10:2475–2484; 1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Currie R. A. NF-Y is associated with the histone ace-tyltransferases GCN5 and P/CAF. J. Biol. Chem. 273:1430–1434; 1998. [DOI] [PubMed] [Google Scholar]
- 19. de Silvio A.; Imbriano C.; Mantovani R. Dissection of the NF-Y transcriptional activation potential. Nucleic Acids Res. 27:2578–2584; 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Dooley K. A.; Millinder S.; Osborne T. F. Sterol regulation of 3-hydroxy-3-methylglutaryl-coenzyme A synthase gene through a direct interaction between sterol regulatory element binding protein and the trimeric CCAAT-binding factor/nuclear factor Y. J. Biol. Chem. 273:1349–1356; 1998. [DOI] [PubMed] [Google Scholar]
- 21. Eckner R.; Arany Z.; Ewen M.; Sellers W.; Livingston D. M. The adenovirus E1A-associated 300-kD protein exhibits properties of a transcriptional coactivator and belongs to an evolutionarily conserved family. Cold Spring Harbor Symp. Quant. Biol. 59:85–95; 1994. [DOI] [PubMed] [Google Scholar]
- 22. Eggers A.; Siemann G.; Blume R.; Knepel W. Gene-specific transcriptional activity of the insulin cAMP-responsive element is conferred by NF-Y in combination with cAMP response element-binding protein. J. Biol. Chem. 273:18499–18508; 1998. [DOI] [PubMed] [Google Scholar]
- 23. Faniello M. C.; Bevilacqua M. A.; Condorelli G.; de Crombrugghe B.; Maity S. N.; Avvedimento V. E.; Cimino F.; Costanzo F. The B subunit of the CAAT-binding factor NFY binds the central segment of the co-activator p300. J. Biol. Chem. 274:7623–7626; 1999. [DOI] [PubMed] [Google Scholar]
- 24. Farina A.; Manni I.; Fontemaggi G.; Tiainen M.; Cenciarelli C.; Bellorini M.; Mantovani R.; Sacchi A.; Piaggio G. Down-regulation of cyclin B1 gene transcription in terminally differentiated skeletal muscle cells is associated with loss of functional CCAAT-binding NF-Y complex. Oncogene 18:2818–2827; 1999. [DOI] [PubMed] [Google Scholar]
- 25. Feldman B.; Poueymirou W.; Papaioannou V. E.; DeChiara T. M.; Goldfarb M. Requirement of FGF-4 for postimplantation mouse development. Science 267:246–249; 1995. [DOI] [PubMed] [Google Scholar]
- 26. Frontini M.; Imbriano C.; diSilvio A.; Bell B.; Bogni A.; Romier C.; Moras D.; Tora L.; Davidson I.; Mantovani R. NF-Y recruitment of TFIID, multiple interactions with histone fold TAF(II)s. J. Biol. Chem. 277:5841–5848; 2002. [DOI] [PubMed] [Google Scholar]
- 27. Gowri P. M.; Yu J. H.; Shaufl A.; Sperling M. A.; Menon R. K. Recruitment of a repressosome complex at the growth hormone receptor promoter and its potential role in diabetic nephropathy. Mol. Cell Biol. 23:815–825; 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Gurtner A.; Manni I.; Fuschi P.; Mantovani R.; Guadagni F.; Sacchi A.; Piaggio G. Requirement for down-regulation of the CCAAT-binding activity of the NF-Y transcription factor during skeletal muscle differentiation. Mol. Biol. Cell 14:2706–2715; 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hewetson A.; Chilton B. S. An Sp1-NF-Y/progester-one receptor DNA binding-dependent mechanism regulates progesterone-induced transcriptional activation of the rabbit RUSH/SMARCA3 gene. J. Biol. Chem. 278:40177–40185; 2003. [DOI] [PubMed] [Google Scholar]
- 30. Hooft v. H.; Li X. Y.; Black D.; Matthes H.; Benoist C.; Mathis D. Co-evolution from yeast to mouse: cDNA cloning of the two NF-Y (CP-1/CBF) subunits. EMBO J. 9:3119–3127; 1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Isaacs R. J.; Harris A. L.; Hickson I. D. Regulation of the human topoisomerase IIalpha gene promoter in confluence-arrested cells. J. Biol. Chem. 271:16741–16747; 1996. [DOI] [PubMed] [Google Scholar]
- 32. Izumi H.; Molander C.; Penn L. Z.; Ishisaki A.; Kohno K.; Funa K. Mechanism for the transcriptional repression by c-Myc on PDGF beta-receptor. J. Cell Sci. 114:1533–1544; 2001. [DOI] [PubMed] [Google Scholar]
- 33. Jackson S. M.; Ericsson J.; Mantovani R.; Edwards P. A. Synergistic activation of transcription by nuclear factor Y and sterol regulatory element binding protein. J. Lipid Res. 39:767–776; 1998. [PubMed] [Google Scholar]
- 34. Jaskelioff M.; Peterson C. L. Chromatin and transcription: Histones continue to make their marks. Nat. Cell Biol. 5:395–399; 2003. [DOI] [PubMed] [Google Scholar]
- 35. Jin S.; Scotto K. W. Transcriptional regulation of the MDR1 gene by histone acetyltransferase and deacetylase is mediated by NF-Y. Mol. Cell. Biol. 18:4377–4384; 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Johnson L. R.; Johnson T. K.; Desler M.; Luster T. A.; Nowling T.; Lewis R. E.; Rizzino A. Effects of B-Myb on gene transcription: Phosphorylation-dependent activity ans acetylation by p300. J. Biol. Chem. 277:4088–4097; 2002. [DOI] [PubMed] [Google Scholar]
- 37. Johnson L. R.; Lamb K. A.; Gao Q.; Nowling T. K.; Rizzino A. Role of the transcription factor Sox-2 in the expression of the FGF-4 gene in embryonal carcinoma cells. Mol. Reprod. Dev. 50:377–386; 1998. [DOI] [PubMed] [Google Scholar]
- 38. Kadonaga J. T. Regulation of RNA polymerase II transcription by sequence-specific DNA binding factors. Cell 116:247–257; 2004. [DOI] [PubMed] [Google Scholar]
- 39. Kelly D.; Kim S. J.; Rizzino A. Transcriptional activation of the type II transforming growth factor-beta receptor gene upon differentiation of embryonal carcinoma cells. J. Biol. Chem. 273:21115–21124; 1998. [DOI] [PubMed] [Google Scholar]
- 40. Lamb K.; Rosfjord E.; Brigman K.; Rizzino A. Binding of transcription factors to widely-separated cis-regulatory elements of the murine FGF-4 gene. Mol. Reprod. Dev. 44:460–471; 1996. [DOI] [PubMed] [Google Scholar]
- 41. Lamb K. A.; Johnson L. R.; Rizzino A. NF-Y binds to the CCAAT box motif of the FGF-4 gene and promotes FGF-4 expression in embryonal carcinoma cells. Mol. Reprod. Dev. 48:301–309; 1997. [DOI] [PubMed] [Google Scholar]
- 42. Lamb K. A.; Rizzino A. Effects of differentiation on the transcriptional regulation of the FGF-4 gene: Critical roles played by a distal enhancer. Mol. Reprod. Dev. 51:218–224; 1998. [DOI] [PubMed] [Google Scholar]
- 43. Lee T. C.; Zhang Y.; Schwartz R. J. Bifunctional transcriptional properties of YY1 in regulating muscle actin and c-myc gene expression during myogenesis. Oncogene 9:1047–1052; 1994. [PubMed] [Google Scholar]
- 44. Li Q.; Herrler M.; Landsberger N.; Kaludov N.; Ogryzko V. V.; Nakatani Y.; Wolffe A. P. Xenopus NF-Y pre-sets chromatin to potentiate p300 and acetylation-responsive transcription from the Xenopus hsp70 promoter in vivo. EMBO J. 17:6300–6315; 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Li X. Y.; Mantovani R.; Hooft v. H.; Andre I.; Benoist C.; Mathis D. Evolutionary variation of the CCAAT-binding transcription factor NF-Y. Nucleic Acids Res. 20:1087–1091; 1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Liberati C.; Ronchi A.; Lievens P.; Ottolenghi S.; Mantovani R. NF-Y organizes the gamma-globin CCAAT boxes region. J. Biol. Chem. 273:16880–16889; 1998. [DOI] [PubMed] [Google Scholar]
- 47. Luger K.; Mader A. W.; Richmond R. K.; Sargent D. F.; Richmond T. J. Crystal structure of the nucleosome core particle at 2.8 A resolution. Nature 389:251–260; 1997. [DOI] [PubMed] [Google Scholar]
- 48. Luster T. A.; Johnson L. R.; Nowling T. K.; Lamb K. A.; Philipsen S.; Rizzino A. Effects of three Sp1 motifs on the transcription of the FGF-4 gene. Mol. Reprod. Dev. 57:4–15; 2000. [DOI] [PubMed] [Google Scholar]
- 49. Luster T. A.; Rizzino A. Regulation of the FGF-4 gene by a complex distal enhancer that functions in part as an enhanceosome. Gene 323:163–172; 2003. [DOI] [PubMed] [Google Scholar]
- 50. Ma Y. G.; Rosfjord E.; Huebert C.; Wilder P.; Tiesman J.; Kelly D.; Rizzino A. Transcriptional regulation of the murine k-FGF gene in embryonic cell lines. Dev. Biol. 154:45–54; 1992. [DOI] [PubMed] [Google Scholar]
- 51. Manni I.; Mazzaro G.; Gurtner A.; Mantovani R.; Haugwitz U.; Krause K.; Engeland K.; Sacchi A.; Soddu S.; Piaggio G. NF-Y mediates the transcriptional inhibition of the cyclin B1, cyclin B2, and cdc25C promoters upon induced G2 arrest. J. Biol. Chem. 276:5570–5576; 2001. [DOI] [PubMed] [Google Scholar]
- 52. Mantovani R. A survey of 178 NF-Y binding CCAAT boxes. Nucleic Acids Res. 26:1135–1143; 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Mantovani R.; Li X. Y.; Pessara U.; Hooft, v. H.; Benoist C.; Mathis D. Dominant negative analogs of NF-YA. J. Biol. Chem. 269:20340–20346; 1994. [PubMed] [Google Scholar]
- 54. Martin G. R. Teratocarcinomas and mammalian embryogenesis. Science 209:768–776; 1980. [DOI] [PubMed] [Google Scholar]
- 55. Marziali G.; Perrotti E.; Ilari R.; Testa U.; Coccia E. M.; Battistini A. Transcriptional regulation of the ferritin heavy-chain gene: the activity of the CCAAT binding factor NF-Y is modulated in heme-treated Friend leukemia cells and during monocyte-to-macrophage differentiation. Mol. Cell. Biol. 17:1387–1395; 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Minucci S.; Botquin V.; Yeom Y. I.; Dey A.; Sylvester I.; Zand D. J.; Ohbo K.; Ozato K.; Scholer H. R. Retinoic acid-mediated down-regulation of Oct3/4 coincides with the loss of promoter occupancy in vivo. EMBO J. 15:888–899; 1996. [PMC free article] [PubMed] [Google Scholar]
- 57. Niswander L.; Martin G. R. Fgf-4 expression during gastrulation, myogenesis, limb and tooth development in the mouse. Development 114:755–768; 1992. [DOI] [PubMed] [Google Scholar]
- 58. Nowling T.; Bernadt C.; Johnson L.; Desler M.; Rizzino A. The co-activator p300 associates physically with and can mediate the action of the distal enhancer of the FGF-4 gene. J. Biol. Chem. 278:13696–13705; 2003. [DOI] [PubMed] [Google Scholar]
- 59. Nowling T. K.; Johnson L. R.; Wiebe M. S.; Rizzino A. Identification of the transactivation domain of the transcription factor Sox-2 and an associated co-activator. J. Biol. Chem. 275:3810–3818; 2000. [DOI] [PubMed] [Google Scholar]
- 60. Okamoto K.; Okazawa H.; Okuda A.; Sakai M.; Muramatsu M.; Hamada H. A novel octamer binding transcription factor is differentially expressed in mouse embryonic cells. Cell 60:461–472; 1990. [DOI] [PubMed] [Google Scholar]
- 61. Ota M.; Eto K.; Ninomiya Y.; Ikeda M. Accumulation of p300 mediates transcriptional repression of simian virus 40 enhancer in undifferentiated F9 embryonal carcinoma cells. Cell Growth Differ. 9:989–997; 1998. [PubMed] [Google Scholar]
- 62. Peng Y.; Jahroudi N. The NFY transcription factor functions as a repressor and activator of the von Willebrand factor promoter. Blood 99:2408–2417; 2002. [DOI] [PubMed] [Google Scholar]
- 63. Peng Y.; Jahroudi N. The NFY transcription factor inhibits von Willebrand factor promoter activation in non-endothelial cells through recruitment of histone deacetylases. J. Biol. Chem. 278:8385–8394; 2003. [DOI] [PubMed] [Google Scholar]
- 64. Peters G.; Brookes S.; Smith R.; Placzek M.; Dick-son C. The mouse homolog of the hst/k-FGF gene is adjacent to int-2 and is activated by proviral insertion in some virally induced mammary tumors. Proc. Natl. Acad. Sci. USA 86:5678–5682; 1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Privalsky M. L. The role of corepressors in transcriptional regulation by nuclear hormone receptors. Annu. Rev. Physiol 66:315–360; 2004. [DOI] [PubMed] [Google Scholar]
- 66. Rappolee D. A.; Sturm K.; Schultz G.; Pedersen R.; Werb Z. Expression of growth factor ligands and receptors in preimplantation mouse embryos. In: Heyner S.; Wiley L., eds. Early embryo development and paracrine relationships. New York: Wiley-Liss; 1990:11–25. [Google Scholar]
- 67. Reith W.; Siegrist C. A.; Durand B.; Barras E.; Mach B. Function of major histocompatibility complex class II promoters requires cooperative binding between factors RFX and NF-Y. Proc. Natl. Acad. Sci. USA 91:554–558; 1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Rizzino A.; Crowley C. Growth and differentiation of embryonal carcinoma cell line F9 in defined media. Proc. Natl. Acad. Sci. USA 77:457–461; 1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Rizzino A.; Kuszynski C.; Ruff E.; Tiesman J. Production and utilization of growth factors related to fibroblast growth factor by embryonal carcinoma cells and their differentiated cells. Dev. Biol. 129:61–71; 1988. [DOI] [PubMed] [Google Scholar]
- 70. Rizzino A.; Orme L. S.; De Larco J. E. Embryonal carcinoma cell growth and differentiation. Production of and response to molecules with transforming growth factor activity. Exp. Cell Res. 143:143–152; 1983. [DOI] [PubMed] [Google Scholar]
- 71. Roder K.; Wolf S. S.; Beck K. F.; Schweizer M. Cooperative binding of NF-Y and Sp1 at the DNase I-hypersensitive site, fatty acid synthase insulin-responsive element 1, located at -500 in the rat fatty acid synthase promoter. J. Biol. Chem. 272:21616–21624; 1997. [DOI] [PubMed] [Google Scholar]
- 72. Ronchi A.; Bellorini M.; Mongelli N.; Mantovani R. CCAAT-box binding protein NF-Y (CBF, CP1) recognizes the minor groove and distorts DNA. Nucleic Acids Res. 23:4565–4572; 1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Rosfjord E.; Lamb K.; Rizzino A. Cryptic promoter activity within the backbone of a plasmid commonly used to prepare promoter/reporter gene constructs. In Vitro Cell Dev. Biol. Anim. 30A:477–481; 1994. [DOI] [PubMed] [Google Scholar]
- 74. Sadowski I.; Bell B.; Broad P.; Hollis M. GAL4 fusion vectors for expression in yeast or mammalian cells. Gene 118:137–141; 1992. [DOI] [PubMed] [Google Scholar]
- 75. Schuettengruber B.; Simboeck E.; Khier H.; Seiser C. Autoregulation of mouse histone deacetylase 1 expression. Mol. Cell. Biol. 23:6993–7004; 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Shi Q.; Gross K. W.; Sigmund C. D. NF-Y antagonizes renin enhancer function by blocking stimulatory transcription factors. Hypertension 38:332–336; 2001. [DOI] [PubMed] [Google Scholar]
- 77. Sinha S.; Maity S. N.; Lu J.; de Crombrugghe B. Recombinant rat CBF-C, the third subunit of CBF/NFY, allows formation of a protein-DNA complex with CBF-A and CBF-B and with yeast HAP2 and HAP3. Proc. Natl. Acad. Sci. USA 92:1624–1628; 1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Sleigh M. J.; Lockett T. J. SV40 enhancer activation during retinoic acid-induced differentiation of F9 embryonal carcinoma cells. EMBO J. 4:3831–3837; 1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Tanaka S.; Kunath T.; Hadjantonakis A. K.; Nagy A.; Rossant J. Promotion of trophoblast stem cell proliferation by FGF4. Science 282:2072–2075; 1998. [DOI] [PubMed] [Google Scholar]
- 80. Tanimoto A.; Chen H.; Kao C. Y.; Moran E.; Sasaguri Y.; Padmanabhan R. Transactivation of the human cdc2 promoter by adenovirus E1A in cycling cells is mediated by induction of a 110-kDa CCAAT-box-binding factor. Oncogene 17:3103–3114; 1998. [DOI] [PubMed] [Google Scholar]
- 81. Thomas M. J.; Seto E. Unlocking the mechanisms of transcription factor YY1: Are chromatin modifying enzymes the key? Gene 236:197–208; 1999. [DOI] [PubMed] [Google Scholar]
- 82. Velcich A.; Delli-Bovi P.; Mansukhani A.; Ziff E. B.; Basilico C. Expression of the K-fgf protooncogene is repressed during differentiation of F9 cells. Oncogene Res. 5:31–37; 1989. [PubMed] [Google Scholar]
- 83. Wilder P. J.; Kelly D.; Brigman K.; Peterson C. L.; Nowling T.; Gao Q. S.; McComb R. D.; Capecchi M. R.; Rizzino A. Inactivation of the FGF-4 gene in embryonic stem cells alters the growth and/or the survival of their early differentiated progeny. Dev. Biol. 192:614–629; 1997. [DOI] [PubMed] [Google Scholar]
- 84. Wright K. L.; Moore T. L.; Vilen B. J.; Brown A. M.; Ting J. P. Major histocompatibility complex class II-associated invariant chain gene expression is up-regulated by cooperative interactions of Sp1 and NF-Y. J. Biol. Chem. 270:20978–20986; 1995. [DOI] [PubMed] [Google Scholar]
- 85. Xu Y.; Banville D.; Zhao H. F.; Zhao X.; Shen S. H. Transcriptional activity of the SHP-1 gene in MCF7 cells is differentially regulated by binding of NF-Y factor to two distinct CCAAT-elements. Gene 269:141–153; 2001. [DOI] [PubMed] [Google Scholar]
- 86. Yoshida T.; Muramatsu H.; Muramatsu T.; Sakamoto H.; Katoh O.; Sugimura T.; Terada M. Differential expression of two homologous and clustered oncogenes, Hst1 and Int-2, during differentiation of F9 cells. Biochem. Biophys. Res. Commun. 157:618–625; 1988. [DOI] [PubMed] [Google Scholar]
- 87. Yuan H.; Corbi N.; Basilico C.; Dailey L. Developmental-specific activity of the FGF-4 enhancer requires the synergistic action of Sox2 and Oct-3. Genes Dev. 9:2635–2645; 1995. [DOI] [PubMed] [Google Scholar]
- 88. Yuan W.; Condorelli G.; Caruso M.; Felsani A.; Giordano A. Human p300 protein is a coactivator for the transcription factor MyoD. J. Biol. Chem. 271:9009–9013; 1996. [DOI] [PubMed] [Google Scholar]
- 89. Yun J.; Chae H. D.; Choy H. E.; Chung J.; Yoo H. S.; Han M. H.; Shin D. Y. p53 negatively regulates cdc2 transcription via the CCAAT-binding NF-Y transcription factor. J. Biol. Chem. 274:29677–29682; 1999. [DOI] [PubMed] [Google Scholar]
- 90. Zhong H.; May M. J.; Jimi E.; Ghosh S. The phos-phorylation status of nuclear NF-kappa B determines its association with CBP/p300 or HDAC-1. Mol. Cell 9:625–636; 2002. [DOI] [PubMed] [Google Scholar]
- 91. Zwicker J.; Gross C.; Lucibello F. C.; Truss M.; Ehlert F.; Engeland K.; Muller R. Cell cycle regulation of cdc25C transcription is mediated by the periodic repression of the glutamine-rich activators NF-Y and Sp1. Nucleic Acids Res. 23:3822–3830; 1995. [DOI] [PMC free article] [PubMed] [Google Scholar]