

Abstract
The subcellular localization of the αNAC coactivator is regulated, but the signaling pathways controlling its nucleocytoplasmic shuttling and coactivation function are not completely characterized. We report here that casein kinase II (CK2) phosphorylated αNAC on several phosphoacceptor sites, especially in an amino-terminal cluster. Deletion or mutation of the clustered CK2 sites induced nuclear accumulation of αNAC in cells. αNAC also localized to the nucleus when endogenous CK2 activity was inhibited by quercetin or 5,6-dichloro-1-β-d-ribofuranosylbenzimidazole (DRB). These observations suggested that phosphorylation by CK2 might play a signaling role in the nuclear export of αNAC. Interestingly, inhibition of the chromosome region maintenance 1 (CRM1) exportin by leptomycin B (LMB) led to accumulation of αNAC in the nucleus. We conclude that CK2 phosphorylation of the N-terminal cluster corresponds to the signal for αNAC’s nuclear export via a CRM1-dependent pathway. Finally, the nuclear accumulation of the protein resulting from the lack of CK2 phosphorylation mediated a slight but significant increase of the αNAC coactivating function on AP-1 transcriptional activity. Thus, αNAC’s exit from the nucleus and capacity to potentiate transcription appear dependent on its phosphorylation status.
Key words: αNAC, Casein kinase II (CK2), c-Jun, CRM1, Leptomycin B, Nuclear export
THE nascent polypeptide associated complex and coactivator alpha (αNAC) gene was first identified as a modulator of translation (46) and purified as a heterodimer with βNAC/BTF3b, previously identified as a transcriptional factor in yeast (19) and higher eukaryotes (52). Moreover, α- and βNAC subunits were shown to enter the nucleus in yeast (13). We characterized the αNAC subunit as being a transcriptional coactivator of the chimeric Gal4-VP16 activator and of c-Jun homodimers in vivo (32,50). αNAC also interacts with the general transcription factor TBP (TATA binding protein) (50). αNAC promotes the interaction between transcription factors bound to DNA and the basal transcriptional machinery, therefore stabilizing the transcription factors on DNA and resulting in enhanced transcription rates. To exert its co-activation function, αNAC must enter the nucleus and we have found that the subcellular localization of the protein is regulated through differential phosphorylation. Indeed, αNAC is a substrate of the integrin-linked kinase (ILK) (37) and of glycogen synthase kinase 3β (GSK3β) (36). Our current model for the potentiation of c-Jun transcription by αNAC is the following: the constitutive phosphorylation of αNAC on residue Thr159 by GSK3β targets the coactivator for degradation by the proteasome (36). The inactivation of GSK3β in response to adhesion and ILK activation (9) would then result in a Thr159-hypophosphorylated αNAC that would become unavailable for proteasome degradation, but would become a substrate for the ILK activity on residue Ser43. The Ser43-phosphorylated αNAC would preferentially interact with c-Jun (21), translocate to the nucleus, and potentiate transcription (32). This model hypothesizes that αNAC must subsequently be exported from the nucleus, and we surmised that this export step could also be modulated by posttranslational modifications. In this report, we further examined the regulation of the subcellular localization of αNAC by one of the major kinases in the cell, casein kinase II (CK2).
CK2 is a constitutively active serine/threonine protein kinase with an α2β2 tetrameric structure. It is found in both the cytoplasm and the nucleus, but was reported to localize predominantly to the nucleus (31). The β subunit is thought to have regulatory properties whereas the α and α′ subunits are catalytic and interact with DNA (10). The DNA binding of the α catalytic subunits in the nucleus abolishes the catalytic activity of the nuclear form of CK2. This loss of DNA binding was observed with some CK2 inhibitors but not with the flavonoid inhibitor quercetin (15). CK2 interacts and phosphorylates a number of proteins such as DNA topoisomerase II (5,8,43), IκBα (2,29,40), as well as transcription factors such as p53 (12,30,31) and c-Jun (28). It also regulates the nuclear import (20,23,48) or export (35) of molecules, and their transcriptional activities (22,24,34,41).
Protein motifs such as nuclear localization signals and nuclear export signals (NES) are involved in the entrance and exit of proteins to and from the nucleus, respectively. This nucleocytoplasmic shuttling of molecules through the nuclear pore complex is energy dependent and requires transporter molecules named importins and exportins [reviewed in (16,33,42)]. Chromosome region maintenance 1 (CRM1) is an export receptor, also named exportin, that binds tightly to the nuclear pore complex. The first evidence of CRM1 as an exportin was shown by using the CRM1 inhibitor leptomycin B (LMB), which blocks the nuclear export of HIV-Rev (47). CRM1 mediates the export of NES-containing proteins (14) and LMB is a useful tool for the identification of CRM1 export substrates (18,27).
In this study, we demonstrated that residues serine 25, 29, 34, and threonine 27, which are conserved among vertebrate αNAC proteins, constitute the major CK2 phosphoacceptor sites both in vitro and in vivo. We then identified these CK2 sites as modulators of the nuclear export of αNAC, via the CRM1-dependent export pathway. We also studied the effects of phosphorylation by CK2 on the αNAC coactivating function and demonstrated that the transcriptional coactivator activity of αNAC was dependent upon phosphorylation by CK2 at the N-terminal phosphoacceptor sites.
MATERIALS AND METHODS
Plasmids and Constructs (Subcloning Details and Vector Maps Available on Request)
The FLAG epitope was inserted into the pSI mammalian expression vector (Promega, Madison, WI) to give the pSI-Flag plasmid. The cDNAs encoding wild-type (wt) or mutated αNAC (see figure legends) were inserted in-frame into pSI-Flag to yield the pSI-NAC-Flag expression vectors.
Full-length αNAC (wt) and mutants cDNAs were subcloned in-frame at their C-termini with the Intein-Chitin binding domain of the pTYB2 expression vector (NEB, Mississauga, ON) to give pTYB2-NAC plasmids.
Cell Culture and Transfection
COS-7 African green monkey kidney cells were maintained in low-glucose DMEM supplemented with 10% fetal bovine serum at 37°C in 5% CO2. All transient transfections were performed using 5 μl/μg of DNA of the GenePorter transfection reagent, according to the manufacturer’s procedure (Gene Therapy System, San Diego, CA).
For immunoprecipitation, COS-7 cells were plated at 3 × 105 cells/60-mm plate, and transiently transfected with 6 μg of pSI-NAC-Flag or mutant. For immunocytochemistry, COS-7 cells were plated at 1.2 × 105 cells/35-mm plate, on gelatin-coated coverslips, and transiently transfected with 0.4 μg of pSI-NAC-Flag, or mutant construct, and 1.6 μg of pBlueScript (Stratagene, La Jolla, CA) or 2 μg of pBlueScript as a control.
Metabolic Labeling
At 48 h posttransfection, the pSI-NAC-Flag transfected COS-7 cells were treated for 1 h with the CK2 inhibitors, quercetin (Sigma, Oakville, ON), at 200 μM, 5,6-dichloro-1-β-d-ribofuranosylbenzimidazole (DRB) (Calbiochem, San Diego, CA), at 180 μM, or vehicle (ethanol) followed by permeabilization with 0.6 U/ml Streptolysin O (Sigma) and labeling for 1 h with 50 μCi of [γ-32P]ATP (Amersham-Pharmacia Canada, Baie d’Urfé, QC), as described by Carter (6), in the presence of inhibitors where indicated in figure legends. The cells were harvested by scraping in 2× lysis buffer (100 mM Tris-Cl, pH 7.4, 300 mM NaCl, 2 mM EDTA, 2 mM EGTA, 2% Triton X-100, 2 mM β-glycerophosphate, 2 mM orthovanadate, 5 mM sodium pyrophosphate) supplemented with 5 μg/ml leupeptin, aprotinin, and pepstatin A and 1 mM PMSF. After centrifugation to pellet cell debris, samples were diluted with H2O to reach 1× lysis buffer and used for immunoprecipitation.
Immunoprecipitation of Flag-Tagged Proteins
The radiolabeled cell lysates were incubated overnight at 4°C with preequilibrated anti-Flag M2 affinity gel according to the manufacturer’s instructions (Sigma). The affinity gel-purified proteins were extensively washed in 1× lysis buffer and resuspended in SDS sample buffer in the absence of DTT. Immunoprecipitates were run on 12% SDS-PAGE. The gel was subsequently dried and exposed for 3 days at −80°C. The intensity of the signals was quantified with the Typhoon PhosphorImager (Amersham-Pharmacia). Protein expression levels were controlled by Western blotting with the anti-NAC antibody (51). The signal was revealed with the ECL+Plus kit (Amersham-Pharmacia) and quantified with the Typhoon PhosphorImager.
Immunocytochemistry
At 24 h posttransfection, the cells transfected with pSI-NAC-Flag wt or mutants were treated for 2 h with quercetin at 200 μM, DRB at 180 μM, LMB at 20 and 50 ng/ml (Dr. M. Yoshida), or the corresponding vehicle where indicated in figure legends. After treatment, the cells were fixed in 4% paraformaldehyde, permeabilized with 0.2% Triton X-100, and the endogenous peroxidase activity was quenched with 1% H2O2. Following blocking with 1% Blocking Reagent (Roche Molecular Biochemicals, Laval, QC) supplemented with 0.2% Tween-20, the cells were incubated with the anti-αNAC antibody (51) or the anti-Flag M2 antibody (Sigma). Secondary antibodies were either a fluorescein isothiocyanate conjugated anti-rabbit IgG antibody or a biotinylated secondary anti-mouse IgG antibody (Vector Lab. Inc., Burlingame, CA). After washes, the antigens were revealed by indirect immunofluorescence or using the avidin biotin peroxidase reagent (Vector Lab. Inc.). The peroxidase staining was revealed with DAB reagent. Results were visualized on a Leica DM-R microscope.
In Vitro Protein Kinase Assays
Recombinant CK2 protein kinase was purchased from NEB. The recombinant proteins, from pTYB2-NAC plasmids, were produced and purified in E. coli following the manufacturer’s procedure (NEB). For in vitro kinase assays, 2 μg of the recombinant proteins was incubated for 15 min at 30°C in CK2 buffer (20 mM Tris-Cl, 50 mM KCl, 10 mM MgCl2, 200 μM ATP, pH 7.5) with 250 units of recombinant CK2 and 5 μCi of [γ-32P]ATP, then resolved by SDS-PAGE on a 12% gel. The dried gel was autoradio-graphed on X-AR film (Kodak, Rochester, NY).
Luciferase Assays
COS-7 cells were seeded at 1.2 × 105 cells/well in six-well plates and transiently transfected the following day using 6 μl/well of the Lipofectamine reagent (Invitrogen Canada Inc., Burlington, ON). Transfections used 300 ng of expression vectors for wild-type αNAC or point mutants as indicated in figure legends, and 300 ng of the pCI-c-Jun expression plasmid (38). The reporter vector (100 ng) was mmp-9 pGL3, which contains the proximal 670 bp of the mmp-9 gene promoter driving luciferase (17). Variations in transfection efficiency were monitored with 40 ng of the pSV6tkCAT reporter (7). The total amount of DNA was completed at 2 μg using the inert pBlue-Script plasmid (Stratagene). Cells were maintained in 0.5% serum throughout and lysates were prepared in the reporter gene assay lysis buffer (Roche Molecular Biochemicals, Laval, QC) 48 h posttransfection. Cell lysate (100 μl) was used for single luciferase reporter assays following the manufacturer’s instructions (Promega). Luciferase activity was measured with a Monolight 2010 luminometer (Analytical Luminescence Laboratory, San Diego, CA). Relative light units were normalized to CAT expression assayed by the CAT Elisa system (Roche Diagnostics, Indianapolis, IN). Under the conditions used, wt αNAC reproducibly increased the transcription of the AP-1-dependent reporter by twofold over the level achieved with c-Jun alone, for an overall induction of sixfold (data not shown). This level of αNAC activity was set at 100% for comparison with the mutant recombinant proteins. Results are mean ± SEM of seven experiments performed in triplicate. Statistical analysis was performed using ANOVA and the Dunnett’s posttest. A value of p < 0.05 was accepted as significant.
RESULTS
αNAC Is a Phosphoprotein In Vivo
The αNAC protein is remarkably conserved among species, and is rich in serine and threonine residues. Several putative phosphorylation sites for known protein kinases (CK2, PKC, PKA) can be identified within the αNAC’s amino acid sequence (Fig. 1A). These 14 putative phosphoacceptor sites were conserved throughout evolution, raising the interesting possibility that they could be targeted by common mechanisms of phosphorylation.
Figure 1.
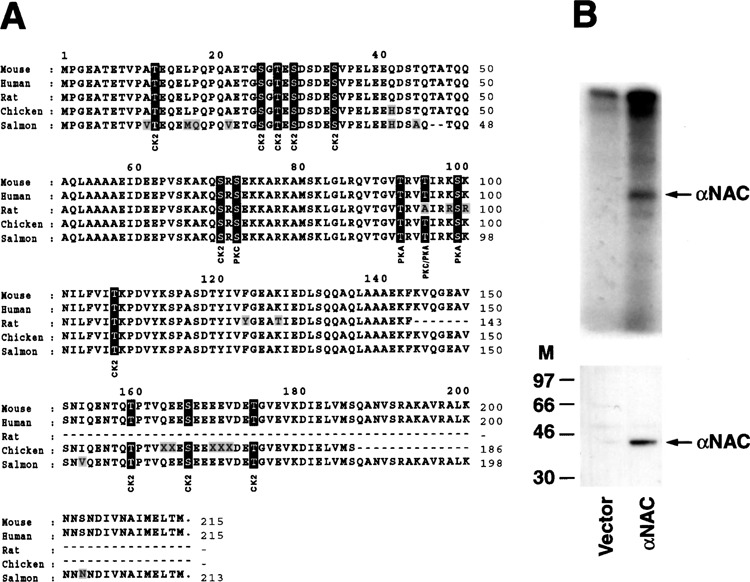
αNAC is a phosphoprotein. (A) Amino acid sequence comparison of the αNAC proteins in vertebrate. αNAC from rat, chicken, and salmon are putative proteins. Amino acid residues 1–215 of mouse αNAC were aligned with αNAC proteins from human, rat, chicken, and salmon using the Clustal algorithm (MegAlign, DNASTAR Inc., Madison, WI). From human to salmon, a 99% to 83% identity was observed between the αNAC proteins. Nonconserved residues are shaded and conserved putative phosphoacceptor sites are boxed. Consensus sites for PKC, PKA, and CK2 are X-S/T-X-R/K, R-X1-2-S/T-X, and X-S/T-X-X-D/E, respectively. (B) αNAC is phosphorylated in COS-7 cells. COS-7 cells, transfected with pSI-NAC-Flag or pBlueScript (vector), were labeled with [γ-32P]ATP, and cellular extracts were immunoprecipitated with anti-Flag beads. Immunoprecipitates were analyzed by immunoblotting with the anti-NAC serum (lower panel) or by autoradiography (upper panel). M, molecular size markers in kDa.
To investigate the phosphorylation status of αNAC, we transfected COS-7 cells with an expression vector for a Flag epitope-tagged αNAC fusion and performed metabolic labeling of the intact cells with [γ-32P]ATP after permeabilization (6). Labeling of phosphoproteins in permeabilized, intact cells is a powerful experimental approach to study protein kinase-catalyzed phosphorylation reactions and identify relevant substrates (6). Radiolabeled cellular lysates were immunoprecipitated with the anti-Flag M2 affinity gel. Autoradiography indicated that the recombinant αNAC protein was expressed and phosphorylated in COS-7 cells (Fig. 1B, lower and upper panels). The immunoprecipitation reaction was specific as anti-GST failed to precipitate labeled proteins (data not shown).
Residues 25, 27, 29, and 34 Are the Major αNAC Phosphorylation Sites
The data presented in Figure 1 confirmed that αNAC is a phosphoprotein in vivo. As 10 out of the 14 putative phosphoacceptor sites are fitting the S/TXXD/E consensus CK2 site, and were conserved in αNAC (Fig. 1A), we examined whether αNAC was a substrate for CK2, one of the major protein kinases in the cell. Several αNAC deletion and point mutants were engineered to determine which residues could serve as phosphoacceptor sites (Fig. 2A).
Figure 2.
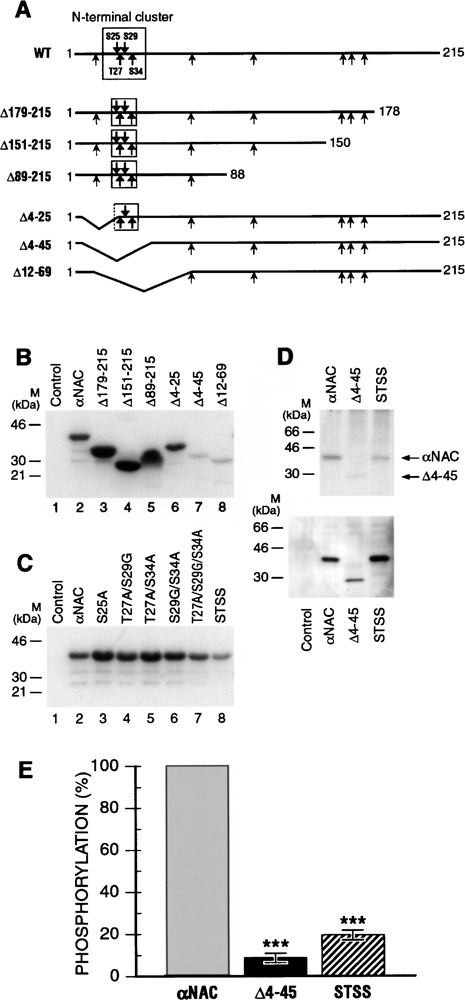
CK2 phosphorylates αNAC in vitro and in vivo. (A) Schematic representation of αNAC illustrating potential CK2 phosphorylation sites. Shown is a schematic diagram of αNAC illustrating putative CK2 residues (arrow), and location of the various serine/threonine-to-alanine/glycine substitutions (bold arrows). The N-terminal and C-terminal deletion mutants of αNAC utilized in this study are also represented. (B) CK2 phosphorylates the αNAC N-terminus in vitro. In vitro CK2 protein kinase assay was performed with recombinant wild-type and deleted mutants of αNAC. The majority of the functional CK2 sites are located in the N-terminal end of the molecule. (C) Identification of the CK2 phosphoacceptor sites cluster. CK2 protein kinase assay was performed with wild-type and point mutants of αNAC. αNAC molecule is especially phosphorylated in the STSS phosphoacceptor sites motif (STSS: quadruple mutant S25A/T27A/S29G/ S34A). (D) CK2 phosphorylates αNAC in vivo. COS-7 cells were transfected with Flag-tagged wt or mutated αNAC expression vectors or pBlueScript (control). After labeling, the cell lysates were immunoprecipitated with anti-Flag beads and analyzed by autoradiography (upper panel) or by immunoblotting with anti-NAC antibody (lower panel). αNAC phosphorylation status was altered by deletion and mutation of the N-terminal cluster of CK2 phosphoacceptor sites. (E) Quantification of the phosphorylation level. The phosphorylation levels of αNAC wt and mutants were quantified with the Typhoon PhosphorImager and wt αNAC level was set at 100%. A significant decrease was observed after deletion (Δ4–45) or mutation (STSS) of the CK2 phosphoacceptor sites. ***p < 0.001.
We produced and purified these proteins in E. coli and the recombinant proteins were used for in vitro protein kinase assays with recombinant CK2 enzyme. These assays revealed that CK2 phosphoacceptor sites were present throughout the protein, but especially in a cluster at the N-terminal end of the molecule, the 4–45 region (Fig. 2B). The threonine and serine residues contained in this cluster were mutated individually or together into alanine or glycine residues. Mutant recombinant proteins were produced, purified, and tested in the in vitro kinase assay with recombinant CK2. The αNAC mutants were differentially affected in their phosphorylation status, and the quadruple mutant (S25A/T27A/S29G/S34A, hereafter referred to as STSS) generated the weakest signal (Fig. 2C). These experiments revealed that the four CK2 phosphoacceptor sites present in the N-terminal cluster were functional in vitro and represented the bulk of CK2 phosphorylation within the molecule.
We then examined the in vivo phosphorylation status of the Δ4–45 and STSS mutant proteins by metabolic labeling, and compared them to that of wt αNAC protein (Fig. 2D). The STSS mutant protein generated a much fainter radioactive signal than wt αNAC. The immunoprecipitated Δ4–45 protein was still phosphorylated, but to a lower level than the STSS protein because it was barely detectable after 32P labeling (Fig. 2D, upper panel). These data indicated that αNAC was phosphorylated by CK2 in vivo and that the N-terminal cluster of CK2 sites was functional in the cells. Immunoblotting performed with anti-NAC antibody (Fig. 2D, lower panel) was used as a control of protein expression to quantify the signals. Quantification performed with a Typhoon PhosphorImager revealed that 80% of the labeled phosphorylation signal was abolished in the STSS mutant (Fig. 2E). The Δ4–45 mutant represented 9% of the wild-type signal, suggesting the presence of additional phosphoacceptor site(s) in this region.
Deletion of the CK2 Phosphoacceptor Sites Leads to Nuclear Accumulation of αNAC
CK2 is one of the kinases known to regulate the nucleocytoplasmic shuttling of proteins. To investigate the role of CK2 phosphorylation on αNAC subcellular localization, we used some of the deletion or site-specific mutants described in Figure 2A. Transient transfections were performed in COS-7 cells with wt or mutated αNAC and the proteins were visualized with the anti-αNAC antibody (51) (Fig. 3). Immunodetection of steady-state expression patterns revealed that wt αNAC had a cytoplasmic and perinuclear expression pattern, but some nuclear staining could also be detected under steady-state conditions (Fig. 3A). In contrast, deletion of the N-terminal end of the protein (mutant Δ4–45) triggered the accumulation of the mutant protein in the nuclei of the cells (Fig. 3B). Identical localization patterns were observed in C1 osteogenic cells (25) (data not shown).
Figure 3.
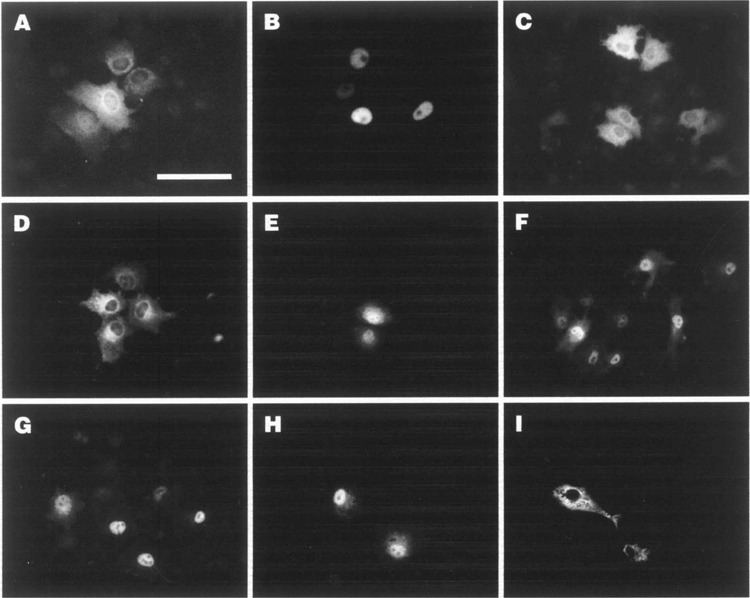
Nuclear accumulation of αNAC-Flag proteins after deletion or mutation of the N-terminal CK2 sites. COS-7 cells were transfected with expression vectors for Flag-tagged wt, deleted, or point mutated forms of αNAC and the overexpressed proteins were detected by indirect immunofluorescence using anti-αNAC antibody. Representative signals are shown. (A) αNAC; (B) Δ4–45; (C) T27A; (D) S29G; (E) S25A; (F) S34A; (G) STSS; (H) S25A/S34A; (I) S25E/S34E. Bar: 100 μM.
We then examined the effects of serine/threonine-to-alanine/glycine substitutions at the CK2 sites on the nuclear accumulation of αNAC (Fig. 3C–I). A wild-type pattern was observed with the T27A mutant (Fig. 3C). As well, the S29G point mutant showed wild-type, cytoplasmic localization patterns (Fig. 3D). Substantial nuclear staining was clearly observed when the serine 25 or serine 34 residues were replaced by alanine residues, but some cytoplasmic staining remained evident (Fig. 3E and F, respectively). The quadruple substitution point mutant STSS, in which the four CK2 sites were mutated, exhibited the highest level of nuclear staining (Fig. 3G). When residues serine 25 and serine 34 were jointly mutated to nonphosphorylable alanine residues (S25A/S34A double mutant), the mutant protein predominantly localized to the nucleus (Fig. 3H). Interestingly, when these residues were mutated to glutamic acid to mimic phosphorylation (S25E/S34E double mutant), the recombinant protein remained cytosolic (Fig. 3I) Thus, the nuclear accumulation of the serine/threonine mutants highlighted a possible important role of the CK2 protein kinase in the regulation of αNAC subcellular localization.
The Inhibition of CK2 Activity Results in αNAC Nuclear Accumulation
CK2 is predominantly present in the nucleus of cells (31) and the flavonoid inhibitor quercetin is known to block its activity (15,39). DRB is also a cell-permeable CK2 inhibitor (10,26). COS-7 cells were transiently transfected with wt αNAC fused to the Flag epitope, then treated with quercetin or DRB for 2 h. After metabolic labeling, the overexpressed proteins were immunoprecipitated with the anti-Flag M2 beads and the signal was analyzed by autoradiography and quantified with a PhosphorImager. Levels of expression of the transfected αNAC protein were normalized by Western blotting (not shown). Inhibition of the endogenous CK2 activity by quercetin induced a significant reduction (55%) of the phosphorylation level of the overexpressed molecule (Fig. 4A). A similar degree of inhibition (60%) was observed following treatment of cells with a different CK2 inhibitor, DRB (Fig. 4A). The reduction in αNAC phosphorylation in vivo following treatment of cells with two different inhibitors of CK2 demonstrated that αNAC was an in vivo substrate of CK2.
Figure 4.
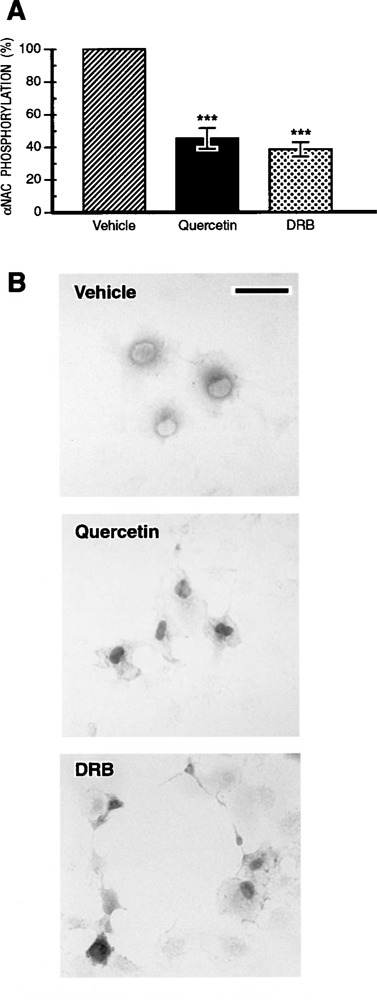
Nuclear accumulation of αNAC after CK2 inhibition. (A) Quercetin or DRB treatment inhibits αNAC phosphorylation in cells. COS-7 cells transfected with pSI-NAC-Flag were treated for 2 h with the CK2 inhibitors, quercetin or DRB, or vehicle, and labeled with [γ-32P]ATP. Cellular extracts were immunoprecipitated with anti-Flag beads. Immunoprecipitates were analyzed by autoradiography and quantified by PhosphorImager analysis. Protein expression was normalized by Western blotting (not shown). ***p < 0.001. (B) Immunocytochemistry was performed in COS-7 cells transiently transfected with the pSI-NAC-Flag expression plasmid. Twenty-four hours posttransfection, the cells were treated for 2 h with vehicle (upper panel), quercetin (middle panel), or DRB (lower panel) and the subcellular localization of αNAC-Flag proteins was revealed after immunostaining with the anti-Flag M2 antibody. Inhibition of the CK2 endogenous activity induced a nuclear accumulation of αNAC in COS-7 cells. Bar: 50 μM.
We further used the CK2 inhibitors quercetin and DRB to investigate the role of CK2 in the control of αNAC subcellular localization (Fig. 4B). The COS-7 cells were transiently transfected with wt αNAC-Flag vector, then treated with quercetin (middle panel), DRB (lower panel), or vehicle (upper panel) for 2 h. Immunocytochemistry was performed with the anti-Flag M2 antibody. These experiments revealed a nuclear accumulation of the recombinant αNAC protein after treatment with quercetin (Fig. 4B, middle panel) or DRB (Fig. 4B, lower panel). Only the patterns observed at the highest doses of inhibitor used are shown, but a progressive, dose-dependent effect was observed for both inhibitors (not shown). Identical results were obtained when C1 osteogenic cells (25) were treated with the inhibitors (not shown). These data demonstrated that the inhibition of CK2 activity induced the nuclear accumulation of αNAC. This inhibition of CK2 activity mimicked the effect of the deletion and the mutation of the N-terminal cluster of CK2 phosphoacceptor sites (Fig. 3). Furthermore, the effect of CK2 inhibitor treatment on αNAC localization in vivo suggested that CK2 controlled the nuclear export of αNAC and thus that phosphorylated serine and threonine residues represented the signal for nuclear export of the molecule.
In Vivo Export of αNAC From the Nucleus
CRM1 is a transporter molecule that exports NES-containing proteins through the nuclear pore complex, and leptomycin B (LMB) is known to specifically inhibit this pathway (27). We investigated whether the CK2-mediated export of αNAC from the nucleus was dependent on the CRM1 pathway. For that purpose COS-7 cells were transiently transfected with the Flag-fused αNAC expression vector and the cells were subsequently treated with LMB 24 h post-transfection. After 2 h of treatment, immunocytochemistry was performed with the anti-αNAC antibody (Fig. 5). Immunostaining revealed that LMB treatment led to the accumulation of αNAC into the nucleus in a dose-dependent manner (Fig. 5). This treatment mimicked the pattern of αNAC localization obtained after deletion or mutation of the CK2 sites (Δ4–45 and STSS), or after quercetin or DRB treatment. We conclude that in vivo phosphorylation of the N-terminal CK2 cluster was the signal for the nuclear export of αNAC and that this export was mediated by the CRM1-dependent pathway.
Figure 5.
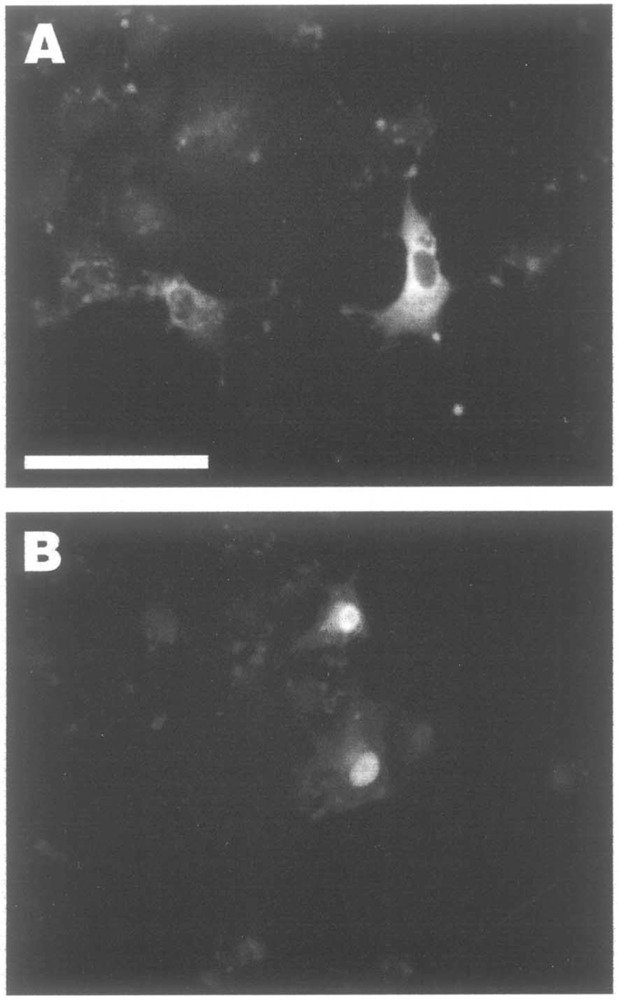
Nuclear export of αNAC by the CRM1-dependent export pathway. Twenty-four hours after transfection with pSI-NAC-Flag, COS-7 cells were treated for 2 h with leptomycin B (LMB), an inhibitor of the CRM1-dependent export pathway, at 20 (A) or 50 ng/ml (B). Bar: 100 μM.
Differential Phosphorylation of the N-Terminal Cluster Modulates αNAC Coactivation Potency
αNAC potentiates the activity of c-Jun homodimers in vivo (32,36,38). Moreover, we demonstrated that the absence of CK2 phosphorylation at the N-terminal end of αNAC mediated its nuclear accumulation. We thus examined whether CK2 phosphorylation could modulate the coactivating function of αNAC. COS-7 cells were transiently cotransfected with an AP-1-dependent reporter plasmid (mmp-9 pGL3) (17) and expression vectors for c-Jun and αNAC. Luciferase reporter assays were performed, and results obtained with wt αNAC or the CK2 phosphoacceptor sites point mutants were compared (Fig. 6). In these experiments, the level of wt αNAC activity was set at 100% for comparison with the mutant recombinant proteins. The experiments were performed seven times in triplicate, and comparable protein expression levels for αNAC and c-Jun were measured using immunoblotting (data not shown).
Figure 6.
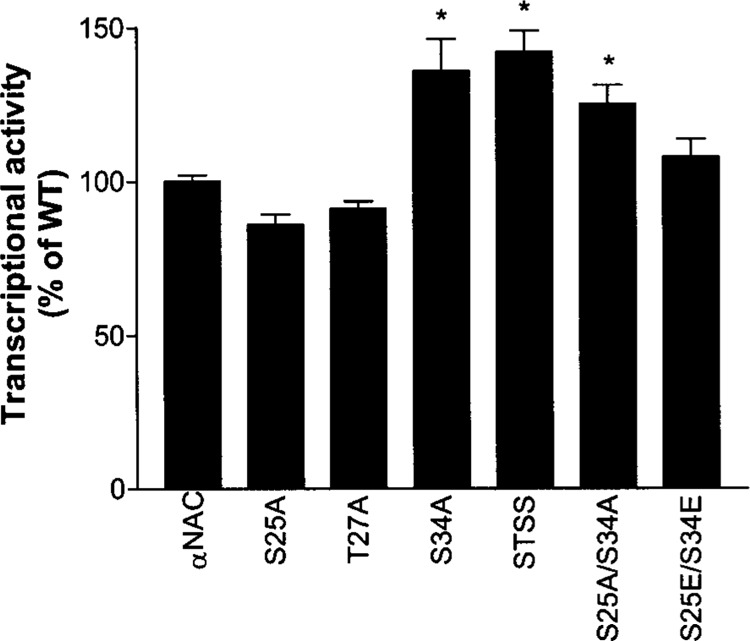
Mutation of the N-terminal CK2 sites modulates αNAC coactivation potency. COS-7 cells were transfected with expression vectors for wild-type (wt) αNAC, or S25A, T27A, S34A, STSS, S25A/S34A, or S25E/S34E mutants, and activator and reporter plasmids as described in Materials and Methods. Single luciferase assays were performed 48 h posttransfection. The expression of the reporter gene measured in the presence of c-Jun and wt αNAC was ascribed a value of 100%. Statistical analysis was performed using ANOVA and the Dunnett’s posttest. *p < 0.05.
The single point mutants S25A and T27A had coactivation potency comparable to wt αNAC. Mutation of residue Ser34 to a nonphosphorylable amino acid, which results in nuclear accumulation (Fig. 3F), led to a significant increase in the coactivation potency of the recombinant protein (Fig. 6). The compound mutants STSS and S25A/S34A that localize to the nucleus (Fig. 3G and H) also displayed increased coactivation activity (Fig. 6). Mutant S25E/S34E had coactivation potency comparable to wt αNAC. These data suggest that differential phosphorylation of αNAC at the N-terminal cluster regulates the ability of αNAC to potentiate AP-1-dependent gene transcription.
DISCUSSION
Although it was established that αNAC is a transcriptional coactivator (32,36,38,50) and that it enters the nucleus (13,36), the precise mechanisms and the identity of the functionally important amino acid residues involved in the regulation of its subcellular localization are just beginning to be unraveled. Accordingly, the central goal of this study was to identify specific signals involved in the regulation of αNAC nuclear export and the protein kinase involved in directly regulating this function.
The sequence of the αNAC protein was highly conserved through evolution, especially in vertebrates, with more than 80% of identity. The C-terminal part of the molecule is the most conserved among species, whereas the N-terminus differs between higher and lower eukaryotes. The acquisition of this N-terminus throughout evolution could correspond to an evolutionary switch emphasizing a different, or more controlled, role of αNAC in vertebrates. Especially, the lack of the highly controlled N-terminus of the molecule in lower eukaryotes such as yeasts could explain the differences observed to date in the functions of αNAC (32,38,45).
As a result of our study, we first identified CK2 as an essential regulator of αNAC subcellular localization. αNAC contains several potential CK2 phosphoacceptor sites in its sequence. Remarkably, the majority of the functional CK2 phosphorylation sites were located in a small cluster at the N-terminus of the molecule. αNAC proteins bearing S/T-to-A/G mutations of the residues from that cluster were phosphorylated less intensely in vitro and in vivo. It appeared that more than one amino acid residue was involved in the regulation of αNAC nuclear localization because the single substitution mutants, S25A and S34A, were not completely localized to the nucleus and still exhibited residual cytoplasmic staining, when compared to the quadruple STSS mutant (Fig. 3). While single alanine/glycine substitution of Thr27 and Ser29 residues alone was without apparent effect on the localization of the protein, the alanine/glycine substitutions of the four (STSS) CK2 phosphoacceptor residues caused a significant nuclear accumulation of αNAC. Mutating two residues of the cluster to glutamic acid residues (mutant S25E/S34E), thus mimicking the effect of phosphorylation, excluded the protein from the nucleus. These data support the conclusion that differential phosphorylation of αNAC by nuclear CK2 regulates the subcellular localization of the coactivator.
Results of metabolic labeling experiments using the Δ4–45 deletion mutant demonstrated that most of the CK2 phosphoacceptor sites are located in the N-terminus of the protein, between residues 4 and 45 (Fig. 2B). Within this domain, five putative phosphoacceptor sites match the CK2 consensus sequence (Fig. 1A). We have mutated each of the five residues, alone and in various combinations. While computer-based analysis identifies Thr12 as a putative CK2 site, it calculates a very low probability for this residue to be a functional site (calculated probability: 0.102). The T12A mutant had wild-type subcellular localization and wild-type coactivating potency (data not shown). Many of the remaining putative phosphoacceptor sites at the C-terminal end were also mutated or deleted without effect on the localization of the protein (data not shown). These observations strongly suggest that the N-terminal cluster of CK2 phosphoacceptor sites we have characterized is the major motif controlling subcellular localization and coactivating activity of αNAC.
The in vitro phosphorylation data raised the possibility that the CK2 protein kinase might be involved in the regulation of αNAC nuclear localization in vivo. We found that inhibition of the endogenous CK2 activity in vivo by the inhibitors quercetin or DRB was sufficient to trigger a nuclear accumulation of αNAC. But how does the phosphorylation status of these serine/threonine residues regulate the subcellular localization of αNAC? One possibility is that the phosphorylation status of these amino acid residues directly affects protein conformation, which in turn regulates the access of αNAC to the nuclear export machinery. The use of leptomycin B, a specific CRM1–nuclear export pathway inhibitor, has provided evidence that αNAC nuclear shuttling was dependent on this export pathway.
Subcellular compartmentalization of αNAC appears to play a major role in the regulation of αNAC function. We have recently shown that αNAC must be phosphorylated by the integrin-linked kinase to enter the nucleus and exert its full coactivating function (37). Similarly, phosphorylation by GSK3β controls the half-life of αNAC (36) and mutation of the GSK3β phosphoacceptor site leads to increased co-activation potency (36). We have found in this study that CK2 phosphoacceptor sites point mutants that accumulate in the nucleus also have increased coactivating activity (Figs. 3 and 6). This observation did not extend to the S25A mutant, perhaps due to the nature of the amino acid substitution that we engineered. Additional substitutions will be required to fully decipher the impact of differential phosphorylation of αNAC by CK2 at residue Ser25.
The CK2 phosphorylated form of αNAC is rapidly sent out of the nucleus by the CRM1-dependent export pathway. The rapid shuttling of αNAC may underlie the way that cells respond to the need for transient AP-1 activation. The intramolecular mechanism of control of the nuclear localization and function that we describe is well suited to allow rapid transit of αNAC between the nucleus and the cytoplasm. Rapid nuclear shuttling allows rapid and transient change in AP-1-dependent gene expression, which is a characteristic of AP-1 responses.
Our findings suggest that a dominant nuclear export over nuclear import contributes to the cytoplasmic localization of αNAC under steady-state conditions. A growing number of proteins have now been shown to undergo regulated nuclear export. In most cases, the mechanism by which export is regulated has not been fully elucidated, but so far many appear to involve phosphorylation (3,4,11,49). Given the number of examples already in the literature, it seems likely that regulation of nuclear export is at least as common as that of nuclear import, and that the steady-state localization of many proteins is largely determined by the relative rates of the two processes.
Once returned to the cytosol, the phosphorylation status of αNAC must be returned to neutral in order for the cycle to initiate again. The residues phosphorylated by CK2 are most certainly dephosphorylated by serine/threonine phosphatase(s) (1,44) that remain to be identified. The identification of these phosphatase enzymes that reverse the effects of the CK2 kinase will constitute an interesting area for future investigation.
ACKNOWLEDGMENTS
This work was supported by a grant from the Shriners of North America. Dr. Minoru Yoshida (University of Tokyo, Japan) kindly provided us with leptomycin B. We used the PhosphorImager from the Centre for Bone and Periodontal Research of McGill University. We thank Mark Lepik and Guylaine Bédard for preparing the figures.
REFERENCES
- 1. Barford D.; Das A. K.; Egloff M. P. The structure and mechanism of protein phosphatases: Insights into catalysis and regulation. Annu. Rev. Biophys. Biomol. Struct. 27:133–164; 1998. [DOI] [PubMed] [Google Scholar]
- 2. Barroga C. F.; Stevenson J. K.; Schwarz E. M.; Verma I. M. Constitutive phosphorylation of I kappa B alpha by casein kinase II. Proc. Natl. Acad. Sci. USA 92:7637–7641; 1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Beals C. R.; Clipstone N. A.; Ho S. N.; Crabtree G. R. Nuclear localization of NF-ATc by a calcineurin-dependent, cyclosporin-sensitive intramolecular interaction. Genes Dev. 11:824–834; 1997. [DOI] [PubMed] [Google Scholar]
- 4. Beals C. R.; Sheridan C. M.; Turck C. W.; Gardner P.; Crabtree G. R. Nuclear export of NF-ATc enhanced by glycogen synthase kinase-3. Science 275:1930–1934; 1997. [DOI] [PubMed] [Google Scholar]
- 5. Cardenas M. E.; Walter R.; Hanna D.; Gasser S. M. Casein kinase II copurifies with yeast DNA topoisomerase II and re-activates the dephosphorylated enzyme. J. Cell Sci. 104:533–543; 1993. [DOI] [PubMed] [Google Scholar]
- 6. Carter N. A. Permeabilization strategies to study protein phosphorylation. In: Ausubel F. M.; Brent R.; Kingston R. E.; Moore D. D.; Seidman J. G.; Smith J. A.; Struhl K., eds. Current protocols in molecular biology 4. New York: John Wiley and Sons; 1997:18.8.1–18.8.19. [Google Scholar]
- 7. Courey A. J.; Tjian R. Analysis of Sp1 in vivo reveals multiple transcriptional domains, including a novel glutamine-rich activation motif. Cell 55:887–898; 1988. [DOI] [PubMed] [Google Scholar]
- 8. Dang Q.; Alghisi G. C.; Gasser S. M. Phosphorylation of the C-terminal domain of yeast topoisomerase II by casein kinase II affects DNA–protein interaction. J. Mol. Biol. 243:10–24; 1994. [DOI] [PubMed] [Google Scholar]
- 9. Delcommenne M.; Tan C.; Gray V.; Rue L.; Woodgett J.; Dedhar S. Phosphoinositide-3-OH kinase-dependent regulation of glycogen synthase kinase 3 and protein kinase B/AKT by the integrin-linked kinase. Proc. Natl. Acad. Sci. USA 95:11211–11216; 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Egyhazi E.; Ossoinak A.; Filhol-Cochet O.; Cochet C.; Pigon A. The binding of the alpha subunit of protein kinase CK2 and RAP74 subunit of TFIIF to protein-coding genes in living cells is DRB sensitive. Mol. Cell. Biochem. 191:149–159; 1999. [PubMed] [Google Scholar]
- 11. Engel K.; Kotlyarov A.; Gaestel M. Leptomycin B-sensitive nuclear export of MAPKAP kinase 2 is regulated by phosphorylation. EMBO J. 17:3363–3371; 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Filhol O.; Baudier J.; Delphin C.; Loue-Mackenbach P.; Chambaz E. M.; Cochet C. Casein kinase II and the tumor suppressor protein P53 associate in a molecular complex that is negatively regulated upon P53 phosphorylation. J. Biol. Chem. 267:20577–20583; 1992. [PubMed] [Google Scholar]
- 13. Franke J.; Reimann B.; Hartmann E.; Kohlerl M.; Wiedmann B. Evidence for a nuclear passage of nascent polypeptide-associated complex subunits in yeast. J. Cell Sci. 114:2641–2648; 2001. [DOI] [PubMed] [Google Scholar]
- 14. Fukuda M.; Asano S.; Nakamura T.; Adachi M.; Yoshida M.; Yanagida M.; Nishida E. CRM1 is responsible for intracellular transport mediated by the nuclear export signal. Nature 390:308–311; 1997. [DOI] [PubMed] [Google Scholar]
- 15. Gatica M.; Jacob G.; Allende C. C.; Allende J. E. DNA inhibits the catalytic activity of the alpha subunit of protein kinase CK2. Biochemistry 34:122–127; 1995. [DOI] [PubMed] [Google Scholar]
- 16. Gorlich D.; Kutay U. Transport between the cell nucleus and the cytoplasm. Annu. Rev. Cell Dev. Biol. 15:607–660; 1999. [DOI] [PubMed] [Google Scholar]
- 17. Gum R.; Lengyel E.; Juarez J.; Chen J. H.; Sato H.; Seiki M.; Boyd D. Stimulation of 92-kDa gelatinase B promoter activity by ras is mitogen-activated protein kinase kinase 1-independent and requires multiple transcription factor binding sites including closely spaced PEA3/ets and AP-1 sequences. J. Biol. Chem. 271:10672–10680; 1996. [DOI] [PubMed] [Google Scholar]
- 18. Henderson B. R.; Eleftheriou A. A comparison of the activity, sequence specificity, and CRM1-dependence of different nuclear export signals. Exp. Cell Res. 256:213–224; 2000. [DOI] [PubMed] [Google Scholar]
- 19. Hu G. Z.; Ronne H. Yeast BTF3 protein is encoded by duplicated genes and inhibits the expression of some genes in vivo. Nucleic Acids Res. 22:2740–2743; 1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hubner S.; Xiao C. Y.; Jans D. A. The protein kinase CK2 site (Ser111/112) enhances recognition of the simian virus 40 large T-antigen nuclear localization sequence by importin. J. Biol. Chem. 272:17191–17195; 1997. [DOI] [PubMed] [Google Scholar]
- 21. Hurtubise M. Interaction of the alphaNAC coactivator with its c-Jun target. M.Sc. thesis, Department of Human Genetics, McGill University; 2003. [Google Scholar]
- 22. Jain N.; Mahendran R.; Philp R.; Guy G. R.; Tan Y. H.; Cao X. Casein kinase II associates with Egr-1 and acts as a negative modulator of its DNA binding and transcription activities in NIH 3T3 cells. J. Biol. Chem. 271:13530–13536; 1996. [DOI] [PubMed] [Google Scholar]
- 23. Jans D. A.; Jans P. Negative charge at the casein kinase II site flanking the nuclear localization signal of the SV40 large T-antigen is mechanistically important for enhanced nuclear import. Oncogene 9:2961–2968; 1994. [PubMed] [Google Scholar]
- 24. Kasahara H.; Izumo S. Identification of the in vivo casein kinase II phosphorylation site within the homeodomain of the cardiac tissue-specifying homeobox gene product Csx/Nkx2.5. Mol. Cell. Biol. 19:526–536; 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kellermann O.; Buc-Caron M. H.; Marie P. J.; Lamblin D.; Jacob F. An immortalized osteogenic cell line derived from mouse teratocarcinoma is able to mineralize in vivo and in vitro. J. Cell Biol. 110:123–132; 1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kim S. J.; Kahn C. R. Insulin regulation of mitogen-activated protein kinase kinase (MEK), mitogen-activated protein kinase and casein kinase in the cell nucleus: A possible role in the regulation of gene expression. Biochem. J. 323:621–627; 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kudo N.; Wolff B.; Sekimoto T.; Schreiner E. P.; Yoneda Y.; Yanagida M.; Horinouchi S.; Yoshida M. Leptomycin B inhibition of signal-mediated nuclear export by direct binding to CRM1. Exp. Cell Res. 242:540–547; 1998. [DOI] [PubMed] [Google Scholar]
- 28. Lin A.; Frost J.; Deng T.; Smeal T.; al-Alawi N.; Kikkawa U.; Hunter T.; Brenner D.; Karin M. Casein kinase II is a negative regulator of c-Jun DNA binding and AP-1 activity. Cell 70:777–789; 1992. [DOI] [PubMed] [Google Scholar]
- 29. McElhinny J. A.; Trushin S. A.; Bren G. D.; Chester N.; Paya C. V. Casein kinase II phosphorylates I kappa B alpha at S-283, S-289, S-293, and T-291 and is required for its degradation. Mol. Cell. Biol. 16:899–906; 1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. McKendrick L.; Milne D.; Meek D. Protein kinase CK2-dependent regulation of p53 function: Evidence that the phosphorylation status of the serine 386 (CK2) site of p53 is constitutive and stable. Mol. Cell. Biochem. 191:187–199; 1999. [PubMed] [Google Scholar]
- 31. Meek D. W.; Simon S.; Kikkawa U.; Eckhart W. The p53 tumour suppressor protein is phosphorylated at serine 389 by casein kinase II. EMBO J. 9:3253–3260; 1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Moreau A.; Yotov W. V.; Glorieux F. H.; St-Arnaud R. Bone-specific expression of the alpha chain of the nascent polypeptide- associated complex, a coactivator potentiating c-Jun-mediated transcription. Mol. Cell. Biol. 18:1312–1321; 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Nakielny S.; Dreyfuss G. Transport of proteins and RNAs in and out of the nucleus. Cell 99:677–690; 1999. [DOI] [PubMed] [Google Scholar]
- 34. Oelgeschlager M.; Krieg J.; Luscher-Firzlaff J. M.; Luscher B. Casein kinase II phosphorylation site mutations in c-Myb affect DNA binding and transcriptional cooperativity with NF-M. Mol. Cell. Biol. 15:5966–5974; 1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Porter C. M.; Havens M. A.; Clipstone N. A. Identification of amino acid residues and protein kinases involved in the regulation of NFATc subcellular localization. J. Biol. Chem. 275:3543–3551; 2000. [DOI] [PubMed] [Google Scholar]
- 36. Quélo I.; Akhouayri O.; Prud’homme J.; St-Arnaud R. GSK3beta-dependent phosphorylation of the alpha-NAC coactivator regulates its nuclear translocation and proteasome-mediated degradation. Biochemistry 43:2906–2914; 2004. [DOI] [PubMed] [Google Scholar]
- 37. Quélo I.; Gauthier C.; Hannigan G. E.; Dedhar S.; St-Arnaud R. Integrin-linked kinase regulates the nuclear entry of the c-Jun coactivator α-NAC and its coactivation potency. J. Biol. Chem. 279:43893–43899; 2004. [DOI] [PubMed] [Google Scholar]
- 38. Queélo I.; Hurtubise M.; St-Arnaud R. αNAC requires an interaction with c-Jun to exert its transcriptional coactivation. Gene Expr. 10:255–262; 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Srivastava A. K. Inhibition of phosphorylase kinase, and tyrosine protein kinase activities by quercetin. Biochem. Biophys. Res. Commun. 131:1–5; 1985. [DOI] [PubMed] [Google Scholar]
- 40. Taylor J. A.; Bren G. D.; Pennington K. N.; Trushin S. A.; Asin S.; Paya C. V. Serine 32 and serine 36 of IkappaBalpha are directly phosphorylated by protein kinase CKII in vitro. J. Mol. Biol. 290:839–850; 1999. [DOI] [PubMed] [Google Scholar]
- 41. Tsutsui H.; Geltinger C.; Murata T.; Itakura K.; Wada T.; Handa H.; Yokoyama K. K. The DNA-binding and transcriptional activities of MAZ, a myc-associated zinc finger protein, are regulated by casein kinase II. Biochem. Biophys. Res. Commun. 262:198–205; 1999. [DOI] [PubMed] [Google Scholar]
- 42. Turpin P.; Ossareh-Nazari B.; Dargemont C. Nuclear transport and transcriptional regulation. FEBS Lett. 452:82–86; 1999. [DOI] [PubMed] [Google Scholar]
- 43. Watabe M.; Nakajo S.; Yoshida T.; Kuroiwa Y.; Nakaya K. Treatment of U937 cells with bufalin induces the translocation of casein kinase 2 and modulates the activity of topoisomerase II prior to the induction of apoptosis. Cell Growth Differ. 8:871–879; 1997. [PubMed] [Google Scholar]
- 44. Wera S.; Hemmings B. A. Serine/threonine protein phosphatases. Biochem. J. 311:17–29; 1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Wiedmann B.; Prehn S. The nascent polypeptide-associated complex (NAC) of yeast functions in the targeting process of ribosomes to the ER membrane [in process citation]. FEBS Lett. 458:51–54; 1999. [DOI] [PubMed] [Google Scholar]
- 46. Wiedmann B.; Sakai H.; Davis T. A.; Wiedmann M. A protein complex required for signal-sequence-specific sorting and translocation. Nature 370:434–440; 1994. [DOI] [PubMed] [Google Scholar]
- 47. Wolff B.; Sanglier J. J.; Wang Y. Leptomycin B is an inhibitor of nuclear export: Inhibition of nucleocytoplasmic translocation of the human immunodeficiency virus type 1 (HIV-1) Rev protein and Rev-dependent mRNA. Chem. Biol. 4:139–147; 1997. [DOI] [PubMed] [Google Scholar]
- 48. Xiao C. Y.; Jans P.; Jans D. A. Negative charge at the protein kinase CK2 site enhances recognition of the SV40 large T-antigen NLS by importin: Effect of conformation. FEBS Lett. 440:297–301; 1998. [DOI] [PubMed] [Google Scholar]
- 49. Yang J.; Bardes E. S.; Moore J. D.; Brennan J.; Powers M. A.; Kornbluth S. Control of cyclin B1 localization through regulated binding of the nuclear export factor CRM1. Genes Dev. 12:2131–2143; 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Yotov W. V.; Moreau A.; St-Arnaud R. The alpha chain of the nascent polypeptide-associated complex functions as a transcriptional coactivator. Mol. Cell. Biol. 18:1303–1311; 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Yotov W. V.; St-Arnaud R. Differential splicing-in of a proline-rich exon converts alphaNAC into a muscle-specific transcription factor. Genes Dev. 10:1763–1772; 1996. [DOI] [PubMed] [Google Scholar]
- 52. Zheng X. M.; Black D.; Chambon P.; Egly J. M. Sequencing and expression of complementary DNA for the general transcription factor BTF3. Nature 344:556–559; 1990. [DOI] [PubMed] [Google Scholar]