

Abstract
Onset of Parkinson’s disease (PD) and Parkinson-like syndromes has been associated with exposure to diverse environmental stimuli. Epidemiological studies have demonstrated that exposure to elevated levels of manganese produces neuropathological changes localized to the basal ganglia, including neuronal loss and depletions in striatal dopamine content. However, understanding the mechanisms associated with manganese neurotoxicity has been hampered by the lack of a good rodent model. Elevated levels of 8-hydroxy-2′-deoxyguanosine (oxo8dG) have been found in brain areas affected in PD. Whether increased DNA damage is responsible for neuronal degeneration or is a mere epiphenomena of neuronal loss remains to be elucidated. Thus, by using mice deficient in the ability to remove oxo8dG we aimed to determine if dysregulation of DNA repair coupled to manganese exposure would be detrimental to dopaminergic neurons. Wild-type and OGG1 knockout mice were exposed to manganese from conception to postnatal day 30; in both groups, exposure to manganese led to alterations in the neurochemistry of the nigrostriatal system. After exposure, dopamine levels were elevated in the caudate of wild-type mice. Dopamine was reduced in the caudate of OGG1 knockout mice, a loss that was paralleled by an increase in the dopamine index of turnover. In addition, the reduction of dopamine in caudate putamen correlated with the accumulation of oxo8dG in midbrain. We conclude that OGG1 function is essential in maintaining neuronal stability during development and identify DNA damage as a common pathway in neuronal loss after a toxicological challenge.
Key words: DNA damage, DNA repair, Manganese, Development, Parkinson’s disease
MANGANESE is an essential element in humans and animals (16). Manganese is a necessary component of glutamine synthetase (30,44) and mitochondrial superoxide dismutase (12,45). Despite the important biological functions, chronic exposure to manganese is detrimental to human health and it is associated with the onset of neurological dysfunction. At the neurochemical level manganese induces loss of striatal dopamine (2), resembling neurochemical changes seen in Parkinson’s disease (PD).
There are several physiological factors that can dramatically affect neurological outcomes due to manganese exposure in the early stages of development. i) Manganese passes from mother to infant across the placenta and in breast milk; moreover, manganese crosses the blood–brain barrier and enters the brain much more easily in infants than in adults (14). ii) The level of absorption shows an age-dependent trend, being highest at birth (99%) and exhibiting a gradual decrease to about 5.5% in adulthood (18,46,49). iii) Animal studies have shown that newborn mice are unable to excrete injected manganese, leading to accumulation in the liver and brain (27). However, the lack of a good animal model has hindered the understanding of molecular events associated with manganese neurotoxicity and the possible mechanisms involved.
Manganese inhibits important enzymes involved in mitochondrial respiration, which will cause leakage of reactive oxygen species (ROS) into the cytosol, ultimately producing free radical damage (29,48). Manganese exposure can also lead to changes in anti-oxidant defense systems such as glutathione, glutathione peroxidase, catalase, and glutathione-S-transferases (25,43). The overall influx of pro-oxidant species and a decrease in antioxidant defenses will likely cause loss of redox homeostasis in affected brain regions, leading to an accumulation of oxidative damage to macromolecules and neuronal loss. It is inferred that damage to DNA by free radicals is more detrimental to neuronal function, because unlike damaged lipids and proteins that can be turned over, DNA restoration depends solely on repair mechanisms (11).
The most frequently studied oxidized base product in DNA is 8-hydroxy-2′-deoxyguanosine (oxo8dG). Elevated levels of oxo8dG have been associated with aging as well as neurodegenerative diseases such as PD and Alzheimer’s disease (17,26,35). The removal of oxo8dG from DNA is initiated by 8-oxoguanine glycosylase 1 (OGG1). Recent studies on the OGG1 knockout mouse reveal accumulation of oxo8dG in an age-dependent manner; however, the mouse exhibits no significant phenotype (13,22,31).
In the present study we characterized the toxic effects of manganese during the early stages of development and determined the role of DNA repair systems in protecting susceptible neuronal populations such as the dopaminergic system of the nigrostriatal pathway. The results suggest that OGG1 activity is essential to protect neurons from the deleterious effects of manganese during early stages of development.
MATERIALS AND METHODS
All materials were purchased from Sigma (St. Louis, MO), unless otherwise indicated. All animal use was conducted in accordance with protocols approved by the Institutional Animal Care and Use Committee.
Manganese Exposure
Mating pairs of 129/SvJ mice wild-type and OGG1 knockout, maintained in a 12-h light/dark cycle, were exposed to manganese (MnCl2: 0, 5, or 10, mg/ml) in the drinking water. At birth, the litters were examined, counted, and weighed. Dams were continuously exposed until the time of weaning on postnatal day (PND) 21. At this time dams were removed and the pups sexed and litter size adjusted to five males. Groups were then maintained on their corresponding manganese dose until PND 30. At this time, cervical dislocation was performed and the brain was extracted. The caudate putamen was dissected and used for neurochemical analysis. The midbrain was also dissected and used for determining gene status (wild-type or OGG1 knockout) and oxo8dG levels.
Dopamine and Metabolites in Caudate Putamen
Levels of dopamine (DA), 3,3-dihydrophenylacetic acid (DOPAC), and homovanillic acid (HVA) in caudate putamen were measured using HPLC with electrochemical detection as previously described (8). Briefly, caudate putamen were weighed after dissection and then homogenized by sonicating in 250 μl of perchloric acid (PCA, 0.05 M) containing 3,4 hydroxybenzylamine (DBA, 31 ng/ml) as an internal standard. Homogenates were centrifuged for 20 min at 14,000 × g, 4°C, and filtered through a 0.45-μm Acro LC 13 filter (Gelman Industries, Ann Arbor, MI). DA, DOPAC, and HVA were measured and quantified by reverse-phase liquid chromatography (HPLC). Sample filtrates were loaded into vials and placed into an ESA Model 546 auto sampler (ESA Inc., Chelmsford, MA) set to 4°C. The mobile phase consisted of water/acetonitrile (9:1, v/v) containing monochloroacetic acid (0.15 M), sodium hydroxide (0.12 M), EDTA (0.60 mM), and sodium octyl sulfate (1.30 mM); the pH of the mobile phase was kept between 3.0 and 3.5. The chromatographic column was an ultrasphere ODS column with a length of 25 cm and internal diameter of 4.6 mm (Beckman Instruments, San Ramon, CA). The flow rate was kept constant during analysis at 1 ml/min, (ESA Model 582 Solvent Delivery Module) and the column eluate was analyzed with an electrochemical detector (ESA Model 5600A CoulArray Detector, 3 ESA Model 6210 four channel electrochemical cells). Potentials of the four coulometric analytical cells of the Coul Array system, placed in series, were as follows: channels 1 through 5 (−50, 0, 25, 100, 200 mV) and channels 6 through 12 (300 mV). The ratio of the peak heights produced by DA and its metabolites (DOPAC and HVA) to the peak height produced by DBA (internal standard) in the samples was used to obtain the caudate levels of DA extrapolated from a calibration curve. This calibration curve was constructed by plotting the ratios of known amounts of DA, DOPAC, and HVA to the internal standard DBA against the ratio of the peak heights, produced by these known amounts, to peak height produced by DBA. Data were recorded, stored, and analyzed on a PC Pentium computer using CoulArray for Windows 32Software (ESA Inc.). Data were expressed as micrograms per gram of wet weight.
Sample Preparation for Analysis of OGG1 Activity and oxo8dG Levels in Midbrain
The midbrain was hand homogenized with buffer containing 20 mM Tris-HCl, pH 8.0, 1 mM EDTA, 1 mM dithiotrietol, 0.5 mM spermine, 0.5 mM spermidine, 50% glycerol, and protease inhibitors. Homogenates were rocked for 30 min after addition of 1/10 vol 2.5 M KCl and spun at 14,000 rpm for 30 min. Then it was stored at −70°C until time of enzymatic assay. The precipitate was saved at −70°C for the analysis of oxidative damage to DNA.
From the supernatant, protein levels are determined using the Bradford method (4). Activity of OGG1 was determined as described elsewhere (6). Briefly, a synthetic probe containing oxo8dG (Trevigen, Gaithersburg, MD) was labeled with 32P at the 5′ end using polynucleotide T4 kinase (Boehringer Mannheim, Germany). The probe used had the following nucleotide sequence, 5′-GAACTAGTGOATCCCCCGGGCTGC-3′ (O = oxo8dG); before the nicking reaction this probe was annealed to its corresponding complementary oligonucleotide. The nicking reaction was carried out using 30 μg of protein extract and the double-stranded labeled probe. The reaction was carried out at 37°C for 2 h and stopped by placing the samples in ice. These time and protein conditions were optimized to obtain values of activity that were well within the linear portion of the kinetics of the enzyme (6). Aliquots of loading buffer containing 90% formamide, 10 mM NaOH, and blue-orange dye (Promega Corp., Madison, WI) were added to each sample. After 5 min of heating at 95°C, samples were chilled and loaded into a polyacrylamide gel (20%) with 7 M urea and 1× TBE, and run at 400 mV for 2 h. Gels were quantified using FLA-3000 Series Fuji Film Fluorescent Image Analyzer and analysis software. The capacity of the extract to remove oxo8dG was expressed as percentage of the cleaved synthetic probe (containing oxo8dG) to the total probe used (densitometric units).
Determination of Oxidative Damage to DNA (oxo8dG Levels)
Procedures for extraction, purification, enzymatic hydrolysis of DNA, and measurement of oxo8dG levels have been previously described (7,23). Briefly, the precipitate from sample preparation (above) was digested with DNAse-free RNAse followed by a digestion with proteinase K. The protein fraction was separated from DNA by three consecutive organic extractions. The DNA was then precipitated by adding two volumes of ethanol (with respect to the aqueous volume) and incubated overnight at −20°C. The purified DNA was prepared for HPLC analysis by resolving it into deoxynucleoside components. The amounts of oxo8dG and 2′-deoxyguanosine (2-dG) were calculated by comparing the peak area of oxo8dG and 2-dG obtained from the enzymatic hydrolysate of the DNA sample to a calibration curve for both compounds. Levels of oxo8dG in the samples were expressed relative to the content of 2-dG [e.g., the molar ratio of oxo8dG/2-dG (fmol oxo8dG/nmol of 2-dG)]. The mobile phase of the HPLC system consisted of 100 mM sodium acetate, pH 5.2, with 5% methanol. Flow rate was kept at 1 ml/min using a Model 582 Solvent Delivery Module (ESA Inc.). DNA was analyzed using a reverse phase YMCbasic HPLC column (4.6 × 150 mm) with a 3-μm particle size (YMC Inc., Wilmington, NC). Oxo8dG and 2-dG were detected by a Model 5600A CoulArray Detector (ESA Inc.) with three model 6210 four-channel electrochemical cells; potentials were set at 175, 200, and 250 V for oxo8dG and at 785, 850, and 890 V for 2-dG. Data were recorded, stored, and analyzed on a PC Pentium computer using CoulArray for Windows 32Software (ESA Inc.). Data are expressed as femtomoles of oxo8dG per nanomoles of 2-dG.
Statistical Analysis
For analysis of the effects of perinatal manganese exposure on DA nd its metabolites in caudate (PND 30), all group values were expressed as mean ± SEM. Between-group differences for striatal concentrations of DA, DOPAC, and HVA in caudate, and oxo8dG levels in midbrain, were evaluated for each manganese exposure level and gene status by two-factor ANOVA. In order to determine the effects of manganese in either wild-type or OGG1 knockout mice, one-way ANOVA was used followed by Student-Newman-Keuls post hoc test to detect differences between manganese groups. The correspondence between oxo8dG levels in midbrain and levels of DA in caudate putamen was done using Pearson’s correlation analysis. The significance level of all evaluations was set at p < 0.05. GraphPad Prism software was used to perform statistical analysis.
RESULTS
Elevated levels of oxo8dG have been presented as evidence of free radical damage associated with the onset of main neurodegenerative diseases (3,17,26,35). However, it is unknown whether elevation in oxo8dG levels represents a step in the process of neurodegeneration or if it is mere epiphenomena associated with neuronal loss. The use of mice lacking OGG1, the main enzyme in the process of recognition and removal of the damaged base, would elucidate this. Figure 1 represents a typical gel for analysis of OGG1 activity in midbrain of mice used for this study.
Figure 1.
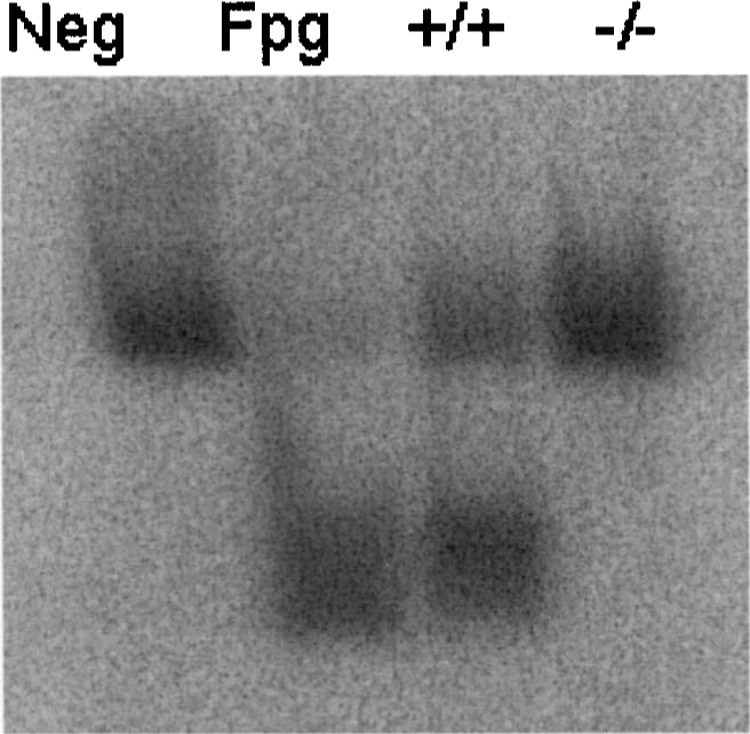
Gel for analysis of OGG1 activity in midbrain of wild-type (+/+) and OGG1 knockout (−/−) mice. Activity of OGG1 enzyme yielded two bands: top band is intact 32P-labeled oligonucleotide, lower band is the 32P-labeled oligonucleotide cleaved by OGG1 and separated by electrophoresis. Lane 1: negative control (no enzyme), lane 2: positive control (Fpg glycosylase enzyme), lane 3: activity in midbrain of wild-type (+/+) mouse, lane 4: activity in midbrain of OGG1 knockout (−/−) mouse.
To determine whether expression of OOG1 can affect the susceptibility of the nigrostriatal pathway to a toxicological challenge with manganese, both wild-type and OGG1 knockout mice were exposed to manganese during development, from conception to PND 30. After manganese exposure, levels of DA and its metabolites were determined in caudate putamen. Figure 2 is a composite of the levels of DA and its main metabolites (DOPAC and HVA) in wild-type and OGG1 knockout mice exposed to manganese. Exposure to manganese altered the levels of DA in both groups of mice. Two-way ANOVA analysis identified a manganese effect [F(2, 38) = 4.45, p < 0.05] and an interaction effect [F(2, 38) = 4.00, p < 0.05]. As expected, OGG1 gene status did not affect DA levels [F(1, 38) = 182, p > 0.05]. Similarly, two-way ANOVA analysis for DOPAC yielded significant effect of manganese [F(2, 38) = 3.46, p < 0.05] and interaction [F(2, 38) = 8.83, p < 0.05]. Analysis of HVA levels did not show a significant effect due to manganese exposure. To determine the degree of change after manganese treatment, data from each gene group were analyzed by one-way ANOVA followed by Student-Newman-Keuls. As denoted by the asterisk in Figure 2, in the wild-type mice, manganese at 5 mg/ml caused an elevation in DA levels in caudate putamen; the 10 mg/ml dose did not produce changes in DA content. No significant difference from control levels was seen for DOPAC and HVA at the two doses of manganese administered for the wild-type mice. In contrast, the OGG1 knockout mice exposed to manganese had a significant drop in striatal DA levels (44% from control), only at the 10 mg/ml level. Levels of DOPAC dropped in the caudate of OGG1 knockout mice in a dose–response manner as a consequence of manganese, and although HVA levels tended to be lower in the manganese group, such differences were not statistically significant.
Figure 2.
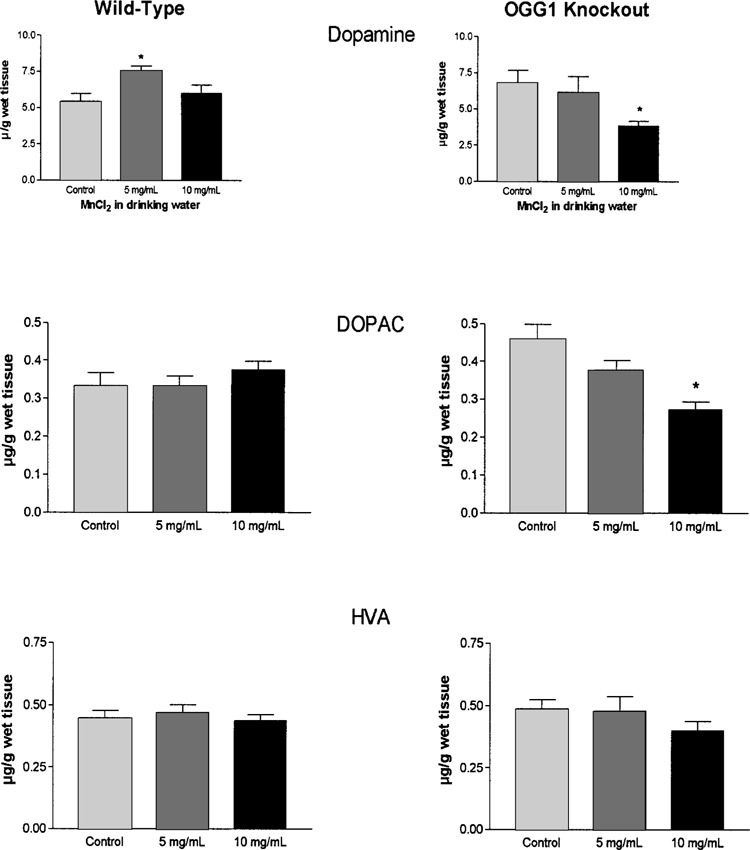
Levels of dopamine, DOPAC, and HVA in caudate putamen of young mice (PND 30) exposed to manganese throughout development. Data expressed as micrograms of dopamine per weight of wet tissue (mean ± SEM). *Significant difference from control (p < 0.05; n = 7–10).
The DA index of turnover, calculated as the ratio of metabolites DOPAC and HVA to DA, was analyzed to determine the level of function in the nigrostriatal system as a consequence of exposure to manganese in both wild-type and OGG1 knockout mice. The increase in DA levels in the wild-type mice at the 5 mg/ml level of manganese exposure corresponded to a drop in the index of turnover (Fig. 3). However, the loss of DA in the OGG1 knockout mice exposed to manganese at the 10 mg/ml level was paralleled by an increase in the index of turnover (Fig. 3).
Figure 3.
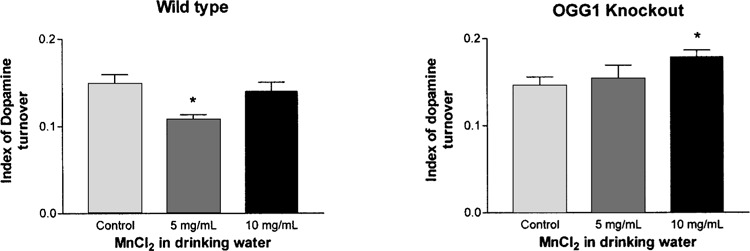
Index of turnover for dopamine in nerve terminals of the nigrostriatal pathway of OGG1 knockout mice. Index of turnover defined as (DOPAC + HVA)/dopamine levels. Data presented as mean ± SEM. *Significant difference from control (p < 0.05; n = 7–10).
In order to elucidate if changes in DA in the caudate were reflective of levels of DNA oxidation in the cell bodies of the midbrain, levels of oxo8dG were analyzed in the midbrain of the same mice exposed to manganese. Table 1 shows levels of oxo8dG as reflective of gene status (wild-type versus knockout) and as a measure of manganese exposure. Oxo8dG levels show a trend of accumulation as a consequence of lack of OGG1 and as a consequence of manganese exposure. However, a statistical significant difference was present only in the 10 mg/ml manganese OGG1 knockout group.
TABLE 1.
Oxo8dG LEVELS IN MIDBRAIN OF PND 30 MICE EXPOSED TO MANGANESE DURING DEVELOPMENT
| Wild-Type (n = 5–7) | OGG1 Knockout (n = 6–8) | |
|---|---|---|
| Control | 23.54 ± 3.30 | 27.90 ± 4.16 |
| MnCl2 5 mg/ml | 32.45 ± 8.03 | 33.16 ± 13.20 |
| MnCl2 10 mg/ml | 29.16 ± 4.77 | 43.80 ± 9.10* |
Oxo8dG levels expressed as femtomoles per nanomoles of 2′-dG
Significantly higher than control level (p < 0.05).
Pearson’s correlations analysis was carried out between catecholamine levels in caudate and oxo8dG levels in midbrain to ascertain if accumulation of oxo8dG at the cell body level is reflective of the degree of effect of manganese to DA levels in caudate. Figure 4 depicts analysis of DA in caudate to its corresponding oxo8dG levels in midbrain for each individual brain analyzed. Such a relationship was evident in the OGG1 knockout mice at the highest exposure to manganese only.
Figure 4.
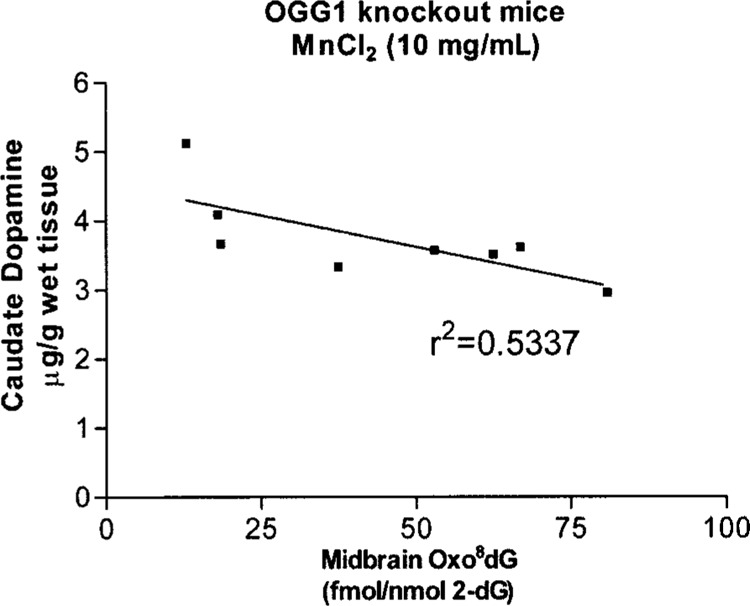
Dopamine levels in caudate putamen in relationship to oxo8dG levels in midbrain. Linear regression analysis indicates that an inverse relationship does exist. Pearson’s correlation analysis (p < 0.05).
DISCUSSION
This is the first report to demonstrate that OGG1 plays an important role in protecting neuronal populations against neurotoxicological challenge. Studies in wild-type mice and rats have shown that manganese is not detrimental to dopaminergic neurons (5,14,32), corroborated by the data from wild-type mice reported in this study. Protocols of exposure to manganese as long as 180 days did not produce loss in DA content; rather, this level of exposure leads to an increase in DA content paralleled by hyperactivity (5,9,32). These studies do not offer evidence of neuropathological changes in the nigrostriatal pathway as a consequence of manganese exposure; however, they do offer evidence that manganese can alter the neurochemistry of the developing nigrostriatal system at levels that cause no effect in the adult (14). Because of the dissimilar outcomes after manganese exposure between humans and rodents, the use of rodents as a model to understand the behavioral changes seen in humans exposed to manganese has been questioned. However, the use of genetic engineering can give new insights into discarded approaches.
OGG1 knockout mice have been found to accumulate oxo8dG in major organs in an age-dependent manner (22,31), and such accumulation is even more dramatic after a toxicological challenge (1). However, neither the age-dependent nor toxin-induced accumulation of DNA damage has been studied in the context of pathological changes. OGG1 knockout mice show no overt physiological deficiencies in aging studies ranging from 2 to 27 months (31). Oxo8dG accumulation in brain has been associated with aging and pathologies involved in neurodegenerative diseases; however, it is not known if OGG1 plays a role in the protection of neurons against free radical damage, or if the accumulation of oxo8dG leads to neuronal demise. One published report failed to detect accumulation in oxo8dG levels as a consequence of manganese exposure during development (5). However, whole brain levels were analyzed rather than levels in specific brain regions affected by manganese neurotoxicity. Our results show increases in oxo8dG levels in midbrain correlate with decreases in DA content and are the first to point to a cause–effect relationship between DNA damage and neuronal dropout. Early studies in wild-type mice identified aging as a factor in the regional specific accumulation of oxo8dG, with regions serving motor function (caudate, midbrain, and cerebellum) showing an age-dependent accumulation of oxo8dG, paralleled by DA loss in caudate and a decline in behavior associated with motor functions (8). Our results demonstrate that a lack of OGG1 allows for higher levels of oxo8dG to accumulate in the mouse midbrain and even higher levels in the manganese-treated mice. However, it is worth noting that oxo8dG levels reflect DNA damage in diverse neuronal populations and nonneuronal cells as well; thus, effects to the dopaminergic neurons in the substantia nigra, the target of manganese’s deleterious effects, could be underestimated.
As with studies of idiopathic and animal models of PD (15,40), manganese exposure to OGG1 knockout mice during development elicits a loss of DA in the striatum with a concomitant response in remaining neurons as evidenced by the elevation in DA index of turnover (Fig. 3). Because of the metabolic pathways involved in DA removal, it is highly likely that an increase in DA turnover will be detrimental to the stability of the remaining neurons. Loss of DA and increased DA turnover is accompanied by a significant rise in the level of oxidized glutathione in brain (41). In addition, compounds that reduce DA turnover are known to be protective against the damaging effects of MPTP, rotenone, and 6-hydroxydopamine (36). These observations confirm that a selective increase in neurotransmitter turnover within nigrostriatal nerve terminals can evoke changes in cellular redox status with detrimental consequences for remaining neurons. In addition, increased DA turnover is suggestive that manganese effects are not merely due to changes in DA biosynthesis but rather that neuronal loss has occurred. This will be confirmed by current studies aimed to quantify neuronal populations in the substantia nigra in the model presented.
The role of a functionally sound DNA repair system in the CNS is underscored by the onset of neurological deficits associated with xeroderma pigmentosum and Cockayne syndrome, where there is an impairment of DNA repair, namely in the removal of oxo8dG (24,34). However, both disorders are characterized by heritable defects, whereas the neurological deficits in PD are sporadic in nature and, despite a predominance of oxidative DNA lesions (oxo8dG) found in postmortem tissues of disease sufferers (35), no genetic deficits in DNA repair have been identified. Such findings suggest that PD may result from subtle changes in DNA repair mechanisms that predispose neuronal populations to the cumulative effects of free radical damage or to the effects of environmental toxicants, predisposing specific neuronal populations to oxidative stressors (20). Relevant to the results of this report, it has been shown that in the context of cancer, OGG1 polymorphism is not a major contributor to cancer susceptibility, but it does alter the impact of environmental factors in cancer development (21). Thus, it remains to be elucidated if similar implications in DNA polymorphisms can be attributed to OGG1 in relation to increased vulnerability in the CNS and the onset of neurological deficits such as PD. Polymorphisms of specific genes have been associated with the etiology and the pathogenesis of PD; such polymorphisms range from metabolizing enzymes (CYP2D6) to mitochondrial gene mutations (10). Such studies and our findings of increased susceptibility to manganese in mice deficient in DNA repair strengthen the hypothesis of PD origin being of a dual nature; a genetic predisposition, if followed by a specific toxicological challenge, will lead to the development of PD (39).
An alternative to the hypothesis of a link between a genetic deficiency and onset of parkinsonism is the possibility that other environmental toxins could affect OGG1 activity, thus affecting the capacity of response of dopaminergic neurons. Recent studies identify that exposure to cadmium can directly inhibit OGG1 activity or even cause a downregulation of its expression (33,47). Similar outcomes may be seen in brains of mice exposed to cadmium, and if such an event occurs concomitant with manganese exposure, a susceptible population of dopaminergic neurons could occur leading to the loss of DA. Therefore, future studies should involve the use of mixtures of metals to elucidate whether or not such an event could play a role in the onset of idiopathic PD.
This report further supports the notion that free radical damage is a key component in the neurotoxicity associated with manganese exposure; specifically, it identifies a putative role for DNA repair in protecting neurons during critical developmental periods. Because it seems that clinical symptoms of PD only appear when striatal DA concentration falls to less than 20% (37), this animal model will be useful to determine if challenges to the nigrostriatal system during development play a role in the onset of neurodegenerative disease later in life. It is important to note that clinical progression in patients with manganese parkinsonism continues even 10 years after cessation of exposure (19). In addition, because of its heavy use in industry, and the increasing use of underground water sources, exposure to manganese is more likely to rise, along with a subsequent increase in risks of neurological effects (38,42).
In conclusion, the present study demonstrates that exposure to manganese during development, in conditions that facilitate accumulation of oxidative damage to DNA, leads to a loss in DA. Moreover, we show that accumulation of DNA damage at the cell body level is representative of the loss of neurotransmitter at the neuronal terminal. Although OGG1 has been extensively studied in the context of cancer, it is surprising the lack of studies aimed to investigate the role of DNA repair in the onset of neurological deficits given the fact that elevated levels of oxo8dG are found in brain regions associated with neuronal loss in specific neurodegenerative diseases. Our results support the hypothesis that oxidative damage to DNA, as determined by the levels of oxo8dG, is a mechanism associated with both the neurotoxic outcome of manganese exposure during development and the neurodegenerative process in PD. Although the use of rodents in models for manganese neurotoxicity has been questioned (28), the use of the OGG1 knockout mice can prove to be a needed tool in understanding the neurotoxic mechanisms associated with human exposure to manganese.
ACKNOWLEDGMENTS
This work was supported by NSF-EPSCoR (EPS-0091995) and NSF-COBRE (5P20RR15583).
REFERENCES
- 1. Arai T.; et al. High accumulation of oxidative DNA damage, 8-hydroxyguanine, in Mmh/Ogg1 deficient mice by chronic oxidative stress. Carcinogenesis 23(12):2005–2010; 2002. [DOI] [PubMed] [Google Scholar]
- 2. Bernheimer H.; et al. Brain dopamine and the syndromes of Parkinson and Huntington. Clinical, morphological and neurochemical correlations. J. Neurol. Sci. 20(4):415–455; 1973. [DOI] [PubMed] [Google Scholar]
- 3. Bogdanov M.; et al. Increased oxidative damage to DNA in ALS patients. Free Radic. Biol. Med. 29(7):652–658; 2000. [DOI] [PubMed] [Google Scholar]
- 4. Bradford M. M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72:248–254; 1976. [DOI] [PubMed] [Google Scholar]
- 5. Brenneman K. A.; et al. Manganese-induced developmental neurotoxicity in the CD rat: Is oxidative damage a mechanism of action? Neurotoxicology 20(2–3): 477–487; 1999. [PubMed] [Google Scholar]
- 6. Cardozo-Pelaez F.; et al. DNA damage, repair, and antioxidant systems in brain regions: A correlative study. Free Radic. Biol. Med. 28(5):779–785; 2000. [DOI] [PubMed] [Google Scholar]
- 7. Cardozo-Pelaez F.; et al. Attenuation of age-dependent oxidative damage to DNA and protein in brain-stem of Tg Cu/Zn SOD mice. Neurobiol. Aging 19(4):311–316; 1998. [DOI] [PubMed] [Google Scholar]
- 8. Cardozo-Pelaez F.; et al. Oxidative DNA damage in the aging mouse brain. Mov. Disord. 14(6):972–980; 1999. [DOI] [PubMed] [Google Scholar]
- 9. Chandra S. V.; Shukla G. S.; Saxena D. K. Manganese-induced behavioral dysfunction and its neurochemical mechanism in growing mice. J. Neurochem. 33(6):1217–1221; 1979. [DOI] [PubMed] [Google Scholar]
- 10. Checkoway H.; et al. Genetic polymorphisms in Parkinson’s disease. Neurotoxicology 19(4–5):635–643; 1998. [PubMed] [Google Scholar]
- 11. Chen Y. H.; Bogenhagen D. F. Effects of DNA lesions on transcription elongation by T7 RNA polymerase. J. Biol. Chem. 268(8):5849–5855; 1993. [PubMed] [Google Scholar]
- 12. Denman R. B.; Wedler F. C. Association-dissociation of mammalian brain glutamine synthetase: Effects of metal ions and other ligands. Arch. Biochem. Biophys. 232(2):427–440; 1984. [DOI] [PubMed] [Google Scholar]
- 13. de Souza-Pinto N. C.; et al. Repair of 8-oxodeoxyguanosine lesions in mitochondrial DNA depends on the oxoguanine dna glycosylase (OGG1) gene and 8-oxoguanine accumulates in the mitochondrial DNA of OGG1-defective mice. Cancer Res. 61(14):5378–5381; 2001. [PubMed] [Google Scholar]
- 14. Dorman D. C.; et al. Neurotoxicity of manganese chloride in neonatal and adult CD rats following sub-chronic (21-day) high-dose oral exposure. J. Appl. Toxicol. 20(3):179–187; 2000. [DOI] [PubMed] [Google Scholar]
- 15. Doudet D. J.; et al. 6-(18F)Fluoro-L-DOPA PET studies of the turnover of dopamine in MPTP-induced parkinsonism in monkeys. Synapse 29(3):225–232; 1998. [DOI] [PubMed] [Google Scholar]
- 16. Freeland-Graves J. H.; Turnlund J. R. Deliberations and evaluations of the approaches, endpoints and sparadigms for manganese and molybdenum dietary recommendations. J. Nutr. 126(9, Suppl.):2435S–2440S; 1996. [DOI] [PubMed] [Google Scholar]
- 17. Gabbita S. P.; Lovell M. A.; Markesbery W. R. Increased nuclear DNA oxidation in the brain in Alzheimer’s disease. J. Neurochem. 71(5):2034–2040; 1998. [DOI] [PubMed] [Google Scholar]
- 18. Garcia-Aranda J. A.; Wapnir R. A.; Lifshitz F. In vivo intestinal absorption of manganese in the rat. J. Nutr. 113(12):2601–2607; 1983. [DOI] [PubMed] [Google Scholar]
- 19. Huang C. C.; et al. Long-term progression in chronic manganism: Ten years of follow-up. Neurology 50(3):698–700; 1998. [DOI] [PubMed] [Google Scholar]
- 20. Khabazian I.; et al. Isolation of various forms of sterol beta-D-glucoside from the seed of Cycas circinalis: Neurotoxicity and implications for ALS-parkinsonism dementia complex. J. Neurochem. 82(3):516–528; 2002. [DOI] [PubMed] [Google Scholar]
- 21. Kim J. I.; et al. hOGG1 Ser326Cys polymorphism modifies the significance of the environmental risk factor for colon cancer. World J. Gastroenterol. 9(5):956–960; 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Klungland A.; et al. Accumulation of premutagenic DNA lesions in mice defective in removal of oxidative base damage. Proc. Natl. Acad. Sci. USA 96(23):13300–13305; 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Laws G. M.; Adams S. P. Measurement of 8-OHdG in DNA by HPLC/ECD: The importance of DNA purity. Biotechniques 20(1):36–38; 1996. [DOI] [PubMed] [Google Scholar]
- 24. Le Page F.; et al. Transcription-coupled repair of 8-oxoguanine: Requirement for XPG, TFIIH, and CSB and implications for Cockayne syndrome. Cell 101(2):159–1571; 2000. [DOI] [PubMed] [Google Scholar]
- 25. Liccione J. J.; Maines M. D. Selective vulnerability of glutathione metabolism and cellular defense mechanisms in rat striatum to manganese. J. Pharmacol. Exp. Ther. 247(1):156–161; 1988. [PubMed] [Google Scholar]
- 26. Lovell M. A.; Gabbita S. P.; Markesbery W. R. Increased DNA oxidation and decreased levels of repair products in Alzheimer’s disease ventricular CSF. J. Neurochem. 72(2):771–776; 1999. [DOI] [PubMed] [Google Scholar]
- 27. Miller S. T.; Cotzias G. C.; Evert H. A. Control of tissue manganese: Initial absence and sudden emergence of excretion in the neonatal mouse. Am. J. Physiol. 229(4):1080–1084; 1975. [DOI] [PubMed] [Google Scholar]
- 28. Newland M. C. Animal models of manganese’s neurotoxicity. Neurotoxicology 20(2–3):415–432; 1999. [PubMed] [Google Scholar]
- 29. Newland M. C.; et al. Visualizing manganese in the primate basal ganglia with magnetic resonance imaging. Exp. Neurol. 106(3):251–258; 1989. [DOI] [PubMed] [Google Scholar]
- 30. Norenberg M. D. Distribution of glutamine synthetase in the rat central nervous system. J. Histochem. Cytochem. 27(3):756–762; 1979. [DOI] [PubMed] [Google Scholar]
- 31. Osterod M.; et al. Age-related and tissue-specific accumulation of oxidative DNA base damage in 7,8-dihydro-8-oxoguanine-DNA glycosylase (Ogg1) deficient mice. Carcinogenesis 22(9):1459–1463; 2001. [DOI] [PubMed] [Google Scholar]
- 32. Pappas B. A.; et al. Perinatal manganese exposure: Behavioral, neurochemical, and histopathological effects in the rat. Neurotoxicol. Teratol. 19(1):17–25; 1997. [DOI] [PubMed] [Google Scholar]
- 33. Potts R. J.; Watkin R. D.; Hart B. A. Cadmium exposure down-regulates 8-oxoguanine DNA glycosylase expression in rat lung and alveolar epithelial cells. Toxicology 184(2–3):189–202; 2003. [DOI] [PubMed] [Google Scholar]
- 34. Reardon J. T.; et al. In vitro repair of oxidative DNA damage by human nucleotide excision repair system: Possible explanation for neurodegeneration in xeroderma pigmentosum patients. Proc. Natl. Acad. Sci. USA 94(17):9463–9468; 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Sanchez-Ramos J.; Overvik E.; Ames B. A marker of oxyradical-mediated DNA damage (8-hydroxy-2′-deoxyguanosine) is increased in nigrostriatum of Parkinson’s disease brain. Neurodegeneration 3:197–204; 1994. [Google Scholar]
- 36. Schapira A. H. Dopamine agonists and neuroprotection in Parkinson’s disease. Eur. J. Neurol. 9(Suppl. 3):7–14; 2002. [DOI] [PubMed] [Google Scholar]
- 37. Seeman P.; Niznik H. B. Dopamine receptors and transporters in Parkinson’s disease and schizophrenia. FASEB J. 4(10):2737–2744; 1990. [DOI] [PubMed] [Google Scholar]
- 38. Sierra P.; et al. Bioaccumulation of manganese and its toxicity in feral pigeons (Columba livia) exposed to manganese oxide dust (Mn3O4). Environ. Res. 79(2):94–101; 1998. [DOI] [PubMed] [Google Scholar]
- 39. Simon D. K.; Lin M. T.; Pascual-Leone A. “Nature versus nurture” and incompletely penetrant mutations. J. Neurol. Neurosurg. Psychiatry 72(6):686–689; 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Sossi V.; et al. Increase in dopamine turnover occurs early in Parkinson’s disease: Evidence from a new modeling approach to PET 18 F-fluorodopa data. J. Cereb. Blood Flow Metab. 22(2):232–239; 2002. [DOI] [PubMed] [Google Scholar]
- 41. Spina M. B.; Cohen G. Dopamine turnover and glutathione oxidation: Implications for Parkinson disease. Proc. Natl. Acad. Sci. USA 86(4):1398–1400; 1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. St-Pierre A.; et al. Bioaccumulation and locomotor effect of manganese dust in rats. Inhal. Toxicol. 13(7):623–632; 2001. [DOI] [PubMed] [Google Scholar]
- 43. Vescovi A.; et al. Interactions of manganese with human brain glutathione-S-transferase. Toxicology 57(2):183–191; 1989. [DOI] [PubMed] [Google Scholar]
- 44. Wedler F. C.; Denman R. B. Glutamine synthetase: The major Mn(II) enzyme in mammalian brain. Curr. Top. Cell Regul. 24:153–169; 1984. [DOI] [PubMed] [Google Scholar]
- 45. Weisiger R. A.; Fridovich I. Mitochondrial superoxide simutase. Site of synthesis and intramitochondrial localization. J. Biol. Chem. 248(13):4793–4796; 1973. [PubMed] [Google Scholar]
- 46. Yokoi K.; Kimura M.; Itokawa Y. Effect of dietary iron deficiency on mineral levels in tissues of rats. Biol. Trace Elem. Res. 29(3):257–265; 1991. [DOI] [PubMed] [Google Scholar]
- 47. Zharkov D. O.; Rosenquist T. A. Inactivation of mammalian 8-oxoguanine-DNA glycosylase by cadmium(II): Implications for cadmium genotoxicity. DNA Repair (Amst). 1(8):661–670; 2002. [DOI] [PubMed] [Google Scholar]
- 48. Zheng W.; Ren S.; Graziano J. H. Manganese inhibits mitochondrial aconitase: A mechanism of manganese neurotoxicity. Brain Res. 799(2):334–342; 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Zlotkin S. H.; Atkinson S.; Lockitch G. Trace elements in nutrition for premature infants. Clin. Perinatol. 22(1):223–240; 1995. [PubMed] [Google Scholar]