

Abstract
The aryl hydrocarbon receptor (AHR) and its DNA binding partner, the aryl hydrocarbon receptor nuclear translocator (ARNT) are basic helix–loop–helix/PAS proteins. The goal of the current study was to determine the extent to which residues R14 and R15 contained within the basic region of the AHR contribute to the DNA binding affinity and stability of the AHR/ARNT heterodimer. Towards this end, we first performed equilibrium binding and dissociation rate analyses using a single dioxin response element (DRE-1). While the K D and B max values obtained from the equilibrium binding analysis were similar for the wild-type AHR (wt AHR) and that containing the substitutions of R14 and R15 with Q residues (Q14Q15 AHR), dissociation rate analyses revealed that the stability of the Q14Q15 AHR DNA binding complex was approximately 10-fold less. Using a two-site DNA binding model, we also found that AHR/ARNT heterodimer does not participate in cooperative binding, as binding of the second dimer appears to be prohibited by occupation of the first. This property was similar regardless of the composition of the amino acids at positions 14 and 15. Finally, reporter assays revealed that the Q14Q15 substitutions severely compromised the ability of the AHR to activate gene expression despite appropriate nuclear localization. The present results revealed that DNA binding stability of the AHR/ARNT heterodimer is an important requirement for its transactivation capabilities and that this stability is governed, in part, by residues R14 and R15 that lie within the basic region of the AHR.
Key words: Aryl hydrocarbon receptor, Basic helix–loop–helix/PAS, DNA binding
THE aryl hydrocarbon receptor (AHR) is a cytosolic transcription factor that, upon binding of ligands such as aliphatic or polyhalogenated aromatic hydrocarbons, localizes to the nucleus where it heterodimerizes with its DNA binding partner, ARNT (aryl hydrocarbon receptor nuclear translocator, hypoxia inducible factor 1β) and upregulates a number of xenobiotic metabolizing genes (6). The recent discoveries that the AHR interacts with the cell cycle regulator Rb (27), upregulates the cell cycle inhibitor p27Kip1 (20,23), and alters in vitro cell mortality (28,29) and that its absence alters cell cycle progression (11) indicate that this signaling pathway is not only important for regulating xenobiotic metabolism, but may also play an important role in regulating cell cycle events.
Both the AHR and ARNT are basic helix–loop–helix(bHLH)/PAS (the Per-ARNT-Sim homology domain) proteins. The mechanisms by which this protein pair regulates gene transcription have been investigated using a number of different approaches. Domain mapping experiments have determined that the N-terminal sequences of both the AHR and ARNT are involved in DNA binding, whereas the C-termini of these proteins elicit transcriptional activation properties (8,15,40). While dimerization appears to be contained within the HLH and PAS domains of both proteins, the PAS domain of the AHR is also involved in mediating ligand binding and its interactions with the heat shock protein, HSP90. More recent observations have indicated that some residues contained within the PAS domain (5,33) and the HLH region (22) of the AHR play critical roles in proper DNA binding of the AHR/ARNT heterodimer. In fact, the presence of the PAS A domain of the AHR, but not ARNT, is critical for maintaining maximum DNA binding affinity and stability of the AHR/ARNT complex (5). In the absence of crystallization analyses of the AHR/ARNT DNA binding complex, these types of qualitative analyses are critical for furthering our understanding of the structural aspects of the DNA binding properties of this protein pair.
Previously, we had shown that substitutions of amino acid residues 36–41 of the murine AHR, but not residues 12–16, with glutamine abolished DNA binding of the AHR/ARNT heterodimer (35). These results led us to conclude that the primary amino acid contacts of the AHR with its DNA recognition site lie within the region bordered by amino acid residues 39–42, and do not involve amino acids 14 and 15. However, our results were somewhat controversial, as other laboratories had performed similar analyses and found that substitution of residue R14 (3,9,16) with either A or K ablated the ability of the AHR/ARNT to bind DNA. On the other hand, substitution of R15, with A either did not appear to alter (9) or yielded a small, albeit insignificant, increase in DNA binding of the AHR, but a significant increase in transactivation (16). Further clouding these functional DNA binding/transactivation analyses was the discovery that residue 14 is critical for nuclear translocation of the AHR (18).
In light of the disparate results described above (3,9,16,35), the objective of the current study was to better characterize the role that residues 14 and 15 of the AHR play in the ability of the AHR/ARNT heterodimer to bind DNA. Our findings indicate that the primary role of the R14R15 residues is in maintaining appropriate stability required for transcriptional activation as substitution of these residues results in a loss of stability of the AHR/ARNT DNA binding complex and a corresponding loss in transcriptional activation. Additional in vitro assays indicate that, unlike other transcription factors, the AHR/ARNT heterodimer does not participate in cooperative DNA binding at least within the genomic contexts employed here. We propose that residues R14 and R15, which reside outside of the region that make critical contacts with DNA (i.e., amino acid residues 34–39) (35), contribute to the formation of a fully functional protein/DNA binding complex by maintaining the appropriate tertiary structure and the optimal protein/protein and/or protein/DNA contacts required for target gene activation.
MATERIALS AND METHODS
Materials
The [35S]methionine (1000 Ci/mmol) and [γ-32P]ATP (3000 Ci/mmol) were from NEN (Boston, MA) and the transcription/translation kit was from Promega (Madison, WI). All other chemicals were from Sigma (St. Louis, MO). The AHR and ARNT antibodies were a gift from Dr. Richard Pollenz (University of South Florida, Gainesville, FL). Purified rabbit IgG was purchased from Sigma (St. Louis, MO).
Oligonucleotides
Oligonucleotides were purchased from Integrated DNA Technologies (Coralville, IA). Given below are the oligonucleotides (DRE in bold letters) that were annealed and radiolabeled for use in gel shift analyses. Oligonucleotides that contained one wild-type DRE sequence (wt DRE-1) are: TCGAGCTGGGGGCATTGCGTGACATAC (OL 17) and TCGAGGTATGTCACGCAATGCCCCCAGC (OL 18). This sequence has been previously determined as the optimum DNA recognition site of the AHR and ARNT complex (34). The oligonucleotides containing the mutated DRE (mt DRE) are: TCGAGCTGGGGGCATTGAATGACATAC (OL 108) and TCGAGGTATGTCAATCAATGCCCCCAGC (OL 109). Oligonucleotides that contained two wild-type DRE sequences (wt 2DRE-11) separated by 11 base pairs are: TCGAGGGCATTGCGTGACATACGCATTGC GTGACATACCA (OL 167) and GATCTGGTATGTCACGCAATGCGTATGTCACGCAATGCCC (OL 168). Those containing two mutated DRE sequences (mt 2DRE-11) are: TCGAGGGCATTGATTGACATACGCATTGATTGACATACCA (OL 169) and GATCTGGTATGTCAATCAATGCGTATGTCAATCAATGCCC (OL 170). Finally, oligonucleotides containing two wild-type DREs separated by 22 base pairs (wt 2DRE-22) are: TCGAGGGCATTGCGTGACATACGCATTACATACGCATTGCGTGACATACCA (OL 261) and TGGTATGTCACGCAATGCGTATGTAATGCGTATGTCACGCAATGCCCTCGA (OL 262). The oligonucleotides used as primers to amplify the AHR cDNAs using the polymerase chain reaction are: forward-GTCGCTCGAGGATGAGCAGCGGCGCCAACATCACC (OL 178) and reverse-GCCAAGCTTACTCTGCACCTTGCTTAGGAATGC (OL 179). Amplification of the AHR cDNA to yield a BamHI fragment that contained the localization sequence from nucleoplasmin (21) to the C-termini was accomplished using OL 113 as the sense primer (GCACTAGTACCATGGAAAGTGGCATGACAGTTTTCC) and OL 298 (GCGGATCCTCATCGCTTCTTCTTCTTTGCCTGTCCTGCCTTCTTCGTTGCTGCGGGTCGCTTACTCTGCACCTTGCTT AGGAATGC) as the antisense primer.
Plasmids
For the purpose of the present study, we will refer to the murine AHR construct, AHR CΔ516 as wt AHR and the AHR construct in which the R14R15 residues have been substituted with Q as Q14Q15 AHR. The generation of these constructs has been described previously (35). Deletion of the C-terminal amino acids of the AHR has been shown to result in an AHR that is capable of dimerizing with ARNT and binding the DRE in a ligand-independent manner. The plasmid used to generate in vitro transcribed ARNT was phuARNT (8). The plasmids used in the analyses of nuclear translocation and transactivation of the full-length wild-type and Q14Q15 AHR were constructed as follows. The full-length AHR constructs (35) were amplified using OL 178 and OL 179 and subcloned into the XhoI, HindIII site of pEGFP-N1 (Clontech, Palo Alto, CA). To insert the localization signal at the C-termini of the constructs, a PCR product was generated using the wt AHR/pEGFP and Q14Q15 AHR/pEGFP as templates and OL 113 and OL 298 as the primers. The resulting BamHI fragment replaced that of the original to generate wt AHRNLS/pEGFP and Q14Q15 AHRNLS/pEGFP. The luciferase reporter plasmids that are regulated by the DREs were generated by inserting two copies of either the annealed wt DRE (OL 17/18) or of the mt DRE (OL 108/109) into the XhoI site of pGL3 (Clontech).
Protein Expression
In vitro expression of the AHR and ARNT constructs was performed using rabbit reticulocyte lysates (Promega) as described previously (8). For verification of protein expression, the translation reactions were performed in the presence of [35S]methionine, and the products were analyzed by SDS-polyacrylamide gel electrophoresis. Quantitation of the expressed proteins was determined by excising the radiolabeled proteins from the gel and scintillation counting. Baculovirus expression and purification of wild-type ARNT were carried out as described previously (4).
Association Equilibrium Analysis
The gel shift assays were performed essentially as described previously (35). The association analyses were performed by adding either increasing concentrations of the probe (Fig. 1) or increasing concentrations of the AHR (Fig. 3). For Figure 1, approximately 1 fmol of in vitro expressed ARNT was incubated with 0.2 fmol of in vitro expressed AHR protein (either wt AHR or Q14Q15 AHR) for 30 min at 30°C. Nonspecific competitor (1 μg salmon sperm DNA) and specific competitor (0.5 fmol of mt DRE) were added and the mixture was incubated at room temperature for 10 min. Following the addition of the indicated concentrations of 32P-labeled probe containing one DRE (OL 17/18) and an incubation for an additional 10 min, the samples were subjected to non-denaturing gel electrophoresis using 0.5 × TBE (45 mM Tris base, 45 mM boric acid, 1 mM EDTA, pH 8.0) as the running buffer (35). The electrophoresis was performed at 4°C. The gels were dried, the radio-labeled bands were excised, and the amount of protein bound to the 32P-labeled DNA probe was quantitated following scintillation counting. The experiments depicted in Figure 3 were performed similarly with the following exceptions. The 35S-labeled reticulocyte lysate-expressed AHR protein was incubated in ascending increments with approximately 2 fmol of baculovirus-expressed ARNT prior to the gel shift analysis and 1 μg of salmon sperm was added. Approximately 0.5 ng (100,000 cpm) of radiolabeled probe containing two DREs (either OL 167/168 or OL 261/262) was added to the AHR/ARNT mixture and incubated for an additional 10 min. The AHR-containing complexes and free probe were quantitated using PhosphorImager analysis and specific binding was expressed as percent of total label in the lane. The statistical analyses, the dissociation constants, K D and B max, were calculated using the Graph-Pad Prism Software (San Diego, CA) using nonlinear regression and the one-site binding equation: Y = B max*X/(K D + X).
Figure 1.

Analysis of DNA binding of AHR variants using DRE-1. Association equilibrium analysis (A, B). The AHR/ARNT complexes were formed following the incubation of approximately 1 fmol of ARNT with either the wt AHR (A) or Q14Q15 AHR (B) proteins that were generated using in vitro transcription/translation reactions. Increasing concentrations of the 32P-labeled probe (DRE-1) were added and the gel shift reactions were performed as described in Materials and Methods. (C) Graphical representation of the gel shift analyses. The K D values were obtained from nonlinear regression analysis. The concentrations of DNA contained within the AHR/ARNT complex (specific binding) were determined using phosphorImager analysis and plotted as a function of the input 32P-labeled probe. The 95% CI of the K D of the wt AHR = 0.63 to 6.27 and that of the B max = 1.44 × 107 to 4.88 × 107. The 95% CI of the K D of the Q14Q15 AHR = 2.58 × 107 to 7.83 × 107 and that of the B max = 2.85 × 107 to 6.23 × 107. The R 2 values were > 0.90. The data are representative of at least three experiments performed in duplicate.
Figure 3.

Use of an oligonucleotide containing two DREs (2DRE-11) requires dimerization of the AHR and ARNT to allow formation of two AHR/ARNT complexes. Gel shift reactions were performed as described in Materials and Methods using either the wt AHR or Q14Q15 AHR and the 32P-labeled DRE-2 as a probe. (A) The gel shift reactions were performed with the wild-type AHR and contained either the anti-ARNT immunoglobulins (lane 2), the anti-AHR immunoglobulins (lane 3), control immunoglobulins (nonspecific rabbit IgG, lane 4, or preimmune serum, lane 5), or excess concentrations of the unlabeled, wt 2DRE-11 (lane 6) or unlabeled, mutated 2DRE-11 (lane 7). I indicates complex I whereas II indicates complex II. (B) The gel shift reactions were performed using the Q14Q15 AHR in the absence (lane 5) or presence of the anti-ARNT immunoglobulin (lane 1), the anti-AHR immunoglobulin (lane 2), or excess concentrations of unlabeled, mutated 2DRE-11 (lane 3) or unlabeled, wild-type 2DRE-11 (lane 5). (C) The gel shift reactions were performed using either the wt 2DRE-11 (lane 1) or wt 1DRE (lane 2) as the probe.
Dissociation Rate Analysis
The dissociation rates of the DNA binding complexes that contained either the wt AHR or Q14Q15 AHR constructs, ARNT, and a single DRE (OL 17/18) were determined using gel shift analysis as described previously (34). Briefly, a 200-fold molar excess of OL 17/18 was added at time 0 and aliquots removed at the indicated time points and applied to a running gel. Half-life (t 1/2) was calculated from the slope of the linear regression curve where t 1/2 = 0.693/k, and k = −2.303 (slope).
Nuclear Translocation and Reporter Assays
For the nuclear translocation studies, CV-1 cells were transiently transfected with either wt AHRNLS/EGFP or Q14Q15 AHRNLS/EGFP using Lipofectamine (Invitrogen). These constructs contain the full-length AHR protein (with either the wild-type or the Q14Q15 substitutions), an additional nuclear localization sequence, and the cDNA of the green fluorescent protein. After the transfection, the cells were cultured for 24 h, treated with either 0.1% DMSO or 1 nM TCDD for 1 h, and visualized using a Nikon TE 2000 Fluorescent microscope.
For the reporter assays, the cells were transiently transfected with the indicated reporter vectors, wt DRE or mt DRE, and the indicated expression vectors, wt AHRNLS/EGFP or Q14Q15 AHRNLS/EGFP, as well as the renilla reporter vector. Twenty-four hours after the transfections, the cells were incubated with either 0.1% DMSO or 1 nM TCDD for 4 h. The cells were harvested and the luciferase and renilla activites were determined using a microplate luminometer (Applied Biosystems).
RESULTS
Association Equilibrium Analysis Using a One-Site Model
As a first approach in understanding the impact of the Q14Q15 substitutions on the function of the AHR, we performed association equilibrium binding studies in which increasing concentrations of the labeled probe were added to mixtures containing fixed amounts of the AHR and ARNT proteins. As shown in Figure 1, substitution of R residues 14 and 15 with that of Q residues resulted in an AHR protein capable of forming a DNA binding complex that displayed a B max (i.e., Q14Q15 AHR = 4.54 × 107 and wt AHR = 3.16 × 107, respectively) and a K D (i.e., Q14Q15 AHR K D = 5.2 nM and wt AHR K D = 3.4 nM) that was not statistically different from that of the wild type AHR.
We then questioned whether the Q14Q15 substitutions of the AHR altered the stability of the AHR/ARNT DNA binding complex. To test this idea, we performed dissociation rate analyses. As shown in Figure 2, we found that the wt AHR/ARNT DNA binding complex is relatively stable (t 1/2 = 28 min). In contrast, substitution with the Q14Q15 residues significantly decreased the stability of this complex (t 1/2 of the Q14Q15 AHR construct = approximately 3.8 min). These results indicate that the R14R15 residues play an important role in maintaining the appropriate DNA binding stability of the AHR/ARNT heterodimer.
Figure 2.
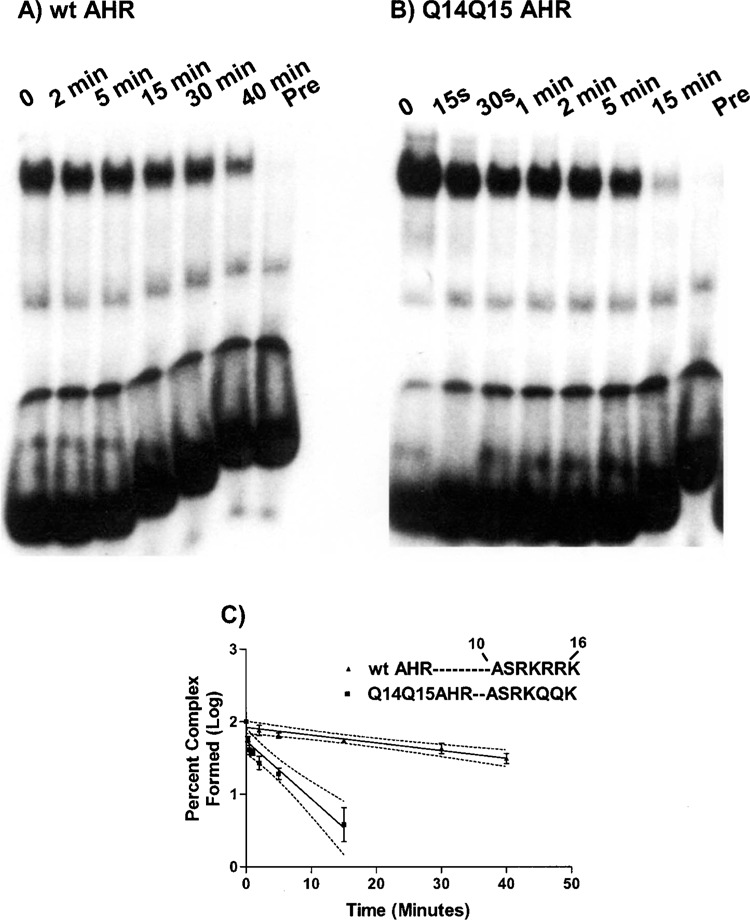
Dissociation analysis of the wt AHR/ARNT and Q14Q15 AHR/ARNT complexes. The AHR/ARNT DNA binding complexes were formed as described in Figure 1 except using 1 ng of 32P-labeled probe(DRE-1). After equilibrium binding had been reached (10 min), excess of unlabeled oligonucleotide was added to the mixture and aliquots were removed at the indicated time points. Pre, the unlabeled oligonucleotide was added prior to the addition of the 32P-labeled probe. A representation of the gel shift analyses of the wt AHR (A) and Q14Q15 AHR (B) are shown. (C) Graphical representation of the gel shift analyses. Each value represents the average of two independent experiments. The 95% confidence intervals of the linear regression slopes are depicted as follows: wt AHR −0.0147 to −0.0067 and Q14Q15 AHR −0.109 to −0.051.
Association Equilibrium Analysis Using a Two-Site Model
Given that regulation of the CYP1A1 promoter by the AHR/ARNT heterodimer involves multiple DREs and that the presence of a second DRE synergistically enhances transactivation of a single DRE (14), we hypothesized that the AHR/ARNT heterodimer may bind DNA in a cooperative manner and that the altered stability of the Q14Q15 AHR may significantly alter its ability to participate in cooperative DNA binding. To test these ideas, we first performed gel shift analyses using an oligonucleotide that contained two dioxin response elements with a spacing of 11 base pairs (wt 2DRE-11). This spacing is based on similar studies used to examine the DNA binding characteristics of the bHLH protein MyoD (39). We reasoned that if the Q14Q15 substitution severely impacted contacts that extended beyond the GCGTG core, this would impair the ability of the Q14Q15 AHR, but not the wt AHR, to participate in cooperative binding.
We first characterized the ability of AHR/ARNT heterodimer to form complexes representative of either occupation of a single recognition site (complex I) or occupation of both recognition sites (complex II). Complex formation of either the wt AHR (Fig. 3A) or Q14Q15 AHR (Fig. 3B) was similar. As shown in Figure 3A (lane 1), two complexes (complexes I and II) of the AHR/ARNT heterodimer are capable of interacting with the oligonucleotide that contains two DREs with the majority of the DNA binding contained within complex I, the lower molecular weight complex. The presence of the AHR and ARNT proteins within each protein–DNA complex was determined using antibodies that recognize either ARNT (Fig. 3A, lane 2 and Fig. 3B, lane 1), or the AHR (Fig. 3A, lane 3 and Fig. 3B, lane 2), but not nonspecific antibodies (Fig. 3A, lanes 4 and 5). Specific binding of complexes I and II to the oligonucleotide containing two sites was demonstrated by the competitive displacement of both complexes using excess oligonucleotides containing wild-type DREs (Fig. 3A, lane 6 and Fig. 3B, lane 4), but not mutated DRE sequences (Fig. 3A, lane 7 and Fig. 3B, lane 3).
We then performed association analyses similar to that described in Figure 1, except using the two-site oligonucleotide (wt 2DRE-11) and increasing concentrations of either the wt AHR (Fig. 4A) or that containing the glutamine substitutions, Q14Q15 AHR (Fig. 4B). Using this type of analysis, cooperative binding would be apparent by a sigmoidal shape to the curve that is representative of the formation of complex II, which is facilitated by the prior formation of complex I (31). As shown (Fig. 4A–C), increasing concentrations of the AHR protein resulted in a significant increase in the formation of complex I and a minimal increase in the formation of complex II. These results indicate that formation of the second AHR/ARNT complex is inhibited by occupation of the first site. This phenomena of “negative cooperativity” was observed when either the wt AHR or Q14Q15 AHR was analyzed.
Figure 4.


Association equilibrium analysis of either the wt AHR or Q14Q15 AHR using either wt 2DRE-11 or wt 2DRE-22. The gel shift analyses were performed as described in Figure 1 and Materials and Methods using increasing concentrations of either wt AHR (A, D) or Q14Q15 AHR (B, E). The gels shown in (A) and (B) were performed using the wt 2DRE-11 as a probe whereas those shown in (D) and (E) were performed using the wt 2DRE-22 as a probe. The graphical representations of these analyses are shown in (C) and (F), respectively. The complexes that contained the AHR and the free probe were quantitated using PhosphorImager analysis to obtain total binding (i.e., that present in complex I + complex II + free probe = total binding). Each AHR-containing complex is expressed as percent of the total. (C) Depiction of the analyses performed using wt2 DRE-11. (D) Depiction of the analyses using wt2 DRE-22. The data are representative of two experiments performed in duplicate.
We then reasoned that perhaps the spacing between the two DRE sites was insufficient for occupation of two AHR/ARNT heterodimers due to steric hindrance and that increasing the spacing to 22 nucleotides might alleviate this negative effect. As shown in Figure 4D–F, interaction with an oligonucleotide containing a spacing of 22 nucleotides was similar to that observed using the wt 2DRE-11 oligonucleotide (Fig. 4A–C) where occupation of the second DRE site was unfavorable when either AHR constructs were used. Interestingly, the ability of the Q14Q15 AHR to form a single heterodimeric complex (i.e., complex I) was less than that of the wt AHR when the longer oligonucleotide was used as a probe (Fig. 4E–F).
The Q14Q15 Substitution Impairs AHR Transactivation
Finally, we questioned whether the decreased affinity and stability of the Q14Q15 AHR construct was sufficient to significantly alter the ability of the AHR/ARNT heterodimer to activate gene transcription. As mentioned previously, this region also has been characterized as important in mediating nuclear localization of the AHR (18). To circumvent the issue of nuclear localization, we generated AHR constructs that were representative of the full-length AHR (i.e., contain the transactivation domain) and expressed both an additional nuclear localization sequence at its C-termini and the green fluorescent protein to aid visualization. As shown in Figure 5 (A and B), the Q14Q15 AHR construct localized to the nucleus in a ligand-independent manner. Examination of the transactivation capability of these two constructs revealed that while the Q14Q15 AHR construct was capable of entering the nucleus (Fig. 5A, B) and dimerizing with the ARNT (35), its ability to activate genes was severely impaired compared to the wt AHR (Fig. 5C).
Figure 5.

Impact of the Q14Q15 substitutions on nuclear translocation and transactivation of the AHR. Full-length AHR constructs containing the Q14Q15 substitutions as well as an additional nuclear localization sequence and GFP at their C-termini were transiently transfected into CV-1 cells. After a 1-h treatment with either DMSO (A) or TCDD (B), the cells were visualized using fluorescent microscopy. (C) CV-1 cells were transiently transfected with the indicated expression plasmids and luciferase reporter plasmids that contained either the wild-type DRE (wtDRE) or mutated DRE (mtDRE). After an overnight incubation, the cells were cultured with either 0.1% DMSO or 1 nM TCDD for 4 h, harvested, and the renilla and luciferase values were determined. The graph is representative of two separate experiments.
DISCUSSION
The major findings of this study are that mutations that lie outside the site of primary amino acid/DNA contacts (35) significantly increase the off-rate between the AHR/ARNT heterodimer and its DNA recognition site and hinder its ability to transactivate genes. We propose that the limited time of contact between the AHR/ARNT heterodimer with its DNA recognition site severely hinders its ability to activate transcription due to insufficient residence time of the DNA-bound complex. This idea is supported by recent studies of the STAT1 protein, where mutations that did not alter the K D of the STAT1/IRF-1 interaction, but greatly increased the off-rate of DNA binding, were incapable of establishing a residency time sufficient to initiate the events required for transcriptional activation (42). As discussed by these authors, one limitation of the gel shift assay is that the kinetics of the protein/DNA interactions may be obscured due to “caging” of the interacting protein–DNA molecules as they enter the pores of the gel. This phenomenon is illustrated in Figures 1 and 2 of this work, where the lower stability of the complex containing the Q14Q15 AHR was detected only when performing the dissociation rate analysis that relies on competitive interactions to occur between the labeled and unlabeled DNA probes in solution. In contrast, the conditions used in performing the association analysis allowed for the detection of a protein–DNA complex that does not appear to have functional consequences.
The importance of R14 of the AHR is further illustrated by its high conservation in all species examined [i.e., from Caenorhadbditis elegans (26) to Homo sapiens (7)]. In contrast, residue 15 may play a more minor role because it is often present as an R residue, but is also found as either Q [Caenorhadbditis elegans (26)] or K [Atlantic tomcod and Oncorhynchus mykiss (1)]. This idea is supported by previous studies that found that substitutions of the R15 residue did not decrease either DNA binding or transactivation of the AHR (3,15,16).
The contribution of the R14R15 residues of the AHR to its DNA binding stability may be a result of participation in 1) contacts with the phosphate backbone, 2) contacts within the AHR/ARNT complex, or 3) the maintenance of a functionally appropriate conformation of the DNA binding complex. The importance of contacts with the DNA phosphate backbone has been shown in studies of the bHLH proteins such as E12 (38). Here, substitutions of residues within the basic region, previously shown by crystallization studies to lack a role in hydrogen bonding with nucleotides, were found to increase the stability of the E12–DNA binding complex in a manner thought to involve nonspecific interactions with the phosphate backbone. However, studies with other HLH proteins have also shown that contacts within the protein dimer play important roles in maintaining stability. This type of interaction has been shown to occur within the HLH motif (i.e., between helix 1 and helix 2) of both USF and Max (12,13). Finally, the R14R15 residues of the AHR may maintain the functionally appropriate conformation of the DNA-bound form of the AHR/ARNT heterodimer. An example of this type of interaction has been detected by crystallographic studies of MyoD, where substitutions of residues 114 and 115 that lie within the basic region of MyoD are thought to displace R111 from its position within the major groove and thereby disrupt the ability of MyoD to activate genes (24). Given that ancillary factors do not appear to significantly contribute to DNA binding of the AHR/ARNT heterodimer (17), we do not anticipate that these residues make contacts with proteins other than the AHR or ARNT.
Information theoretic analysis of the associations formed among bHLH proteins reveals that the basic region of the AHR is likely to be involved in numerous protein–protein contacts (2). This mathematical approach examines patterns of sequence diversity within a class of proteins to explore how sequence variability and properties of individual amino acids dictate protein structure. Based on these predictions, the basic region of the AHR is highly likely to form contacts with either its own HLH or PAS regions or those of ARNT. In fact, the impairment of such contacts may be the mechanism(s) underlying the recent observations that substitutions of residues that lie either within the HLH (22) or PAS domain (33) of the AHR severely impact the ability of the AHR/ARNT heterodimer to bind DNA.
The prototypical target gene of the AHR/ARNT heterodimer is CYP1A1, which within the murine CYP1A1 promoter contains four copies of the DRE [see (41) for review]. It has been shown previously that these four binding motifs interact synergistically where each of the four recognition sites of the AHR/ARNT equally contribute to enhancer function (14). Although synergistic activation of transcription is typically thought to involve an increase in the accessibility to additional binding sites via protein–protein interactions that result in histone modification and chromatin remodeling (32), another mechanism that may come into play is cooperative DNA binding (31,36). Cooperative DNA binding is often facilitated by protein–protein interactions and/or DNA looping, which allows the binding of one DNA binding complex at a single site to enhance the binding of a DNA binding complex at additional, distant sites. For example, the cooperativity of NFAT and the Fos-Jun heterodimer, which bind adjacent, low-affinity binding sites within the interleukin-2 promoter, is driven by formation of polar interactions and extensive protein–protein interactions as well as alterations in protein–DNA interactions that include a shift in nucleotides contacted by an R residue of NFAT (36). Given that the AHR/ARNT heterodimer synergistically activates transcription in vivo (14) and is capable of inducing a bend in DNA indicative of DNA looping (10), we hypothesized that the AHR/ARNT heterodimer is capable of participating in cooperative DNA binding and that the R14R15 residues within the AHR may play a role in this activity. While our results (Figs. 3 and 4) performed using a high-affinity consensus DNA recognition site indicate that the AHR/ARNT heterodimer does not exhibit cooperative DNA binding, the possibility remains that cooperative DNA binding by the AHR/ARNT is like that of the androgen receptor and dependent on the context of the DNA sequences (30).
Analysis of the ability of the AHR/ARNT heterodimer to interact with two DREs revealed that negative modulation is involved in the formation of the second complex, complex II (Fig. 4). The negative impact observed upon binding of the AHR/ARNT complex to the second DRE site is in contrast to that observed with the nuclear hormone receptors (25,37) and other bHLH proteins, such as MyoD (30). In these systems, DNA binding using a two-site model was found to be cooperative where occupation of the dimers at the first site enhances occupation at the second site. The results presented in Figure 4 are also unexpected due to the previous observations that gene activation by the AHR/ARNT heterodimer appeared to proceed in a cooperative manner (14). Taken together, these results indicate either that the in vitro binding approaches do not appropriately represent events that occur in vivo, that additional proteins are required to mediate the cooperative effects of the AHR/ARNT heterodimer observed in vivo, or that the gene activation cooperatively arises from a non-DNA binding mechanism.
In summary, we have identified amino acids residing within the basic region of the AHR that play an important role in maintaining stability of the AHR/ARNT heterodimer and, in this manner, can dictate its ability to activate genes. Substitutions of these amino acids with glutamine residues alter the ability of the AHR/ARNT heterodimer to bind DNA by decreasing thermodynamic stability of the complex.
ACKNOWLEDGMENTS
We thank Dr. Pollenz for the AHR and ARNT antibodies (University of South Florida Medical Center) and Dr. Steven Post and Dr. Peter Spielmann for advice on the binding analyses. This work was supported by the National Institute of Environmental Health, grants ES 08088 (H.I.S.) and ES-07266 (S.C.W.).
REFERENCES
- 1. Abnet C. C.; Tanguay R. L.; Hahn M. E.; Heideman W.; Peterson R. E. Two forms of aryl hydrocarbon receptor type 2 in rainbow trout (Oncorhynchus mykiss). Evidence for differential expression and enhancer specificity. J. Biol. Chem. 274:15159–15166; 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Atchley W. R.; Wollenberg K. R.; Fitch W. M.; Terhalle W.; Dress A. W. Correlations among amino acid sites in bHLH protein domains: An information theoretic analysis. Mol. Biol. Evol. 17:164–178; 2000. [DOI] [PubMed] [Google Scholar]
- 3. Bacsi S. G.; Hankinson O. Functional characterization of DNA-binding domains of the subunits of the heterodimeric aryl hydrocarbon receptor complex imputing novel and canonical basic helix–loop–helix protein–DNA interactions. J. Biol. Chem. 271:8843–8850; 1996. [DOI] [PubMed] [Google Scholar]
- 4. Chan W. K.; Chu R.; Jain S.; Reddy J. K.; Bradfield C. A. Baculovirus expression of the Ah receptor and Ah receptor nuclear translocater. Evidence for additional dioxin responsive element-binding species and factors required for signaling. J. Biol. Chem. 269:26464–26471; 1994. [PubMed] [Google Scholar]
- 5. Chapman-Smith A.; Lutwyche J. L.; Whitelaw M. L. Contribution of the Per/Arnt/Sim (PAS) domains to DNA dinding by the basic helix-loop-helix PAS transcriptional regulators. J. Biol. Chem. 279:5353–5362; 2004. [DOI] [PubMed] [Google Scholar]
- 6. Denison M. S.; Nagy S. R. Activation of the aryl hydrocarbon receptor by structurally diverse exogenous and endogenous chemicals. Annu. Rev. Pharmacol. Toxicol. 43:309–334; 2003. [DOI] [PubMed] [Google Scholar]
- 7. Dolwick K. M.; Schmidt J. V.; Carver L. A.; Swanson H. I.; Bradfield C. A. Cloning and expression of a human Ah receptor cDNA. Mol. Pharmacol. 44:911–917; 1993. [PubMed] [Google Scholar]
- 8. Dolwick K. M.; Swanson H. I.; Bradfield C. A. In vitro analysis of Ah receptor domains involved in ligand-activated DNA recognition. Proc. Natl. Acad. Sci. USA 90:8566–8570; 1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Dong L.; Ma Q.; Whitlock J. P. Jr. DNA binding by the heterodimeric Ah receptor. Relationship to dioxin-induced CYP1A1 transcription in vivo. J. Biol. Chem. 271:7942–7948; 1996. [DOI] [PubMed] [Google Scholar]
- 10. Elferink C. J.; Whitlock J. P. Jr. 2,3,7,8-Tetrachlorodibenzo-p-dioxin-inducible, Ah receptor-mediated bending of enhancer DNA. J. Biol. Chem. 265:5718–5721; 1990. [PubMed] [Google Scholar]
- 11. Elizondo G.; Fernandez-Salguero P.; Sheikh M. S.; Kim G.-Y.; Fornace A. J.; Lee K. S.; Gonzalez F. J. Altered cell cycle control at the G2/M phases in aryl hydrocarbon receptor-null embryo fibroblast. Mol. Pharmacol. 57:1056–1063; 2000. [PubMed] [Google Scholar]
- 12. Ferre-D’Amare A. R.; Prendergast G. C.; Ziff E. B.; Burley S. K. Recognition by Max of its cognate DNA through a dimeric b/HLH/Z domain. Nature 363:38–45; 1993. [DOI] [PubMed] [Google Scholar]
- 13. Ferre-D’Amare A. R.; Prognonec P.; Roeder R. G.; Burley S. K. Structure and function of the b/HLH/Z domain of USF. EMBO J. 13:180–189; 1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Fisher J. M.; Denison M. S.; Whitlock J. P. Jr. Organization and function of a dioxin-responsive enhancer. J. Biol. Chem. 265:9676–9681; 1990. [PubMed] [Google Scholar]
- 15. Fukunaga B. N.; Probst M. R.; Reisz-Porszasz S.; Hankinson O. Identification of functional domains of the aryl hydrocarbon receptor. J. Biol. Chem. 270:29270–29278; 1995. [DOI] [PubMed] [Google Scholar]
- 16. Fukunaga B. N.; Hankinson O. Identification of a novel domain in the aryl hydrocarbon receptor required for DNA binding. J. Biol. Chem. 271:3743–3749; 1996. [DOI] [PubMed] [Google Scholar]
- 17. Henry E. C.; Rucci G.; Gasiewicz T. A. Purified to homogenetity of the heteromeric DNA-binding form of the aryl hydrocarbon receptor from rat liver. Mol. Pharmacol. 46:1022–1027; 1994. [PubMed] [Google Scholar]
- 18. Ikuta T.; Eguchi H.; Tachibana T.; Yoneda Y.; Kawajiri K. Nuclear localization and export signals of the human aryl hydrocarbon receptor. J. Biol. Chem. 273:2895–2904; 1998. [DOI] [PubMed] [Google Scholar]
- 19. Karchner S. I.; Powell W. H.; Hahn M. E. Identification and functional characterization of two highly divergent aryl hydrocarbon receptors (AHR1 and AHR2) in the teleost Fundulus heteroclitus. Evidence for a novel subfamily of ligand-binding basic helix loop helix-Per-ARNT-Sim (bHLH-PAS) factors. J. Biol. Chem. 274:33814–33824; 1999. [DOI] [PubMed] [Google Scholar]
- 20. Kolluri S. K.; Weiss C.; Koff A; Gottlicher M. P27(Kip1) induction and inhibition of proliferation by the intracellular Ah receptor in developing thymus and hepatoma cells. Genes Dev. 13:1742–1753; 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lees M. L.; Whitelaw M. L. Multiple roles of ligand in transforming the dioxin receptor to an active basic helix–loop–helix/PAS transcription factor complex with the nuclear protein Arnt. Mol. Cell. Biol. 19:5811–5822; 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Levine S. L.; Petrulis J. R.; Dubil A.; Perdew G. H. A tetratricopeptide repeat half-site in the aryl hydrocarbon receptor is important for DNA binding and transactivation potential. Mol. Pharmacol. 58:1517–1524; 2000. [DOI] [PubMed] [Google Scholar]
- 23. Levine-Fredman A.; Chen L.; Elferink C. J. Cytochrome P4501A1 promotes G1 phase cell cycle progression by controlling aryl hydrocarbon receptor activity. Mol. Pharmacol. 65:461–469; 2004. [DOI] [PubMed] [Google Scholar]
- 24. Ma P. C.; Rould M. A.; Weintraub H.; Pabo C. O. Crystal structure of MyoD bHLH domain-DNA complex: Perspectives on DNA recognition and implications for transcriptional activation. Cell 7:451–459; 1998. [DOI] [PubMed] [Google Scholar]
- 25. Massaad C.; Coumoul X.; Sabbah M.; Garlatti M.; Redeuilh G.; Barouki R. Properties of overlapping EREs: synergistic activation of transcription and cooperative binding or ER. Biochemistry 37:6023–6032; 1998. [DOI] [PubMed] [Google Scholar]
- 26. Powell-Coffman J. A.; Bradfield C. A.; Wood W. B. Caenorhabditis elegans orthologs of the aryl hydrocarbon receptor and its heterodimerization partner the aryl hydrocarbon receptor nuclear translocator. Proc. Natl. Acad. Sci. USA 95:2844–2849; 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Puga A.; Xia Y.; Elferink C. Role of the aryl hydrocarbon receptor in cell cycle regulation. Chem. Biol. Interact. 141:117–130; 2002. [DOI] [PubMed] [Google Scholar]
- 28. Ray S. S.; Swanson H. I. Alteration of keratinocyte differentiation and senescence by the tumor promoter dioxin. Toxicol. Appl. Pharmacol. 192:131–145; 2003. [DOI] [PubMed] [Google Scholar]
- 29. Ray S. S.; Swanson H. I. Dioxin-induced immortalization of normal human keratinocytes and silencing of p53 and p16INK4a. J. Biol. Chem. 279:27187–27193; 2004. [DOI] [PubMed] [Google Scholar]
- 30. Reid K. J.; Hendy S. C.; Saito J.; Sorensen P.; Nelson C. C. Two classes of androgen receptor elements cooperativity through allosteric interactions. J. Biol. Chem. 276:2943–2952; 2001. [DOI] [PubMed] [Google Scholar]
- 31. Senear D. F.; Ross J. B. A.; Laue T. M. Analysis of protein and DNA-mediated contributions to cooperative assembly of protein-DNA complexes. Methods 16:3–20; 1998. [DOI] [PubMed] [Google Scholar]
- 32. Stein G. S.; Lian J. B.; van Wijnen A. J.; Stein J. L.; Javed A.; Montecino M.; Zaidi S. K.; Young D.; Choi J.-Y.; Gutierrez S.; Pockwinse S. Nuclear microenvironments support assembly and organization of the transcriptional regulatory machinery for cell proliferation and differentiation. J. Cell. Biochem. 91:287–302; 2004. [DOI] [PubMed] [Google Scholar]
- 33. Sun W.; Zhang J.; Hankinson O. A mutation in the aryl hydrocarbon receptor (AHR) in a cultured mammalian cell line identifies a novel region of AHR that affects DNA binding. J. Biol. Chem. 272:31845–31854; 1997. [DOI] [PubMed] [Google Scholar]
- 34. Swanson H. I.; Chan W. K.; Bradfield C. A. DNA binding specificities and pairing rules of the Ah receptor, ARNT, and SIM proteins. J. Biol. Chem. 270:26292–26302; 1995. [DOI] [PubMed] [Google Scholar]
- 35. Swanson H. I.; Yang J. H. Mapping the protein/DNA contact sites of the Ah receptor and Ah receptor nuclear translocator. J. Biol. Chem. 271:31657–31665; 1996. [DOI] [PubMed] [Google Scholar]
- 36. Travers A. Activation by cooperating conformations. Curr. Biol. 8:616–618; 1998. [DOI] [PubMed] [Google Scholar]
- 37. Tsai S. Y.; J-Tsai M.; O’Malley B. W. Cooperative binding of steroid hormone receptors contributes to transcriptional synergism at target enhancer elements. Cell 57:443–448; 1989. [DOI] [PubMed] [Google Scholar]
- 38. Vitola S. J.; Wang A.; Sun W.-H. Substitution of basic amino acids in the basic region stabilizes DNA binding by E12 homodimers. Nucleic Acids Res. 24:1921–1927; 1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Weintraub H.; Davis R.; Locksho D.; Lassar A. MyoD binds cooperatively to two sites in a target enhancer sequence: Occupancy of two sites is required for activation. Proc. Natl. Acad. Sci. USA 87:5623–5627; 1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Whitelaw M. L.; Gustafsson J. A.; Poellinger L. Identification of transactivation and repression functions of the dioxin receptor and its basic helix–loop–helix/PAS partner factor Arnt: Inducible versus constitutive modes of regulation. Mol. Cell. Biol. 14:8343–8355; 1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Whitlock J. P. Jr. Induction of cytochrome P4501A1. Annu. Rev. Toxicol. 39:103–125; 1999. [DOI] [PubMed] [Google Scholar]
- 42. Yang E.; Henriksen M. A.; Schaefer O.; Zakharova N.; Darnell J. E. Dissociation time from DNA determines transcriptional function in a STAT1 linker mutant. J. Biol. Chem. 277:13455–13462; 2002. [DOI] [PubMed] [Google Scholar]