

Abstract
Miniature inverted-repeat transposable elements (MITEs) are widespread and abundant in both plant and animal genomes. Despite the discovery and characterization of many MITE families, their origin and transposition mechanism are still poorly understood, largely because MITEs are nonautonomous elements with no coding capacity. The starting point for this study was P instability factor (PIF), an active DNA transposable element family from maize that was first identified following multiple mutagenic insertions into exactly the same site in intron 2 of the maize anthocyanin regulatory gene R. In this study we report the isolation of a maize Tourist-like MITE family called miniature PIF (mPIF) that shares several features with PIF elements, including identical terminal inverted repeats, similar subterminal sequences, and an unusual but striking preference for an extended 9-bp target site. These shared features indicate that mPIF and PIF elements were amplified by the same or a closely related transposase. This transposase was identified through the isolation of several PIF elements and the identification of one element (called PIFa) that cosegregated with PIF activity. PIFa encodes a putative protein with homologs in Arabidopsis, rice, sorghum, nematodes, and a fungus. Our data suggest that PIFa and these PIF-like elements belong to a new eukaryotic DNA transposon superfamily that is distantly related to the bacterial IS5 group and are responsible for the origin and spread of Tourist-like MITEs.
Transposable elements (TEs) have been divided into two classes according to their transposition intermediate. Class 1 (RNA) elements transpose by means of an RNA intermediate and most have either long terminal repeats (LTR-retrotransposons) or terminate at one end with a poly(A) tract (LINEs and SINEs). Class 2 (DNA) elements transpose by means of a DNA intermediate and usually have terminal inverted repeats (TIRs). In eukaryotes, class 2 families, such as the maize Ac/Ds or the Drosophila P elements, consist of autonomous and nonautonomous members. Autonomous elements encode transposase that binds to cis-acting sequences residing in the terminal regions of both autonomous and nonautonomous elements to catalyze their transposition (for review, see ref. 1). Nonautonomous elements usually arise from autonomous elements by point mutations and/or internal deletion(s). Integration of most TEs results in a duplication of the target site, so that each element is flanked by a target site duplication (TSD) of conserved length and sometimes sequence (1).
Miniature inverted-repeat transposable elements (MITEs) are a recently described group of TEs that have been found in a wide range of plants and animals (2–10). In plants, the majority of characterized MITE families can be divided into two groups based on similarity of their TIRs and TSDs: there are Tourist-like MITEs and Stowaway-like MITEs. Despite the abundance of MITEs in many genomes (≈2% of Caenorhabditis elegans and ≈6% of rice), their origin and transposition mechanism remains poorly understood (11–13). All MITE families have a suite of common structural features including high copy number (≈500–10,000 per haploid genome), conserved within-family length (<500 bp), and sequence and target site preference. The fact that many MITE families share their TIRs, TSDs, and, in one case, even internal sequences with larger TEs encoding transposases has been interpreted to mean that MITEs originated from autonomous DNA elements (6, 9, 10, 14, 15).
To date, no MITE family has been shown to be actively transposing. In the absence of activity, it has been difficult to determine how MITEs are generated and how they attain such high copy numbers. For this reason the focus of this study is an actively transposing family of class 2 elements from maize called P instability factor (PIF). PIF elements were first discovered as six independent insertions into exactly the same site in intron 2 of the maize R gene (Fig. 1a; ref. 16). These six elements inserted in both orientations and fell into two structural classes, referred to as PIF-6 (5.2 kb) and PIF-12 (2.3 kb). Of particular interest was the finding that PIF was related to a 364-bp MITE-like sequence that appeared to have inserted into another maize TE (16). In this study we demonstrate that this 364-bp sequence is the founding member of a Tourist-like MITE family called miniature PIF (mPIF). In addition to their sequence similarities, mPIF and PIF elements insert into a sequence-specific 9-bp palindrome. The structure of the PIF family was further investigated through the isolation of several family members including the putative autonomous PIF element (PIFa). PIFa-like elements were identified by database searches in rice and Arabidopsis, as well as nematodes and a fungus. These data provide evidence for a superfamily of elements that may be responsible for the amplification of Tourist-like MITEs in the genomes of plants and animals.
Figure 1.

Target site preference of PIF and mPIF elements. (a) Six PIF elements inserted independently into exactly the same position in the second intron of the maize R gene (16). Triangles represent PIF TIRs and black rectangles represent exons 2 and 3 of R. (b) Consensus extended target site derived from a comparison of the sequences flanking 30 mPIF elements. (c) Consensus extended target site derived from a comparison of the sequences flanking 14 PIF elements. Gray rectangles indicate the trinucleotide duplicated on element insertion (the TSD). Numbers represent the percentage of times that a nucleotide appeared at that position.
Materials and Methods
Genetic Stocks, DNA Extraction, and Library Construction.
All strains were derived from the maize inbred W22.
r-sc:124Y2902 is a derivative of R-sc:124 (R allele conferring pigmentation of aleurone, embryo, and coleoptile) with a 2.3-kb PIF insertion in the second intron of the Sc component (16) causing loss of kernel pigmentation. Excision restores kernel pigmentation.
r-g:14qs131 is a derivative of R-r:standard that contains only the P component (R gene that confers pigmentation of roots, coleoptiles, seedling leaf tips, and anthers). Insertion of a 5.2-kb PIF into intron 2 (16) eliminates pigmentation in these tissues while element excision restores color.
Stable 2 is a PIF-inactive strain homozygous for r-sc:124Y2902 (provided by J. Kermicle, Univ. of Wisconsin), derived as follows: a PIF-active strain homozygous for r-sc:124Y2902 was crossed to a strain homozygous for r-r (R allele conditioning colorless kernels and colored plants) and several resulting ears were found to have very few or no spotted or solidly pigmented kernels, indicating low or no PIF activity. Seeds from each ear were grown and self-pollinated, and PIF-inactive strains homozygous for the r-sc:124Y2902 chromosomes obtained. Stable 2 is one such strain that lost PIF activity, as no spotted kernels were observed above background when it was self-pollinated. However, spotted kernels were readily observed at normal frequency when Stable 2 was crossed to strains with PIF activity.
Strain R is a PIF-active strain homozygous for the r-g:14qs131 allele.
Plant DNA was extracted from young leaves as described (17). The small insert genomic library was constructed from strain R as described (18).
Generation of a Population Segregating for PIF Activity.
Stable 2 (r-sc:124Y2902, PIF-inactive, see above) was crossed with strain R (homozygous for r-g:14qs131, PIF-active) to produce a population of plants called SR (PIF-active). Spotted kernels from this population (due to somatic excision of the PIF element from r-sc:124Y2902) were grown and crossed to Stable 2 to obtain a population (called SRS) segregating for PIF activity. Fifteen SR and 28 SRS plants were generated from spotted kernels and 13 Stable 2 plants were generated from unpigmented kernels. DNA was extracted from young leaves and analyzed by transposon display (see below).
Transposon Display and Recovery of Gel Bands.
TD was performed as described (19) with the following modifications. PIF-specific PCR primers (PR1, PR2, PF1, and PF2) were derived from the PIF subterminal sequences to specifically amplify the flanking sequences of PIF but not mPIF elements (primer sequences available on request). The primer combinations used were: PR2 and MseI+0 for 5′ end preselective amplification, PR1 (labeled with 33P) and MseI+0 for 5′ end selective amplification, PF2 and BfaI+0 for 3′ end preselective amplification, PF1 (labeled with 33P) and BfaI+0 for 3′ end selective amplification. The final annealing temperature was 55°C (PCR cycle parameters available on request). Radioactive PCR products were recovered from polyacrylamide gels as described (http://tto.biomednet.com) and amplified by PCR with the same primers and under the same conditions as those used for the respective TD selective amplifications.
PCR Amplification and Sequencing of PIF Elements.
PIF0.4, PIF1.1, PIF1.6, and PIF1.7 were amplified from total genomic DNA by PCR using Taq DNA polymerase (Perkin–Elmer) with primers derived from the PIF subterminal sequences such that they would not amplify mPIF elements. Longer PIF elements were amplified by using Elongase (GIBCO/BRL) under conditions that favor the production of long products (20) with primers derived from PIF sequences internal to the PIF0.4 deletion breakpoints (Fig. 2). Amplification of the PIFa element used primers derived from flanking genomic sequences (PCR cycle parameters and primer sequences available on request).
Figure 2.

Schematic representation of the structure of the PIF transposon family. Elements are named according to their length and are drawn to scale. Only one element from each subfamily is shown. PIF5.2 is previously described as PIF-6 and PIF2.3 is 98% identical to PIF-12 (16). Black triangles represent TIRs. Green and red rectangles represent the terminal sequences conserved in all elements (see text). Open rectangle indicates the fact that the internal region of PIF5.2 was not sequenced. Dark blue, pink, brown, light green, purple, and light blue rectangles represent internal regions unique to each subfamily. Yellow rectangle represents the internal region of mPIF.
PCR products were cloned by using the TA Cloning Kit according to manufacturer's instructions (Invitrogen). All sequencing reactions were performed by the Molecular Genetics Instrumentation Facility of the Univ. of Georgia. The sequences for 32 mPIFs and PIFa were deposited in the GenBank database (accession nos. AF416298–AF416329 and AF412282, respectively).
Computational Analysis.
GenBank database searches were performed with the various blast servers available from the National Center for Biotechnology Information (http://www.ncbi.nlm.nih.gov). The gene structures of PIFa and PIF-like elements were predicted by the NETGENE2 (http://www.cbs.dtu.dk; ref. 21), NETSTART 1.0 (http://www.cbs.dtu.dk; ref. 22), and FGENESH (http://genomic.sanger.ac.uk/gf/gf.html; ref. 23) programs. Protein sequences were obtained from GenBank or by conceptual translation of predicted genes, and aligned by using CLUSTALX (24). Phylogenetic analysis was carried out with PAUP* Version 4.0b8 (25), using both the neighbor-joining and maximum parsimony methods with default parameters.
Results and Discussion
mPIF Is a MITE Family.
Several features of the previously identified 364-bp PIF-related sequence including short TIRs and a 3-bp TSD rich in A and T residues were reminiscent of MITEs. Southern blot analysis confirmed that this sequence was highly repetitive in maize but not in sorghum or rice (data not shown). To estimate the copy number of related elements in the maize genome and to isolate more copies for analysis, a genomic library (average insert size 1.5 kb) was prepared from maize inbred line W22 and screened with the 364-bp sequence. The hybridization of 369 plaques of 1.1 × 105 screened (representing ≈1.6 × 105 kb or ≈6% of the genome) provided an estimate of ≈6 × 103 copies of this sequence per haploid genome (369/6% = 6,150). In contrast, the copy number of the larger PIF elements was estimated by Southern blot analysis to be ≈25 (W. B. Eggleston, unpublished data).
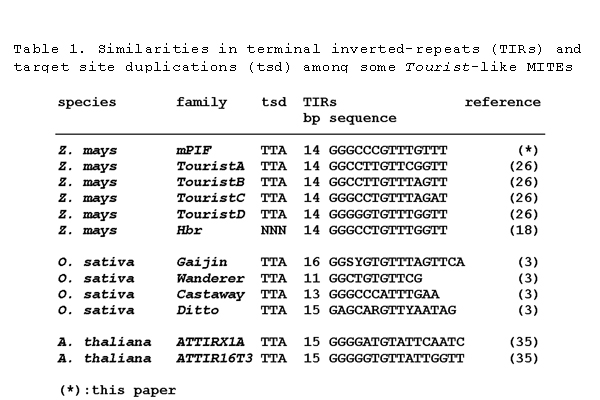
Thirty-two of the 369 positive plaques were randomly chosen for further analysis. Thirty of the 32 contained complete elements that were, on average, 358 bp, had perfect 14-bp TIRs, and displayed over 90% sequence identity. All elements were rich in A and T residues (71%) and had no significant coding capacity. Twenty-eight of the 30 full-length elements were flanked by a conserved 3-bp TSD (TTA/TAA). We named this new MITE family miniature PIF (mPIF). The 32 mPIF genomic sequences were deposited in GenBank (accession nos. AF416298–AF416329). Based on the TSD and TIR sequences, mPIF can be classified as a typical Tourist-like MITE family (see Table 1, which is published as supporting information on the PNAS web site, www.pnas.org; ref. 26). Comparison between the consensus mPIF sequence and previously characterized PIF elements (16) reveals identical TIRs and similar subterminal sequences extending for ≈100 bp from the termini (overall similarity of ≈70%). The most internal 150 bp of mPIF elements was not related to PIF elements.
Identical Extended Target Site Preference for mPIF and PIF Elements.
The insertion of six of the larger PIF elements into exactly the same site in the R gene prompted us to examine whether mPIF and PIF insertion sites were conserved beyond the TSD. Comparison of the sequences flanking the 30 full-length mPIF elements revealed remarkable conservation of an extended 9-bp target site centered on the TSD (Fig. 1b). Significantly, this sequence matches the insertion site in the R gene.
To determine whether the larger PIF elements have the same target site preference, sequences flanking some of the other ≈25 PIF elements in the genome were recovered by using the TD procedure. TD is a modification of the Amplified Fragment Length Polymorphism procedure (19, 27) that generates PCR products anchored in a transposon and a flanking restriction site (see Materials and Methods). To this end, PCR primers were designed to amplify genomic sequences flanking PIF (and not mPIF) termini. Approximately 50 PCR products, 25 from each end, were displayed after gel electrophoresis (Fig. 3). This corresponds to about 25 PIF elements, which is in agreement with the prior copy number determination (W. B. Eggleston, unpublished data). A total of 14 PCR products were recovered, sequenced, and used to derive a consensus target site that was found to be identical for both mPIF and PIF elements (Fig. 1c).
Figure 3.

Transposon display (TD) analysis of a population segregating for PIF activity. Only a subset of the population analyzed by TD is shown. PIF TD was carried out from both the 5′ end (left half of gel) and the 3′ end (right half of gel). Arrowheads indicate PCR products that cosegregate with activity. Open arrowhead indicates PCR products that did not cosegregate with activity in other plants (not shown). SR, plants heterozygous for the autonomous PIF element; Stable 2, plants without PIF activity; SRS, PIF-active plants from the cross between SR and Stable 2 (see Materials and Methods for details); M, 30- to 330-bp molecular weight marker. A schematic representation indicating the positions of the PCR primers is also shown. Arrows represent PCR primers and stars indicate primers labeled with 33P; black rectangles represent BfaI or MseI adapters and hatched rectangles represent terminal regions conserved in all sequenced PIF elements.
Extended target site preference has been reported for several bacterial transposons (28, 29) and there is evidence that some eukaryotic class 2 elements may have some preference beyond the TSD (30, 31). However, to our knowledge, PIFs and mPIFs display the longest and most specific target site preference ever documented among eukaryotic class 2 TEs. Additional support for the existence of a specific 9-bp insertion site comes from the fact that the sequences flanking mPIF elements judged to have inserted most recently (based on highest sequence identity to the mPIF consensus and insertion site polymorphism among maize strains) are most similar to the consensus target sequence (data not shown). What is particularly surprising is that despite targeting such a specific insertion site, mPIF elements still managed to attain a higher copy number than virtually all other characterized class 2 elements. Given that a 9-bp sequence is expected to occur, on average, about once in 250 kb, ≈10,000 copies of this sequence are predicted to be in the maize genome. It is remarkable that most of these sites may be occupied by mPIF elements.
Structure of PIF Family Members.
Because target site preference has been shown, in a few cases, to be a function of the transposase (28, 32), the existence of a common 9-bp target for both mPIF and PIF elements strongly suggests that their transposition reactions are catalyzed by the same or a closely related transposase. For this reason, it was thought that isolation of additional PIF elements might lead to the isolation of the autonomous element responsible for the origin and amplification of both mPIF and PIF elements.
The two PIF elements at the R locus (PIF5.2 and PIF2.3) are nonautonomous elements that only share their terminal sequences (Fig. 2; ref. 16). To isolate additional PIF family members, PCR primers derived from PIF sequences internal to the TIRs were used to amplify genomic DNA. Primers were designed to amplify PIF but not mPIF elements. The predominant PCR product was of 483 bp and was found to be a deletion derivative of a longer PIF element (PIF0.4). Three other products of 1.1 kb, 1.6 kb, and 1.7 kb were also cloned and sequenced. To isolate longer elements that may not have competed successfully in the initial PCR reactions, primers derived from sequences internal to the deletion breakpoint of PIF0.4 were used, along with PCR conditions that favor the production of longer products. This procedure led to the isolation of eight additional PIF elements ranging from ≈1.1 kb to ≈5.2 kb, of which four were completely sequenced. All of the elements (except PIF0.4) are highly conserved (>90%) in their terminal regions; however, the internal sequences are dissimilar and serve to distinguish distinct subfamilies (Fig. 2). Unfortunately, none of these elements were considered autonomous, because computer analysis failed to detect significant coding capacity or any similarity to known transposases.
Isolation of the PIFa Element.
A genetic approach to isolate an autonomous PIF element was used, involving the application of transposon display to a population segregating for PIF activity (see Materials and Methods). Genomic DNA from plants grown from spotted kernels (+PIF activity) and colorless kernels (−PIF activity) were analyzed by using primers facing outward from the PIF termini (Fig. 3). Only one product from each end cosegregated with PIF activity. The sequences derived from these products were used to design PCR primers from the genomic sequences adjacent to the PIF termini (20 bp and 25 bp from the 5′ and 3′ termini, respectively) and used in a single reaction to amplify genomic DNA (Fig. 4a). One product of 3.7 kb was amplified from the PIF active but not the PIF inactive plants, thus confirming that the cosegregating TD products were derived from sequences flanking the same element (designated PIFa; Figs. 2 and 4a).
Figure 4.

PIFa is present in PIF-active plants but absent from PIF-inactive plants. Agarose gels of PCR products are shown. A + or − indicates the presence or absence, respectively, of PIF activity in the strains used for genomic DNA isolation. (a) Amplification of the entire PIFa element by using primers derived from flanking genomic sequences. A 3.7-kb product was obtained from PIF-active (SR, lane 1, and SRS, lane 3), but not from PIF-inactive (Stable 2, lane 2) plants (see Materials and Methods for strain designations). (b) Amplification of genomic DNA from the PIFa insertion site. Products of the appropriate size (≈900 bp) were obtained from PIF-active plants that have served as the progenitors for the PIF-inactive Stable 2 (lanes 3–6), but not from 12 Stable 2 plants grouped into two pools of six each (lanes 1 and 2). (c) PCR amplification of an internal region of PIFa not present in any other sequenced PIF element. Products of appropriate size (≈1.3 kb) were obtained from SR (lane 1), as well as two SRS plants that do not have PIFa at this locus (SRS15 and SRS31, lanes 2 and 3), suggesting that PIFa has transposed but may still be present in the genome. No product was obtained from a pool of 14 Stable 2 plants (lane 4). Arrows represent the positions of PCR primers, triangles represent TIRs, and lines represent PIFa flanking sequences. M, molecular weight marker.
Additional evidence for the cosegregation of PIFa with PIF activity was obtained by carrying out amplification reactions with different primer pairs. Primers derived from the internal region of PIFa and from sequences flanking the PIFa insertion site should amplify a 900-bp product if PIFa is at the locus. A product of this size was obtained from four PIF active strains that had served as parents for progeny without PIF activity where, presumably, PIFa had been lost following meiosis (Fig. 4b, lanes 3–6). The absence of PIFa from PIF-inactive plants is indicated by the failure to amplify a 900-bp product from 12 plants whose DNA was grouped into two pools of six (Fig. 4b, lanes 1 and 2). Finally, two of the 28 PIF-active SRS plants did not have PIFa at the original locus, as determined by TD, possibly because PIFa had transposed to another site in the genome. To test whether this was the case, PCR primers were designed from an internal region of PIFa that is not present in other PIF elements. Amplification of DNA from these two active plants along with 14 inactive plants (derived from parents heterozygous for PIFa; see Materials and Methods) confirmed the presence of PIFa in the former but not the latter (Fig. 4c). This result also demonstrated that the loss of PIFa correlated with the loss of PIF activity.
PIF Is Member of a Superfamily of DNA Transposons.
The sequence of PIFa revealed a 3,728-bp element that, like all other PIF elements, contains the conserved terminal regions (Fig. 2). The central 2.5 kb, not found in other PIF elements, contains two ORFs longer than 100 aa (Fig. 5a). Only the first ORF (313 aa) produced significant hits (E value > 10−15, with sequences from Arabidopsis and rice BACs and with two Sorghum bicolor entries) when used as a query in TBLASTN searches with translated sequences (complete list available on request). Amino acid identities among these sequences range from 25–50% (45–65% similarity) over 100–250 aa tracts. Further TBLASTN searches with some plant products and multiple iterations with PSI-BLAST also uncovered significant similarity with two putative proteins from C. elegans, one from its close relative C. briggsae, and one from the basidiomycete fungus F. neoformans (see Fig. 5 legend for accession nos.). Finally, limited but significant homology was detected with several transposases encoded by bacterial insertion sequences of the IS5 group (see Fig. 6, which is published as supporting information on the PNAS web site; ref. 29).
Figure 5.

The PIF-IS5 superfamily of transposons. (a) Structure and coding capacity of PIFa and several PIF-like elements. ORFs larger than 100 aa are schematically depicted as hatched rectangles. The predicted intron/exon structure is shown, as is the putative initiation codon (indicated by i). TIRs are represented by black triangles. Rectangles shaded in gray represent ORFs sharing significant similarity (i.e., PIF-like transposases). Other ORFs are not related, although the At-PIF2 downstream gene can encode a protein that has several paralogs in the Arabidopsis genome. However, these paralogous sequences are not associated with a PIF-like transposase (data not shown). In addition, Os-PIF1 and Cb-PIF1 contain nested insertions of a variety of repetitive sequences, thus making it difficult to unambiguously determine element length. For this reason, the length shown for these PIF-like elements is approximate. Species, GenBank accession numbers, and coordinates are: Zm-PIFa, Z. mays AF412282; Os-PIF1, Oryza sativa AC025098, 101769–109139; Os-PIF2, O. sativa AP01111, 2889–7665; At-PIF2, Arabidopsis thaliana TM021B04, 16996–21224; CbPIF1, Caenorhabditis briggsae AC090524, 69398–71455; Fn-PIF1, Filobasidiella neoformans AC068564, 3620–6989. (b) Putative TSD and terminal inverted-repeats (TIRs, size in bp) of PIF-like elements. (c) Phylogenetic relationship of putative PIF-like proteins and IS5 transposases. The unrooted tree was constructed with the neighbor-joining method from a clustalx alignment, which includes the complete product conceptually translated from the largest ORF of Zm-PIFa (313 aa), various eukaryotic homologs identified by database searches and several representatives of the IS5 group of transposases (ref. 29; see Fig. 6). Bootstrap values (1,000 replicates) support the grouping of the plant, nematode, and bacterial proteins. Ce-PIF2 is identical to the product recently reported as the Tc8.1 putative transposase by Le et al. (2001). Species and GenBank accession numbers are: At-PIF1, A. thaliana AB017067; Ce-PIF1, C. elegans CEF57G4; Ce-PIF2, C. elegans CELF14D2; IS5, Escherichia coli J01735; ISL2, Lactobacillus helveticus X77332; IS702, Calothrix sp. X60384; IS470, Streptomyces lividans AB032065; IS493, S. lividans M28508.
The evolutionary relationship among these proteins was analyzed by aligning the translated product from the complete PIFa ORF (313 aa) with other PIF-like putative proteins identified by database searches and generating phylogenetic trees. A CLUSTALW multiple alignment (Fig. 6) revealed several well conserved amino acid blocks, most notably among the plant products. Both the neighbor-joining and parsimony methods produced trees with similar topologies (Fig. 5c). Bacterial transposases and eukaryotic homologs group separately, whereas plant and nematodes products form distinct monophyletic clades within the eukaryotic sequences. Nonetheless, branch lengths between and within kingdoms indicate that there is extensive diversity in this protein superfamily (Fig. 5c).
To determine whether the PIF-like coding sequences were part of TEs, sequences flanking these hits were searched for structural features reminiscent of transposons. Several Arabidopsis and rice ORFs, as well as the C. briggsae ORF, are flanked by inverted repeats (IRs) that share significant sequence similarity with the maize PIF TIRs (Fig. 5b). In addition, these IRs, like PIF TIRs, are flanked by a direct repeat of the TTA trinucleotide. Furthermore, BLAST searches reveal that each of these PIF-like elements belongs to a repeat family in their respective genomes (called At-PIF, Os-PIF, and Cb-PIF, respectively) where they display high intrafamily sequence similarities (>90%). Interestingly, many PIF-like family members are short internally deleted copies of homogeneous size that resemble mPIF and other MITEs (see Fig. 7, which is published as supporting information on the PNAS web site). All of these MITEs are Tourist-like in that they possess TIRs similar to some of the previously described Tourist elements and are flanked by a 3-bp A/T-rich sequence that is probably the TSD.
Several features shared by PIF and PIF-like elements strongly suggest that together they represent a new superfamily of eukaryotic DNA transposons that arose from a common ancestor. These features include their homologous coding regions, as well as TIRs of similar length and sequence shared by all plant PIF-like elements. In addition, all of the PIF-like elements identified in this study generate a 3-bp TSD and, in all but one case, the duplication is TTA (it is AAT for the F. neoformans element). Consensus extended target sites cannot be derived for the PIF-like elements because of the small number of elements identified by database searches. However, because the length and sequence of the TSDs are functions of the transposase (28, 29, 33, 34), the similarities noted among the PIF-like elements suggest that their transposases are related not only evolutionarily, but also functionally.
As mentioned above, coding regions shared by PIF-like elements are also related to the transposases encoded by the IS5 group of bacterial insertion sequences (Fig. 5c). Interestingly, many IS5 elements also create 3-bp TSDs on insertion [e.g., subgroups ISL2, IS427, and IS1031 (29)] and some display a preference for TNA targets (e.g., subgroup IS1031). Moreover, IS1031A from Acetobacter xylinum has an extended target preference for the motif TCTNAR, with TNA being duplicated (29). This consensus matches that of PIF elements. Taken together, these data support the view that PIF-like elements belong to a new eukaryotic DNA transposon superfamily that is distantly related to the bacterial IS5 group.
PIF-like elements belong to the same superfamily as Harbinger, a previously identified sequence that was discovered as part of an extensive search for repeats in the Arabidopsis genome (35). Our database searches indicate that Harbinger represents only one of the multiple PIF lineages present in the Arabidopsis genome (C.F., unpublished data). Kapitonov and Jurka (35) also reported similarities between the putative transposase of Harbinger and several hypothetical proteins from rice, sorghum, and C. elegans, as well as the transposases of IS5 elements. Based on these similarities, they proposed to classify Harbinger as a member of a new superfamily of DNA transposons. However, in their study, only Harbinger was characterized as a “bona fide” transposable element (i.e., with TIRs and other features of DNA elements). More recently, one of the putative IS5-related transposases identified by Kapitonov and Jurka (35) in C. elegans was shown to be part of a transposable element associated with Tourist-like MITE family members (36). Our results extend these findings by showing that IS5-related TE families are present in diverse eukaryotic organisms, including maize, rice, C. briggsae, and a fungus. Because the maize PIF was the first family identified in eukaryotes (16) and the only one with demonstrated activity, we propose to name this new superfamily of DNA transposons the PIF-IS5 superfamily.
Conclusions
The origin and spread of MITEs throughout plant and animal genomes largely remains a mystery despite the characterization of many MITE families and the availability of thousands of MITE sequences. A major reason for this is that MITEs are nonautonomous elements with no significant coding capacity. As such, associations between MITE families and potentially autonomous elements have, until this study, been restricted to computer-assisted searches for larger related elements in genomes that are completely sequenced like A. thaliana or C. elegans (15, 35–37). We call this the “bottom-up” approach because the sequences of nonautonomous family members are used as queries to identify potentially autonomous family members. The major limitation of this approach is that nothing is known of the genetic activity of the larger elements and hence of the entire TE family.
In contrast, the starting point for this study was PIF, an active class 2 TE family. Similarity between PIF elements and a 364-bp sequence led to the discovery of mPIF, a Tourist-like MITE family, the discovery of an unprecedented 9-bp palindromic target sequence for PIF and mPIF elements, and the identification of the putative autonomous PIFa, which encodes a transposase that is related to transposases encoded by other TEs in plant, animal, and bacterial genomes. We call this the “top-down” approach because a family of genetically active elements was used to identify a MITE family. The association of a MITE family with a genetically active system should ultimately furnish the biochemical tools necessary to address, experimentally, the larger questions regarding the origin and spread of MITEs.
Supplementary Material
Acknowledgments
We thank Dr. Jerry Kermicle for providing the maize strain Stable 2 and Drs. Kelly Dawe and Michael Scanlon for helpful discussions. This work was supported by a grant from the National Institutes of Health (to S.R.W.) and a Gant-in-Aid from Virginia Commonwealth University (to W.B.E.).
Abbreviations
- MITE
miniature inverted-repeat transposable element
- TE
transposable element
- TD
transposon display
- TIRs
terminal inverted repeats
- TSD
target site duplication
Footnotes
References
- 1.Capy P, Bazin C, Higuet D, Langin T. Dynamics and Evolution of Transposable Elements. Austin, TX: Landes; 1998. [Google Scholar]
- 2.Bureau T E, Wessler S R. Plant Cell. 1992;4:1283–1294. doi: 10.1105/tpc.4.10.1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bureau T E, Ronald P C, Wessler S R. Proc Natl Acad Sci USA. 1996;93:8524–8529. doi: 10.1073/pnas.93.16.8524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Casacuberta E, Casacuberta J M, Puigdomenech P, Monfort A. Plant J. 1998;16:79–85. doi: 10.1046/j.1365-313x.1998.00267.x. [DOI] [PubMed] [Google Scholar]
- 5.Morgan G T. J Mol Biol. 1995;254:1–5. doi: 10.1006/jmbi.1995.0593. [DOI] [PubMed] [Google Scholar]
- 6.Oosumi T, Garlick B, Belknap W R. J Mol Evol. 1996;43:11–18. doi: 10.1007/BF02352294. [DOI] [PubMed] [Google Scholar]
- 7.Tu Z. Proc Natl Acad Sci USA. 1997;94:7475–7480. doi: 10.1073/pnas.94.14.7475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Izsvak Z, Ivics Z, Shimoda N, Mohn D, Okamoto H, Hackett P B. J Mol Evol. 1999;48:13–21. doi: 10.1007/pl00006440. [DOI] [PubMed] [Google Scholar]
- 9.Feschotte C, Mouchès C. Gene. 2000;250:109–116. doi: 10.1016/s0378-1119(00)00187-6. [DOI] [PubMed] [Google Scholar]
- 10.Tu Z. Proc Natl Acad Sci USA. 2001;98:1699–1704. doi: 10.1073/pnas.041593198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Surzycki S A, Belknap W R. Proc Natl Acad Sci USA. 2000;97:245–249. doi: 10.1073/pnas.97.1.245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tarchini R, Biddle P, Wineland R, Tingey S, Rafalski A. Plant Cell. 2000;12:381–391. doi: 10.1105/tpc.12.3.381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mao L, Wood T C, Yu Y, Budiman M A, Tomkins J, Woo S, Sasinowski M, Presting G, Frisch D, Goff S, et al. Genome Res. 2000;10:982–990. doi: 10.1101/gr.10.7.982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Smit A F A, Riggs A D. Proc Natl Acad Sci USA. 1996;93:1443–1448. doi: 10.1073/pnas.93.4.1443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Feschotte C, Mouchès C. Mol Biol Evol. 2000;17:730–737. doi: 10.1093/oxfordjournals.molbev.a026351. [DOI] [PubMed] [Google Scholar]
- 16.Walker E L, Eggleston W B, Demopulos D, Kermicle J, Dellaporta S L. Genetics. 1997;146:681–693. doi: 10.1093/genetics/146.2.681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.McCouch S R, Kochert G, Yu Z H, Khush G S, Coffman W R, Tanksley S D. Theor Appl Genet. 1988;76:815–829. doi: 10.1007/BF00273666. [DOI] [PubMed] [Google Scholar]
- 18.Zhang Q, Arbuckle J, Wessler S R. Proc Natl Acad Sci USA. 2000;97:1160–1165. doi: 10.1073/pnas.97.3.1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Casa A M, Brouwer C, Nagel A, Wang L, Zhang Q, Kresovich S, Wessler S R. Proc Natl Acad Sci USA. 2000;97:10083–10089. doi: 10.1073/pnas.97.18.10083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Marillonnet S, Wessler S R. Genetics. 1998;150:1245–1256. doi: 10.1093/genetics/150.3.1245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hebsgaard S M, Korning P G, Tolstrup N, Engelbrecht J, Rouze P, Brunak S. Nucleic Acids Res. 1996;24:3439–3452. doi: 10.1093/nar/24.17.3439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pedersen A C, Nielsen H. Plant Mol Biol. 1997;5:226–233. [PubMed] [Google Scholar]
- 23.Salamov A A, Solovyev V V. Genome Res. 2000;10:516–522. doi: 10.1101/gr.10.4.516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Thompson J D, Gibson T J, Plewniak F, Jeanmougin F, Higgins D G. Nucleic Acids Res. 1997;25:4876–4882. doi: 10.1093/nar/25.24.4876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Swofford D L. PAUP*: Phylogenetic Analysis Using Parsimony (and other methods) Sunderland, MA: Sinauer; 1999. [Google Scholar]
- 26.Bureau T E, Wessler S R. Proc Natl Acad Sci USA. 1994;91:1411–1415. doi: 10.1073/pnas.91.4.1411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vos P, Hogers R, Bleeker M, Reijans M, van de Lee T, Hornes M, Frijters B A, Pot J, Peleman J, Kuiper M, Zabeau M. Nucleic Acids Res. 1995;23:4407–4414. doi: 10.1093/nar/23.21.4407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Craig N L. Annu Rev Biochem. 1997;66:437–474. doi: 10.1146/annurev.biochem.66.1.437. [DOI] [PubMed] [Google Scholar]
- 29.Mahillon J, Chandler M. Microbiol Mol Biol Rev. 1998;62:725–774. doi: 10.1128/mmbr.62.3.725-774.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ketting R F, Fischer S E, Plasterk R H. Nucleic Acids Res. 1997;25:4041–4047. doi: 10.1093/nar/25.20.4041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liao G C, Rehm E J, Rubin G M. Proc Natl Acad Sci USA. 2000;97:3347–3351. doi: 10.1073/pnas.050017397. . (First Published March 14, 2000; 10.1073/pnas.050017397) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pribil P A, Haniford D B. J Mol Biol. 2000;303:145–159. doi: 10.1006/jmbi.2000.4135. [DOI] [PubMed] [Google Scholar]
- 33.Beall E L, Rio D C. Genes Dev. 1997;11:2137–2151. doi: 10.1101/gad.11.16.2137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Plasterk R H A, Izsvák Z, Ivics Z. Trends Genet. 1999;15:326–332. doi: 10.1016/s0168-9525(99)01777-1. [DOI] [PubMed] [Google Scholar]
- 35.Kapitonov V V, Jurka J. Genetica. 1999;107:27–37. [PubMed] [Google Scholar]
- 36.Le Q H, Turcotte K, Bureau T. Genetics. 2001;158:1081–1088. doi: 10.1093/genetics/158.3.1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Le Q H, Wright S, Yu Z, Bureau T. Proc Natl Acad Sci USA. 2000;97:7376–7381. doi: 10.1073/pnas.97.13.7376. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}