

Abstract
Transposable elements (TEs) are a major driver of plant genome evolution. A part from being a rich source of new genes and regulatory sequences, TEs can also affect plant genome evolution by modifying genome size and shaping chromosome structure. TEs tend to concentrate in heterochromatic pericentromeric regions and their proliferation may expand these regions. Here, we show that after the split of melon and cucumber, TEs have expanded the pericentromeric regions of melon chromosomes that, probably as a consequence, show a very low recombination frequency. In contrast, TEs have not proliferated to a high extent in cucumber, which has small TE-dense pericentromeric regions and shows a relatively constant recombination rate along chromosomes. These differences in chromosome structure also translate in differences in gene nucleotide diversity. Although gene nucleotide diversity is essentially constant along cucumber chromosomes, melon chromosomes show a bimodal pattern of genetic variability, with a gene-poor region where variability is negatively correlated with gene density. Interestingly, genes are not homogeneously distributed in melon, and the high variable low-recombining pericentromeric regions show a higher concentration of melon-specific genes whereas genes shared with cucumber and other plants are essentially found in gene-rich chromosomal arms. The results presented here suggest that melon pericentromeric regions may allow gene sequences to evolve more freely than in other chromosomal compartments which may allow new ORFs to arise and eventually be selected. These results show that TEs can drastically change the structure of chromosomes creating different chromosomal compartments imposing different constraints for gene evolution.
Keywords: heterochromatin, recombination, transposon, genetic variability
Introduction
Transposable elements (TEs) are mobile genetic elements present in the genome of virtually all organisms. They account for a variable fraction of all plant genomes, from the low 3.12% in Utricularia gibba (Ibarra-Laclette et al. 2013) to up to 85% in maize (Springer et al. 2009). This high variability of TE content highlights the dynamics of plant genome size during evolution which is, at least in part, the consequence of the capacity of TEs to proliferate and invade genomes over short periods of time. Good examples of a rapid genome size increase due to TE proliferations are the genome of the rice relative Oryza australiensis, which doubled its genome size in 3 Ma due to the amplification of a small number of retrotransposon families (Piegu et al. 2006) and the expansion of the pepper genome due to the accumulation of elements of the Del subgroup of Ty3/Gypsy retrotransposons over 7.5–20 Ma (Park et al. 2012).
Transposable elements are a rich source of new genes and regulatory sequences and have been recognized as important players in plant genome evolution (Lisch 2013), although their mutagenic capacity is also a threat for the genomes they inhabit. As a consequence, genomes have developed sophisticated mechanisms to control them (Fultz et al 2015). In addition, TEs tend to accumulate in heterochromatic regions of the genome where the gene density is low (Contreras et al. 2015). This biased distribution of TEs is the result of different forces. On the one hand, although some TEs such as DNA transposons or different members of the Copia retrotransposon superfamily target genic regions for integration, other retrotransposons, for example most of those belonging to the Gypsy superfamily, target heterochromatic regions for insertion (Contreras et al. 2015). On the other hand, selection tends to eliminate deleterious insertions, concentrating TE insertions in regions where gene density is low (Sigman and Slotkin 2016). Moreover, the rate of elimination of TEs by intra or interelement recombination is lower in the heterochromatic repetitive regions because these regions often show a lower recombination rate (Zamudio et al. 2015). As a consequence, TEs are usually not homogeneously distributed and tend to accumulate in pericentromeric regions and other heterochromatic regions of the chromosomes.
This nonuniform distribution of TEs also influences the distribution of other chromosomal features. Indeed, as TEs are the main target of silencing mechanisms, which control their activity tightly (Bennetzen and Wang 2014; Ito and Kakutani 2014) TEs location is associated with heterochromatic epigenetic marks (Ito and Kakutani 2014). Consequently, the epigenetic silencing of the TEs in the heterochromatin reinforces the heterochromatic state of these regions (Bierhoff et al. 2014), which is essential for the normal functioning of these important chromosomal regions (Dernburg et al. 1996). In addition, the concentration of TEs in pericentromeric regions may help centromeres to resist microtubule tension during mitosis and meiosis (Freeling et al. 2015) and retrotransposon insertion into the centromeres contributes to the rapid evolution of these structures (Han et al. 2016), which is important for the evolution of the species as a whole.
We have previously shown that in melon (Cucumis melo L.) the pericentromeric regions have a high density of TEs and show a very low recombination rate which is accompanied by higher nucleotide diversity as compared with the euchromatic regions where TE density is low and gene density is high (Sanseverino et al. 2015). In order to get more insight into the possible evolutionary consequences of the relationship among TE accumulation, recombination frequency and genetic variability in melon, we extended this analysis and compared the melon genome to that of its close relative cucumber (Cucumis sativus L.). Our results show that after the split of these two Cucumis species, some 10 Ma (Sebastian et al. 2010), whereas cucumber has maintained relatively uniform chromosomes, in terms of recombination, genetic variability and gene distribution, melon chromosomes have evolved two very different compartments, with drastically different recombination frequencies, variability and gene type distribution.
Materials and Methods
Data Retrieval and Annotation
Publicly available fasta sequences and GFF3 annotation files from melon genome version 3.5.1 and cucumber Gy14 were retrieved from Melonomics (http://melonomics.net/) and Phytozome (http://phytozome.jgi.doe.gov), respectively. Cucumber scaffolds were assembled in pseudomolecules based on a previously published, high resolution genetic map (Yang et al. 2013). Fasta files from 20 cucumber varieties (Qi et al. 2013) and seven melon varieties (Sanseverino et al. 2015) were also fetched. Total transposable element annotation in melon and cucumber was performed with REPET package v2.2 (Flutre et al. 2011). Briefly, de novo TE detection was carried out with the TEdenovo pipeline from the REPET package using default parameters and step 8, corresponding to the clustering of consensus, was excluded. To obtain the reference TE annotations, the TEannot pipeline from the REPET package was run using WUBLAST 2.0 (Washington University—BLAST, http://blast.wustl.edu/), sensitivity for BLASTER of 2 (BLR_sensitivity: 2) and steps four and five, corresponding to the SSR detection, were excluded. Annotations shorter than 200 bp were discarded. A total of 168,008 potential TE sequences were identified in the melon genome. These sequences were classified whenever possible into the two major TE classes filtering out overlapping TEs from different classes (5% of the annotated TE sequences). A total of 79,612 potential TE sequences were identified in cucumber. These sequences were classified whenever possible into the two major TE classes filtering out overlapping TEs from different classes (0.5% of the annotated TE sequences). In order to look for TE families shared between the two genomes the melon TE consensus sequences obtained from TEdenovo were used to annotate cucumber using TE annot, and the cucumber TE consensus sequences obtained from TEdenovo were used to annotate melon using TE annot. In both cases, no sequence showing sufficient similarity using the default parameters was detected.
Recombination, Variability, and Correlations Comparison between Melon and Cucumber
Recombination maps were obtained from reference tables of physical (expressed in megabases) versus genetic distances (expressed in centiMorgans). We used a cubic spline method implemented in spline function in R to calculate the slope per window (that is, the derivative) and their curves were plotted for each of the chromosomes as previously described (Argyris et al. 2015). Statistics for melon and cucumber were obtained in 100 and 500 kb windows, respectively. The regions without recombination data available were masked in the analysis. Fastaconvtr and mstatspop software (both available by the authors at http://bioinformatics.cragenomica.es/numgenomics/people/sebas/software/software.html) were used to estimate the silent (synonymous plus noncoding), synonymous, nonsynonymous variability (Gojobori and Nei 1986), coding and other noncoding positions using the GTF annotation file, and also to calculate nucleotide diversity estimates (Watterson 1975; Tajima 1983) considering position with missing values as described (Ferretti et al. 2012). In order to estimate the association between any two variables, we used the R-environment (http://www.rproject.org) to calculate Kendall rank association values and their probabilities. Following Sanseverino et al. (2015), we calculated the mean of the variable located on the y axis on 100 separated bins for the variable located on the x axis. In case of calculating partial correlation analyses, we assumed data followed a normal distribution, so we compared the residuals of the variables in relation to a third variable to account. Correlation and significance statistics were obtained using the Pearson method.
Gene-Poor and Gene-Rich Compartment Setting
We studied if the gene density per window was linearly associated with levels of nucleotide variability or with recombination rate. We analyzed two different models: 1) a linear model using all data or 2) a model combining two linear models calculated from the division of the whole sample into two subsets of gene density (low and high). For the second model, the position that divided the low and high density was estimated using the following method: after sorting the 500 kb size windows by its number of coding positions, we divided this data in two groups (low and high gene density). We started from the low gene density group having 50 windows (and the rest in the highest group), then, the next divisions increased the size of the low density group by one window each time, until having a minimum of 50 windows in the high coding density group (and the rest in the lowest group). For each division, we calculated the sum of the RSS (Residual Sum of Squares) for the high and low groups as a proxy to fit the best parameter of the model, considering a Gaussian distribution for each group. The position with the lowest RSS (summing the RSS of low and high groups) was defined as the best parameter for the model with two groups (equivalent to a maximum likelihood selection of the parameter). In order to infer the best model (that is, a bimodal model with separation between low and high gene densities, and an unimodal model), we calculated the Akaike Information Criterion (AIC) for each model and compared them using a chi-square test with one degree of freedom.
Statistical Analyses
The statistical significance of the differences of distributions of nucleotide diversity and the ratios nonsynonymous versus synonymous variability in different compartments were performed using the Kolmogorov–Smirnov nonparametric test (Sokal and Rohlf 1995).
Orthology and Homology Analysis
Orthologous relationships between melon and cucumber were obtained from PhylomeDB4 (Huerta-Cepas et al. 2014) and they were filtered out (tagged as “Excluded”) when: 1) Multiple relationships were found in one-to-one orthologous pairs; 2) Orthologous genes were not found in one or both annotation files due to annotation updates; or 3) at least one gene for each pair overlapped with another gene. Additional orthology data with other available species was also retrieved from PhylomeDB4 in order to annotate melon genes with no orthologous relationship with cucumber genes but with orthologues in other species (tagged as “Other ortho”).
Additionally, melon proteins homology against all plant sequences was obtained with blastp against a user-constructed database of GenBank plant sequences (Viridaeplantae) obtained with NCBI’s eutils (https://eutils.ncbi.nlm.nih.gov/entrez/eutils/). Blastp results were grouped in several categories (see supplementary table 1, Supplementary Material online. Syntenic maps were produced with SyMAP (Soderlund et al. 2011) for chromosome 1 in melon and chromosome 7 in cucumber.
Quality Analysis of Nonorthologous Gene-Poor Melon Proteins
Quality of chosen melon predicted proteins was estimated with several parameters: blastp hits against plant databases (see above), previously published orthology data (Garcia-Mas et al. 2012), transposable element overlap with genes, GO terms, presence–absence variation, gene length, exon number per gene, first residue in protein sequence and partial gene annotation. Domain prediction was performed with hmmscan (Finn et al. 2011) with Pfam database (Finn et al. 2014), and filtered by a minimum value of e-value <1 and i-value <1. Melon gene expression was obtained from available public data [http://melonet-db.agbi.tsukuba.ac.jp]. Intrinsic Structural Disorder (ISD) was computed as previously described (Wilson et al. 2017). Briefly, ISD was calculated with IUPred (Dosztányi et al. 2005) for every main isoform (i.e., longest) protein sequence with no cysteines. Intergenic control “proteins” were obtained with the available perl script intergenic_control_sequence_generator.pl (https://github.com/MaselLab) to obtain sequences of similar length and environment as the neighboring proteins.
Results and Discussion
TEs Have Expanded Melon Pericentromeric Regions after the Cucumber-Melon Lineages Split
Melon and cucumber are related species that diverged from a common ancestor some 10 Ma (Sebastian et al. 2010). Although the number of chromosomes has been reduced in cucumber after the split of both species (Yang et al. 2014), both genomes are highly colinear. However, the melon genome is 23% bigger than that of cucumber (Arumuganathan and Earle 1991). A previous analysis suggested that the increase of the melon genome size was at least in part due to a higher recent accumulation of TEs (Garcia-Mas et al. 2012). However, this was based in a rough comparison of partial TE annotations of both genomes. The quality of the genome sequence and assembly may introduce a certain bias in the annotation of TEs, but the use of different methods and thresholds for TE annotation is recognized as the main problem for comparing TE populations between different genomes (Hoen et al. 2015). In order to reevaluate the difference proposed in TE content between melon and cucumber, we annotated both melon and cucumber genomes with the same TE annotation pipeline, the REPET package (Flutre et al. 2011), by using the same parameters and thresholds. Forty-four percent of the melon genome sequence was annotated as TE-related whereas only 27% of the cucumber genome was annotated as TE-related (table 1). Since a different quality of the genome assembly may bias the comparison, we analyzed the TE content of the unassembled fraction of both the melon and cucumber genomes. The unassembled fraction is similar for both genomes (15.75% for melon and 13.6% for cucumber), and in both cases the TE content is similar to that of the assembled genome (43.4% for melon and 32.7% for cucumber). In summary, the results presented here confirm that, indeed, TEs have accumulated in the melon genome to a greater extent than in that of cucumber. Moreover, the analysis of the annotated TEs reveals a low level of sequence similarity between melon and cucumber elements, indicating that the vast majority of TE families are not shared between melon and cucumber, and probably amplified after the split from their common ancestor, which is compatible with our previous analysis that suggested a peak of LTR retrotransposon amplification in melon 2 Ma (Garcia-Mas et al. 2012).
Table 1.
Summary Statistics of REPET Transposable Element Annotation in Melon and Cucumber Gy14
| Class | Order | Superfamily | Code | Melon |
Cucumber Gy14 |
||
|---|---|---|---|---|---|---|---|
| # of Copies | % of Genome | # of Copies | % of Genome | ||||
| I | LTR | None | RLX | 74,161 | 23.44 | 20,469 | 8.34 |
| I | LINE | LINE | RIX | 11,913 | 2.64 | 5,088 | 1.97 |
| I | SINE | SINE | RSX | 391 | 0.04 | 360 | 0.08 |
| I | DIRS | None | RYX | 4,212 | 1.65 | 1,490 | 0.50 |
| I | Not classified | RXX | 42,555 | 7.42 | 48,683 | 14.69 | |
| II | TIR | None | DTX | 21,383 | 7.11 | 1,347 | 0.66 |
| II | Rolling | Helitron | DHX | 1,699 | 0.30 | 1,067 | 0.35 |
| II | Rolling | Maverick | DMX | 729 | 0.43 | 491 | 0.15 |
| II | Crypton | Crypton | DYX | 183 | 0.05 | 0 | 0 |
| II | Not classified | DXX | 2,015 | 0.45 | 170 | 0.05 | |
| NonClass.a | None | None | XXX | 290 | 0.06 | 27 | 0.01 |
| Total | 159,531 | 43.60 | 79,192 | 26.81 | |||
nonclassified TE sequences.
In both melon and cucumber, TEs are nonhomogeneously distributed along chromosomes. TEs concentrate in pericentromeric regions whereas distal parts of the chromosomes usually show a relative low density of TEs (supplementary fig. 1, Supplementary Material online). This accumulation of TEs in the pericentromeric regions is more obvious in melon. As already suggested by our previous analyses (Garcia-Mas et al. 2012; Sanseverino et al. 2015), although the gene-rich chromosomal arms have a similar size in melon and cucumber (the size of the regions where gene coverage is higher than TE coverage is 87 Mb in melon and 119 Mb in cucumber), the TE-rich pericentromeric regions are much larger in melon (the size of the regions where TE coverage is higher than gene coverage is 268 Mb in melon and 52.4 Mb in cucumber), suggesting that TEs have expanded these regions during the recent evolution of the melon genome (supplementary fig. 1, Supplementary Material online). This expansion may have engulfed additional genes into TE-dense regions (regions where the coverage of annotated TEs is higher than that of genes). Indeed, the fraction of genes in TE-dense regions is much higher in melon than in cucumber (52.9% vs. 20.2%). This can be seen clearly when comparing cucumber chromosome 7 with melon chromosome 1, which are the only two chromosomes that maintained complete synteny after the disploidy process that the cucumber genome suffered during its recent evolution (Yang et al. 2014). Only 37% of the cucumber genes in chromosome 7 are in TE-dense regions, whereas 53% of the melon genes in chromosome 1 are located within these regions. A synteny analysis of these two chromosomes shows that whereas most orthologous pairs lay in gene-dense or TE-dense regions in both species, in 28% of the cases (354 of 1,261pairs) the cucumber genes are located in a gene-dense region and their melon counterparts lay in a TE-dense region (fig. 1). Some of these changes of compartment are due to small inversions and reorganizations of the melon-cucumber equivalent chromosomal regions since the split of both species (see fig. 1). However, this analysis suggests that in many cases the proliferation of TEs and the expansion of the pericentromeric regions may have captured melon genes that were probably close to these regions in the ancestor genome, whereas the orthologous genes in cucumber have remained in gene-dense regions.
Fig. 1.
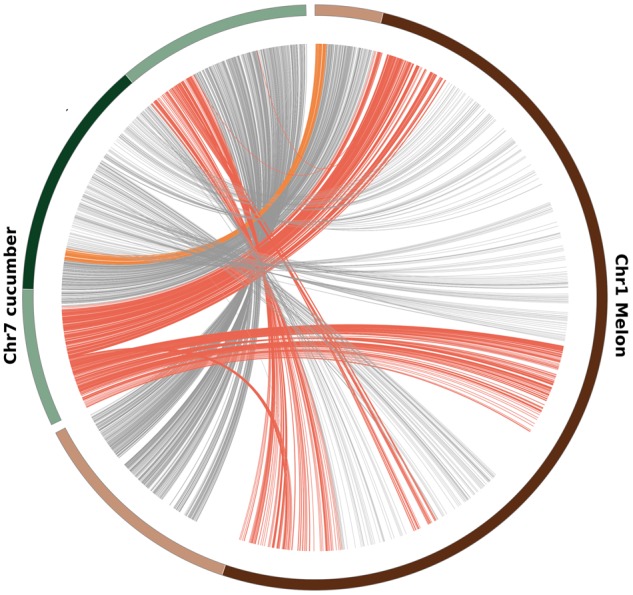
—Synteny analysis of melon chromosome 1 (brown) and cucumber chromosome 7 (green) based on melon-cucumber orthologous genes. Chromosomal regions where the density of TEs is higher than that of genes are shown as dark brown or green boxes for melon and cucumber, respectively. Red lines connect orthologous genes located in a gene-dense region in cucumber and in a TE-dense region in melon. Orange lines connect orthologous genes located in a gene-dense region in melon and in a TE-dense region in cucumber. Grey lines connect orthologous genes located in a gene-dense region or in TE-dense region in both genomes.
Recombination Is Suppressed in Pericentromeric Regions in Melon but Not in Cucumber
We have already shown that in melon, recombination is positively correlated with gene density. Recombination is high in melon chromosomal arms whereas is almost completely suppressed in pericentromeric regions (Sanseverino et al. 2015). Genomes often show a negative correlation between recombination rate and TE density, although the strength of this association is variable across TE types and species (Kent et al. 2017). Here, we analyzed the distribution of recombination frequency along chromosomes in cucumber and compared it with that of melon. Interestingly, recombination is not correlated with gene density in cucumber and is relatively constant along chromosomes, not being suppressed in the pericentromeric regions (fig. 2b and d and supplementary fig. 2b, Supplementary Material online), in sharp contrast with what happens in melon (fig. 2a and c andsupplementary fig. 2a, Supplementary Material online).
Fig. 2.

—Recombination in chromosome 1 of melon (left) and chromosome 7 of cucumber (right). Correlation of gene density versus recombination rate in melon (a) and cucumber (b). Red spots represent the mean of the recombination rate on 100 separated bins with respect to gene density. Distribution of recombination rate along the chromosome in melon (windows of 500 kb) (c) and cucumber (windows of 100 kb) (d). The red line indicates cumulative recombination rate. The grey bars on top indicate TE-dense regions.
Recombination is usually suppressed at highly methylated repetitive regions in order to prevent problems during meiosis leading to erratic chromosomal events (Zamudio et al. 2015). The large TE-dense pericentromeric regions of melon seem to have an important effect on the recombination frequency in these regions. This is not the case of cucumber where the TE-dense pericentromeric region is much smaller. Interestingly, in other compact genomes with small pericentromeric regions such as Arabidopsis, the suppression of recombination is limited to the centromere and does not extend to the pericentromeric regions (Melamed-Bessudo et al. 2016), the frequency of recombination being essentially constant along chromosomes. In contrast, bigger genomes with larger TE-rich pericentromeric regions, such as Arabis alpina (Willing et al. 2015), tomato (Demirci et al. 2017) or many cereals (Melamed-Bessudo et al. 2016; Mascher et al. 2017), present an almost complete suppression of recombination in the pericentromeric regions. Here we see, in line with what has been seen in Arabis alpina (Willing et al. 2015), that the expansion of the pericentromeric regions in melon, due to the accumulation of TEs, correlates with the establishment of a large nonrecombining region. Although the cause-effect relationship is not yet established, the coincidence in time of TE expansion, TE silencing and suppression of recombination suggests a link between these phenomena (Kent et al. 2017; Maside et al. 2005). To what extent the length of the region and the TE density may be determinant for recombination suppression in pericentromeric regions has not yet been analyzed, but it is tempting to hypothesize that there is a threshold above which the size and the TE density of these regions triggers recombination suppression.
Gene Density and Gene Nucleotide Diversity along Chromosomes
Recombination affects genetic variability by allowing different alleles to be selected independently. In general, when recombination is too low to avoid interference between genes, high levels of directional selection (positive or negative) will result in a sweep to surrounding regions and a general loss of variability. Therefore, in pericentromeric regions where recombination is low, a low degree of genetic variability is expected. This is what has been seen in some plant species, for example in barley (Baker et al. 2014) and soybean (Du et al. 2012). However, in other cases, pericentromeric regions show higher genetic diversity than more distal regions in spite of having a lower recombination rate, as shown in rice (Flowers et al. 2012), populus (Wang et al. 2016) and eucalyptus (Gion et al. 2016). This has been explained by a nonhomogeneous distribution of gene density and selection (Flowers et al. 2012). Indeed, regions with low gene density may not present enough functional positions (that may result in selective sweeps) to show an effect of selection, allowing neutral variation to accumulate.
We have previously shown that in melon the low recombining pericentromeric regions show higher variability (Sanseverino et al. 2015). In order to get more insight on this phenomenon we have analyzed here in more detail these relationships in melon and cucumber. Our results show that in cucumber, where recombination frequency is independent of gene density and is essentially constant along chromosomes, both synonymous and nonsynonymous genetic variability are independent of gene density and only the silent variability is correlated (negatively) with gene density (fig. 3), which may be explained by the presence among silent positions of regulatory elements under selective pressure, which should be more frequent in gene-dense regions. The constant and relatively high level of recombination throughout cucumber chromosomes may be enough to avoid that selection of certain alleles may result in long selective sweeps. As a consequence genetic variability is independent of gene density, which, in addition, is relatively homogeneously distributed along chromosomes.
Fig. 3.

—Correlation of gene density versus nonsynonymous (a, b), synonymous (c, d), and silent (e, f) variability in melon (left) and cucumber (right). Red dots indicates the mean of the nonsynonymous variability on 100 separated bins with respect to gene density.
In contrast, in melon the three different types of genetic variability, silent, synonymous and nonsynonymous, show a more complex pattern of correlation with gene density (fig. 3). Melon chromosomes can be split in two regions of frequency, which may lead to a nonuniform pattern of variability. Selective sweeps may occur in regions of high gene density, whereas variability would remain high in regions were gene density is low.
The analysis of the correlation of the genetic variability, and in particular that of nonsynonymous variability, with gene density suggests a bimodal pattern (supplementary fig. 3, Supplementary Material online). Indeed, the difference between the Akaike Information Criterion (AIC) for a bimodal pattern versus an unimodal pattern is 275.09 (P value ≪ 1e-12) using a chi-square distribution of 1dgf. A regression analysis assuming a bimodal pattern allowed us to define a gene density threshold that divides the genome in a relatively gene-rich region where there is no correlation between gene variability and gene density (hereafter, gene-rich), and a relatively gene-poor region where the variability is negatively correlated with gene density (hereafter gene-poor).
Melon-Specific Genes in Gene-Poor Regions Show Higher Variability
The nonsynonymous nucleotide diversity in gene-poor and gene-rich regions shows significantly different distributions (P value < 2.2 e−16) and the mean value is nearly double in the former (πnsyn = 0.0043 vs. πnsyn = 0.0022) (fig. 4a). As already explained, the reason could be a higher presence of essential genes in gene-rich regions that could result in a higher frequency of selective sweeps.
Fig. 4.

—Violin plots showing the distribution of nonsynonymous variability in gene-rich (right) and gene-poor (left) regions for all genes (a), orthologous genes (b), and nonorthologous genes (c). The mean (dot), and the standard error intervals (vertical line) are also depicted for each category. All distributions are truncated at variability 0.02 (outliers are not shown).
Gene variability is expected to differ among different genes. For this reason we decided to analyze the variability of melon specific genes and that of orthologous genes between melon and cucumber. To this end, we performed an orthology analysis between melon and cucumber and analyzed separately the variability of melon orthologous and nonorthologous genes in gene-rich and gene-poor regions (table 2). Interestingly, although the variability of melon orthologous genes has a more similar distribution (P value = 5.37 e−5) and similar mean values in both chromosomal compartments (fig. 4b), nonorthologous genes show a very different distribution (P value < 2.2 e−16) and a very different mean value (πnsyn = 0.0092 gene-poor; πnsyn = 0.0056, gene-rich) (fig. 4c).
Table 2.
General Statistics for Melon Genes Depending on Orthology and Gene-Density Region Localization
| Gene-Rich | Gene-Poor | ||
|---|---|---|---|
| Orthologs | Many-to-Many | 539 (75.2) | 178 (24.8%) |
| Many-to-One | 855 (83.3%) | 168 (16.4%) | |
| One-to-Many | 320 (89.9%) | 36 (10.1%) | |
| One-to-One | 12,275 (91.3%) | 1,142 (8.5%) | |
| Other | 1,196 (83.9%) | 228 (16%) | |
| No Orthologs | 3,374 (61.7%) | 2,091 (38.2%) | |
| Excluded genes | 2,083 (80.4%) | 503 (19.4%) | |
| Total | 20,642 (82.5%) | 4,346 (17.4%) | |
Note.—Frequency computed per orthology category (rows). Partial genes (i.e., those genes that span both gene-rich and gene-poor region) are not shown. One-to-one: a single gene in melon has a single orthologue in cucumber; one-to-many: a single gene in melon shows orthology to several genes in cucumber; many to one: several genes in melon show orthology to a single gene in cucumber; many-to-many: several genes in melon show orthology to several genes in cucumber.
These results suggest that the melon genome has different chromosomal compartments that impose different evolutionary constraints upon gene sequences. In order to further explore this possibility we analyzed the ratio of nonsynonymous over synonymous variability in the two chromosomal compartments. The nonsynonymous/synonymous ratio distribution of variability for nonorthologous genes shows a different profile in the two chromosomal compartments. Indeed, the peak of the nonsynonymous/synonymous ratio for nonorthologous genes is smoother and is shifted to more positive values in the case of gene-poor regions, which may suggest relaxation of the purifying selection (fig. 5a). This trend can also be seen for genes orthologous between melon and cucumber where, although the graphs of both chromosomal compartments peak at the same position (−0.5), the distribution for gene-poor regions is slightly shifted towards more positive values (fig. 5b).
Fig. 5.
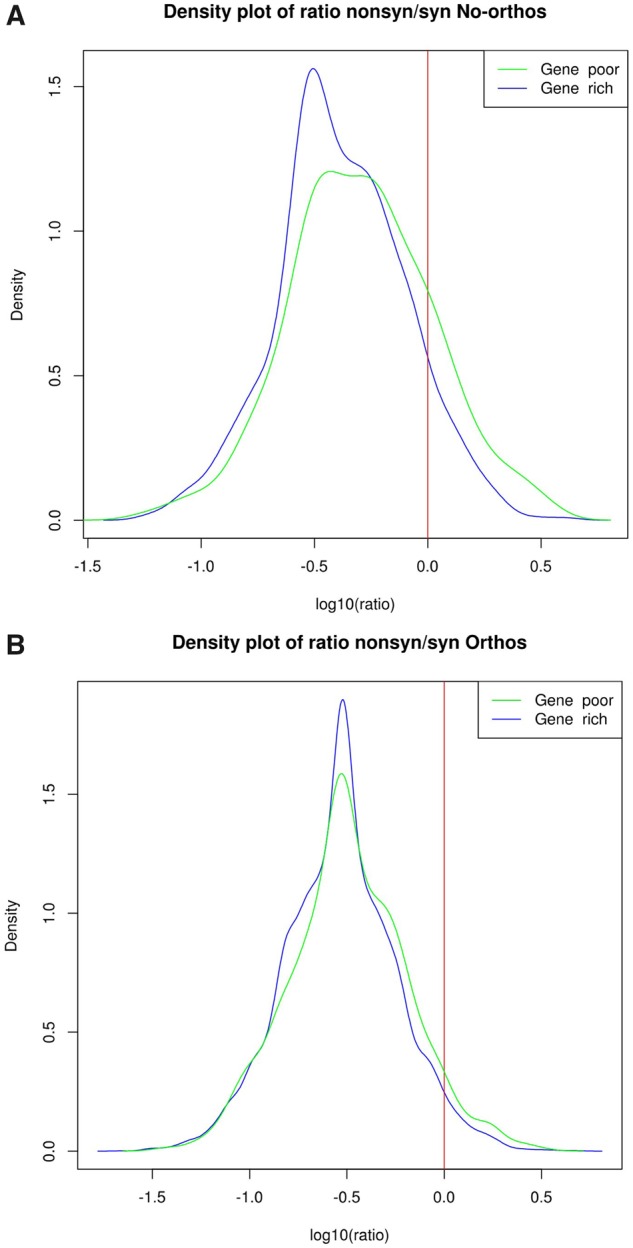
—Density plot of the ratio nonsynonymous/synonymous variability in nonorthologous genes (a, top) and orthologous genes (b, bottom). Genes are distinguished between those located in gene-poor or in gene-rich regions.
Gene-Poor Regions Concentrate Melon-Specific Genes
Our results show that melon genes in different chromosomal regions show different nucleotide variability suggesting that they evolved differently. Interestingly, our results also show that this effect is more striking for melon genes that do not have an orthologous counterpart in cucumber. We therefore decided to analyze the distribution of melon-cucumber orthologous and nonorthologous genes in the gene-rich and gene-poor regions here defined.
Pseudogenes and TE-related ORFs can be miss-annotated as genes and this could introduce a bias in our analysis if these miss-annotations were more frequent for a particular gene type (orthologous, nonorthologous) or in a particular gene compartment, as it is the case for TEs. In order to avoid TE-related miss-annotated genes interfering in the analysis, we searched for sequence similarities of the peptides potentially encoded by the annotated genes against plant databases. Only 1% of the nonorthologous genes in gene-poor regions showed significant similarity to TE-related proteins (supplementary table 1, Supplementary Material online), suggesting that TE-related proteins are not introducing a bias in our analysis. Moreover, the analysis of a number of parameters of these ORFs in gene-poor regions suggest that they indeed correspond to potential protein-coding genes (supplementary table 1, Supplementary Material online).
Our analysis indicates that whereas the vast majority of the orthologous genes between cucumber and melon are located in gene-rich regions and only 10% of them are located in gene-poor regions, the distribution of the nonorthologous melon genes is very different, with 38% of them in gene-poor regions (table 2). This suggests that whereas the most conserved and relatively old genes concentrate in gene-rich regions, gene-poor regions may contain more recent genes with less conserved functions. Interestingly, different patterns can be identified also among genes classified as orthologous. There are a number of possible relationships of orthology, depending on whether the orthologous pairs are single genes or belong to multigene families. Most orthologous relationships between melon and cucumber genes are one-to-one, meaning that a single gene in melon has an orthologous relationship with only one gene in cucumber. These genes, orthologous one-to-one, are essentially located in gene-rich regions (91.3%). There are very few cases where a single gene in melon shows orthology to several genes in cucumber (one-to-many), which is not surprising as the genome size and the number of genes is smaller in cucumber. The melon genes involved in the few one-to-many orthologous relationships are also essentially located in gene-rich regions (89.9%). Interestingly the distribution of genes involved in many-to-one and many-to-many orthologous relationships is less skewed with only 83.3% of the former and 75.2% of the later in gene-rich regions. The 1,023 melon genes involved in a many-to-one relationship of orthology with cucumber are grouped in 508 orthology groups. 56 of them included genes in both gene-rich and gene-poor regions. The analysis of these groups shows that in 41 cases (73%) the melon sequence more closely related to that of cucumber is located in a gene-rich region, whereas in only 20 cases (35.7%) the sequence less related to the cucumber sequence is found in this compartment (supplementary table 2, Supplementary Material online). This reinforces the idea that melon genes that have maintained a close identity to their cucumber counterparts are essentially located in the gene-rich compartment, whereas the more divergent sequences are frequently located in the gene-poor compartment.
In order to complete our analysis we have reanalyzed a phylogenomic analysis of all melon protein-coding genes in 22 sequenced plant species previously reported (Garcia-Mas et al. 2012) to determine how genes that appeared at different evolutionary times are distributed between the gene-poor and gene-rich regions as defined here. The orthology and paralogy relationships across the 24,885 protein-coding genes were previously classified in four categories: 1) “All”: widespread genes found in all plant species and nonplant outgroups, 2) “InSplit”: widespread plant-specific genes that are found in at least 20 of the 23 plant species, 3) “Specific”: Species-specific genes with no detectable homologs in other species, and 4) “Others”: genes without a clear pattern (Garcia-Mas et al. 2012). The results presented in table 3 show that close to 90% of the genes present in plant and nonplant genomes (“All”) are found in gene-rich regions, and that this high percentage decreases for plant-specific genes (85.8%, “InSplit”) and even more for melon specific genes (57.9%). The 42.1% of the melon specific genes in gene-poor regions represents a significant enrichment of this class when compared with the other three categories.
Table 3.
General Statistics for Melon Genes Depending on Orthology from Garcia-Mas et al. (2012) and Gene-Density Region Localization
| Gene-Rich | Gene-Poor | |
|---|---|---|
| Alla | 5,698 (89.8%) | 640 (10.1%) |
| InSplit (Plant-specific) | 9,813 (85.8%) | 1,609 (14.1%) |
| Specific (Melon-specific) | 2,190 (57.9%) | 1,592 (42.1%) |
| Other | 2,844 (85.8%) | 470 (14.2%) |
| Total | 20,545 | 4,311 |
Note.—Frequency computed per orthology category (rows). Partial genes (i.e., those genes that span both gene-rich and te-rich region) are not shown.
Including H. sapiens, P. falciparium, D. melanogaster, C. elegans, and S. cerevisiae.
Our results therefore show that melon genes are not uniformly distributed along chromosomes and that whereas conserved genes tend to be located in the distal gene-rich regions, melon specific genes are frequently found in the TE-rich pericentromeric chromosomal regions. Several reports have shown that plant genes can be distributed nonuniformly in chromosomes and that pericentromeric TE-regions can accumulate particular gene types. However, the type of genes accumulated seems to be different in different genomes. For example, it has been recently shown that the long heterochromatic pericentromeric regions of tomato are enriched in tomato-specific genes, and in particular in those related to fruit ripening, whereas genes found in all plants are depleted from these regions (Jouffroy et al. 2016). In contrast, a recent analysis of the barley genome shows that genes related to defense responses and reproductive processes, which are supposed to be more species-specific, concentrate in the short distal parts of the chromosomes whereas genes related to translation or cellular respiration, among others, which are supposed to be shared by all organisms, concentrate in the pericentromeric regions (Mascher et al. 2017). This is also what is found in the wheat chromosome 3B, which concentrates stress-related genes in the distal parts of the chromosome (Choulet et al. 2014). Thus, whereas long TE-rich pericentromeric regions seem in all cases to show suppressed recombination, the effect this has on gene variability, as discussed above, and on gene distribution could be very different among different plant species.
As already discussed, the level of variability depends on both the strength of selection and the frequency of recombination. The relatively high variability of the low recombining pericentromeric regions in melon could be the result of the limited number of selective positions in this region. In other words, a relaxed purifying selection in low gene density and low recombinant regions could allow for the maintenance of neutral or slightly deleterious mutations without a significant effect on the general fitness of the organism. An analysis of the size and number of exons of genes located in the two chromosomal compartments shows that genes located in TE-rich regions are in general smaller than those located in gene-rich regions (mean length of 1,861 vs. 2,952 nt) and contain a lower number of exons (mean number of exons 3.54 vs. 6.47) (supplementary fig. 4, Supplementary Material online). This suggests that TE-rich regions may contain more gene fragments, which are probably decaying genes. However, these ORFs may also correspond to new genes arising in these regions of relaxed purifying selection. Natural proteins have a structure that shows a higher level of intrinsic structural disorder (ISD) than random sequences (Yu et al. 2016). This higher structural disorder probably allows natural proteins to increase functional diversity and to stay soluble and avoid aggregating, which would be an important hazard for the cell (Yu et al. 2016). Interestingly, it has been recently shown that young genes have an exaggerated gene-like structure and show higher structural disorder than older genes (Wilson et al. 2017). We therefore decided to analyze the ISD of the predicted nonorthologous genes located in TE-rich regions and compare them to orthologous genes in gene-rich regions and random sequences of the same length and composition. Our results show that the nonorthologous genes of the TE-rich regions have the highest ISD value (fig. 6), suggesting that an important fraction of them are protein-coding genes and may indeed be younger than the genes in gene-rich regions.
Fig. 6.
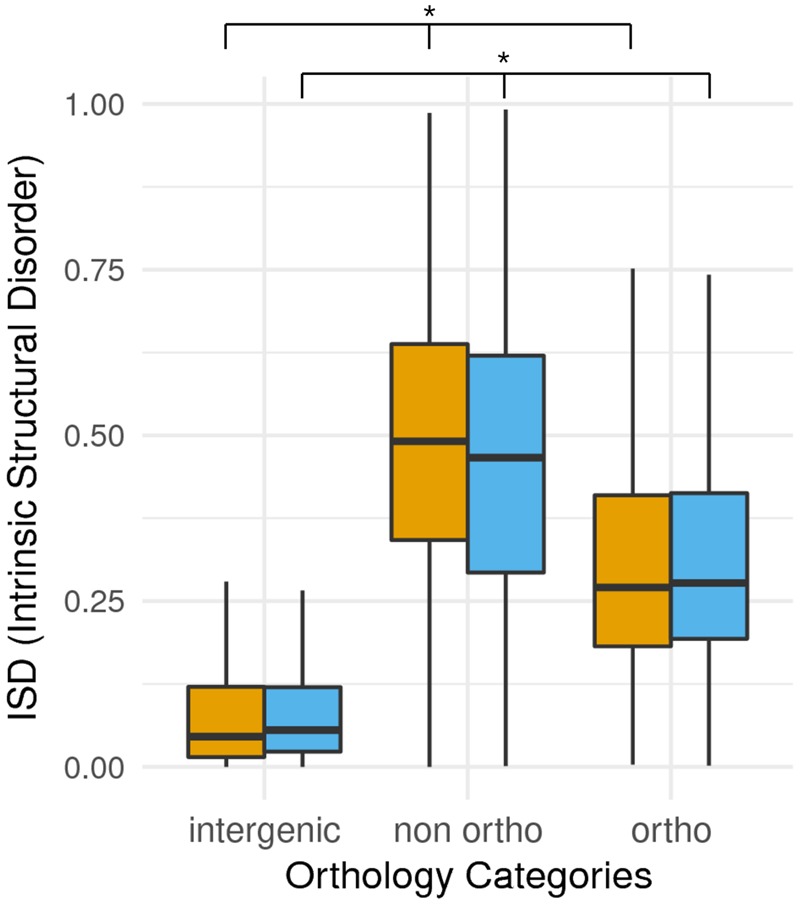
—Intrinsic Structural Disorder (ISD) for melon intergenic regions, melon proteins without ortholog in cucumber and proteins with orthologous relationship in cucumber. Each category is divided into genes (or sequences in the case of intergenic control regions) located in gene-poor (orange) or gene-rich (blue) regions. Kolmogorov–Smirnov tests confirmed significant different distributions for gene-rich genes (or intergenic sequences) in each category and, on the other hand, for gene-poor genes (or intergenic sequences) in each category (P value < 0.01 in all cases).
Concluding Remarks
Here, we show that after the split of melon and cucumber, some 10 Ma, TEs have expanded the pericentromeric regions of melon chromosomes that, probably as a consequence, show a very low recombination frequency. In contrast, TEs have not proliferated to a high extent in cucumber, which has small TE-dense pericentromeric regions and shows a relatively constant recombination rate along chromosomes. Our results therefore suggest that TEs can drastically change the structure of chromosomes creating two very different chromosomal compartments defined by a high or a low recombination frequency. This highlights that TEs, and in particular their proliferation, may have important consequences for gene and genome evolution beyond their more obvious mutagenic capacity.
These large nonrecombinogenic regions impose particular constraints on gene evolution. This translates in some cases, such as in barley, in a low variability and a concentration of housekeeping genes in these regions, whereas in other cases such as melon, this correlates with a concentration of high variability and species-specific genes. The reasons for this difference could be multiple, including a different chromosomal structure in the ancestor, or a difference in the strength of selection. Our results suggest that the melon pericentromeric regions may contain a number of selective positions too low to induce a decrease of genetic variability, most mutations having a neutral or slightly deleterious effect. Moreover, we also show that the genes located in these regions show features of young genes. This suggests that melon pericentromeric regions may allow gene sequences to evolve more freely than in other chromosomal compartments and that this may allow new genes or gene domains to appear and eventually be selected.
Supplementary Material
Supplementary data are available at Genome Biology and Evolution online.
Supplementary Material
Acknowledgments
This work was supported by Ministerio de Economia y Competitividad grants AGL2013-43244-R and AGL2016-78992-R to J.C., Ministerio de Economia y Competitividad grant AGL2015-64625-C2-1-R to J.G.-M. and Centro de Excelencia Severo Ochoa 2016–2020, and the CERCA Programme/Generalitat de Catalunya to J.C., J.G.-M. and S.R.-O.
Literature Cited
- Argyris JM, Pujol M, Martín-Hernández AM, Garcia-Mas J.. 2015. Combined use of genetic and genomics resources to understand virus resistance and fruit quality traits in melon. Physiol Plant. 1551:4–11. [DOI] [PubMed] [Google Scholar]
- Arumuganathan K, Earle ED.. 1991. Nuclear DNA content of some important plant species nuclear DNA content material and methods. Plant Mol Biol Rep. 93:208–218. [Google Scholar]
- Baker K, et al. , 2014. The low-recombining pericentromeric region of barley restricts gene diversity and evolution but not gene expression. Plant J. 796:981–992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennetzen JL, Wang H.. 2014. The contributions of transposable elements to the structure, function, and evolution of plant genomes. Annu Rev Plant Biol. 65:505–530. [DOI] [PubMed] [Google Scholar]
- Bierhoff H, Postepska-Igielska A, Grummt I.. 2014. Noisy silence: non-coding RNA and heterochromatin formation at repetitive elements. Epigenetics 91:53–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choulet F, et al. , 2014. Structural and functional partitioning of bread wheat chromosome 3B. Science 3456194:1249721. [DOI] [PubMed] [Google Scholar]
- Contreras B, Vives C, Castells R, Casacuberta JM.. 2015. The impact of transposable elements in the evolution of plant genomes: from selfish elements to key players In: Pontarotti P, editor. Evolutionary biology: biodiversification from genotype to phenotype. Cham: Springer International Publishing; p. 93–105. [Google Scholar]
- Demirci S, et al. , 2017. Distribution, position and genomic characteristics of crossovers in tomato recombinant inbred lines derived from an interspecific cross between Solanum lycopersicum and Solanum pimpinellifolium. Plant J. 893:554–564. [DOI] [PubMed] [Google Scholar]
- Dernburg AF, Sedat JW, Hawley RS.. 1996. Direct evidence of a role for heterochromatin in meiotic chromosome segregation. Cell 861:135–146. [DOI] [PubMed] [Google Scholar]
- Dosztányi Z, Csizmók V, Tompa P, Simon I.. 2005. The pairwise energy content estimated from amino acid composition discriminates between folded and intrinsically unstructured proteins. J Mol Biol. 3474:827–839. [DOI] [PubMed] [Google Scholar]
- Du J, et al. , 2012. Pericentromeric effects shape the patterns of divergence, retention, and expression of duplicated genes in the paleopolyploid soybean. Plant Cell 241:21–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferretti L, Raineri E, Ramos-Onsins S.. 2012. Neutrality tests for sequences with missing data. Genetics 1914:1397–1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finn RD, et al. , 2014. Pfam: the protein families database. Nucleic Acids Res. 42:D1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finn RD, Clements J, Eddy SR.. 2011. HMMER web server: interactive sequence similarity searching. Nucleic Acids Res. 39(Web Server issue):W29–W37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flowers JM, et al. , 2012. Natural selection in gene-dense regions shapes the genomic pattern of polymorphism in wild and domesticated rice. Mol Biol Evol. 292:675–687. [DOI] [PubMed] [Google Scholar]
- Flutre T, Duprat E, Feuillet C, Quesneville H.. 2011. Considering transposable element diversification in de novo annotation approaches. PLoS One 61:e16526.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freeling M, Xu J, Woodhouse M, Lisch D.. 2015. A solution to the c-value paradox and the function of junk DNA: the genome balance hypothesis. Mol Plant 86:899–910. [DOI] [PubMed] [Google Scholar]
- Fultz D, Choudury SG, Slotkin RK.. 2015. Silencing of active transposable elements in plants. Curr Opin Plant Biol. 27:67–76. [DOI] [PubMed] [Google Scholar]
- Garcia-Mas J, et al. , 2012. The genome of melon (Cucumis melo L.). Proc Natl Acad Sci U S A. 10929:11872–11877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gion J-M, et al. , 2016. Genome-wide variation in recombination rate in Eucalyptus. BMC Genomics 17:590.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gojobori T, Nei M.. 1986. Relative contributions of germline gene variation and somatic mutation to immunoglobulin diversity in the mouse. Mol Biol Evol. 32:156–167. [DOI] [PubMed] [Google Scholar]
- Han J, et al. , 2016. Rapid proliferation and nucleolar organizer targeting centromeric retrotransposons in cotton. Plant J. 886:992–1005. [DOI] [PubMed] [Google Scholar]
- Hoen DR, et al. , 2015. A call for benchmarking transposable element annotation methods. Mob DNA 61:13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huerta-Cepas J, Capella-Gutiérrez S, Pryszcz LP, Marcet-Houben M, Gabaldón T.. 2014. PhylomeDB v4: zooming into the plurality of evolutionary histories of a genome. Nucleic Acids Res. 42(Database issue):D897–D902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibarra-Laclette E, et al. , 2013. Architecture and evolution of a minute plant genome. Nature 4987452:94–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito H, Kakutani T.. 2014. Control of transposable elements in Arabidopsis thaliana. Chromosom Res. 222:217–223. [DOI] [PubMed] [Google Scholar]
- Jouffroy O, et al. , 2016. Comprehensive repeatome annotation reveals strong potential impact of repetitive elements on tomato ripening. BMC Genomics 171:624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent TV, Uzunović J, Wright SI.. 2017. Coevolution between transposable elements and recombination. Philos Trans R Soc B. 372:20160458.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisch D. 2013. How important are transposons for plant evolution?. Nat Rev Genet. 141:49–61. [DOI] [PubMed] [Google Scholar]
- Mascher M, et al. , 2017. A chromosome conformation capture ordered sequence of the barley genome. Nature 5447651:427–433. [DOI] [PubMed] [Google Scholar]
- Maside X, Assimacopoulos S, Charlesworth B. 2005. Fixation of transposable elements in the Drosophila melanogaster genome. Genet. Res. 85:195–203. [DOI] [PubMed] [Google Scholar]
- Melamed-Bessudo C, Shilo S, Levy AA.. 2016. Meiotic recombination and genome evolution in plants. Curr Opin Plant Biol. 30:82–87. [DOI] [PubMed] [Google Scholar]
- Park M, et al. , 2012. Evolution of the large genome in Capsicum annuum occurred through accumulation of single-type long terminal repeat retrotransposons and their derivatives. Plant J. 696:1018–1029. [DOI] [PubMed] [Google Scholar]
- Piegu B, et al. , 2006. Doubling genome size without polyploidization: dynamics of retrotransposition-driven genomic expansions in Oryza australiensis, a wild relative of rice. Genome Res. 1610:1262–1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi J, et al. , 2013. A genomic variation map provides insights into the genetic basis of cucumber domestication and diversity. Nat Genet. 4512:1510–1515. [DOI] [PubMed] [Google Scholar]
- Sanseverino W, et al. , 2015. Transposon insertion, structural variations and SNPs contribute to the evolution of the melon genome. Mol Biol Evol. 3210:2760–2774. [DOI] [PubMed] [Google Scholar]
- Sebastian P, Schaefer H, Telford IRH, Renner SS.. 2010. Cucumber (Cucumis sativus) and melon (C. melo) have numerous wild relatives in Asia and Australia, and the sister species of melon is from Australia. Proc Natl Acad Sci U S A. 10732:14269–14273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sigman MJ, Slotkin RK.. 2016. The first rule of plant transposable element silencing: location, location, location. Plant Cell 282:304–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soderlund C, Bomhoff M, Nelson WM.. 2011. SyMAP v3.4: a turnkey synteny system with application to plant genomes. Nucleic Acids Res. 3910:e68.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sokal RR, Rohlf FJ.. 1995. Biometry: the principles and practice of statistics in biological research, 3rd edn.New York: W.H. Freeman and Co. [Google Scholar]
- Springer NM, et al. , 2009. Maize inbreds exhibit high levels of copy number variation (CNV) and presence/absence variation (PAV) in genome content. PLoS Genet. 511:e1000734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tajima F. 1983. Evolutionary relationship of DNA sequences in finite populations. Genetics 1052:437–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Street NR, Scofield DG, Ingvarsson PK.. 2016. Natural selection and recombination rate variation shape nucleotide polymorphism across the genomes of three related populus species. Genetics 2023:1185–1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watterson GA. 1975. On the number of segregating sites in genetical models without recombination. Theor Popul Biol. 72:256–276. [DOI] [PubMed] [Google Scholar]
- Willing E-M, et al. , 2015. Genome expansion of Arabis alpina linked with retrotransposition and reduced symmetric DNA methylation. Nat Plants 12:14023. [DOI] [PubMed] [Google Scholar]
- Wilson BA, Foy SG, Neme R, Masel J.. 2017. Young genes are highly disordered as predicted by the preadaptation hypothesis of de novo gene birth. Nat Ecol Evol. 16:0146.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L, et al. , 2014. Next-generation sequencing, FISH mapping and synteny-based modeling reveal mechanisms of decreasing dysploidy in Cucumis. Plant J. 771:16–30. [DOI] [PubMed] [Google Scholar]
- Yang L, et al. , 2013. A 1,681-locus consensus genetic map of cultivated cucumber including 67 NB-LRR resistance gene homolog and ten gene loci. BMC Plant Biol. 13:53.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu JF, et al. , 2016. Natural protein sequences are more intrinsically disordered than random sequences. Cell Mol Life Sci. 7315:2949–2957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamudio N, et al. , 2015. DNA methylation restrains transposons from adopting a chromatin signature permissive for meiotic recombination. Genes Dev. 2912:1256–1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.