


Abstract
Primate-specific NBL2 macrosatellite is hypomethylated in several types of tumors, yet the consequences of this DNA hypomethylation remain unknown. We show that NBL2 conserved repeats are close to the centromeres of most acrocentric chromosomes. NBL2 associates with the perinucleolar region and undergoes severe demethylation in a subset of colorectal cancer (CRC). Upon DNA hypomethylation and histone acetylation, NBL2 repeats are transcribed in tumor cell lines and primary CRCs. NBL2 monomers exhibit promoter activity, and are contained within novel, non-polyA antisense lncRNAs, which we designated TNBL (Tumor-associated NBL2 transcript). TNBL is stable throughout the mitotic cycle, and in interphase nuclei preferentially forms a perinucleolar aggregate in the proximity of a subset of NBL2 loci. TNBL aggregates interact with the SAM68 perinucleolar body in a mirror-image cancer specific perinucleolar structure. TNBL binds with high affinity to several proteins involved in nuclear functions and RNA metabolism, such as CELF1 and NPM1. Our data unveil novel DNA and RNA structural features of a non-coding macrosatellite frequently altered in cancer.
INTRODUCTION
Somatic global genomic hypomethylation is frequent in almost all cancer types (1–5). Most of the tumor-associated somatic hypomethylation reflects demethylation of tandem and interspersed repetitive DNA elements (6–13). Although repetitive sequences account for at least half of the human genome, they were long considered ‘junk’ DNA, with no functionality (14). However, growing evidence suggests they play diverse regulatory roles and are critical for 3D nuclear organization (15). A causal link between DNA hypomethylation and tumorigenesis, in part by promoting DNA rearrangements, has been described in cancers, cell lines and mouse models (12,16–23). How demethylation of DNA repeats contributes to chromosomal instability and cancer is not well understood, but several hypotheses have been proposed. For instance, as many chromatin regulators bind to repeat-rich pericentromeric and centromeric heterochromatin (24–28), cancer-associated hypomethylation is likely to affect their binding dynamics and thus their local and genome-wide functions (5). Also, recent reports show that hypomethylation of satellite DNA repeats may lead to their aberrant transcription in several cancers (29,30), but the effects of these transcripts are mostly unknown.
Satellite DNA is characterized by tandem repeats classified into micro-, mini- and macro-satellites depending on their repeat unit size. While micro- and mini- satellite repeat units range from a few to 10 bp and from 10 to hundreds of bp respectively, macrosatellite units can be of several kb. All satellite sequences cover ∼3% of the human genome (31), and macrosatellite repeats (MSRs) occupy strategic locations, such as telomeres, pericentromeric regions, or adjacent to protein coding genes. Thus, among repeat classes, MSRs can be regarded as unique regulatory DNA structures. In somatic cells the majority of MSRs are organized into heterochromatin.
Some MSRs were shown to play fundamental roles in the organization of the genome and development of diseases (32). In particular, reduced copy number of polycomb-bound D4Z4 macrosatellite repeats is directly linked to the development of facioscapulohumeral dystrophy (FSHD) (33,34). Lower amounts of polycomb repressive complex and consequently heterochromatin loosening, allow transcription of a lncRNA, DBE-T, from D4Z4 repeats. In turn, DBE-T recruits Ash1L in cis, resulting in aberrant upregulation of adjacent genes (35). Thus, lncRNA DBE-T acts as a mediator of the epigenetic switch occurring at the D4Z4 locus upon contraction of the repeat array.
Aside from D4Z4, the X-linked DXZ4 MSR has also been studied in detail. DXZ4 plays a critical role in X chromosome inactivation and produces long non-coding transcripts, which seem to be developmentally regulated and may contribute to heterochromatin formation at DXZ4 (36). However, other known MSRs are vaguely explored. Many are poorly annotated, sequenced, and assembled in the reference genome (32). Little to nothing is known about the transcriptional deregulation of MSRs in cancer.
In our previous studies we proposed a ‘wear and tear model’ hypothesizing that DNA hypomethylation accumulates during aging by the unavoidable loss of methylation copy fidelity during successive DNA replication rounds (37). We also described a pericentromeric repeat element hypomethylated in colorectal, ovarian, and breast cancers (37,38). The repeat, initially characterized as SST1 element, was later identified as identical to NBL2, a 1.4 kb, primate-specific MSR discovered more than 20 years ago due to its cancer-specific hypomethylation (39–42). Recently, we observed that ‘severe’ NBL2 demethylation in 7% of colorectal cancers (CRC) did not follow the ‘wear and tear model’ because it did not increase with the age of the cancer patients. Further analysis associated NBL2 demethylation with genomic damage, but also with the presence of p53 mutations in a subset of CRC (38). Our results also showed that helicase HELLS (also known as PASG, LSH and SMARCA6) contributes to maintain SST1/NBL2 sequences methylated.
In continuation of this previous work, we have determined here the location of NBL2 MSR in the linear genome and three-dimensional nuclear space. We report that NBL2 is transcribed into lncRNAs, referred as TNBL (Tumor-associated NBL2 transcript), in CRC and provide structural insights of this novel macrosatellite repeat-derived lncRNA.
MATERIALS AND METHODS
NBL2 in silico genomic analysis
NBL2 distribution analysis was performed by aligning NBL2 consensus sequence (made by aligning 21 monomers from chromosome 21) to the human genome version 38 (hg38) using the Exonerate software program (43). Each aligned region was confirmed to belong to an SST1 region using the “rmsk” table from the UCSC Genome Browser. Ideograms were generated using the online visualization tool PhenoGram (44).
Mammalian cell culture
Colorectal, ovarian and breast cancer cell lines were obtained from ATCC and cultured according to recommended protocols. Human-rodent somatic cell hybrid cell lines were purchased from Coriell Institute and cultured according to recommended culturing conditions. During AZA treatments media was changed every 24 h and fresh media with AZA was added. TSA was added 16–20 h before cell harvest. Optimal drug concentrations and time periods for each cell line were: HCT116 and DKO 0.5 μM AZA (72 h) and 0.3 μM TSA; Caco-2, MDA-MB231 and A2780-ADR 3 μM AZA (96 h) and 0.5 μM TSA; TOV112D 3 μM AZA (72 h) and 0.3 μM TSA. Somatic cell hybrids were treated with 3 μM AZA for 96 h and 0.3 μM TSA 16–20 h before harvest. Chromosome 21 somatic cell hybrid was treated for 72 h with AZA.
Actinomycin D (ActD) was used at final concentration of 5 μg/ml. LS174T and DKO were harvested at 0, 0.5, 1, 2, 4 and 6 h after adding ActD. For RNA FISH purposes, ActD was incubated 30′ for LS174T and DKO cells, and 4.5 h for clone 12 and 48.
Metaphase FISH
Lymphocyte cells from a healthy donor (46XX) were processed with HANABI Metaphase Chromosome Harvester (ADS Biotec) in the Cytogenetic's platform of the Hospital Germans Trias i Pujol. The probe for DNA FISH was labeled with Biotin-dCTP (Life Technologies) by amplifying an internal region of NBL2 with NBL2 Primer set 1 F and Primer set 2 R. Conditions of amplification were as follows: 95°C 2′; 40 cycles of 95°C 45″, 60°C 45″ and 72°C 1′. A mixture of 150 ng probe, 7.5 μg Cot-1 (Life technologies) and 10 μg salmon sperm (Life technologies) was ethanol precipitated. Pellet was dissolved in 20 μl hybridization buffer (Sigma). Metaphase chromosome spreads were spotted onto glass slides. After RNase A treatment (200 μg/ml in PBS, 1 h at 37°C), denaturation was performed with 70% formamide/2× SSC for 10′ at 75°C. Slides were washed in PBS and dehydrated through an ice-cold EtOH increasing gradient (70%, 90% and 100% for 3′ each). PCR probe mix was heated at 85 °C for 5′ to separate DNA strands and applied to the slides. A coverslip was placed on top of the probe, sealed with parafilm and incubated in a humid chamber at 37°C overnight (ON). Next day, preparations were submitted to stringency washes at 45°C: two washes in 50% formamide/0.5× SSC for 15′, two washes of 5′ in 0.5× SCC and two washes in 0.5× SSC/0.1% Tween-20 for 5′ each. Signal detection was performed with Tyramide Signal Amplification kit (Life Technologies). Slides were blocked with 1% BSA in PBS for 30′, and incubated with 100 μl Streptavidin antibody diluted in 1% BSA/PBS (1:100, Life Technologies) for 30′ at RT. Finally, preparations were washed in PBS, incubated with 100 μl Tyramide labeled with Alexa Fluor 488 (1:1000) 5–10′ and washed 3× in PBS. Slides were mounted with Vectashield medium containing DAPI H-1200 (Vector Laboratories).
RNA extraction
RNA extraction was performed with miRNAeasy kit (Qiagen) or Maxwell LEV Simply RNA tissue kit (Promega) following manufacturer's instructions. Extraction with miRNAeasy kit was followed by DNaseI treatment (DNA-free kit, Ambion) according to manufacturer's instructions. Maxwell is an automated system for RNA extraction that includes DNaseI treatment. Each sample was rigorously tested for DNA contamination by qPCR after each extraction (see below). Accurate assessment of RNA quality from CRC patients was done with Bioanalyzer (RNA Nano Assay: 25–500 ng/μl).
cDNA synthesis and qPCR analysis
Reverse transcription was performed with SuperScript® III or IV First-Strand Synthesis System (Thermo Fisher Scientific) with random hexamers (or oligo(dT) when indicated) on 0.2–3 μg of RNA. For strand-specific reverse transcription 2 pmol NBL2 forward or reverse primer were added. Relative expression was determined by quantitative polymerase chain reaction (qPCR) using SYBR Green I master mix (Thermo Fisher Scientific) according to manufacturer's instructions. Conditions of amplification were as follows: 95°C 10′; 45 cycles of 95°C 15″, 60°C 20″ and 72°C 25″. Expression levels were normalized using PUM1 in cell lines, PUM1 and BETA2 in CRC patients, GAPDH in somatic cell hybrids. A list of primers used in qPCR analyses is summarized in Supplementary Table S1. Their efficiencies were compared to ensure analysis by the comparative Ct method. Relative expression data was analyzed comparing the Ct values of the gene of interest with Ct values of the reference gene for every sample. We used the formula 2ΔΔCt, ΔΔCt being the difference between the Ct of the RNA of interest and the Ct of the housekeeping gene. Duplicates or triplicates were made for each sample and primer set.
RNA fractionation
Protocol for RNA fractionation was performed as described in (35). Cells were pelleted and lysed with 175 μl/106 cells of cold Buffer 1 (50 mM Tris–HCl pH 8.0, 140 mM NaCl, 1.5 mM MgCl2, 0.5% NP-40, 2 mM Vanadyl Ribonucleoside Complex) and incubated 5′ on ice. Lysed cells were centrifuged 2′ at 4°C and 300 g. Supernatant (cytoplasmic fraction) and pellet (nuclear fraction) were kept on ice and used for RNA extraction. In case of further fractionating to nuclear soluble and nuclear insoluble fractions, pellet was lysed with 175 μl/106 cells of cold Buffer 2 (50 mM Tris–HCl pH 8.0, 500 mM NaCl, 1.5 mM MgCl2, 0.5% NP-40, 2 mM Vanadyl Ribonucleoside Complex) and kept on ice for 5′. Lysate was centrifuged 2′ at 4°C and 16 300 g. The supernatant corresponds to the nuclear-soluble fraction. The remaining pellet corresponds to the nuclear-insoluble fraction. Total RNA from aqueous solutions (cytoplasmic and nuclear-soluble fractions) was extracted with Qiagen miRNAeasy kit, whereas the chromatin-associated fraction was processed with the same kit according to instructions for pellet extractions.
RNase R treatment
Treatment was performed essentially as described by Memczak and colleagues (45). Briefly, total RNA from LS174T cells was incubated with 3 U of RNase R (Epicentre Biotechnologies) per μg of RNA for 15′ at 37°C. RNA was purified with MEGAclear™ clean-up kit (Ambion), followed by cDNA synthesis with SuperScript IV and qPCR.
Chromatin Immunoprecipitation (ChIP)
ChIP was performed with ChIP-IT® Express Enzymatic Magnetic Chromatin Immunoprecipitation Kit and Enzymatic Shearing Kit (version F5, Active Motif), according to instructions. Cells were crosslinked with 1% formaldehyde for 10′ at RT, quenched with glycine (0.125 M) for 5′ at RT, washed 2× with ice-cold PBS, scraped and pelleted by centrifugation. Chromatin was enzymatically digested (MnaseI) for 8′ at 37°C. Equal amounts of chromatin (100 μg) were diluted to 500 μl with IP buffer and incubated with 3 μg of desired antibody (H3K9ac (07-352, Millipore), total H3 (ab1791, Abcam), control IgG (ab46540, Abcam)) ON at 4°C on a rotation wheel. DNA-antibody complexes were recovered by adding protein A/G magnetic beads (Millipore) for 2 h. After two washes with low salt buffer, 2 with high salt buffer, 1 with LiCl buffer and a final wash with TE (10 mM Tris pH 8.1, 1 mM EDTA), immunoprecipitated DNA was eluted, reverse crosslinked at 65°C ON, treated with RNAse A (10 μg/ml) and proteinase K (200 μg/ml), and purified by phenol chloroform extraction and ethanol precipitation. Air-dried pellet was dissolved in 50 μl of MilliQ water and 2.5 μl were used for qPCR following the same conditions described in qPCR analysis. Comparative Ct method relative to input material was used to analyze the relative enrichment of histone marks at selected genome regions.
Northern blots
We used DIG Northern Starter Kit from Roche for probe preparation and chemiluminiscence detection according to instructions. All buffers were prepared in RNAse-free, DEPC treated ultrapure water (MiliQ H2O), and treated with DEPC, when possible.
To prepare probe template, cDNA from HCT116 AZA/TSA treated cells was amplified with NBL2 specific primers containing a T7 or T3 overhang in one of the primers, depending on the desired direction of in vitro transcription (IVT). IVT and DIG labeling were performed with MaxiScript (Ambion) and DIG-11-UTP (Roche) according to instructions. The efficiency of DIG incorporation and concentration of the RNA probe was analyzed by dot blotting serial dilutions of the prepared probe and control probe of known concentration (DIG Northern Starter kit, Roche). We assured all RNA samples were not contaminated with gDNA by qPCR. Samples were prepared by mixing 16 μg of total RNA with an equal volume of 2× RNA loading buffer (Ambion loading buffer II mixed with formaldehyde (Sigma) to final 8% formaldehyde), denatured at 67°C for 10′, 94°C for 1.5′ and chilled on ice for 1′. Denatured RNA samples were loaded on the denaturing agarose gel (1.1% agarose (Roche), 6.6% formaldehyde (Sigma) in 1× MOPS) and run at 80 V for ∼6 h in 1× MOPS (20 mM MOPS (Sigma), 5 mM sodium acetate (Sigma), 2 mM EDTA (Ambion)). RNA was transferred onto a positively charged nylon membrane (Roche) via capillary transfer ON. The following day, the membrane was briefly washed in 2× SSC (0.3 M NaCl (Sigma), 0.03 M sodium citrate (Sigma), pH 7), and crosslinked by baking at 120°C for 30′. RNA integrity and transfer efficiency was assessed by staining the membrane with methylene blue. Membrane was prehybridized in ULTRAhyb® Ultrasensitive Hybridization Buffer (Ambion) for 1 h at 68°C. Probe was denatured by diluting 1:10 000 in ULTRAhyb® Ultrasensitive Hybridization Buffer at 68°C for 1′. Hybridization was performed ON at 68°C in a glass roller bottle. The following day, membranes were washed 2× in low stringency buffer (2× SSC, 0.1% SDS) for 5′ at RT, followed by 2 high stringency washes (0.1× SSC, 0.1% SDS) at 68°C for 15. Membrane was washed in Washing buffer (0.1 M maleic acid, 0.15 M NaCl, pH 7.5, Tween-20 0.3% (v/v)) and blocked in Blocking solution for 30′. Antibody solution containing anti-digoxigenin-AP (1:10 000) was incubated 30′ at RT. Membrane was washed 2× for 15′ in washing buffer, and equilibrated in detection buffer (0.1 M Tris–HCl, 0.1 M NaCl, pH 9.5) for 5′. Chemiluminiscence reagent CDP-Star was added, and the membrane was exposed to an autoradiography film (Sigma).
DNA extraction and bisulfite sequencing
DNA from pelleted cells was isolated with Maxwell Cell DNA Purification kit. One microgram of DNA was transformed with bisulfite using EZ DNA Methylation™ kit (Zymo Research). One hundred fifty nanogram of bisulfite-converted DNA was amplified with primers (designed considering C to T conversion) specific for an internal region of NBL2 with FastStart High Fidelity PCR System (Roche) as described previously (38). PCR products were cloned into TA vector (Stratagene) and minipreps from ∼10–20 colonies were sanger sequenced using T7 or T3 primer. Sequences were aligned using CLC software. Alignments were exported to BiQ Analyzer program to determine methylation of each CpG site.
Dual luciferase reporter assay
The insert of interest was obtained by PCR using a high-fidelity polymerase (FastStart High Fidelity, Roche) with primers having a restriction enzyme sequence overhang. Fragments were designed to have different cloning sites on each primer to ensure directionality of the cloning. Fragments shorter than 2 kb were amplified using the following conditions: 95°C 2′; 40 cycles of 95°C 30′, 59°C 30′, 72°C 2′; 72°C 4′. Longer fragments were amplified according to the following conditions: 95°C 2′; 10 cycles of 95°C 30″, 59°C 30″, 68°C 4′ 30″; 30 cycles of 95°C 30″, 59°C 30″, 68°C 4′ 30″ adding 20″ each additional cycle; 72°C 7′. PCR products were gel purified, cloned into pGL3-Basic plasmid (Promega) and sequenced.
The insert and pGL3-Basic plasmid were digested with two restriction enzymes (KpnI, HindIII or NheI; New England Bioloabs) according to instructions. After digestion, the plasmid was dephosphorylated using alkaline phosphatase. Plasmid and insert were ligated using T4 ligase (NEB) in 1:3 ratio. Minipreps were prepared with NucleoSpin Plasmid EasyPure kit (Macherey-Nagel). Plasmids were sequenced to ensure the correct sequence and directionality of the insert.
DKO and LS74T cells were plated in a 12-well plate in order to be 40–50% confluent at the time of transfection. For each transfection sample, the complexes were prepared as follows: 50 ng pRL-CMV and 800 ng pGL3-insert of interest were diluted in 100 μl medium without serum. Polyethylenimine (PEI) was diluted in 100 μl medium without serum. Solutions were incubated for 5′ at RT. The diluted DNA was combined with diluted PEI, and incubated for 15′ at RT and finally 800 μl medium without serum were subsequently added to the mixture. Meanwhile, cells were washed 2× with PBS. 200 μl of the mixture were added to each well. Media was changed with 1 ml medium + serum after 5–6 h. Twenty four hours post-transfection, luciferase reading was performed with Dual-Luciferase® Reporter Assay System kit from Promega.
NBL2 3D DNA FISH/NPM1 immunofluorescence
3D DNA FISH was performed mainly as described in (46), with changes in the detection system. The same probe was used as for metaphase FISH. All washing steps were performed at RT, if not otherwise noted. Briefly, coverslips were coated with poly-l-lysine (10 μg/ml) for 1 h. Following coating, cells were seeded at desired confluence. On the day of FISH, coverslips were washed 2× with PBS and fixed in 4% paraformaldehyde for 10′, quenched in 0.1 M Tris–Cl, pH 7.4 for 10′ and permeabilized for 10′ in 0.1% saponin/0.1% Triton X-100 in PBS. After permeabilization, cells were washed 2 × 5′ in PBS, and incubated in 20% glycerol/PBS for at least 20′. Subsequently, coverslips were subjected to three freeze/thaw cycles in liquid nitrogen, washed 2 × 5′ in PBS and incubated in 0.1 M HCl for 30′, followed by 5′ wash in PBS. Coverslips were subjected to second round of permeabilization in 0.5% saponin/0.5% Triton X-100/PBS for 30′, washed 2× in PBS and equilibrated in 50% formamide/2× SSC for at least 10′. 10 μl probe premix was spotted on a glass slide, and the coverslip was gently placed on the drop. Slides were heated for 2′ at 78°C in order to denature simultaneously gDNA and probe, and hybridization was left in a humid chamber ON at 37°C. The following day coverslips were subjected to stringency washes: 50% formamide/2× SSC 15′ at 45°C; 0.2× SSC 15′ at 63°C; 2× SSC 5′ at 45°C; 2× SSC 5′ at RT; short rinse in PBS. Signal development was performed with Tyramide Signal Amplification kit (Life technologies) as described in the Metaphase FISH section. If continuing with immunofluorescence, cells were rinsed briefly in PBS, and permeabilized in PBT (PBS 0.1% Tween-20) for 15′. Cells were blocked for 30′ in 5% BSA in PBT. Cells were incubated with the primary antibody for NPM1 (1:100) in PBT 5% BSA for 1 h, washed 3 × 5′ in PBT and incubated with secondary antibody anti-mouse Alexa Fluor 594 (1:100) in PBT 5% BSA. Cells were washed 3× in PBS and mounted with Vectashield mounting medium containing DAPI H-1200 (Vector Laboratories).
Single molecule RNA FISH
Single molecule RNA FISH was performed as described in (47). A set of 48 custom tiled oligonucleotides labeled with Quasar 570 targeting NBL2 transcript were designed with LGC Biosearch Technologies' Stellaris online RNA FISH probe designer (Stellaris® Probe Designer version 4.2) and produced by LGC Biosearch Technologies. As controls for proper hybridization to nuclear and cytoplasm RNAs, two pre-designed probe sets labeled with Quasar 570 for GAPDH and MALAT1 (LGC Biosearch Technologies) were used.
After coating glass coverslips with poly-l-lysine in PBS (10 μg/ml) cells were seeded at desired confluence. On the day of RNA FISH, coverslips were washed 2× with RNase-free PBS and fixed with 3.7% formaldehyde in PBS for 10′ at RT. After fixation, coverslips were washed 2× with PBS and immersed in ice-cold 70% EtOH supplemented with 100 U/ml Superase RNase inhibitor (Ambion). Coverslips were left in 70% EtOH on 4°C up to 6 h. Coverslips were briefly washed with 2 ml of Wash buffer A (LGC Biosearch Technologies) at RT for 5′. In the case of RNase A treated controls, prior washing with Wash buffer A cells were incubated in 2 ml of RNase A (200 μg/ml in PBS, 1 h at 37°C). Next, cells were hybridized with 100 μl hybridization buffer (LGC Biosearch Technologies) containing corresponding probe (1:100) ON at 37°C in a humid chamber. The following day cells were washed with 1 ml of wash buffer A for 30′ at 37°C, followed by another wash with wash buffer A containing Hoechst DNA stain (1:1000, Sigma-Aldrich) for 30′ at 37°C. For Stimulated Emission Depletion (STED) microscopy, nuclear stain was Picogreen instead of Hoechst. Before mounting on a glass slide, cells were briefly (2–5′) washed in 1 ml of wash buffer B (LGC Biosearch Technologies). Cells were mounted with ProlongGold (Life Technologies). Slides were left to curate overnight at 4°C before proceeding to image acquisition.
Coverslips intended for immunofluorescence and RNA FISH were processed in the same way as described above, with the following changes: (i) hybridization buffer contained 1:100 dilution of NBL2 probe and 1:100 dilution of primary antibody (NPM1, Abcam; SAM68, Santa Cruz) and (ii) the first wash with wash buffer A after ON hybridization contained 1:200 diluted anti-mouse or anti-rabbit secondary antibody labeled with Alexa Fluor 488 (Abcam). For SAM68 intended for STED, secondary antibody labeled with Abberior STAR 635 was used instead of Alexa Fluor 488, and no DNA staining was applied.
NBL2 RNA/DNA FISH
Cells were seeded on poly-L-lysine coated 8-well glass Millicell EZ SLIDE. TNBL single molecule FISH was carried out as described in the previous section. At least 100 nuclei were positioned and their coordinates were saved on widefield fluorescence microscope Olympus ScanR, with a 40× 0.75NA UPlan FL N objective and a Hamamatsu Orca-ER camera. Subsequent DNA FISH was performed according to the protocol previously described (48). Probe sequence, probe preparation and probe signal detection was the same as in the 3D DNA FISH approach. NBL2 DNA FISH was imaged on the same positions used to sample TNBL RNA. Alignment of images and observed drift correction was done with FIJI plugin MultiStackReg v1.45. DAPI was used as a reference to correct the drift that occurred between the two imaging.
Microscopy and image analysis
Metaphase FISH images were obtained with wide-field fluorescence microscope Olympus with 100×/1.3NA UPlanFL N objective. 3D NBL2 DNA FISH/NPM1 immunofluorescence zeta stacks were acquired with Zeiss Axio Observer Z1 inverted confocal microscope using 63x/1.4NA DIC M27 objective. Images of Alexa fluor 488 were taken with 488 nm excitation and emission detection between 498 and 584 nm. Pixel size was set to 80 nm. Images of Alexa fluor 594 were taken with 594 nm excitation and emission detection between 604 and 704 nm. 3D stacks were subsequently deconvolved using the Huygens deconvolution software (Scientific Volume Imaging, SVI).
Single molecule RNA FISH and single molecule RNA FISH/NPM1 immunofluorescence zeta stacks were obtained with Zeiss Axio Observer Z1 wide-field fluorescence microscope using 63x/1.4NA DIC M27 objective. 3D stacks were deconvolved using the Huygens deconvolution software (Scientific Volume Imaging, SVI).
Confocal and STED images of NBL2 RNA FISH were obtained on a Leica TCS SP8 STED 3X (Leica Microsystems, Mannheim, Germany) on a DMI8 stand using a 100×/1.4NA HCS2 PL APO objective and a pulsed supercontinuum light source. The STED settings were chosen to retain most part of lateral resolution and at the same time provide some improvement in the axial dimension (20% 3D STED). Accordingly, multichannel 3D super-resolution stacks were acquired at a pixel size of 26 nm and a z-step of 110 nm. Zoom was set at 5 in order to approximately fill the image with one nucleus. Image size was set at 880 × 880 pixels, but whenever the nucleus shape and/or orientation was favorable, image was cut in y to speed up the acquisition. Scanner speed was set at 1000 Hz in bidirectional mode. Confocal images of PicoGreen were taken with 488 nm excitation and emission detection between 495 and 552 nm without integration and average. Images of Quasar 570 were taken with 556 nm excitation and emission detection between 562 and 650 nm. For STED imaging, a continuous depletion laser at 660 nm was added at 100% intensity. The channels of 3D stacks were taken sequentially in a stack by stack acquisition mode. 3D stacks were deconvolved using the Huygens deconvolution software (Scientific Volume Imaging, SVI) for confocal and STED modes using shift correction to account for drift during stack acquisition.
Confocal and STED images of NBL2 RNA FISH and SAM68 immunofluorescence were taken on the same microscope using a 93 × 1.3NA Glyc HC CS2 PL APO objective with a motorized correction collar and a pulsed supercontinuum light source ('white light laser'). Given the strong 3D component in the structures of interest, the STED settings were chosen to maximize the resolution improvement in the axial dimension and at the same time to retain some improvement in the lateral dimension (70% 3D STED). Accordingly, multichannel 3D super-resolution stacks were acquired at a pixel size of 23 nm and with a z-step of 68 nm. Pinhole diameter was set at 1 Airy Unit (AU) for the confocal channels and at 0.6 AU for the STED channels. Zoom was set at 4 in order to approximately fill the image with one nucleus. Image size was set at 1384 × 1384 pixels, but whenever the nucleus shape and/or orientation was favorable, image was cut in y to speed up the acquisition. Scanner speed was set at 700 Hz in bidirectional mode. The correction collar position was adjusted in every imaged field to ensure the image quality throughout the whole imaged volume. Images of Quasar 570 were taken as described previously. For STED imaging, a pulsed depletion laser at 775 nm was added with a 300 ps pulse delay at 50% intensity. Confocal images were taken without integration and with 2× frame average; STED images were taken without integration and 3× frame average. Images Abberior 635 were taken with 645 nm excitation and emission detection between 652 and 750 nm. For STED imaging, a pulsed depletion laser at 775 nm was added with 300 ps pulse delay at 30% intensity. Confocal images were taken without integration and with 2× frame average; STED images were taken with 2× line integration and 3–4× frame average. The channels of 3D stacks were taken sequentially in a stack by stack acquisition mode. Once acquired, 3D stacks were deconvolved and corrected for drift as described previously. Confocal channels were acquired at the beginning of the sequences to act as a reference for the drift correction of STED channels.
TNBL RNA FISH and SAM68 immunostaining 3D rendering was performed with Imaris software. Coordinates (x, y, z) for each generated surface in Imaris were exported and used to calculate the distances between centers of mass. Xtension Kiss and Run was used to generate closest exterior surface distances.
RNA affinity purification (RNA-AP)
RNA-AP was performed according to published protocols from our collaborators (49). Briefly, 100–200 ng gel-purified NBL2 monomer PCR product with T7 overhang was used as a template for in vitro transcription (IVT) per 20 μl of IVT reaction. IVT was performed ON according to instructions (MEGAscript kit, Ambion) and Biotin-labeled using Biotin-16-UTP (Epicentre). Probes were ethanol precipitated and dissolved in water.
Cell pellet was resuspended in total protein lysis buffer (20 mM Tris pH 7.4, 150 mM NaCl, 1.5 mM MgCl2, 2 mM DTT, 0.5% NP-40, 0.5% Na-deoxycholate, Complete EDTA-free protease inhibitor (Roche), 100 U/ml RiboLock (Thermo Scientific)). Protein extracts were pre-cleared with Streptavidin Sepharose beads (GE Healthcare) for 3 h at 4°C rotating on a wheel, and supplemented with 0.1 mg/ml yeast tRNA and 100 U/ml RiboLock.
To block the beads, Streptavidin Sepharose Beads were washed 2× with 500 μl of WB-100 (20 mM HEPES pH7.9, 100 mM of KCl, 10 mM MgCl2, 0.01% NP-40 and 1 mM of DTT) and blocked at 4°C for 2.5 h with blocking buffer (1 mg/ml of BSA, 200 μg/ml Glycogen, 200 μg/ml yeast tRNA (Sigma) and wash buffer-100). Beads were washed 3× with 500 μl of WB-300 (20 mM HEPES pH 7.9, 300 mM of KCl, 10 mM MgCl2, 0.01% NP-40 and 1 mM of DTT), transferred to a new tube and stored in WB-150 until the preparation of protein extracts.
50 pmol refolded in vitro transcribed RNA (5′ at 65°C; 15′ at RT, then transfer to ice) was added to pre-cleared protein extracts supplemented with tRNA & RiboLock and incubated 1h at 4°C on a rotating wheel. Afterward, 50 μl of blocked beads were added to RNA-protein mix and incubated for 1.5 h at 4°C on a rotating wheel. Protein-RNA-beads complexes were washed 5× with 1 ml WB-150 (20 mM HEPES pH 7.9, 150 mM of KCl, 10 mM MgCl2, 0.01% NP-40 and 1 mM of DTT) for 5′ at 7 rpm and 4°C on a rotating wheel. After the last wash, beads were transferred to a fresh tube.
Proteins were eluted from the beads with 150 μl elution buffer (50 μg/ml RNase A in WB-150) for 15′ on ice. Eluted proteins were precipitated with ice-cold acetone ON at –20°C, pelleted, washed with 80% EtOH. Dry pellet was resuspended in 30 μl 2× SDS sample buffer. 15 μl was run on SDS–PAGE gel. After silver staining the gel, band extraction of differentially enriched proteins and mass spectrometry was performed at Core Facility for Mass Spectrometry & Proteomics at ZMBH in Heidelberg, Germany. Data was viewed and interpreted using Scaffold software.
Western blots
Pulled-down protein samples (5 μl) were size separated on SDS-polyacrylamide gel (9%) for 100′ at 120 V and transferred onto a PVDF membrane by electroblotting (75′ at 400 mA). The membrane blocked with 5% non-fat dry milk ON at 4°C. Membranes were subjected to immunoblotting with the desired primary antibody (CELF1 1/2000 (sc-20003, Santa Cruz); NPM1 1/2000 (ab10530, Abcam); RUVBL2 1/3500 (SAB4200194, Sigma); XRCC5 1/2000 (ab79391, Abcam); PTBP1 1/1000 (ab133734, Abcam); NONO 1/300 (sc-376804, Santa Cruz); PLRG1 1/200 (sc-376729, Santa Cruz) in TBST (0.5% Tween-20) for 4–5 h at RT. Blots were incubated with Horse Radish Peroxidase-conjugated secondary antibody (Polyclonal Goat Anti-mouse Immunoglobulin/HRP (Dako Denmark); or Polyclonal Goat Anti-rabbit Immunoglobulin/HRP (Dako Denmark)) diluted 1:10 000 in 5% milk in TBST for 1 h at RT, washed 3× TBST and finally Pierce® ECL western Blotting Substrate was added. Membranes were exposed to an autoradiography film (Sigma) inside a cassette and developed.
UV crosslink and RNA immunoprecipitation
Protocol was carried out mainly as described in (50) with changes in the UV crosslink step according to Cabianca et al. (35), and other minor changes. After washing the cells (LS174T NBL2 low expressing and high expressing clone) 2× with ice-cold PBS, 10 ml of ice-cold PBS was added and cells were UV crosslinked 2× with 100 000 μJ/cm2 with an interval of 1′ between two irradiations.
Cell pellet was lysed on ice in lysis buffer (0.5% NP40, 0.5% Na-deoxycholate, 300 U/ml superase inhibitor (Ambion), protease inhibitor (Roche) in PBS, pH 7.9) and put on rotation (25′ at 4°C). 30 U of Turbo DNase (Ambion) were added to the samples, incubated 15′ at 37°C, and centrifuged 15′ at 11 000 g at 4°C. Supernatant was transferred into a new tube and passed through a 45 μm filter (Milipore). For each RNA IP 500 μg of lysate was dissolved in lysis buffer to 200 μl and precleared with 25 μl of Dynabeads Protein G. 2 μg of antibody was added (CELF1 sc-20003, Santa Cruz; NPM1 ab10530, Abcam; RUVBL2 SAB4200194, Sigma; PLRG1 sc-376729, Santa Cruz) and incubated ON at 4°C on a rotation wheel. Ten percent of precleared supernatant was saved as input. The following day, 50 μl of pre-washed Dynabeads G were added to each tube and left on rotation for an additional hour. Beads were washed 3× (5′ at 4°C) with PBS supplemented with 1% NP40, 0.5% Na Deoxycholate, additional 150 mM NaCl (final 300 mM), and 1:200 Superase inhibitor (Ambion). Beads were resuspended in 100 μl of PBS + 10× DNase buffer and 3 U of Turbo DNase (Ambion) were added. Samples were incubated 30′ on rotation at 37°C. Beads were washed 3× (5′ at 4°C) with 1% NP40, 0.5% Na Deoxycholate, 10 mM EDTA, additional 150 mM NaCl (total 300 mM), 1:200 Superase inhibitor (Ambion) in PBS. RNA was eluted in 100 μl of elution buffer (100 mM Tris–HCl (pH 7.5), 50 mM NaCl, 10 mM EDTA, 100 μg Proteinase K, 0.5% SDS) for 30′ at 55°C, with shaking. After, samples were centrifuged at 16 100 g at RT. The supernatant was used for RNA extraction with Maxwell LEV Simply RNA tissue kit (Promega). cDNA synthesis and qPCR were performed as usual.
RESULTS
Pericentromeric NBL2 repeats from acrocentric chromosomes localize in the perinucleolar region
In the unlocalized GL000193.1 contig of the human reference genome hg37, the 1.4 kb NBL2 macrosatellite unit is repeated in tandem 21 times (Supplementary Figure S1A). Each repeat starts with a GA-rich sequence and CA dinucleotide repeats of variable length. These 21 NBL2 repeats share between 90 and 99% identity except for the truncated repeat 21 (Supplementary Figure S1B). To facilitate assessing the consequences of NBL2 hypomethylation we set out to determine more precisely its genomic location by PCR on human-rodent somatic cell hybrids. All NBL2-specific primer sets amplified NBL2 sequences from chromosomes 9, 13, 14, 15 and 21. Of note, four of these chromosomes are acrocentric (Figure 1A). Less robust signals with lower intensity or from less primer pairs came from chromosomes 7, 12, 20 and Y, suggesting that these chromosomes contain fewer copies or more divergent NBL2 repeats (Figure 1A).
Figure 1.

NBL2 distribution in the human genome. (A) NBL2 PCR analysis on DNA from human-rodent somatic cell hybrids containing a single human chromosome isolated in a rodent background. Top: in pink are represented the regions amplified by four NBL2 specific primer sets located across the repeat unit. Bottom: PCR products of each primer set from genomic DNA of somatic cell hybrids containing the indicated human chromosome. (B) NBL2 DNA FISH on metaphase spreads from leukocytes of a healthy donor (46XX). Hybridization signals were detected close to the centromeres in the short arms of acrocentric chromosomes 13, 14, 15 and 21 (n = 30). NBL2 in green, DAPI in grey. (C) Ideogram showing genome-wide distribution of NBL2 repeats in the human genome version hg38. 301 hits were color-grouped according to their similarity to NBL2 consensus sequence. Acrocentric chromosomes positive for NBL2 by PCR (A) and FISH (B) are highlighted by a colored box. (D) NBL2 DNA FISH (green) and NPM1 immunofluorescence (magenta) on interphase nuclei (grey) of HCT116 cell line. 3D view (on the right) shows NBL2 repeats from acrocentric chromosomes located in the perinucleolar region. On average, each nucleus contained 8 NBL2 FISH signals, with 92% of detected signals located adjacent to nucleolus (n = 30 nuclei).
Quantitative PCR (qPCR) with primer sets amplifying a smaller region of NBL2 indicated that acrocentric chromosomes 13, 14, 15 and 21 carry NBL2 in a higher copy number (table in Supplementary Figure S1C). Chromosomes 13, 14, 15 and 21 also shared DNA sequences adjacent to the NBL2 repeat cluster, which allowed to roughly estimate the NBL2 relative abundance between these chromosomes. In human-rodent somatic cell hybrids, chromosome 13 had the highest copy number of NBL2 repeats, followed by chromosome 21 and 15 (graphic in Supplementary Figure S1C).
Next, we studied the NBL2 location by performing DNA FISH on metaphase chromosomes from lymphocytes of a healthy donor. Hybridization signals were detected close to centromeres in the short arm of chromosome 21, and any two of the PCR-positive chromosomes 13, 14 or 15, difficult to distinguish by DAPI staining due to their similarity (Figure 1B). These results extend previous findings (39,51) and add a more precise information on NBL2 localization and quantitative variation.
While performing this study, contig GL0000193.1 was incorporated into genome built hg38 and mapped to chromosome 21. In silico analysis performed using a consensus NBL2 sequence obtained from the 21 monomers revealed a total of 301 hits (Figure 1C). Most conserved copies (80–100% similarity to the NBL2 consensus) are located in the pericentromeric regions of chromosomes 7, 9, 12, 20 and 21 (Figure 1C, green circles), partially coinciding with our PCR results on human-rodent somatic cell hybrids. More divergent copies are placed in the pericentromeric regions of chromosomes 1, 7, 9, 12, 16, 17, 18, 20, 22 and Y; and in the long arm of chromosomes 4 and 19. Some chromosomes carry homogenous tandem arrays, while other chromosomes have tandem arrays of NBL2 with variable levels of similarity. The absence of mapped NBL2 repeats from acrocentric chromosomes 13, 14 and 15 (Figure 1C), in contrast with our experimental data (shown in Figure 1A, 1B and Supplementary Figure S1C) highlights the fact that DNA repeats are still poorly assembled in the reference genome.
To study the spatial distribution of NBL2 repeats on interphase nuclei, we performed NBL2 DNA FISH with the same probe used for the metaphase FISH (Figure 1B), followed by immunofluorescence against nucleophosmin (NPM1), a nucleolus marker. Most detected dots (92%) associated with the nucleolus in HCT116 CRC cells (Figure 1D). More precisely, 3D visualization showed that NBL2 repeats from acrocentric chromosomes were located in the perinucleolar region (Figure 1D, right).
NBL2 is transcribed, when de-regulated by DNA demethylation and histone acetylation
DNA methylation at CpG sites is a potent repressor of gene expression. NBL2 sequences have high GC content (62%, Supplementary Figure S1A), a high observed to expected CpG ratio (82%) and are highly methylated in normal somatic cells (52). Thus, we hypothesized that hypomethylation of non-coding NBL2 sequences could lead to their expression. We analyzed expression of NBL2 by RT qPCR in CRC cell lines. NBL2 expression was almost undetectable in CRC cell lines harboring highly methylated NBL2 (Caco-2, HCT116) whereas hypomethylated NBL2 (in LS174T cells) were transcribed (Figure 2A). Treatment with the DNA hypomethylating agent 5-aza-2′-deoxycytidine (AZA) resulted in increased NBL2 transcription in both methylated cell lines, in a time-dependent manner (Figure 2B).
Figure 2.

DNA methylation and histone deacetylation maintain NBL2 repressed. (A) Left: NBL2 methylation assessed by bisulfite sequencing in 3 CRC cell lines. Right: relative NBL2 expression analyzed by RT qPCR in CRC cell lines. (B) Left: NBL2 expression analyzed by RT qPCR in HCT116 cell line untreated (NT) and treated with AZA for 72h. Right: NBL2 expression analyzed by RT qPCR in Caco-2 cell line untreated (NT) and treated with AZA in a 5-day time course experiment. (C) Upper left: NBL2 expression assessed by RT qPCR in HCT116 cell line untreated (NT), treated with AZA, treated with AZA followed by TSA, and treated with TSA only. Upper right: relative abundance of NBL2 transcript compared to PUM1 levels (assigned as 1 in each sample) within each treatment group. Lower left: NBL2 methylation assessed by bisulfite sequencing in each treatment group. Each dot represents the average methylation of one NBL2 bisulfite converted sequence. Lower right: H3K9ac chromatin immunoprecipitation at NBL2 loci. Enrichment levels were normalized with H3 total levels at NBL2 loci. (D) Top left: NBL2 methylation assessed by bisulfite sequencing in HCT116 and DKO. Each dot represents the average methylation of one NBL2 bisulfite converted sequence. Top right: NBL2 methylation in HCT116 and DKO represented by a lollipop graph. Each line represents the internal region of 1 bisulfite converted NBL2 cloned DNA molecule. White and black circles: unmethylated and methylated CpGs, respectively. Lower left: RT qPCR of NBL2 in HCT116 untreated, DKO untreated, DKO treated with AZA and DKO treated with TSA. Lower right: relative abundance of NBL2 transcripts compared to PUM1 levels (assigned as 1 in each sample). All qPCRs show relative levels obtained using comparative Ct method and normalized with PUM1.
In contrast, treatment with the histone deacetylase inhibitor Trichostatin A did not affect NBL2 transcript levels by itself but synergized with AZA treatment in both HCT116 and Caco-2 cells (Figure 2C, upper panel, Supplementary Figure S2A). Accordingly, increased transcription in AZA/TSA treated cells was associated with increased acetylation of histone H3 at lysine 9 (H3K9ac) at NBL2 sequences (Figure 2C, bottom right). Collectively, these data show that DNA methylation and histone deacetylation maintain NBL2 repressed, with DNA methylation as a dominant suppressor mark. The same result was observed in ovarian (A2780-ADR, TOV112D) and breast (MDA-MB231) cancer cell lines (Supplementary Figure S2B–D) showing the generalization of the repression mechanism.
NBL2 transcripts were compared with the levels of the housekeeping gene PUM1 (Pumilio RNA Binding Family Member 1), frequently used to normalize expression in CRC cell lines. NBL2 RNA levels were comparable to the levels of PUM1 mRNA (in Caco-2 cells, Supplementary Figure S2A middle), or even surpass them tenfold (HCT116 cells, Figure 2C, top right). Similar profile was observed in ovarian and breast cancer cell lines (Supplementary Figure S2B–D) again showing a general pattern of behavior in human cancer cells.
The relationship between NBL2 DNA methylation and acetylation in regulating its expression was analyzed in HCT116 DNMT1 and DNMT3b double knockout cells (DKO) that have strongly reduced global DNA methylation (53). NBL2 repeats were almost completely unmethylated and transcribed in DKO cells compared with HCT116 cells, thus excluding that the overexpression originated from a non-specific effect of AZA (Figure 2D). While AZA did not further increase expression, TSA-induced levels of NBL2 expression were higher than in HCT116 AZA/TSA treated cells (compare NBL2 expression in Figure 2D with 2C).
NBL2 repeats are also transcribed in primary CRC specimens
To test whether expression also co-occurs with NBL2 hypomethylation in primary cancers, we analyzed two groups of primary CRC and matched normal tissues (Figure 3). The results showed NBL2 over-expression in 3 out of 37 primary CRC (8.1%), resembling the frequency of severe NBL2 hypomethylation (7%) that we previously described (38). One tumor (727T, Figure 3, group 1) did not show severe hypomethylation of NBL2, suggesting that somatic NBL2 DNA hypomethylation and overexpression are related, but not in a linear dependent manner. However, we cannot rule out that increasing sequencing depth would unveil completely unmethylated sequences in tumor 727. Disruption of histone deacetylation at moderately hypomethylated NBL2 elements may induce strong NBL2 expression. Since NBL2 RNA is detected upon DNA demethylation in cancer, we designated these transcripts TNBL (Tumor-associated NBL2 transcript).
Figure 3.

NBL2 somatic DNA hypomethylation predisposes for NBL2 overexpression in CRC. NBL2 expression analyzed in 2 CRC groups. Group 1 (24 tumor and matching normal tissue samples) analyzed for NBL2 expression by RT qPCR (top) and methylation by bisulfite sequencing (bottom). Three representative cases are shown: two cases (727 and 33) with NBL2 overexpression in the tumor and one case (700) with no detectable NBL2 expression. Group 2 (13 tumor and matching normal tissue samples) analyzed for NBL2 expression by RT qPCR (top) and NBL2 DNA methylation by bisulfite sequencing (bottom). In methylation graphs each dot represents average methylation of an internal region of 1 NBL2 bisulfite converted sequence. Red dots for tumor, grey dots for normal. Pie chart shows the percentage of patients expressing NBL2 considering both groups (n = 37). Statistical analysis was not performed due to low number of samples.
TNBL is a nuclear, non-polyA, stable, long non-coding RNA
TNBL was amplified from random primed cDNA but not from oligo(dT), similarly to non-polyA 18S rRNA, indicating that TNBL lacks a polyA tail (Figure 4A). TNBL was enriched in reverse transcriptase reactions primed with an NBL2-specific forward oligonucleotide and not with a reverse primer, implying that transcription occurs in the antisense direction of the repeat (Supplementary Figure S3A). TNBL was enriched in the nuclear fraction, more specifically in the nuclear insoluble fraction (Figure 4B and Supplementary Figure S3B).
Figure 4.

TNBL is a long, non-polyA, nuclear and stable transcript. (A) TNBL RT qPCR of oligo(dT) or random primed cDNA from HCT116 (left) and Caco-2 (right) AZA and TSA treated cells. Control RNAs: PUM1 as a polyA containing RNA and 18S rRNA as a non polyA containing RNA. (B) Relative enrichment (in %) of TNBL, XIST and PUM1 transcripts in nuclear insoluble, nuclear soluble and cytoplasmic RNA fractions from AZA and TSA treated Caco-2 cells analyzed by RT-qPCR. (C) TNBL stability relative to GAPDH RNA measured by RT qPCR at several time points in LS174T cells treated with Actinomycin D (ActD) during a 6 h time course using two NBL2 specific primer sets (1 and 3). PUM1 and MALAT1 were used as controls of mature RNA, and 45s rRNA of pre-mature RNA. (D) Left: TNBL sense probe 1 Northern blot on RNA from untreated (NT), AZA and AZA/TSA treated Caco-2, HCT116 and TOV112D cells. RNA from mouse C2C12 myoblasts treated with AZA/TSA was used as a negative control. Middle: TNBL sense probe 2 Northern blot on RNA from HCT116 untreated (NT) and AZA/TSA treated, DKO untreated and TSA treated. Negative control: chromosome 9 human-rodent somatic cell hybrid treated with AZA/TSA. Right: TNBL sense probe 2 Northern blot on RNA from LS174T cells. Sample integrity and equal loading was assessed with 18s and 28s rRNA methylene blue staining. (E) NBL2 expression analyzed with RT qPCR in human rodent somatic cell hybrids untreated (NT), AZA treated, and AZA/TSA treated. (F) Northern blot on RNA from chromosome 13, 14, 15 and 21 human rodent somatic cell hybrids untreated and AZA/TSA treated. Sample integrity and equal loading was assessed with 28s rRNA methylene blue staining. (G) Scheme of DNA fragments cloned into pGL3-basic vector driving luciferase gene expression (LUC). Promoter activity was analyzed in DKO cells. NBL2 array is represented 3′ to 5′. 16 different constructs were analyzed: 10 different NBL2 monomer sequences (1.4 kb each); a monomer 21 (928 bp, truncated monomer); a monomer 21 with adjacent transposon elements (TE1: L1MD2-MLT1D); and four fragments encompassing distinct transposon element upstream regions: TE1 (L1MD2-MLT1D), TE2 (MLT1D-MER70-int), TE3 (LTR7-MER70A), TE4 (LTR7). The graph shows relative luciferase activity of tested fragments compared to empty vector. P = 3.9E–03, as evaluated by Mann-Whitney-Wilcoxon test.
In mammalian cells, a major step triggering mRNA decay is the removal of the 3′-poly(A). TNBL lacks poly(A), therefore it could represent an unstable run-through RNA. To determine TNBL stability, cells expressing TNBL endogenously (LS174T and DKO) were treated with the transcription inhibitor Actinomycin D (ActD). TNBL stability showed a similar profile to that of mature RNA of PUM1, lncRNA MALAT1, and GAPDH (Figure 4C, Supplementary Figure S3C) in contrast with the pre-mature form of ribosomal RNA (45S rRNA) that was depleted after 30 min of ActD treatment. This result shows that TNBL is a stable transcript and that the regions monitored by RT qPCR form part of a mature RNA molecule. Next, we analyzed the TNBL length by Northern blots on RNA from CRC (Caco-2, HCT116, DKO) and ovarian cancer (TOV112D) cell lines treated with AZA and AZA/TSA, and in LS174T that contains unmethylated NBL2. Antisense probes did not give a hybridization signal (not shown) confirming the antisense direction of transcription. Two sense probes, designed against two different regions of the NBL2 repeat (Supplementary Figure S3D), hybridized against two high molecular weight RNAs of approximately 13 and 10 kb, and a lower of 4.2 kb (Figure 4D). These bands were absent in Caco-2, HCT116 and TOV112D untreated cells (all having methylated NBL2), and increased after treatment with AZA and AZA/TSA, coincident with the RT qPCR results. Furthermore, relative enrichment of those bands between the cell lines after AZA/TSA treatment is consistent with RT-qPCR data, with Caco-2 levels being the lowest. Similar band patterns were displayed by untreated LS174T, showing that the transcripts induced by AZA/TSA treatment corresponded to the endogenous transcripts.
Three observations suggest that these transcripts include complete NBL2 monomers. First, both probes designed at opposing ends of the NBL2 monomer detected similar patterns of the differently sized transcripts (Figure 4D). Second, entire NBL2 monomers were amplified from cDNA with two different primer pairs (Supplementary Figure S3E). Third, PCR with primers amplifying the join region of adjacent monomers indicated that more than one repeat unit may be included in at least one of the two major transcripts (Supplementary Figure S3E). This ‘cross-boundary’ PCR together with TNBL’s high stability and lack of polyA tail, could be explained by a circular RNA molecule. However, TNBL was not resistant to treatment with exonuclease RNAse R, behaving different from a circular RNA control (hCIRC 1 (45)) and similar to non-circular RNAs such as PUM1 and GAPDH (Supplementary Figure S3G).
TNBL originates from chromosomes 13, 14, 15 and 21
To determine the origin of TNBL detected by RT qPCR and Northern blot approaches, cell hybrids found positive for NBL2 DNA amplification (9, 12, 13, 14, 15, 20, 21, Y) plus several negative controls (hybrids containing chromosomes 1, 11 and X) were treated with AZA and AZA/TSA, and TNBL expression was measured by RT qPCR. Chromosome 21 showed the highest expression of TNBL upon demethylation and histone acetylation, followed by chromosomes 15, 14 and 13 (Figure 4E). Of note, none of the primers, which also recognized NBL2 from chromosomes 9, 12, 20 and Y (Figure 1A), amplified any RNA from these chromosomes (Figure 4E). Furthermore, and consistent with the RT qPCR data, chromosome 9 that contains several NBL2 copies intermingled with NBL2-like sequences (Figure 1C), did not give rise to any band detectable with a TNBL specific Northern blot probe (Figure 4D, probe 2). Northern blots on hybrids expressing TNBL upon induction with AZA/TSA showed that both transcripts of 13 and 10 kb originate from all four acrocentric chromosomes (Figure 4F). The absence of band 4.2 kb together with its localization close to the 28S rRNA, questions whether this is a real transcript or could represent an artifact due to impaired migration of degraded transcripts, an issue that will have to be determined. H3K9ac chromatin immunoprecipitation on untreated, AZA and AZA/TSA hybrid cells containing chromosome 21 confirmed the enrichment of histone acetylation at NBL2 repeats upon AZA/TSA treatment (Supplementary Figure S3F). Collectively, these results show that the regulation of TNBL expression from each of the four chromosomes harboring the highest copy number of NBL2 macrosatellites is conserved in the rodent cellular environment and that TNBL can originate from chromosomes 13, 14, 15 and 21. Whether chromosomes harboring NBL2-like sequences (for instance non-pericentromeric repeats at chromosome 4), are expressed under normal conditions or in cancer environment was beyond the scope of this study and remains to be analyzed.
NBL2 repeats contain promoter activity
The strong induction upon AZA and AZA/TSA treatments, combined with the expression of transcripts of precise size, suggested the existence of a specific transcription start site and a strong promoter. There is a cluster of truncated retrotransposons spanning 10 kb at 3′ of the annotated NBL2 MSR of chromosome 21 (see scheme in Figure 4G, Supplementary Figure S1A). As these TEs are located 5′ of the observed transcriptional direction, we decided to analyze their potential promoter activity by luciferase reporter assays. Several fragments containing these transposable elements, and 11 distinct NBL2 monomers from chromosome 21 were individually cloned and tested in DKO (Figure 4G). These truncated forms of retrotransposons did not elicit >2-fold promoter activity compared with the empty vector. On the other hand, NBL2 monomers consistently yielded higher promoter activities than TEs, with the highest activity from some monomers only 3-fold lower than the strong CMV positive control promoter. The average luciferase activity of all monomers was significantly higher than that of TEs (P = 3.9E–03, Mann-Whitney-Wilcoxon Test). These results suggest that some NBL2 monomers harbor internal functional promoters.
TNBL accumulates as a perinucleolar aggregate at NBL2 loci
To analyze TNBL localization in the three-dimensional nuclear space, we performed single molecule RNA FISH. HCT116 nuclei showed upregulation of TNBL signals upon AZA treatment, which were further enhanced by AZA/TSA (Supplementary Figure S4A). TNBL appeared widely distributed in the nucleus with several focal, bright signals. Quantification of TNBL signals showed 0 to 2 dots per nucleus in untreated HCT116 cells, while AZA and AZA/TSA treated HCT116 cells showed 2–30, and 7–75 dots per nucleus, respectively.
In TNBL endogenously expressing cells LS174T and DKO, TNBL appeared exclusively nuclear, preferentially as an aggregate located in the perinucleolar region (Figure 5A for LS174T, Supplementary Figure S5A for DKO). TNBL aggregates were more abundant in LS174T than in DKO. In both cell lines, TNBL aggregates could not be resolved well using wide-field or confocal microscopy. To better resolve the structure of the aggregates, we used Stimulated Emission Depletion (STED) super-resolution microscopy. The results show that these aggregates contain high amounts of TNBL molecules (Figure 5B and Supplementary Figure S5B). TNBL was also dispersed across the nucleus, more evident in LS174T cells that express higher amounts of TNBL compared with DKO (compare Figure 5B with Supplementary Figure S5B).
Figure 5.

TNBL is a perinucleolar, stable RNA. (A) Maximum intensity projections of TNBL RNA FISH (yellow) and NPM1 immunofluorescence (magenta) in LS174T cells. TNBL forms clusters adjacent to the nucleolus, with typically one or two clusters per nucleus. All of the clusters were located in the perinucleolar region (n = 50 nuclei). DNA shown in grey. Zeta stacks were acquired with a wide-field microscope and deconvolved with Huygens deconvolution software. Scale bar, 5 μm. (B) Maximum intensity projections of confocal and stimulated emission depletion (STED) imaging of TNBL RNA FISH (yellow) in LS174T cells. DNA in blue. Scale bar, 5 μm. (C) Maximum intensity projections of TNBL RNA FISH in untreated LS174T cells and treated with Actinomycin D (ActD) for 30 min and 4.5h. Zeta stacks were acquired with a wide-field microscope. TNBL in yellow, DNA in blue. On the right: quantification of clustered TNBL (labelled as C) and dispersed TNBL (labelled as D) in all treatment conditions. 4.5h treatment with Actinomycin D results in total depletion of TNBL clusters and significant increase in dispersed TNBL signals (***P ≤ 0.001, as evaluated by unpaired t-test versus 30′ ActD or NT; n = 50 nuclei). (D) NBL2 DNA/RNA FISH. NBL2 in magenta, TNBL in yellow. Bar plot (n = 70 nuclei) shows the percentage of NBL2 loci co-localizing with TNBL clusters (N–C), and the percentage of TNBL clusters co-localizing with NBL2 loci (C–N). Nucleus border is outlined with a dashed line. (E) TNBL during mitosis detected by RNA FISH in LS174T high expressing clones. TNBL in yellow, DNA in gray. Scale bar, 5 μm.
To analyze whether TNBL aggregates represent nascent or mature transcripts, RNA FISH was performed on DKO and LS174T cells treated with the RNA polymerase inhibitor actinomycin D (ActD). After 30 minutes of treatment TNBL aggregates were still present, indicating they were accumulations of mature transcripts (Figure 5C, Supplementary Figure S5C and D). Numerous TNBL transcripts were present during mitosis even though cells were treated with ActD, emphasizing TNBL stable nature (Supplementary Figure S5D, red arrow). Longer periods of ActD treatment (4.5 h) disrupted TNBL perinucleolar aggregates, releasing multiple TNBL transcripts that remained exclusively nuclear, suggesting that active transcription was necessary for aggregate formation.
In contrast with TNBL, the NBL2 MSR loci from chromosomes 13, 14, 15 and 21 were not clustered in the nucleus (Supplementary Figure S5E). Thus, TNBL could be expressed only from one chromosome, or from several acrocentric chromosomes, but accumulating in only one region. To address this issue, we performed NBL2 DNA/RNA FISH (Figure 5D). TNBL aggregate co-localized with one NBL2 loci in 90% of instances (bar plot in figure 5D, n = 70, quantified in 7 randomly selected fields). In contrast, from the eight NBL2 loci consistently detected by our FISH approach in interphase nuclei (corresponding to the two alleles of chromosomes 13, 14, 15 and 21) only around 20% co-localized with the TNBL aggregate. These results demonstrate that TNBL aggregates only accumulate around a subset of NBL2 loci, probably those that originate the transcript. Additional fainter TNBL signals (not aggregates) are dispersed in the nuclei and do not co-localize with NBL2 loci (Figure 5D).
All together, these results demonstrate that TNBL has a consistent location in the nucleus, with the vast majority accumulating at a subset of NBL2 loci, forming densely packed aggregates.
As controls, MALAT1 and GAPDH RNA FISH were coupled with NPM1 immunofluorescence (Supplementary Figure S6A), and confirmed the specificity of TNBL RNA FISH and immunostaining approach, showing MALAT1 characteristic localization in the nucleus (54), and GAPDH in the cytoplasm.
TNBL expression during mitosis
LS174T cells showed TNBL heterogeneity with some cells harboring high TNBL levels visible by RNA FISH. We monitored the behavior of TNBL expression during mitosis in clones expressing homogenously high TNBL levels (Figure 5E). When chromatin started to condense in early prophase, the TNBL aggregate was still present. However, in late prophase, the aggregate disassembled and multiple transcripts were released into the cytoplasm of the dividing cell. This again shows the high quantity of TNBL molecules retained within the aggregate. This process coincides with the timing of disassembly of the nuclear envelope and most of nuclear bodies such as the nucleolus (55). TNBL transcripts were present during metaphase outside the chromatin fraction and were transferred into daughter cells in anaphase. Once chromatin started to decondensate at the end of telophase, the majority of TNBL was located in the nucleus.
To summarize, these results show that TNBL is frequently detected as clusters of mature stable RNAs that transiently disaggregate during mitosis and after prolonged inhibition of RNA polymerases.
TNBL aggregate accumulates tightly adjacent to the SAM68 nuclear body
Since TNBL forms aggregates in the perinucleolar region, we investigated whether it associates with the two nuclear bodies described in the perinucleolar region: the perinucleolar compartment (PNC), consisting mainly of PTBP1 and CELF1 RNA binding proteins (56,57), and the SAM68 nuclear body (SNB), enriched in SAM68 RNA binding protein, among others (58,59). Both PNC and SNB appear preferentially in tumor cells and rarely in normal cells and their occurrence has been positively linked with the metastatic capacity (60,61). We performed TNBL RNA FISH in concert with CELF1 and SAM68 immunofluorescence in LS174T cells. CELF1 PNCs were not detectable by immunofluorescence (data not shown), while formation of SNBs was detected in all nuclei (Figure 6A). In addition to form SNBs, SAM68 is also diffusely distributed in the nucleus (lighter magenta staining outside SNBs in Figure 6A, B, D) in accordance with previous reports (58). The results showed that the TNBL aggregate consistently localized adjacent to SNB (Figure 6A).
Figure 6.
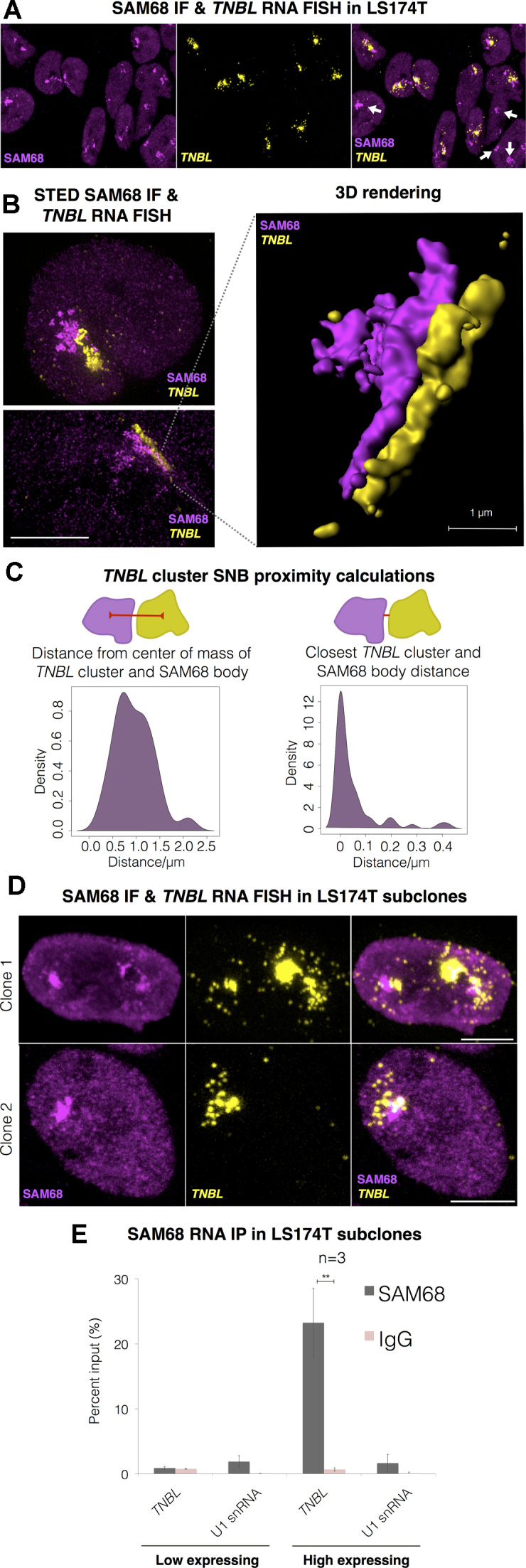
TNBL forms aggregates adjacent to SAM68 perinucleolar body (SNB). (A) Maximum intensity projection of SAM68 IF (magenta) and TNBL RNA FISH (yellow) confocal imaging in LS174T. SNBs (intense magenta) are located adjacent to TNBL clusters. White arrows: SNBs that do not co-localize with TNBL. (B) Left: SAM68 IF and TNBL RNA FISH STED images, top: both structures in an irregular round form, bottom: elongated form. Scale bar, 5 μm. Right: 3D rendering of the bottom left image showing SNB and TNBL aggregate in a lateral interaction through the entire length of the structures. Scale bar, 1 μm. (C) Proximity calculations between SNB and TNBL cluster in STED imaging (n = 54). Left: distances between SNB and TNBL clusters centers of mass. Right: closest exterior distances between SNB and TNBL clusters, median value 0 μm and maximum measured distance at 0.42 μm. (D) Maximum intensity projection of TNBL RNA FISH (yellow) and SAM68 IF confocal imaging in TNBL high expressing LS174T clones. Scale bar, 5 μm. (E) SAM68 UV crosslink RNA IP in high and low TNBL expressing LS174T clones. TNBL and negative control U1 snRNA were monitored by RT qPCR. Error bars indicate s.d., n = 3, **P ≤ 0.01, as evaluated by unpaired t-test vs. IgG.
By STED microscopy TNBL and SAM68 structures appeared in a tight lateral interaction and seemed to have similar shapes. Most of the times both structures had an irregular round form (Figure 6B, top image). In some instances, they acquired an elongated form, and in those cases both structures seemed specular images (Figure 6B, bottom image). 3D rendering illustrated their interaction in the proximal boundaries, and the same starting and ending point of the lateral interaction (Figure 6B). Proximity calculations revealed that mass centers between TNBL clusters and SNBs were maintained within a 0.23–2.15 μm distance range, with a median value of 0.92 μm (Figure 6C left graph). Furthermore, the majority of the closest exterior distances between TNBL aggregates and SNB surfaces were 0 μm (28 out of 54 analyzed surfaces), with little to no overlap, while the maximum distance measured was 0.42 μm (Figure 6C, right graph). The maximum resolution achieved with our STED settings was approximately 40 nm lateral and 100 nm axial. Based on this analysis we conclude that the TNBL-SNB interaction is robust. These structures did not seem to imbricate into each other but rather they appeared to have opposite surfaces possibly adjoined by a linker structure.
In two TNBL high expressing clones TNBL-SAM68 aggregates were also present (Figure 6D). In clone 1 that showed more than one SNB per nucleus, all and each of the TNBL aggregates were located adjacent to SNBs (Figure 6D upper panel) showing an apparent interdependence. However, SNBs were not always accompanied by TNBL clusters (Figure 6A white arrows). GAPDH and lncRNA MALAT1 control RNA FISH coupled with SAM68 immunofluorescence, reinforced the specificity of the TNBL distribution adjacent to SAM68 bodies (Supplementary Figure S6B).
In RNA immunoprecipitation of UV-crosslinked cells, TNBL appeared enriched in SAM68 pull-downs compared to the negative control RNA U1 snRNA (Figure 6E). This result further supports the close interaction between TNBL aggregates and SAM68-enriched SNBs.
TNBL interacts with CELF1 and NPM1
To identify additional proteins interacting with TNBL we used RNA affinity purification (RNA-AP) coupled to protein identification by mass spectrometry. Biotin-labeled and unlabeled NBL2 monomer-derived in vitro transcripts (IVTs) were incubated with total protein extracts from three different CRC cell lines. As negative controls, two IVTs of the lncRNA MALAT1 of the same size and GC content were used.
A number of protein bands specifically enriched in NBL2 IVT pull-downs were observed after gel electrophoresis and silver staining (Figure 7A). By visual inspection, we selected bands on the gel differential between NBL2 IVT and MALAT1 IVT controls in three different cell lines (DKO TSA, Caco-2 and LS174T). Bands 1 to 16 numerically labelled on top of the RNA-AP gel (arrows in Figure 7A) indicate consistent bands present in all three cell lines, which serve as biological replicates, that were not enriched in MALAT1 IVT control pull downs. The gel was cut in slices containing these differential bands (arrows in Figure 7A) and each band from two cell lines was analysed in order to have a biological replica of the mass spectrometry. After mass spectrometry analysis of each slice, known contaminants and proteins of inappropriate size were excluded. Since TNBL is only present in the nucleus, we also discarded cytoplasmic proteins. After these exclusions, the remaining proteins were ranked according to their prevalence in the respective band (by peptide count). Gel bands indicated in Figure 7B contained the top 20 ranked proteins, which included many candidates in the category of DNA repair, chromatin remodeling and RNA metabolism, such as stability and splicing (Figure 7C).
Figure 7.

TNBL interacts with CELF1 and NPM1. (A) Polyacrylamide gel with size-separated proteins from RNA-affinity purification (RNA-AP). RNA-AP was performed with protein extracts from 3 CRC cell lines (DKO TSA treated cells, Caco-2 and LS174T). Probes: NBL2 monomer in vitro transcript (IVT) unlabeled (NBL2 U) as a background control, NBL2 monomer IVT biotin-labeled (NBL2 L), 2 biotin-labeled negative control IVTs corresponding to different regions of MALAT1 of the same size and GC content as NBL2 IVT (C1 and C2). Bands indicated with red arrows were cut and subjected to mass spectrometry in replicas. Band indicated with a blue arrow was pulled down only with MALAT1 IVT 2. U stands for unlabeled, L for labeled. (B) Proteins with most peptide count within each band. (C) Functional ontology of proteins enriched with NBL2 RNA-AP. (D) Western blot validation of proteins pulled down with RNA-AP. NBL2 IVT specifically interacts with NPM1 and CELF1 compared with negative control IVTs. (E) UV crosslink and RNA IP with antibodies against CELF1 (left) and NPM1 (right) in LS174T TNBL high expressing clone. TNBL enrichment was monitored by RT-qPCR with two primer sets (1 and 3). Enrichment of JunD, MALAT, PUM1 and U1 snRNA was monitored to control background precipitation. Error bars indicate s.d., CELF1 RNA IP n = 7, NPM1 RNA IP n = 3. ***P ≤ 0.001, as evaluated by unpaired t-test versus IgG.
Western blots of the same affinity purifications confirmed the enrichment of the NPM1 and CELF1 with NBL2-monomer derived IVT and not with the control MALAT1 IVTs (Figure 7D), underscoring the specificity of these interactions. Some of these factors were not validated (XRCC5 or PTBP1), while others such as RUVBL2 or PLRG1 also precipitated with MALAT1 IVT. Thus, from these data, CELF1 and NPM1 were the most specific and robust interactors of NBL2 IVT. Furthermore, from UV-crosslinked cell lysates, both CELF1 and NPM1 co-immunoprecipitated with TNBL compared to negative controls (MALAT1, PUM1 and U1 snRNA). JunD was used as a positive control to determine the strength of these interactions (62), however in our cellular model JunD does not seem to bind CELF1 (Figure 7E).
DISCUSSION
The impact of DNA hypomethylation in the generation of chromosomal abnormalities has been described in mice and human tumors (7,8,10–13), but exactly how this happens is unknown. DNA hypomethylation of NBL2 MSR is a recurrent event of low to intermediate frequency in many types of cancer, but understanding its functional output has been hindered because of incomplete mapping of NBL2 MSRs in the reference genome (32). Here, we mapped the major locations of NBL2 MSR to four of five human acrocentric chromosomes. We found that hypomethylation of NBL2 MSR is tightly associated with its expression into lncRNAs, that we globally designated as TNBL.
Internal promoters of NBL2 MSRs are likely to drive expression of TNBL in cancer
Based on our results we propose a model where NBL2 loci are tightly repressed by a synergy between DNA methylation and histone deacetylation, with DNA methylation being the sealing repressive mark. In normal colon epithelia, high DNA methylation maintains NBL2 from acrocentric chromosomes 13, 14, 15 and 21 repressed (Figure 8). This model also includes our previous work (38), where we showed that in normal colon cells highly methylated NBL2 loci are also enriched in the repressive histone mark H3K9me3, whereas upon NBL2 hypomethylation in cancer, H3K9me3 is reduced. The present study shows that in these circumstances, TNBL expression is induced albeit at lower levels, and only when acetylated histones are present at hypomethylated NBL2, TNBL accumulates in high quantity. Taken into consideration the co-dependency between DNA methylation and H3K9me3 (63) we cannot rule out that loss of H3K9me3 could also contribute to de-repression of NBL2 loci. Thus, TNBL levels originating from hypomethylated NBL2 may vary between different cells, and depend on NBL2 DNA hypomethylation, H3K9 methylation and histone acetylation degrees, and the chromosome affected. RT qPCR and Northern blot (Figure 4E and F, respectively) on somatic cell hybrids indicate that chromosomes 21 (followed by chromosome 15) are the ones producing higher amounts of TNBL. Given that DNA/RNA FISH data shows that TNBL aggregates co-localize with a subset of NBL2 loci (Figure 5D), we reason that one or both NBL2 MSR alleles of chromosome 21 or 15 originate TNBL aggregates.
Figure 8.

Model for TNBL expression in CRC. Acrocentric chromosomes 13, 14, 15 and 21 contain tandem arrays of NBL2 MSR, repressed by DNA methylation and histone deacetylation in normal colon epithelia. Upon NBL2 DNA hypomethylation TNBL lncRNA is expressed at moderate levels. Subsequent increase of histone acetylation results in high TNBL expression. TNBL location is nuclear where it mostly forms aggregates in the perinucleolar region close to NBL2 loci. These aggregates interact with SNB creating cancer-specific perinucleolar structures. Outside of the perinucleolar aggregates, TNBL is widely dispersed in the nucleus. Whether TNBL selective binding to NPM1, SAM68 and CELF1 proteins, which are involved in genome organization, splicing regulation and mRNA stability respectively, could impair their functionalities and potentially impact nuclear architecture and cell behavior is at the moment unknown.
Previous work detected NBL2 RNAs in B cell lines from ICF syndrome patients and two neuroblastoma cell lines (64). However, due to the lack of a detectable promoter, NBL2 transcripts were disregarded and considered as run-throughs from unknown nearby promoters (51). Our results rule out a run-through transcript, since NBL2 repeats are consistently transcribed into long transcripts of a defined length, and we provide evidence that NBL2 MSR promote their own transcription by harboring internal promoter activity, an aspect of MSRs reported before (reviewed in (32)). We considered a group of transposon elements (TE), located adjacent to NBL2 repeats and upstream of TNBL transcriptional direction, as the initial suspects for promoting TNBL expression, since they share features of putative promoters for some lncRNAs (65). However, these TEs did not elicit promoter activity in luciferase assays. Still, we cannot rule out that these TEs may act as enhancers modulating NBL2 transcription in vivo. In silico analysis revealed that monomer repeats contain more than 30 consensus sequences for transcriptional factors, but their functional implication is under investigation.
Four acrocentric chromosomes contain NBL2 MSRs in higher copy numbers and share their proximal DNA sequences. NBL2 polymorphisms and copy number variation between those chromosomes could be the determining factor in NBL2 promoter activity and consequently TNBL transcriptional upregulation. Yet, the size of TNBL originating from different chromosomes appears the same or very similar, indicating that the transcriptional unit is conserved in all four acrocentric chromosomes and may not strictly depend on NBL2 repeat copy number. Northern blots consistently showed two high molecular weight bands that could be the result of a splicing or processing event, or the presence of two different transcriptional start sites in different monomers. In either case, both transcripts contain at least one repeat unit. Moreover, the fact that endogenous (in LS174T) and drug-induced (AZA and AZA/TSA treated Caco-2, HCT116, TOV112D and DKO TSA cells) TNBL transcripts show similar profiles in Northern blot assays, reinforces the structured and not random nature of this transcript. Long read RNA sequencing may eventually characterize precisely the sequence of TNBL transcripts.
TNBL expression appears to be an aberrant characteristic of cancer cells. However, we cannot exclude the possibility that NBL2 and NBL2-like sequences may be expressed in a limited time window during normal development or in response to physiological signals. In support of this, blast searches against databases of expressed sequence tags (EST) in normal tissues such as thymus and pregnant uterus, and also in teratoma cultures undergoing neuronal differentiation, returned hits of the 1.4 kb NBL2 monomer with high similarity. We could not determine the transcriptional start site based on these blast analyses, and 5′ and 3′ RACE experiments failed despite being attempted on several occasions with various approaches, possibly due to the repetitive nature of this RNA.
TNBL forms part of a cancer-specific perinucleolar body
TNBL aggregates are in close proximity to SAM68 nucleolar bodies (SNB), and our measurements assessing the type of interaction indicate that a linker molecule may exist between both structures. In light of DNA/RNA FISH data, showing that TNBL aggregates co-localize with NBL2 loci, we wonder whether the nm-scale space between TNBL aggregate and SNB would be sufficient to include chromatin fibers that in the interphase nucleus are 10 nm thick (66). SAM68 bodies only appear in tumor cells and depend on transcription (58). Although not all SNBs were associated with high quantity of TNBL, the TNBL-SAM68 aggregate structure represents a novel and distinct type of cancer-specific perinucleolar structure.
The perinucleolar compartment can be considered as a sort of ‘heterochromatin factory’, that contains mainly silent rDNA clusters and satellite repeats from the short arm of acrocentric chromosomes (67–69). NBL2 MSR location in the short arms of four acrocentric chromosomes, coincident and relatively close to the location of rDNA clusters, uni and tri-dimensionally, raises the question of whether NBL2 MSR is structurally or functionally related to rDNA. However, our data also shows distinctive features between NBL2–rDNA and TNBL–rRNA, and thus the two seem to be only coincidentally related.
TNBL interacts with the RNA processing machinery
The high amount of TNBL transcripts may act as molecular sponges for splicing factors, other RNA species, or merely distort nuclear organization. In addition to highly dense TNBL aggregates in the perinucleolar region, TNBL transcripts are dispersed in the nucleus where they do not co-localize with NBL2 loci. This opens the possibility that TNBL transcripts migrate and interact with chromatin or other nuclear macromolecules, thus acting in trans. Whether dispersed TNBL signals reflect an active movement of molecules to exhibit their trans-related function or merely passive diffusion from the aggregate is unknown.
CELF1, a widely distributed cytoplasmic and nuclear protein with multiple functions vital for proper mRNA metabolism (70), was one of the main TNBL interactors in the in vitro pull-down assay (RNA-AP). The CELF1-TNBL binding is intriguing since CELF1 has also been implicated in multiple diseases including cancer (71,72). Moreover, CELF1 forms part of cancer-specific perinucleolar bodies known as the perinucleolar compartment (60,73,74). However, in LS174T cells we did not detect formation of CELF1 perinucleolar bodies, neither a significant overlap between TNBL RNA FISH and CELF1 immunofluorescence signals, probably due to sensitivity limits of both techniques. Since TNBL was immunoprecipitated with CELF1 in a robust manner, we hypothesize that CELF1-TNBL interaction may occur in non-perinucleolar regions where TNBL is less abundant, but still present (Figure 8).
It seems unlikely that the aberrant expression of TNBL we observed in some CRC cells could be phenotypically inconsequential. TNBL could be functionally relevant in cancer by sequestering and affecting the function of proteins and/or nucleic acids, as we suggest in the model of Figure 8. The transcriptional process itself at NBL2 loci rather than the RNA molecule, could also passively disrupt nuclear organization. This has been suggested as a mechanism (Cat's cradle model) to explain how lncRNAs may impact nuclear structure (75). Our work started with the aim to identify the mechanisms that could explain the historically observed linkage between DNA demethylation and DNA instability in cancer. Whether TNBL may be part of this link and contribute to nuclear dysfunction by yet unknown mechanisms remains to be addressed.
Overall, our data discloses several novel structural features of macrosatellites and their lncRNAs with potential implications in genome stability and CRC pathogenesis. TNBL high expression, high stability, peculiar distribution in the nucleus, and binding to a group of RNA regulatory proteins, indicates it might have an impact in cancer cell phenotype. TNBL aberrant expression reflects a cancer-specific change in the epigenetic landscape occurring in a subset of colorectal tumors. Our work sets the basis for future studies to test whether TNBL expression could be a biomarker of a subset of CRCs with distinct vulnerabilities exploitable for therapeutic intervention.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Mireia Jordà for useful advices on Northern blot procedure, Miguel Angel Peinado and Mar Muñoz for providing DKO cells and RNA and DNA samples from CRC patients. We thank the whole team from CRG Advanced Light Microscopy unit for their support, especially Xavier Sanjuan Samarra for helping in setting up STED imaging. We thank Anna Lladó and Sébastien Tosi from Advanced Digital Microscopy core facility of IRB for the support in RNA/DNA FISH image position overlap. Fixed solutions of metaphase chromosomes were provided by Isabel Granada from the Cytogenetics laboratory in Hospital Germans Trias i Pujol (Catalan Institute of Oncology).
Authors’ Contributions: G.D., M.P. and S.V.F. designed the experimental plan, analyzed the data and wrote the manuscript. G.D. performed the experimental work; J.B.1 assisted in NBL2 metaphase FISH and provided technical and intellectual input, J.B.2 assisted in bisulfite sequencing and RT-qPCRs; J.K.S. performed bisulfite analysis on some primary tumors; A.R. and S.D. contributed to RNA-AP; S.A. assisted in bioinformatic analysis; M.B. contributed with experimental design, tools and intellectual input; M.P. and S.V.F. supervised the project.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Spanish Ministry of Health Plan Nacional de I+D+I, ISCIII, FEDER [FIS PI09/02444, PI12/00511, PI15/01763 to M.P.]; Agència de Gestió d’Ajuts Universitaris i de Recerca [2017-SGR-529 to M.P., 2017-SGR-305 to M.B.]; Ministerio de Economía y Competitividad [BFU2015-66559-P to M.B.]; Instituto de Salud Carlos III PIE16/00011 to M.B. and S.V.F]; Fundación Olga Torres [F.O.T. to S.V.F]; G.D. is supported by FPU fellowship ‘beca de formación de profesorado universitario’ [FPU12/06605] Ministerio de Educación Cultura y Deporte. Funding for open access charge: Spanish Ministry of Health Plan Nacional de I+D+I, ISCIII, FEDER [PI15/01763 to M.P.]; Fundación Olga Torres [PIE16/00011 to S.F.].
Conflict of interest statement. None declared.
REFERENCES
- 1. Diala E.S., Hoffman R.M.. Hypomethylation of HeLa cell DNA and the absence of 5-methylcytosine in SV40 and adenovirus (type 2) DNA: analysis by HPLC. Biochem. Biophys. Res. Commun. 1982; 107:19–26. [DOI] [PubMed] [Google Scholar]
- 2. Feinberg A.P., Vogelstein B.. Hypomethylation distinguishes genes of some human cancers from their normal counterparts. Nature. 1983; 301:89–92. [DOI] [PubMed] [Google Scholar]
- 3. Gama-Sosa M.A., Slagel V.A., Trewyn R.W., Oxenhandler R., Kuo K.C., Gehrke C.W., Ehrlich M.. The 5-methylcytosine content of DNA from human tumors. Nucleic Acids Res. 1983; 11:6883–6894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Feinberg A.P., Vogelstein B.. Hypomethylation of ras oncogenes in primary human cancers. Biochem. Biophys. Res. Commun. 1983; 111:47–54. [DOI] [PubMed] [Google Scholar]
- 5. Ehrlich M. DNA hypomethylation in cancer cells. Epigenomics. 2009; 1:239–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ehrlich M. DNA methylation in cancer: too much, but also too little. Oncogene. 2002; 21:5400–5413. [DOI] [PubMed] [Google Scholar]
- 7. Ehrlich M., Woods C.B., Yu M.C., Dubeau L., Yang F., Campan M., Weisenberger D.J., Long T., Youn B., Fiala E.S. et al. Quantitative analysis of associations between DNA hypermethylation, hypomethylation, and DNMT RNA levels in ovarian tumors. Oncogene. 2006; 25:2636–2645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hoffmann M.J., Schulz W.A.. Causes and consequences of DNA hypomethylation in human cancer. Biochem. Cell Biol. 2005; 83:296–321. [DOI] [PubMed] [Google Scholar]
- 9. Weisenberger D.J., Campan M., Long T.I., Kim M., Woods C., Fiala E., Ehrlich M., Laird P.W.. Analysis of repetitive element DNA methylation by MethyLight. Nucleic Acids Res. 2005; 33:6823–6836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Narayan A., Ji W., Zhang X.Y., Marrogi A., Graff J.R., Baylin S.B., Ehrlich M.. Hypomethylation of pericentromeric DNA in breast adenocarcinomas. Int. J. Cancer. 1998; 77:833–838. [DOI] [PubMed] [Google Scholar]
- 11. Qu G., Dubeau L., Narayan A., Yu M.C., Ehrlich M.. Satellite DNA hypomethylation vs. overall genomic hypomethylation in ovarian epithelial tumors of different malignant potential. Mutat. Res. 1999; 423:91–101. [DOI] [PubMed] [Google Scholar]
- 12. Qu G.Z., Grundy P.E., Narayan A., Ehrlich M.. Frequent hypomethylation in Wilms tumors of pericentromeric DNA in chromosomes 1 and 16. Cancer Genet. Cytogenet. 1999; 109:34–39. [DOI] [PubMed] [Google Scholar]
- 13. Rodriguez J., Vives L., Jordà M., Morales C., Muñoz M., Vendrell E., Peinado M.A.. Genome-wide tracking of unmethylated DNA Alu repeats in normal and cancer cells. Nucleic Acids Res. 2008; 36:770–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ohno S. So much ‘junk’ DNA in our genome. Brookhaven Symp. Biol. 1972; 23:366–370. [PubMed] [Google Scholar]
- 15. Shapiro J.A., von Sternberg R.. Why repetitive DNA is essential to genome function. Biol. Rev. Camb. Philos. Soc. 2005; 80:227–250. [DOI] [PubMed] [Google Scholar]
- 16. Ji W., Hernandez R., Zhang X.Y., Qu G.Z., Frady A., Varela M., Ehrlich M.. DNA demethylation and pericentromeric rearrangements of chromosome 1. Mutat. Res. 1997; 379:33–41. [DOI] [PubMed] [Google Scholar]
- 17. Wong N., Lam W.C., Lai P.B., Pang E., Lau W.Y., Johnson P.J.. Hypomethylation of chromosome 1 heterochromatin DNA correlates with q-arm copy gain in human hepatocellular carcinoma. Am. J. Pathol. 2001; 159:465–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Schulz W.A., Elo J.P., Florl A.R., Pennanen S., Santourlidis S., Engers R., Buchardt M., Seifert H.-H., Visakorpi T.. Genomewide DNA hypomethylation is associated with alterations on chromosome 8 in prostate carcinoma. Genes. Chromosomes Cancer. 2002; 35:58–65. [DOI] [PubMed] [Google Scholar]
- 19. Kokalj-Vokac N., Almeida A., Viegas-Péquignot E., Jeanpierre M., Malfoy B., Dutrillaux B.. Specific induction of uncoiling and recombination by azacytidine in classical satellite-containing constitutive heterochromatin. Cytogenet. Cell Genet. 1993; 63:11–15. [DOI] [PubMed] [Google Scholar]
- 20. Chen R.Z., Pettersson U., Beard C., Jackson-Grusby L., Jaenisch R.. DNA hypomethylation leads to elevated mutation rates. Nature. 1998; 395:89–93. [DOI] [PubMed] [Google Scholar]
- 21. Gaudet F., Hodgson J.G., Eden A., Jackson-Grusby L., Dausman J., Gray J.W., Leonhardt H., Jaenisch R.. Induction of tumors in mice by genomic hypomethylation. Science. 2003; 300:489–492. [DOI] [PubMed] [Google Scholar]
- 22. Eden A., Gaudet F., Waghmare A., Jaenisch R.. Chromosomal instability and tumors promoted by DNA hypomethylation. Science. 2003; 300:455. [DOI] [PubMed] [Google Scholar]
- 23. Howard G., Eiges R., Gaudet F., Jaenisch R., Eden A.. Activation and transposition of endogenous retroviral elements in hypomethylation induced tumors in mice. Oncogene. 2008; 27:404–408. [DOI] [PubMed] [Google Scholar]
- 24. Bozhenok L., Wade P.A., Varga-Weisz P.. WSTF-ISWI chromatin remodeling complex targets heterochromatic replication foci. EMBO J. 2002; 21:2231–2241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Craig J.M., Earle E., Canham P., Wong L.H., Anderson M., Choo K.H.A.. Analysis of mammalian proteins involved in chromatin modification reveals new metaphase centromeric proteins and distinct chromosomal distribution patterns. Hum. Mol. Genet. 2003; 12:3109–3121. [DOI] [PubMed] [Google Scholar]
- 26. Nan X., Tate P., Li E., Bird A.. DNA methylation specifies chromosomal localization of MeCP2. Mol. Cell. Biol. 1996; 16:414–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Cammas F., Oulad-Abdelghani M., Vonesch J.-L., Huss-Garcia Y., Chambon P., Losson R.. Cell differentiation induces TIF1beta association with centromeric heterochromatin via an HP1 interaction. J. Cell Sci. 2002; 115:3439–3448. [DOI] [PubMed] [Google Scholar]
- 28. Fodor B.D., Shukeir N., Reuter G., Jenuwein T.. Mammalian Su(var) genes in chromatin control. Annu. Rev. Cell Dev. Biol. 2010; 26:471–501. [DOI] [PubMed] [Google Scholar]
- 29. Eymery A., Horard B., El Atifi-Borel M., Fourel, Berger F., Vitte A.-L., Van den Broeck A., Brambilla E., Fournier A., Callanan M. et al. A transcriptomic analysis of human centromeric and pericentric sequences in normal and tumor cells. Nucleic Acids Res. 2009; 37:6340–6354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ting D.T., Lipson D., Paul S., Brannigan B.W., Akhavanfard S., Coffman E.J., Contino G., Deshpande V., Iafrate A.J., Letovsky S. et al. Aberrant overexpression of satellite repeats in pancreatic and other epithelial cancers. Science. 2011; 331:593–596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Treangen T.J., Salzberg S.L.. Repetitive DNA and next-generation sequencing: computational challenges and solutions. Nat. Rev. Genet. 2013; 13:36–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Dumbovic G., Forcales S.-V., Perucho M.. Emerging roles of macrosatellite repeats in genome organization and disease development. Epigenetics. 2017; 12:515–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. van Deutekom J.C., Wijmenga C., van Tienhoven E.A., Gruter A.M., Hewitt J.E., Padberg G.W., van Ommen G.J., Hofker M.H., Frants R.R.. FSHD associated DNA rearrangements are due to deletions of integral copies of a 3.2 kb tandemly repeated unit. Hum. Mol. Genet. 1993; 2:2037–2042. [DOI] [PubMed] [Google Scholar]
- 34. Bodega B., Ramirez G.D.C., Grasser F., Cheli S., Brunelli S., Mora M., Meneveri R., Marozzi A., Mueller S., Battaglioli E. et al. Remodeling of the chromatin structure of the facioscapulohumeral muscular dystrophy (FSHD) locus and upregulation of FSHD-related gene 1 (FRG1) expression during human myogenic differentiation. BMC Biol. 2009; 7:41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Cabianca D.S., Casa V., Bodega B., Xynos A., Ginelli E., Tanaka Y., Gabellini D.. A Long ncRNA links copy number variation to a Polycomb/Trithorax epigenetic switch in fshd muscular dystrophy. Cell. 2012; 149:819–831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Figueroa D.M., Darrow E.M., Chadwick B.P.. Two novel DXZ4-associated long noncoding RNAs show developmental changes in expression coincident with heterochromatin formation at the human (Homo sapiens) macrosatellite repeat. Chromosom. Res. 2015; 23:733–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Suzuki K., Suzuki I., Leodolter A., Alonso S., Horiuchi S., Yamashita K., Perucho M.. Global DNA demethylation in gastrointestinal cancer is age dependent and precedes genomic damage. Cancer Cell. 2006; 9:199–207. [DOI] [PubMed] [Google Scholar]
- 38. Samuelsson J., Dumbovic G., Polo C., Moreta C., Alibés A., Ruiz-Larroya T., Giménez-Bonafé P., Alonso S., Forcales S.-V., Perucho M.. Helicase Lymphoid-Specific enzyme contributes to the maintenance of methylation of SST1 pericentromeric repeats that are frequently demethylated in colon cancer and associate with genomic damage. Epigenomes. 2017; 1:2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Thoraval D., Asakawa J., Wimmer K., Kuick R., Lamb B., Richardson B., Ambros P., Glover T., Hanash S.. Demethylation of repetitive DNA sequences in neuroblastoma. Genes. Chromosomes Cancer. 1996; 17:234–244. [DOI] [PubMed] [Google Scholar]
- 40. Nagai H., Kim Y.S., Yasuda T., Ohmachi Y., Yokouchi H., Monden M., Emi M., Konishi N., Nogami M., Okumura K. et al. A novel sperm-specific hypomethylation sequence is a demethylation hotspot in human hepatocellular carcinomas. Gene. 1999; 237:15–20. [DOI] [PubMed] [Google Scholar]
- 41. Choi S.H., Worswick S., Byun H.-M., Shear T., Soussa J.C., Wolff E.M., Douer D., Garcia-Manero G., Liang G., Yang A.S.. Changes in DNA methylation of tandem DNA repeats are different from interspersed repeats in cancer. Int. J. Cancer. 2009; 125:723–729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Igarashi S., Suzuki H., Niinuma T., Shimizu H., Nojima M., Iwaki H., Nobuoka T., Nishida T., Miyazaki Y., Takamaru H. et al. A novel correlation between LINE-1 hypomethylation and the malignancy of gastrointestinal stromal tumors. Clin. Cancer Res. 2010; 16:5114–5123. [DOI] [PubMed] [Google Scholar]
- 43. Slater G., Birney E., Box G., Smith T., Waterman M., Altschul S., Gish W., Miller W., Myers E., Lipman D. et al. Automated generation of heuristics for biological sequence comparison. BMC Bioinformatics. 2005; 6:31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Wolfe D., Dudek S., Ritchie M.D., Pendergrass S.A.. Visualizing genomic information across chromosomes with PhenoGram. BioData Min. 2013; 6:18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Memczak S., Jens M., Elefsinioti A., Torti F., Krueger J., Rybak A., Maier L., Mackowiak S.D., Gregersen L.H., Munschauer M. et al. Circular RNAs are a large class of animal RNAs with regulatory potency. Nature. 2013; 495:333–338. [DOI] [PubMed] [Google Scholar]
- 46. Bolland D.J., King M.R., Reik W., Corcoran A.E., Krueger C.. Robust 3D DNA FISH using directly labeled probes. J. Vis. Exp. 2013; 78:e50587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Raj A., van den Bogaard P., Rifkin S.A., van Oudenaarden A., Tyagi S.. Imaging individual mRNA molecules using multiple singly labeled probes. Nat. Methods. 2008; 5:877–879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Namekawa S.H., Lee J.T.. Detection of nascent RNA, single-copy DNA and protein localization by immunoFISH in mouse germ cells and preimplantation embryos. Nat. Protoc. 2011; 6:270–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Gutschner T., Hämmerle M., Eissmann M., Hsu J., Kim Y., Hung G., Revenko A., Arun G., Stentrup M., Gross M. et al. The noncoding RNA MALAT1 is a critical regulator of the metastasis phenotype of lung cancer cells. Cancer Res. 2013; 73:1180–1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Jeon Y., Lee J.T.. YY1 Tethers Xist RNA to the inactive X nucleation center. Cell. 2011; 146:119–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Nishiyama R., Qi L., Tsumagari K., Weissbecker K., Dubeau L., Champagne M., Sikka S., Nagai H., Ehrlich M.. A DNA repeat, NBL2, is hypermethylated in some cancers but hypomethylated in others. Cancer Biol. Ther. 2005; 4:440–448. [DOI] [PubMed] [Google Scholar]
- 52. Miranda T.B., Jones P.A.. DNA methylation: The nuts and bolts of repression. J. Cell. Physiol. 2007; 213:384–390. [DOI] [PubMed] [Google Scholar]
- 53. Rhee I., Bachman K.E., Park B.H., Jair K.-W., Yen R.-W.C., Schuebel K.E., Cui H., Feinberg A.P., Lengauer C., Kinzler K.W. et al. DNMT1 and DNMT3b cooperate to silence genes in human cancer cells. Nature. 2002; 416:552–556. [DOI] [PubMed] [Google Scholar]
- 54. Hutchinson J.N., Ensminger A.W., Clemson C.M., Lynch C.R., Lawrence J.B., Chess A.. A screen for nuclear transcripts identifies two linked noncoding RNAs associated with SC35 splicing domains. BMC Genomics. 2007; 8:39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Cooper G.M., Hausman R.E.. The cell: a molecular approach. 2007; 4th editionWashington, DC: ASM Press. [Google Scholar]
- 56. Ghetti A., Piñol-Roma S., Michael W.M., Morandi C., Dreyfuss G.. hnRNP I, the polypyrimidine tract-binding protein: distinct nuclear localization and association with hnRNAs. Nucleic Acids Res. 1992; 20:3671–3678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Huang S. Review: perinucleolar structures. J. Struct. Biol. 2000; 129:233–240. [DOI] [PubMed] [Google Scholar]
- 58. Chen T., Boisvert F.M., Bazett-Jones D.P., Richard S.. A role for the GSG domain in localizing Sam68 to novel nuclear structures in cancer cell lines. Mol. Biol. Cell. 1999; 10:3015–3033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Mannen T., Yamashita S., Tomita K., Goshima N., Hirose T.. The Sam68 nuclear body is composed of two RNase-sensitive substructures joined by the adaptor HNRNPL. J. Cell Biol. 2016; 214:45–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Huang S., Deerinck T.J., Ellisman M.H., Spector D.L.. The perinucleolar compartment and transcription. J. Cell Biol. 1998; 143:35–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Kamath R.V., Thor A.D., Wang C., Edgerton S.M., Slusarczyk A., Leary D.J., Wang J., Wiley E.L., Jovanovic B., Wu Q. et al. Perinucleolar compartment prevalence has an independent prognostic value for breast cancer. Cancer Res. 2005; 65:246–253. [PubMed] [Google Scholar]
- 62. Edwards J.M., Long J., De Moor C.H., Emsley J., Searle M.S.. Structural insights into the targeting of mRNA GU-rich elements by the three RRMs of CELF1. Nucleic Acids Res. 2013; 41:7153–7166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Du J., Johnson L.M., Jacobsen S.E., Patel D.J.. DNA methylation pathways and their crosstalk with histone methylation. Nat. Rev. Mol. Cell Biol. 2015; 16:519–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Kondo T., Bobek M.P., Kuick R., Lamb B., Zhu X., Narayan A., Bourc’his D., Viegas-Pequignot E., Ehrlich M., Hanash S.M.. Whole-genome methylation scan in ICF syndrome: hypomethylation of non-satellite DNA repeats D4Z4 and NBL2. Hum Mol Genet. 2000; 9:597–604. [DOI] [PubMed] [Google Scholar]
- 65. Kelley D., Rinn J.. Transposable elements reveal a stem cell-specific class of long noncoding RNAs. Genome Biol. 2012; 13:R107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Fussner E., Ching R.W., Bazett-Jones D.P.. Living without 30nm chromatin fibers. Trends Biochem. Sci. 2011; 36:1–6. [DOI] [PubMed] [Google Scholar]
- 67. McStay B., Grummt I.. The epigenetics of rRNA Genes: From molecular to chromosome biology. Annu. Rev. Cell Dev. Biol. 2008; 24:131–157. [DOI] [PubMed] [Google Scholar]
- 68. Sullivan G.J., Bridger J.M., Cuthbert A.P., Newbold R.F., Bickmore W.A., McStay B.. Human acrocentric chromosomes with transcriptionally silent nucleolar organizer regions associate with nucleoli. EMBO J. 2001; 20:2867–2874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Németh A., Conesa A., Santoyo-Lopez J., Medina I., Montaner D., Péterfia B., Solovei I., Cremer T., Dopazo J., Längst G.. Initial genomics of the human nucleolus. PLoS Genet. 2010; 6:e1000889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Vlasova I.A., Bohjanen P.R. Posttranscriptional regulation of gene networks by GU-rich elements and CELF proteins. RNA Biol. 2008; 5:201–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Choi W.T., Folsom M.R., Azim M.F., Meyer C., Kowarz E., Marschalek R., Timchenko N.A., Naeem R.C., Lee D.A.. C/EBPbeta suppression by interruption of CUGBP1 resulting from a complex rearrangement of MLL. Cancer Genet. Cytogenet. 2007; 177:108–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Chettouh H., Fartoux L., Aoudjehane L., Wendum D., Clapéron A., Chrétien Y., Rey C., Scatton O., Soubrane O., Conti F. et al. Mitogenic insulin receptor-A is overexpressed in human hepatocellular carcinoma due to EGFR-mediated dysregulation of RNA splicing factors. Cancer Res. 2013; 73:3974–3986. [DOI] [PubMed] [Google Scholar]
- 73. Timchenko L.T., Miller J.W., Timchenko N.A., DeVore D.R., Datar K.V., Lin L., Roberts R., Caskey C.T., Swanson M.S.. Identification of a (CUG)n triplet repeat RNA-binding protein and its expression in myotonic dystrophy. Nucleic Acids Res. 1996; 24:4407–4414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Huang S., Deerinck T.J., Ellisman M.H., Spector D.L.. The dynamic organization of the perinucleolar compartment in the cell nucleus. J. Cell Biol. 1997; 137:965–974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Melé M., Rinn J.L.. ‘Cat's cradling’ the 3D genome by the act of LncRNA transcription. Mol. Cell. 2016; 62:657–664. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.