


Abstract
Thymine DNA glycosylase (TDG) is a DNA repair enzyme that excises a variety of mismatched or damaged nucleotides (nts), e.g. dU, dT, 5fC and 5caC. TDG is shown to play essential roles in maintaining genome integrity and correctly programming epigenetic modifications through DNA demethylation. After locating the lesions, TDG employs a base-flipping strategy to recognize the damaged nucleobases, whereby the interrogated nt is extruded from the DNA helical stack and binds into the TDG active site. The dynamic mechanism of the base-flipping process at an atomistic resolution, however, remains elusive. Here, we employ the Markov State Model (MSM) constructed from extensive all-atom molecular dynamics (MD) simulations to reveal the complete base-flipping process for a G.T mispair at a tens of microsecond timescale. Our studies identify critical intermediates of the mispaired dT during its extrusion process and reveal the key TDG residues involved in the inter-state transitions. Notably, we find an active role of TDG in promoting the intrahelical nt eversion, sculpturing the DNA backbone, and penetrating into the DNA minor groove. Three additional TDG substrates, namely dU, 5fC, and 5caC, are further tested to evaluate the substituent effects of various chemical modifications of the pyrimidine ring on base-flipping dynamics.
INTRODUCTION
Human genome is constantly under detrimental threats from cytotoxic and mutagenic DNA damages (1). Efficient corrections of these DNA lesions are therefore critical for maintaining genome integrity and preventing premature aging and cancer (2). The DNA damages caused by base oxidation, deamination and alkylation can be restored by base excision repair (BER) pathways (3–6). Thymine DNA glycosylase (TDG), as one member of the uracil DNA glycosylase (UNG) superfamily (7), is involved in the first-line defense to excise a variety of DNA lesions through the BER pathway. TDG recognizes the T·G and U·G mispairs caused by deamination of cytosine and 5-methylcytosine (5mC). In addition, TDG also excises several chemically damaged bases through an active DNA demethylation process, e.g. 5-formylcytosine (5fC) and 5-carboxycytosine (5caC) generated by ten-eleven-translocation (Tet) proteins (8–10). These epigenetic modifications performed by TDG are critical for programming embryonic development in mice (11–18).
TDG adopts a universal base-flipping strategy to extrude the damaged nt from the DNA helical stack and recognize the flipped base through the active site residues (see Figure 1A), as also characterized in other DNA repair/modify enzymes (19–21). The base-flipping of DNA nt is involved in a number of critical biological processes, including epigenetic control of gene expression and DNA repair etc., and thus subjects to extensive experimental (22–26) and computational investigations (27–30). The spontaneous base-flipping from naked DNA duplex has been studied using imino proton exchange assay or fluorescence probes (e.g. 2-aminopurine or 2-AP). The estimated lifetime of an intrahelical A·T pair ranges from a few to tens of milliseconds (ms) depending on its varied locations in DNA and the sequence context (26,31,32), whereas for the G·C pair the intrahelical lifetime is significantly increased. In addition, the presence of the T·G mispair can promote the base-flipping process comparing to the canonical DNA with an intrahelical lifetime ∼1 ms (33). Moreover, free energy calculations have been performed to investigate the base-flipping mechanisms for both canonical and damaged DNA. A qualitative agreement has been reached suggesting that C and T exhibit comparable flipping energetics along either the minor or major groove, while the major groove path is more favored for larger purine bases (29,34–36). For damaged DNA nts, on the other hand, such as uracil in U·G and thymine in T·G, they have a considerable lower flipping barrier than that of their canonical counterparts (30,37,38), which likely facilitates the repair proteins to rapidly locate the lesions.
Figure 1.

(A) A schematic illustration of the base-flipping process exerted by TDG during DNA repair. The lesion searching is initiated by protein-DNA interaction to form the Interrogation Complex (IC), followed by penetration of an intercalated residue Arg275 of TDG into the DNA minor groove, which results in formation of the Recognition Complex (RC). (B) The modeled structure of the IC with the key motifs highlighted in a zoomed-in image, in which the T·G mispair is colored in green/magenta, and the TDG active site residues Tyr152 and Asn191 are shown in gray sticks; the intercalation loop (Ser271–Arg281) is highlighted in blue and the intercalated residue Arg275 is shown in blue sticks; the gating region (Ile136 to Phe159) is shown in cyan. (C) The minimized structure of the RC with the mismatched dT nt flipped into the TDG active site.
It can be expected that the DNA-binding proteins would have profound influences on the base-flipping dynamics by altering the flipping energetics and DNA structures. In particular, what strategies the DNA glycosylases use to search for and interrogate the DNA lesions have been a focus of many structural (21,39–42), biochemical (43–49), NMR (23) and single-molecule spectroscopic studies (47,50–55). One of the most extensively studied DNA glycosylase is uracil DNA glycosylase (UDG), which is structurally related to TDG (56,57). The former NMR studies suggest that UDG captures the damaged bases in a passive scenario whereby the extrahelical bases emerge spontaneously by thermal motions of DNA chains as UDG is searching for the lesions (23,58), while an active role of UDG has also been proposed by other biochemical studies (46,48). In addition, single-molecule investigations on bacterial formamidopyrimidine–DNA glycosylase (Fpg or MutM) that corrects the 8-oxoguanine(oxoG)·C damage indicate an active interrogation of the damaged bases through a key intercalation loop of MutM (47,51). Their results are supported by several crystal structures of the MutM–DNA complex in which an oxoG or normal G base is trapped in diverse forms, from intrahelical (39,59,60), partially (61) to fully extruded states (60,62). Likewise, both bacterial 3-methyladenine (3mA) glycosylase (AlkA) and MutY adenine DNA glycosylase that specifically excise 3mA and A (from A·oxoG lesion), respectively, have been trapped targeting to an intrahelical base pair (bp), in complex with either undamaged (for AlkA) (63) or damaged DNA (for MutY) (40). Taken together, the above static structures of different glycosylases suggest an active role of the enzyme in promoting the base extrusion initiated from an intrahelical state. Moreover, for the HhaI cytosine-5 methyltransferase (M.HhaI), both isotope labeling assay and free energy calculations indicate an active role of the protein in extruding the cytosine via the major groove (27,64,65).
Comparing to the above systems, limited experimental studies have been conducted for TDG regarding its lesion-searching and substrate recognition mechanisms. Recently, by employing atomic force microscopy (AFM) and fluorescence assay (66), Buechner et al. demonstrate that regardless of the specific or non-specific DNA chains, the TDG binding can trap DNA in two major conformations, with DNA bending angle of ∼30° and ∼60°, respectively. The former is considered as a searching complex (SC) in which the DNA conformation adopts an innate structure of naked DNA prior to TDG binding. The latter is a RC, a predominate state in the presence of TDG. Comparison of these two states suggests an active role of TDG in sculpting the DNA backbone (66). Moreover, site-directed mutagenesis and biochemical studies performed by A.C. Drohat and his coworkers also shed lights on the critical roles of several TDG residues (e.g. Arg275, Ala145, and Asn191) in substrate recognition and/or enzymatic catalysis, and provide structural basis for the TDG specificity (67–72).
Furthermore, crystallographic studies have brought about structural insights into the mechanisms of target interrogation and recognition for TDG (69,73–77). Based on one recently resolved RC structure where the mispaired DNA nt is flipped into the TDG active site (74), the interrogations of the flipped bases can be pictured as consecutive motions of the enzyme exerted on DNA. As a result, TDG is shown to bend the DNA chains and compress the DNA backbone, giving rise to a DNA bending angle of ∼40° (see Figure 1C for the minimized structure). The above ‘phosphate pinch’ is thought to facilitate the base-flipping process (21,76). Meanwhile, the residue Arg275 from an intercalation loop can penetrate into the void space left by the flipped nt, which potentially prevents the extrahelical base from returning back to DNA helix. On the other hand, Maiti et al. captured one TDG conformation bound to an undamaged DNA, viewed as an interrogation complex (IC) in which the inspected nt adopts an intrahelical state and Arg275 lies within the minor groove (76). Notably, the DNA bending angle in the above IC is ∼25°, which is smaller comparing to that of the RC (74). Taken together, in line with the previous AFM work (66), the cryptographic observations seem to support an active role of TDG in sculpturing the DNA structure.
Although the previous experimental studies have provided profound insights into the structural basis of TDG associated with its biological function, the atomistic-level revelation of the base-flipping dynamics initiated from an intrahelical state to a completely flipped state remains unknown and experimental characterization of the above process is still challenging due to limited spatiotemporal resolutions. In this work, by constructing a Markov State Model (MSM) based on extensive all-atom molecular dynamics (MD) simulations accumulated to ∼30 microseconds (μs), we reveal the dynamics of the complete base-flipping process for the T·G mispair in the presence of TDG, and provide atomistic-level details of how TDG sculpts the DNA structure, i.e. bending the DNA backbone, penetrating and widening the DNA groove. Moreover, additional MD simulations were performed for naked DNA aiming to reveal the specific role of TDG in recognizing the lesions, thereby an active role of TDG in promoting the base-flipping process is proposed. Finally, we extended our studies to other TDG substrates (including dU, 5fC and 5caC) in order to comparatively investigate the dynamics of the base-flipping process for different target nts with varied chemical modifications.
MATERIALS AND METHODS
Modeling the IC and RC of the TDG–DNA system bearing one T·G mispair
We constructed the RC based on one crystal structure of the TDG–DNA complex containing 24 DNA bps with one flipped 2′-fluoroarabino analogue of dU (pdb ID: 5hf7) (74). We replaced the Fluorine atom with H atom and added one methyl group on the 5-site of the pyrimidine ring of the flipped nt. This final model of RC was then subjected to energy minimization to relieve local steric clashes (see Figure 1C). Next, to model the IC, we first extracted the double-stranded DNA chains from the above RC and modeled an intrahelical T·G-containing DNA duplex (see the Supplementary Figure S1A for the energy minimized DNA structure). Next, starting from the above naked DNA conformation, we performed one 7-ns MD simulation to relax the DNA backbone by constraining two ends of the DNA chains (see below for the details of the MD setup). The last snapshot of the above MD trajectory was then used for modeling the IC (see the Supplementary Figure S1A). That is, the TDG conformation from the crystal structure 5hf7 was directly modeled to the above obtained DNA conformation by superimposing the two ends of the DNA backbone. Since the intercalation loop from the above TDG conformation is in an penetrated form, we then adopted the intercalation loop from another crystal structure of TDG (residues Ser273 to Arg281 from PDB structure 2rba) bound to a non-specific DNA and transplanted the loop conformation to the above TDG–DNA model (see Supplementary Figure S1A). Finally, we minimized the final complex energetically (see Figure 1B). Based on the above IC and RC, we next performed TMD simulations to obtain initial base-flipping pathways.
Notably, we did not use the complete TDG structure from 2rba because the TDG in 5hf7 was solved using a longer protein construct containing 29 additional N-terminal residues (TDG82–308) compared to the TDG from 2rba (TDG111–308) (see Supplementary Figure S2). These additional N-terminal residues have been shown to increase the substrate binding affinity and also TDG activity (74). Therefore, the TDG from 5hf7 resembles more to the full-length TDG than the TDG from 2rba. In addition, 5hf7 was solved at a higher resolution (1.54 Å) than 2rba (2.79 Å) using improved crystallization methods, which can provide more accurate side-chain locations and interaction networks between TDG and DNA/waters. Moreover, the binding interfaces between TDG and DNA are also quite different between 5hf7 and 2rba. In the former, the TDG can form more direct contacts with the DNA backbones, e.g. through five positively charged residues, namely Lys240, Lys246, Lys232, Arg275, and Arg110 (see Supplementary Figure S3B), compared to that in 2rba in which only one of the above contacts is present (see Supplementary Figure S3A). Taken together, 5hf7 provides more information regarding the TDG–DNA binding interfaces and also gives a more complete and accurate 3D structure of TDG.
Obtaining initial base-flipping pathways using TMD simulations
Based on the above IC and RC, we performed TMD to derive initial base-flipping pathways for the mispaired dT nt by constraining the backbone P atoms of two DNA ends using a force constant of 500 kcal/mol/Å2 (see Supplementary Figure S4A). All the TMD simulations were performed using AMBER package (78). The amber force field 99SB with PARMBSC0 correction was used to describe the TDG–DNA system (79–82). After comparing the structures of IC and RC, we can see that not only the DNA backbones needed to be morphed, but the TDG structure also requires a conformational change in order to ensure the overall TDG–DNA complex can be well fitted to RC after TMD simulation (see Supplementary Figure S5). Therefore, the targeting regions were selected as several discontinuous regions, including 16 P atoms of several DNA nts locating at the middle part of the DNA chain; the heavy atoms of the mispaired dT nt and one of its adjacent dA nt; the heavy atoms of the intercalated residue Arg275; the Cα atoms of the TDG residue from Cys276 to Glu303 (see Supplementary Figure S4A). The modeled IC was served as the starting conformation and was targeted to the RC within 100 ps TMD simulations using a pulling force constant of 1 and 5 kcal/mol/Å2, respectively. The TMD simulations captured a base-flipping pathways along the minor groove (see Supplementary Figure S6A), and resulted in a well convergence of the TDG–DNA structure to the targeted RC, with the relative RMSD values all <2 Å for the regions of DNA backbones, Arg275 and all TDG Cα atoms (see Supplementary Figure S6B-C).
In addition, to examine how varied targeting regions might affect the TMD results, we designed three additional TMD setups (see Supplementary Figure S7A): (i) all the above chosen nucleic acids atoms, and no TDG atoms was used (setup A); (ii) the selected atoms in setup A plus the heavy atoms of the key intercalated residue Arg275 (setup B); (iii) the selected atoms in setup B plus all TDG Cα atoms (setup C). For each setup, we used two different force constants to perform the TMD simulations (1 and 5 kcal/mol/Å2). The TMD results indeed show that the base-flipping follows two paths for different TMD setups. In setup A, the major-groove path is favored while the minor-groove path is dominant for the other two TMD setups (see Supplementary Figure S7B). Nevertheless, in setup A, since we did not choose any TDG atoms, the final TMD conformation deviates significantly from the targeted RC with the relative RMSD values >4 Å for both R275 and TDG Cα atoms (see Supplementary Figure S8B). In contrast, inclusion of the R275 into the targeting regions substantially drives the TDG structure much closer to the RC (see Supplementary Figure S8B). Likewise, in setup C, the final TMD structure demonstrates nearly the same TDG–DNA conformation as the targeted RC (with RSMD difference <0.5 Å, see Supplementary Figure S8B). In short, the TDG structure apparently has profound impacts on the base-flipping pathways. It is also noteworthy that the intercalated residue Arg275 plays a critical role in the base-flipping process, as evidenced by former experimental studies (70), therefore, Arg275 is indispensable for the TMD simulations.
Taken together, the original TMD setup has provided reasonable base-flipping pathways. We thus performed three parallel TMD simulations using a force constant of 5 kcal/mol/Å2. Each trajectory was initiated with randomly generated velocities and was saved at a time interval of 1 ps. The obtained three base-flipping pathways resemble to each other very well and the final snapshot from each TMD trajectory only has a RMSD difference of ∼0.5 Å to the RC for the targeting regions (see the Supplementary Figure S4B). Finally, in addition to the minimized IC and RC, we extracted 50 conformations from one of the above three TMD simulations at an equal time interval of 2 ps. We thus collected 52 TDG–DNA complexes along the base-flipping pathway, which served as the input structures for the subsequent unbiased MD simulations.
Shooting unbiased MD simulations
We totally conducted two rounds of MD simulations. In the first round, we performed 20 ns MD simulations starting from each of the above 52 selected TDG–DNA complexes (see Supplementary Figure S9A). All the MD simulations were performed using Gromacs-4.6 package (83–85), and the AMBER99sb force field with PARMBSC0 corrections for nucleic acid were employed to describe the TDG–DNA system (79–82). The TDG–DNA complex was centered in a triclinic box filled with 17283 SPC water molecules (86). 98 Na+ and 49 Cl− were added in the water box by randomly replacing the solvent waters to neutralize the system and ensure an ionic concentration of 0.15 M. The final system contains 56935 atoms. The cutoff distance for the Van der Waals and short-range electrostatic interactions were set to 12 Å. The long-range electrostatic interactions were treated using the Particle-Mesh Ewald (PME) summation method (87). The LINCS algorithm was used to constrain all the chemical bonds (88). For each configuration, the steepest decent method was employed to minimize the structure, followed by a 500 ps restrained NVT MD simulations by constraining all the heavy atoms of the system. Then, the system temperature was gradually increased from 50 K to 310 K within 200 ps and kept at 310 K using the velocity rescaling thermostat (89). Finally, the whole system was subject to 20 ns NVT MD simulations at 310 K.
Moreover, after projecting the MD conformations onto the same two reactions coordinates as used in the main text Figure 5D (see below), a clear conformational gap exists between the intrahelical and extrahelical state regions (see Supplementary Figure S9B), suggesting that the chosen 52 conformations from the TMD simulations were not sufficient to cover the complete base-flipping path. Therefore, to enhance the samplings around the above gap regions, we chose 36 extra conformations from one of the above 52 trajectories and performed additional 20-ns MD simulations for each conformation (see the circled region in Supplementary Figure S9B). As shown in Supplementary Figure S9B, these additional MD simulations substantially improved the samplings along the base-flipping pathways. Therefore, in the first round, we collected a total of 88 20-ns MD trajectories. Since the first round of MD simulations were initiated from the TMD simulations with external forces exerted on the system, we then performed the second round of MD simulations to eliminate the introduced energy biases. To select the input structures for the second round of MD simulations, for each of the 1st-round 88 MD trajectories, we discarded all the conformations of the first 10 ns simulation dataset and only kept the last 10 ns MD conformations.
Figure 5.

TDG is actively involved in the base-flipping process. (A) Five random TDG–DNA conformations from the S2 state were chosen and superimposed, with only DNA chains shown. (B) Histograms of RMSD of the mispaired dT nt relative to the IC for the DNA system with (in red) and without TDG (in black). In the background, the fluctuations of the RMSD values calculated based on the MSM are represented by colored boxes for S1 (in gray) and S2 (in pink). (C) Four points, namely P1-P4, are used to define the pseudodihedral for the base-flipping process. P1 is the center of mass (COM) of the two base pairs adjacent to the mismatched bp (in orange); P2 and P3 are the COMs of two phosphate groups connecting to the flipped base, respectively; P4 is the COM of the six-member pyrimidine ring of the flipped nt. (D) Free energy profile of the MD conformations projected onto two reaction coordinates: the pseudodihedral defined in (C) and the COM distance between the dT nt (O2 and N3 atoms) and its opposite dG nt (N1 and O6 atoms). The corresponding values calculated for the naked DNA conformations from the 3 μs straigtfoward MD trajectory are plotted in black dots.
Then, we performed geometric clustering for the above truncated first-round MD dataset (a total of 1000 × 88 = 88, 000 MD snapshots) by firstly decomposing the high-dimensional configurations onto low-dimensional space using the time-structure independent component analysis (tICA) implemented in the MSMbuilder-3.3 package (90–93). tICA is shown to be able to efficiently capture the slowest dynamics for certain conformational changes of biomolecules and has become a popular tool for high-dimensional decomposition when constructing the MSM (94–98). Here, we chose 1014 distance pairs between the following atoms as the metrics for the tICA (see Supplementary Figure S10A):
Heavy base atoms of the mispaired dT nt — Heavy base atoms of the opposed dG nt
Heavy base atoms of the mispaired dT nt — Cα atoms of the TDG residues Ser271 to Arg281
Heavy base atoms of the mispaired dT nt — Cα atoms of the TDG residues ILE136 to Phe159
Heavy base atoms of the mispaired dT nt — Cα atoms of the TDG residues Gly188 to Ile203
Heavy base atoms of the mispaired dT nt — heavy side-chain atoms of the residue Tyr152
Heavy base atoms of the mispaired dT nt — heavy side-chain atoms of the residue Asn191
Heavy base atoms of the mispaired dT nt — heavy side-chain atoms of the residue Arg275
Heavy base atoms of the opposed dG nt — Cα atoms of the TDG residues Ser271 to Arg281
Heavy base atoms of the opposed dG nt — heavy side-chain atoms of the residue Arg275
Heavy side-chain atoms of the residue Arg275 — Cα atoms of the TDG residues Thr197 to Ile203
Heavy side-chain atoms of the residue Arg275 — Cα atoms of the TDG residues Tyr152 to Phe159
Next, by constructing a time-lagged correlation matrix, we obtained the top four slowest tICs, onto which each MD conformation was then projected. Next, based on the above low-dimensional dataset, we classified the projected conformations into 100 clusters using the K-centers algorithm implemented in the MSMbuilder package, and chose the center conformation for each cluster as the representative structure (94,99). We then conducted the second-round MD simulations by running three parallel 100-ns MD simulations starting from each of the above 100 center conformations with different initial velocities (in total of 300 100-ns MD trajectories). In order to construct a connected transition probability matrix, we deleted 15 of the above 300 MD trajectories. Finally, we collected a total of 285 100-ns MD trajectories with an aggregated simulations time of ∼30 μs for the subsequent MSM construction.
MSM construction and validation
MSM discretizes the phase space into many metastable states, i.e. based on the geometric differences, so that within each state the transitions among different conformations are relatively faster than the inter-state transitions (97,99–104). One can first construct the transition probability matrix T (TPM) in which each entry Tij represents the transition probability of microstate i to microstate j after a certain lag-time τ. The chosen lag-time τ should be long enough so that no internal barrier exists between conformations within each microstate. Therefore, each transition after the time τ from any state will only depend on the current state but not the states visited before (a Markovian property). In this way, one can evolve the states using the following equation to a long timescale dynamics of interest:
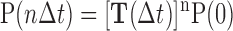 |
where the P(0) is the state distributions at time 0 and the P(nΔt) is the state distributions after time of nΔt. The stationary distributions of each state and the kinetic information for each state-to-state transition can be calculated by solving the eigen-functions of the T. In addition, the implied timescale can be calculated using the following equation:
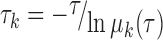 |
where the τ is the lag time used to construct the TPM, μk is the kth eigenvalue of T. The implied timescale curves can be plotted against different τ values. One can then determine the markovian time Δt after which the implied timescale curves start to level off. The kinetic information, i.e. the transition time between two sets of conformations, can be readily deduced from the implied timescale curves, thereby the timescale for the slowest transitions can be estimated.
Here, we adopted a splitting and lumping procedure to construct the MSM. We first decomposed all the MD conformations into various numbers of microstates using the same decomposition methods as described above (tICA dimension reduction followed by K-centers clustering, see Supplementary Figure S10A). Next, to visualize the key intermediate states involved in the base-flipping process, we further lumped one of the MSMs at the microstate level into a six-state kinetic model using the PCCA+ algorithm (105). The detailed procedure is described below.
Splitting the MD conformations into various numbers of microstates
We evaluated the discretization effects on the kinetic properties by constructing several MSMs using different correlation lag-time for tICA and different number of microstates. In specific, we performed tICA at three different correlation lag-time: 20, 30, and 40 ns, and under each lag-time, we grouped the MD conformations into 500, 600, 700, and 800 microstates, respectively. For each case (a total of 12 sets of parameter) we constructed a MSM and calculated the corresponding implied timescales. The results show that the slowest timescale all converges at ∼100 μs, indicating the model is robust to the choices of different number of microstates and correlation lag-times (see Supplementary Figure S11). Finally, to reveal the key intermediate states during the base-flipping process, we further lumped the 500-state MSM built at the correlation lag-time of 20 ns into 6-state kinetic model using the PCCA+ algorithm (105).
Convergence test of the MSM using different simulations datasets
In order to prove that our MD simulations are sufficient to construct a reliable MSM, we evaluated the convergence of the MSM by choosing different subsets from the complete simulations dataset. In specific, we extracted six subsets of the complete dataset with different accumulated simulation times by truncating each MD simulation into varied lengths, namely 14 μs (50 ns × 285), 17 μs (60 ns × 285), 20 μs (70 ns × 285), 23 μs (80 ns × 285), 26 μs (90 ns × 285) and 29 μs (100 ns × 285), respectively. Then, for each subset data, we decomposed the conformations into 500 microstates using the K-centers clustering method, and constructed a 500-state MSM. To exam whether the sampling is converged or not, we projected the MD conformations from each subset onto the same top two tICs. As shown in Supplementary Figure S12, all datasets demonstrate similar free energy landscapes and no extra metastable state appears while increasing the simulation time, indicating that the current MD simulations are well enough to explore the major intermediate states of the flipped nt. Next, to ensure that the kinetics is also converged, we plotted the implied timescale curves for each above subset data. The results clearly suggest that the slowest dynamics involved in the base-flipping process all converge to a timescale of ∼100 μs (see Supplementary Figure S13), indicating that our model is well constructed and capable of extracting reliable kinetic information.
Calculations of the mean first passage time (MFPT) and the stationary distributions
To extract both the thermodynamic and kinetic properties from the MSM, we built a long Monte Carlo (MC) simulations based on the 500-state MSM through a random walk starting from any microstate, and we randomly chose a number within 0–1 and then determined the next transition at a time step of 20 ns according to the corresponding transition probability from the TPM. We finally built a 10 ms long MC trajectory, which is long enough to equilibrate all metastable states, thereby the stationary distributions and the MFPT can be readily calculated. In order to estimate the standard errors, we adopt a bootstrapping strategy to randomly select 285 trajectories from the original trajectory list 100 times (with replacement and duplication). Then, for each bootstrapping, a new MC trajectory was generated and the corresponding thermodynamic and kinetic properties were calculated. Finally, by averaging over all the 100 bootstrappings, we calculated the means and the corresponding standard errors.
MD setup for naked DNA duplex
To construct the IC, we extracted the DNA duplex from the above RC and modeled an intrahelical T·G-containing DNA duplex (see the Supplementary Figure S1A for the energy minimized DNA structure). Then, the above naked DNA conformation was solvated in the SPC water box and 54 Na+ ions were added to neutralize the nucleic acid. The AMBER99sb force field with PARMBSC0 corrections was used to describe the DNA. The final structure was subjected to energy minimization using the steepest decent method, followed by 200-ps restrained MD simulation by constraining all the heavy atoms of nucleic acid. Then, after increasing the temperature from 50 to 310 K within 200 ps, we performed a 7-ns MD simulation to relax the DNA backbones with the two DNA ends constrained.
On the other hand, to evaluate the role of the TDG in stabilizing the partially flipped nt, we randomly chose five TDG–DNA complexes from the S2 state obtained by the MSM. S2 was selected because we proposed that the S2 state was the first checkpoint for the TDG residue Arg275 to recognize the flipped base by forming the cation-π interactions. From each of the above 5 candidates, we removed the TDG and only kept the DNA chains for the subsequent MD simulations. For each naked DNA conformation, we performed one unbiased 100-ns NVT MD simulation at 310K using the same MD setup as described above. In the end, the last 50-ns simulation data for each MD trajectory was used for the final analysis.
MD setup for comparative investigations on different TDG substrates
In addition to the dT nt that is the main focus of the current work, we further extended our studies to three other TDG substrates, namely dU, 5fC and 5caC. Based on our MSM, the entry of the flipped dT nt into the TDG active site occurs in the S4→S5 transition, which also limits the base eversion dynamics (from S1 to S5). We thus chose the S4→S5 transition as a checkpoint to comparatively evaluate kinetic properties of the base-flipping dynamics for different nts. In specific, based on the tICA projection (see Figure 2A), we randomly selected seven conformations of the TDG–DNA complex from the TS region for the S4→S5 transition (see the circled area in Supplementary Figure S14). Next, to model the complexes for different bps for each of above seven conformations, we replaced the T·G mispair with U·G, 5fC·G and 5caC·G bp, respectively. Finally, starting from each modeled complex, we solvated the structure in the SPC water box and added appropriate number of Na+ and Cl− ions to keep the ionic strength at 0.15 M. The amber force fields of three TDG substrates, including dU, 5fC and 5caC, were generated based on the corresponding amber parameters of dT and dC nts. The complete model was then subject to the energy minimization using the steepest decent method, followed by 200-ps restrained MD simulation by constraining all the heavy atoms of the solute (protein and nucleic acid). We finally performed 100-ns unbiased NVT MD simulation at 310 K after increasing the temperature from 50 to 310 K within 200 ps. For each of the collected MD trajectories (in total of 28), we kept the last 50-ns simulation data for the final analysis.
Figure 2.

The MSM captures six metastable states during the base-flipping process of mispaired dT nt. (A) Free energy projections of all the MD conformations onto the two slowest tICs, the location of each macrostate is labeled (from S1 to S6). (B) A scatter plot with each MD conformation mapped onto the two slowest tICs. The snapshots that belong to the same macrostate are shown in a same color: S1 (black), S2 (orange), S3 (green), S4 (blue), S5 (cyan), S6 (red). In addition, the 6-state kinetic network derived from the MSM is provided, with the size of each circle roughly proportional to the corresponding equilibrium population: 18.8 ± 3.7% (S1); 4.0 ± 1.2% (S2); 25.2 ± 4.7% (S3); 11.1 ± 2.3% (S4); 11.3 ± 2.5% (S5); 29.7 ± 9.2% (S6) and the MFPT for the forward transition is also provided above each arrow. (C) Selected representative conformation for each macrostate (S1–S6). The structure was randomly selected from the most populated microstate for each macrostate. Key residues that interact with the dT nt (in green) are shown in sticks. Several key hydrogen bonds are highlighted with dashed lines. Refer to Figure 1 for other representations.
RESULTS
Structural features of the IC and RC for the TDG–DNA system
To study the complete base-flipping process for the T·G mispair, we firstly constructed two TDG–DNA complexes containing one extrahelical (fully flipped) or intrahelical T·G mispair, namely RC and IC, respectively. The RC was directly built from one recently resolved crystal structure of the TDG–DNA complex (pdb ID: 5hf7, see Figure 1C). The IC was built by firstly constructing a DNA duplex containing one wobbled T·G bp based on the above RC, followed by modeling the TDG–DNA complex based on one TDG conformation bound to non-specific DNA (pdb ID: 2rba, see Figure 1B, Supplementary Figure S1A, and the Materials and Methods section for more details of the model construction). Finally, the modeled IC and RC were subjected to energy minimization to relieve the steric clashes.
Comparisons of the above minimized RC and IC demonstrate major structural differences in the intercalation loop region (Ser271–Arg281) and DNA shape. In IC, the intercalated residue Arg275 is lying along the DNA minor groove and forms electrostatic interactions with the mispaired dT backbone P–O− (see Figure 1B). The intrahelical T·G mispair does not form a perfect wobble bp in this minimized structure, nevertheless, a restored wobble structure can be observed from the following unbiased MD simulations (see next section). On the other hand, for RC, the mispaired dT nt is extruded into the TDG active site, and the intercalated residue Arg275 is penetrated into the void space in DNA generated by the flipped nt (see Figure 1C). Several motifs from TDG, including the intercalation loop (Ser271–Arg281), Pro-rich loop (Tyr152–Asn157), Gly–Lys loop (Gly231–Lys232) and water-activating loop (Leu139–Gly142), contribute to compress the DNA backbone. In addition, the penetration of the Arg275 into the DNA helical stack leads to an increase of the minor groove widths of the T·G mispair and its adjacent bp from 8.4 to 12.2 Å and 7.0 to 10.1 Å, respectively (see Supplementary Figure S1B). Moreover, the roll angle between the two bps adjacent to the mismatched site increases from ∼9° to ∼40° after the Arg275 penetration, suggesting a severe bending of the DNA backbone exerted by TDG. Finally, the IC and RC were employed as the starting structures for the subsequent Targeted MD (TMD) to obtain initial base-flipping pathways. All three parallel TMD simulations indicate that the minor groove is the dominant base extrusion pathway. We then performed extensive unbiased MD simulations to construct the MSM (see Materials and Methods section for more details of the TMD setup and MSM construction).
Complete base-flipping dynamics in TDG–DNA complex revealed by MSM
By constructing the MSM from extensive MD trajectories (accumulated simulations time of ∼30 μs), we reveal the complete base-flipping dynamics of the mismatched dT nt from the IC to RC at an atomistic detail. The MSM reveals six metastable states of the dT nt during its base-eversion process, namely S1–S6 states (see Figure 2A and B). Among which, the S1 and S6 states correspond to the IC and RC, respectively. In S1, the T·G mispair forms a wobble bp and the intercalated residue Arg275 resides within the DNA minor groove by forming direct electrostatic interactions with the mispaired dT backbone P–O− (see Figures 2C and 3B). In contrast, in S6, the dT nt is fully extruded from the DNA duplex and reaches into the TDG active site (see Figure 2C). In addition, the intercalated residue Arg275 penetrates into the DNA minor groove and forms direct contacts with the backbones of the flipped nt and its two neighboring DNA nts (see Figure 4B). In addition, the Arg275 forms one hydrogen bond (HB) with the Pro198 backbone C=O, which locks the DNA chain in a completely flipped conformation (see Figure 2C). On the other hand, the flipped thymine forms several polar contacts with the TDG residues directly (i.e. Asn191 and Ile139) or through bridging waters (i.e. His151), and forms nonpolar contacts with the residues Tyr151 and Ala145 (see Figure 3B).
Figure 3.

Structural analysis for each metastable state involved in the base-flipping process. (A) Distances between the heavy atoms of the mismatched dT nt and the Cα of each residue from two TDG motifs: the intercalation loop (S271–R281) and the gating region (I136–F159). For each state, the average value over all the conformations belong to that state is plotted. (B) Highlighted interactions between the mimatched dT nt and its surrounding residues/waters. The same representative conformations from Figure 2 are used here, in which the intercalation loop (S271–R281) and the gating region (I136–F159) are shown in blue and cyan, respectively.
Figure 4.

Penetration of the intercalated residue Arg275 into the DNA helix widens the groove width and bends the DNA backbone. In S1 (A), Arg275 is lying along the minor groove and forms electrostatic interactions with the backbone P–O− group of the mispaired dT nt. In S6 (B), however, Arg275 is tightly locked by the pinched DNA backbone by interacting with the backbone P–O− groups of three DNA nts and Pro198 backbone C=O group. (C) The distance between the Arg275 CZ atom and Pro198 O atom was measured for the S1 and S6 states. For each measurement, the mean value (in black) was averaged over all the microstates that belong to the same macrostate, and the corresponding standard error (in blue) was calculated. (D) To measure the minor groove width, two bps were selected: the mispaired T·G and one of its adjacent bp (denoted as d1 and d2, respectively). (E) The calculated d1 and d2 for the S1 and S6 states. (F) To measure the bending of the DNA backbone exerted by TDG, the roll angle between the two bps adjacent to the mispaired T·G bp was calculated for S1 and S6. The mean and corresponding errors are obtained in the same way as that in Figure 4C.
In addition to S1 and S6, four additional metastable states are clearly identified in the MSM, namely S2–S5 (see Figure 2A and B). Using the transition path theory (106,107), we find the dominant base-flipping path follows S1→S2→S3→S4→S5→S6 (see Figure 2B). The S1 state initiates the base-flipping process by firstly transiting to the S2 state where the dT nt unstacks with its adjacent bases and forms cation-π interactions with the TDG residue Arg275 (see Figure 3A and B). This newly discovered interaction suggests a potential role of the intercalated residue Arg275 in recognizing and stabilizing the partially flipped nt in the early base-flipping process. In addition, the thymine can form HBs with the residues Ser273 and Cys276 from the intercalation loop (see Figure 3B). The further transition of the dT nt from S2 to S3 results in forming π-π stacking with the residue Tyr152 and two additional HBs with residues Asn157 and Gly142 through its O4 and O2 atoms (see Figures 2C and 3). Notably, the subsequent transition of the dT nt from S3 to S4 can establish the first native contact observed in RC, namely the HB between the thymine O2 atom and Ile139 backbone N–H (see Figure 3B). Moreover, the thymine O4 atom forms HB with the side-chain of TDG residue Ser272, and the π–π stacking between the thymine and Tyr152 in S3 switches to a T-shape stacking in S4 (see Figure 3B). Interestingly, the S4→S5 transition of the dT nt can restore the π–π stacking between the thymine and Tyr152 by inserting into the TDG active site and form most of the native contacts with the residues Asn191, Ala145, Tyr152, and Ile139, as observed in RC. Notably, up to S5, the residue Arg275 remains out of the penetration site and lies in the DNA minor groove by forming polar contacts with the DNA backbone P–O−. The complete invasion of the intercalation loop into the minor groove is accomplished after the S5→S6 transition (see Figure 2C), resulting in forming stable interactions with the residue Pro198 backbone C=O and DNA backbone P–O− groups (see Figure 4A–C).
Taken together, the overall base-flipping process exerted by TDG consists of two separate conformational changes: one is eversion of the mispaired dT nt during the S1→S5 transitions, the other is penetration of the intercalated residue Arg275 into the DNA minor groove during the S5→S6 transition. Consistently, the first tIC, representing the slowest dynamics component, exhibits the largest positive correlation with the distance changes between the flipped dT O2 atom and the Cα atom of the active site residue Gly138, and negatively correlates with the distance between two respective atoms from the mispaired dT and opposing dG (see Supplementary Figure S3B), which provide two potential reaction coordinates to describe the complete base-flipping process. Furthermore, the above two distances have the largest correlations with the first tIC comparing to other tICs (see Supplementary Figure S15), confirming that the changes of the above two distances are indeed tightly coupled with the slowest conformational changes.
Our MSM demonstrates that the overall base-flipping dynamics takes place at tens to hundreds of μs during which the penetration of the residue Arg275 into the helical stack is the rate-limiting step, occurring at ∼26 μs, comparing to the <10 μs dynamics in other transitions (see Figure 2B). The rationale for the above slow dynamics is likely due to the energy penalties cost by desolvation of Arg275 prior to the penetration and the side-chain twisting of Arg275 restrained by the narrow space of the DNA helical stack. Importantly, penetration of Arg275 into the minor groove gives rise to a significant increase of the minor groove width by ∼2 Å at the mispaired site and its adjacent bp (see Figure 4D and E). Meanwhile, the TDG exerts a severe DNA bending with the roll angle changed from ∼18° in S1 to ∼28° in S6 (see Figure 4F), suggesting an active role of TDG in sculpturing the DNA structures.
TDG stabilizes the partially flipped nt at the early stage of the base-flipping
As described before, the intercalated residue Arg275 is shown to directly interact with the partially flipped dT nt in S2 by forming a cation–π recognition (see Figure 3B). In order to further exam the specific role of Arg275 and more importantly, to reveal whether the recognition on the partially flipped dT by TDG actively drives further base flipping dynamics, we performed additional MD simulations for the naked DNA duplex (without TDG binding) starting from the DNA conformations derived from the S2 state. Note that the input structures are not chosen from the S1 state because the dynamics of the spontaneous base flipping from an intrahelical state (S1) is a relatively slow process (ms or longer) such that it is impossible to be directly observed using regular MD simulations (24,33). Instead, we randomly chose five TDG–DNA complexes from the S2 state with an attempt to see and compare how the partially flipped nt would behave in the presence and absence of TDG (see Figure 5A). Based on each selected TDG–DNA conformation, we then performed one 100 ns MD simulations for both TDG–DNA complex and the corresponding naked DNA conformation (in total of ten 100-ns MD trajectories), and kept the last 50 ns conformations of each MD trajectory for the final analyses.
Our results indicate that in the presence of TDG, the partially flipped dT nt can be stabilized in an extrahelical form, and most of the MD conformations are structurally similar to the S2 state, exhibiting a relative RMSD value of ∼7 Å comparing to the S1 state (see Figure 5B). Strikingly, some MD conformations move even further away from the S1 state, with the relative RMSD value reaching up to 15 Å. In sharp contrast, after removing the TDG, the halfway flipped base can quickly retract to the DNA helix stack in all the sampled DNA conformations, which resembles the S1 state with a relative RMSD difference of ∼2.5 Å (see Figure 5B), suggesting that the extrahelical dT nt in naked DNA is unstable and prone to return back to the DNA helix. Thus, the above results suggest that the intercalated residue Arg275 is critical to stabilize the partially flipped nt in the S2 state, which in turn, actively facilitates the subsequent base eversion.
As noted above, direct observation of the spontaneous base-flipping using computation simulations is still challenging due to the relatively slow dynamics (24). Despite of the difficulties, we performed one 3 μs straightforward long-time MD simulation for the naked DNA duplex extracted from the IC. As expected, the T·G mispair remains within the DNA duplex during the whole simulation time, as indicated from the calculated distance between the T·G mispair as well as the pseudodihedral angle for the base-flipping as defined in former studies (24,108) (see Figure 5C). In specific, the COM distance between the dT nt (O2 and N3 atoms) and its opposite dG nt (N1 and O6 atoms) is ∼3 ± 1 Å, and the pseudodihedral for the flipped dT nt is ∼19 ± 7° (see Figure 5D). Therefore, the naked DNA conformations sampled from the 3 μs MD simulation all reside within the S1 state rather than the flipped or partially flipped state that have the corresponding distance >7 Å and the pseudodihedral less than –50° (see Figure 5D). Moreover, the roll angle between the two bps adjacent to the mismatched site is ∼18 ± 8°, which is also very close to the value observed in the S1 state from the MSM (see Figure 4F). Taken together, the simulation for the naked DNA conformation suggests that the T·G mispair remains in an intrahelical state within the 3 μs period, and the mispaired DNA region adopts a bent conformation that can be potentially recognized by TDG, as also suggested by recent AFM studies (66).
Comparisons of base-flipping dynamics for different TDG substrates
In addition to the dT nt, TDG is also capable of excising several other damaged bases, such as dU, 5fC and 5caC (8,71,72). To probe the base-flipping mechanisms for these three alternative TDG substrates, we conducted additional MD simulations for a comparative study. To construct the initial models for varied nts, from the T·G simulation dataset, we firstly selected seven random TDG–DNA conformations near the transition state (TS) between the S4 and S5 states (see Figure 2A and Supplementary Figure S14). It is noteworthy that the S4→S5 transition is not only the final step of the base-flipping motion of the dT nt, but also the slowest step compared to other base extrusion transitions (see Figure 2B). Therefore, the TS for the S4→S5 transition serves as an appropriate checkpoint to evaluate the flipping mechanisms for different target nts. Then, we designed three additional damaged bps, namely U·G, 5fC·G, and 5caC·G, while replaced the original T·G bp with each of these bps. Finally, starting from each of the seven TS conformations for each above bp system, we performed one 100 ns MD simulations and collected a total of 28 (4 × 7 conformations) 100-ns MD trajectories for the final analyses.
The results show that, for the T·G system, the TDG–DNA structure adopts two major states, one locates around the TS region and the other remains at the S4 state, with a relative RMSD value of ∼2.3 and 4 Å, respectively, comparing to one completely flipped reference structure (see Figure 6A). One conformation chosen at the RMSD value of ∼2.3 Å clearly shows that the dT nt is not fully flipped into the TDG active site yet (see Figure 6B). In specific, the dT O2 atom forms one HB with the Ile139 backbone N-H, whereas no direct contact between the thymine and other TDG residues, i.e. Tyr152 and Asn191, is observed. Instead, two water molecules can form HBs with the polar groups of thymine. In addition, the 5-methyl group of dT nt can form nonpolar contacts with the residues Ala145 and Pro153 (see Figure 6B). Interestingly, the U·G system demonstrates a different dynamic behavior comparing to that of the T·G. In particular, most of the MD conformations reach to a completely flipped state with an average RMSD value of ∼1 Å relative to the reference structure, and only a small fraction of MD conformations transits to the S4 state (see Figure 6A). As shown in one selected structure (see Figure 6B), the uracil can establish at least three HBs with the TDG residues, Ile139, Tyr152, and Asn191 through its polar groups, and forms π-π stacking with the Tyr152 side-chain. Therefore, comparing to the dT nt, the dU nt appears to reach to the TDG active site faster, which is likely due to less steric hindrance the uracil endures during the base eversion process owing to the lack of the 5-methyl group.
Figure 6.

Comparisons of the base-flipping process for different TDG substrates: dT, dU, 5fC, and 5caC. (A) Histogram plots of the RMSD of the flipped nt relative to the minimized RC for each substrate. The starting structures are chosen near the transition state (TS) regions between S4 and S5 and are labeled with black dashed line in each plot. In the background, the fluctuations of the RMSD values calculated based on the MSM are represented by colored boxes for S4 in blue and S5 in cyan. (B) Representative conformations for each of the four substrates. For dT nt, one conformation near the TS is selected. For other three bases, the flipped state is used.
On the other hand, both the 5fC·G and 5caC·G systems show very different dynamical and structural properties from the G·dT/dU systems owning to the presence of the cytosine ring. Strikingly, all the conformations transit to the fully flipped state in the 5fC·G system (see Figure 6A), showing a strong tendency to reach the TDG active site. Structural comparison shows an obvious HB-shift from the dT/dU O4 atom to the formyl group of 5fC paring with the Tyr152 backbone N–H (see Figure 6B). Moreover, one water molecule bridging the 5fC N4 atom and residue His151 can be observed. In addition, the formyl group of 5fC can form nonpolar contacts with the residues Ala145 and Pro153, and the key residue Tyr152 tightly stacks with the 5fC. In contrast, in the 5caC·G system, several distinct states can be observed, consisting of the fully flipped state (most populated), TS, and the states remaining at S4 (see Figure 6A). Structural examination of one representative flipped state shows that the presence of the carboxylate group in 5caC attracts several water molecules, one of which forms HB with the residue His151. In addition, the 5caC N3 atom can form one HB with the residue Asn191 (see Figure 6A). Notably, the HB between the O2 atom on the pyrimidine ring and the residue Ile139 is present in all the four systems. Taken together, dU, 5fC and 5caC nts appear to exhibit faster binding kinetics to TDG than the dT nt, which likely in turn, facilitate the catalysis. Moreover, comparing with the dU and 5fC nts that show similar binding tendencies toward TDG, the 5caC nt appears less likely to reach to the TDG active site.
DISCUSSIONS
TDG is shown to adopt a base-flipping strategy to extrude the target base from the DNA helix to its active site, as revealed from the crystal structures (74,76). However, due to the lack of the information regarding the recognition of TDG on undamaged DNA and the static nature of the crystallographic structures, the dynamics of the complete base flipping process at an atomistic level remain unclear. Here, by employing extensive MD simulations combined with the MSM construction, we demonstrate the complete base flipping dynamics of the mispaired dT nt exerted by TDG on a tens of microsecond timescale. Our MSM reveals six metastable state of the mispaired dT nt during its base-flipping process. Particularly, we highlight a critical role of one intercalated residue Arg275 in stabilizing the partially flipped nt (S2) and then penetrating into the DNA minor groove to finally lock the complex in a fully flipped state, which kinetically limits the overall base-flipping process. Notably, the stepwise nature of the base-flipping process observed in TDG has also been captured in other DNA repair/modify enzymes using biochemical (e.g. for UDG and M.HhaI) (48,64,109) or computational (e.g. for O6-alkylguanine-DNA alkyltransferase) (110) approaches. In particular, Stivers and his coworkers, employing both site-mutagenesis and fluorescence assays (48,109), demonstrate that the conformational change of UDG involving the penetration of the intercalation loop into the minor groove takes place after the base-flipping process. The finding appears quite in line with our observations in TDG. Moreover, we propose an active role of TDG in promoting the extrusion of the intrahelical dT nt, as also suggested from previous AFM studies (66). Finally, our comparative studies illustrate distinctive dynamic and structural features for various TDG substrates.
Crystallographic studies have obtained several TDG–DNA complexes where the targeting nt is in either intrahelical (e.g. 2rba) or extrahelical states (e.g. 5hf7 etc.), and the trapped substrates includes U (e.g. 5hf7), C (e.g. 2rba), 5fC (e.g. 5t2w), and 5caC (e.g. 3uo7). To compare these crystal structures with the possible metastable state captured by MSM, we firstly measured the bending angles for seven TDG–DNA structures that contain an flipped yet not cleaved nt except for 2rba that contains a TDG-product complex. As shown in Supplementary Figure S16A, the calculated roll-angle value ranges from 37° to 45°, which is larger than the value for the S6 state reported in current study (28 ± 9°). One possible explanation for the above discrepancy is that all these crystal structures were intentionally designed as an uncleavable form by using either substrate analogs or TDG variants, which may lead to some local structural distortions to prevent the catalysis. In addition, we further projected each crystal structure onto the same energy landscape as shown in Figure 5D for the targeting nt. It clearly shows that all the structures can be assigned to the S6 state except for 2rba that resides in the S1 state region (see Supplementary Figure S17).
Notably, further examinations of the binding interfaces between TDG and DNA show distinct structural features for different crystal structures. In specific, five important contacting points between TDG and DNA backbones, formed by positively charged residues Lys240, Lys246, Lys232, Arg275, and Arg110, can be observed in 5hf7 and 5t2w, as well as in the S6 state reported here (see Supplementary Figure S16B). In contrast, only one of the above contacts (formed by Lys232) is observed in the non-specific complex of 2rba (see Supplementary Figure S3A), which is significantly different from the S1 state where all the above interactions are present (see Supplementary Figure S3C). Moreover, the extended N-terminal region of TDG in 5hf7 (residues 85–110) sterically occludes the potential binding site of the second TDG, as observed in 2rba (see Supplementary Figure S18). Therefore, the second TDG adjacent to the non-specific binding TDG in 2rba is likely to alter the binding mode between TDG and DNA, resulting in a different DNA orientation from that in S1 (compare Supplementary Figure S3A and C). For other specific complexes, one key interaction between the N-terminal residue Arg110 and DNA backbone is missing compared to 5hf7 and 5t2w but other interactions are kept (see Supplementary Figure S16B). Taken together, the non-specific complex in 2rba demonstrates quite different structural features from the S1 state in terms of the TDG–DNA binding interfaces. Whereas the 5hf7 and 5t2w resemble to the S6 state.
TDG actively promotes the damaged base extrusion
Whether the repair enzyme actively promotes the base-flipping or passively traps the extrahelical nt has been a subject of many structural (39,40,63), biochemical (48), NMR (23) and single-molecule studies (47,51,66). One way to answer this question is to directly compare the flipping rate of the damaged nt with and without protein binding, as evidenced by previous imino proton exchange assay for UDG (23). In this study, by probing and comparing the kinetics of the S1→S2 transition with and without TDG, we try to provide further insights into the substrate recognition mechanisms. Our MSM shows that the S1→S2 transition occurs at ∼2 μs (see Figure 2B). In comparisons, the former NMR studies have indicated that the lifetime of the intrahelical T:G mispair is ∼1 ms (33), which is about 103-fold slower than that in the presence of TDG, suggesting an active rather than a passive role of TDG in promoting the base-flipping process (see Figure 7). The subsequent transitions starting from the S2 state involve the compression of the DNA backbone and eversion of the target nt into the active site, which are also actively promoted by TDG. The active role of other DNA repair/modify proteins in promoting the base flipping have also been demonstrated by former biochemical or crystallographic studies (40,45,48,63,64,111). For example, the key intercalation residues, e.g. the Leu125 in AlkA (63) and Tyr88 in MutY (40), are shown to be capable of recognizing the intrahelical lesion bases though the intercalation domains have not penetrated into the DNA minor groove yet, implying an active role of the glycosylase in promoting the base extrusion.
Figure 7.

A schematic illustration of the proposed base-flipping mechanism by TDG.
Consistently, our control simulations for the naked DNA conformations indicate that the T·G mispair can remain in an intrahelical state within at least 3 μs (see Figure 5D). More interestingly, the bending angle of the above naked DNA is ∼18±8°, which is very close to the value observed for the S1 state in the presence of TDG (see Figure 4F). This result suggests that the naked DNA chain that contains a T:G mispair adopts a bent conformation, which can be directly recognized by TDG to initiate the base-flipping. For the undamaged naked DNA, on the other hand, it is predominantly in an unbent form, as indicated by the recent AFM studies (66), which likely prevents the initial recognition by TDG. Nevertheless, since the non-specific DNA chain can also transiently explore a bent conformation (i.e. with a bending angle of ∼30°) (66), TDG may still recognize the bent non-specific DNA conformation and even perform the catalysis, which explains the experimental observation that the TDG exhibits weak but still detectable activities against the matched G:C pair (72). Another scenario for TDG to recognize the undamaged DNA is that TDG firstly binds to the unbent DNA and then actively bend the DNA chain to a conformation that can facilitate the base-flipping process (i.e. the S1 state), which, however, appears energetically unfavorable.
TDG promotes the damaged base extrusion via the minor groove
Whether the damaged base extrudes along the minor or major groove in the presence of the DNA repair/modify proteins is the subject of many theoretical studies (27,28,37,59,111–113). For the bacterial MutM that excises the 8-oxoG (or Fpg), former free energy calculations suggest a base-flipping path along the DNA minor groove, with an overall extrusion barrier of only ∼4 kcal/mol (59). It is also worth to note that the key intercalated residue in Fpg is also an Arginine (i.e. Arg109 in E. coli, and Arg112 in B. stearothermophilus) though the global protein fold is different from TDG. Whereas for the cytosine 5-methyltransferase that methylates the 5-site of the cytosine ring, the base extrusion via the major groove has been shown to be more favorable than the minor groove pathway (27). In TDG or its structurally related systems, however, the detailed base-flipping pathway remained unclear. Here, our TMD simulations captured a base extrusion path via the DNA minor groove in the presence of TDG. Importantly, a key cation-π recognition between the intercalated residue Arg275 and the partially flipped dT nt observed in S2 can stabilize the early base-flipping intermediate and facilitate further base extrusion into the active site. Consistently, former site-directed mutagenesis studies have suggested that the substitution of Arg275 with other residues, i.e. R275A and R275L, can significantly reduce the lesion rates for T and BrU but has very small effects for U and FU that are comparatively smaller and contain a less stable N-glycosylic bond (70), suggesting the potential role of Arg275 in promoting the base-flipping. Nevertheless, the counterpart residue of Arg275 in UDG is replaced by a Leucine (i.e. Leu272 in human UDG), which is apparently distinct from Arginine in terms of the chemical structures and properties. In this regard, the above cation-π interaction observed in TDG will not be fulfilled by the Leucine in UDG. Notably, substitution of Arg275 with Leu leads to a reduced TDG activity against T and BrU compared to the wild type TDG (70), suggesting that the base-flipping mechanisms may vary between UDG and TDG although they share a very similar structural fold.
TDG actively bends the DNA backbone and widens the minor groove width
Our studies show that the invasion of the TDG intercalation loop into the DNA minor groove significantly widens the minor groove width by ∼2 Å, and bends the DNA backbone by ∼10° on average (see Figure 4E and F). The results are very similar to the crystallographic observations for the TDG structures bound with damaged or undamaged DNA chains (74,76). In specific, the TDG is shown to bend the undamaged DNA backbone by ∼25°, with the intercalated residue Arg275 lying along the minor groove of the DNA duplex (76). In contrast, in RC, the damaged DNA helix is bent by ∼43° with the residue Arg275 penetrated into the DNA minor groove. Notably, the recent AFM work demonstrated that TDG could stabilize DNA in two major conformations with an average DNA bending angle of ∼30° and ∼60°, regardless of specific or non-specific DNA sequences (66). These reported values are larger than the values calculated here (19 ± 8.8° versus 28.2 ± 9.2°). The above discrepancy is likely caused by several factors. Firstly, it is noteworthy that the ways to measure the bending angles by former experiments and current work are different. Here, we evaluated the DNA bending by calculating the roll angle between the two bps adjacent to the mispaired bp, which, apparently, cannot be resolved using the single-molecule methods. Moreover, it is also worth to note that our system only contains 24 DNA bps, which is far shorter than the DNA substrates that are used in the experiments (>1000 bp). Finally, the N-terminal region of TDG (residues 82–106) has been shown to play an critical role in tightening DNA binding (74). Unfortunately, due to its disordered nature, we are not able to take this region into account in this study.
The identified base-flipping pathway warrants further experimental tests
Here, we identified a complete base-flipping pathway for the mispaired dT nt exerted by TDG, our MSM indicates that the dominant flipping process follows the sequential transitions in the order of S1→S2→S3→S4→S5→S6. Notably, along the dominant path, several previously unidentified TDG residues are found to be critical in stabilizing the key intermediate state of the flipped and partially flipped dT nt. In specific, the residues Ser273 and Cys276 in S2, Gly142 and Asn157 in S3, Ser273 and Ser272 in S4 can directly form HBs with the polar groups of the thymine through their side chain or backbone atoms (see Figure 3). Therefore, these abovementioned residues can be subject to future mutagenesis tests to further evaluate their specific roles in the base-flipping process. One potential experimental attempt is to design a mutant TDG that may capture a partially flipped base, e.g. by substitutions of certain residues to block the early base-flipping path. Interestingly, one recent experimental study shows that the A145G mutant TDG exhibits higher glycosylase activity than the wt TDG (69). Based on our model, the A145G mutation is likely to relieve some steric clashes between the 5-methyl group of thymine and the Ala145 during the S4→S5 transition, thereby facilicates the base-flipping process.
Effects of chemical modifications of the pyrimidine ring on the base-flipping dynamics
Finally, we compared the base-flipping dynamics for different TDG substrates based on the dominant path identified for the dT nt, under an assumption that different substrates share the same base-flipping pathway. We also assume that the S5→S6 transition (the Arg275 penetration step) is decoupled from the S1 to S5 transitions (base-flipping), therefore, the dynamics of the S5→S6 transition are similar for different nts. We thus selected the S4→S5 transition for the comparative studies because this transition is the slowest step during the base-eversion process (from S1 to S5).
Compared with dU, 5fC, and 5caC nts, dT nt seems to exhibit the lowest flipping rate, which likely in turn slows down the overall catalytic cycling. This result is consistent with the former experimental observation that TDG has a higher catalytic activity against dU than dT (69). In addition, dU and 5fC appear to have comparable flipping kinetics since most of the conformations for dU and all for 5fC starting from the TS regions transit to the fully flipped states (see Figure 6A). dU, lack of one methyl group comparing to dT, experiences less steric clashes with the TDG residues, e.g. Ala145, while entering into the active site. The finding is consistent with former experimental results that the larger size of the 5-substituent on the thymine has detrimental effects on the lesions rates by TDG (71,72). For 5fC, on the other hand, owing to its unique structural feature of the cytosine ring, exhibits a distinct interaction network with the TDG residues compared to dT (see Figure 6A). In contrast, 5caC can explore several different conformational regions though the fully flipped state prevails. Notably, compared to other three nts, 5caC carries one negative charge, which leads to its strong interactions with the solvent waters. Therefore, the desolvation can be one of the main energy penalties to prevent 5caC from entering into the crowded active site of TDG, implying a relatively lower cleavage rate for 5caC than that for 5fC, as also indicated in previous studies (68). Although our theoretical results are in line with some of the experimental observations (69,70), it should be noticed that different nts may have varied base-flipping pathways. Therefore, the TS region we selected for the S4→S5 transition may also be different for various bases. A systematic investigation for each base system similar to the T·G mispair studied here is required in order to sufficiently address the above issue.
Supplementary Material
ACKNOWLEDGEMENTS
We acknowledge the computational support from the Beijing Computational Science Research Center (CSRC) and the π supercomputer at Shanghai Jiao Tong University.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Pujiang Talent Project of Shanghai [17PJ1403600 to L.T.D.]; Startup funding from Shanghai Center for Systems Biomedicine, Shanghai Jiao Tong University [WF220441503]; NSFC [11775016, 11635002 to J.Y.]. Funding for open access charge: Pujiang Talent Project of Shanghai.
Conflict of interest statement. None declared.
REFERENCES
- 1. Hoeijmakers J.H. Genome maintenance mechanisms for preventing cancer. Nature. 2001; 411:366–374. [DOI] [PubMed] [Google Scholar]
- 2. Tomasetti C., Li L., Vogelstein B.. Stem cell divisions, somatic mutations, cancer etiology, and cancer prevention. Science. 2017; 355:1330–1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lindahl T. Instability and decay of the primary structure of DNA. Nature. 1993; 362:709–715. [DOI] [PubMed] [Google Scholar]
- 4. McCullough A.K., Dodson M.L., Lloyd R.S.. Initiation of base excision Repair: Glycosylase mechanism and structure. Annu. Rev. Biochem. 1999; 68:255–285. [DOI] [PubMed] [Google Scholar]
- 5. Bauer N.C., Corbett A.H., Doetsch P.W.. The current state of eukaryotic DNA base damage and repair. Nucleic Acids Res. 2015; 43:10083–10101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. David S.S., O'Shea V.L., Kundu S.. Base-excision repair of oxidative DNA damage. Nature. 2007; 447:941–950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Cortázar D., Kunz C., Saito Y., Steinacher R., Schär P.. The enigmatic thymine DNA glycosylase. DNA Repair. 2007; 6:489–504. [DOI] [PubMed] [Google Scholar]
- 8. Wu X., Zhang Y.. TET-mediated active DNA demethylation: mechanism, function and beyond. Nat. Rev. Genet. 2017; 18:517–534. [DOI] [PubMed] [Google Scholar]
- 9. Maiti A., Drohat A.C.. Thymine DNA glycosylase can rapidly excise 5-formylcytosine and 5-carboxylcytosine: potential implications for active demethylation of CpG sites. J. Biol. Chem. 2011; 286:35334–35338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Guo J., Su Y., Zhong C., Ming G.L., Song H.. Hydroxylation of 5-methylcytosine by TET1 promotes active DNA demethylation in the adult brain. Cell. 2011; 145:423–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Drohat A.C., Coey C.T.. Role of base excision ‘Repair’ enzymes in erasing epigenetic marks from DNA. Chem. Rev. 2016; 116:12711–12729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Bellacosa A., Drohat A.C.. Role of base excision repair in maintaining the genetic and epigenetic integrity of CpG sites. DNA Repair. 2015; 32:33–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Song C.X., Szulwach K.E., Dai Q., Fu Y., Mao S.Q., Lin L., Street C., Li Y., Poidevin M., Wu H.. Genome-wide profiling of 5-formylcytosine reveals its roles in epigenetic priming. Cell. 2013; 153:678–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Shen L., Wu H., Diep D., Yamaguchi S., D’Alessio A.C., Fung A., Zhang K., Zhang Y.. Genome-wide analysis reveals TET- and TDG-dependent 5-methylcytosine oxidation dynamics. Cell. 2013; 153:692–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. He Y.F., Li B.Z., Li Z., Liu P., Wang Y., Tang Q., Ding J., Jia Y., Chen Z., Li L. et al. . Tet-Mediated formation of 5-carboxylcytosine and its excision by TDG in mammalian DNA. Science. 2011; 333:1303–1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ito S., Shen L., Dai Q., Wu S.C., Collins L.B., Swenberg J.A., He C., Zhang Y.. Tet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine. Science. 2011; 333:1300–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Cortellino S., Xu J., Sannai M., Moore R., Caretti E., Cigliano A., Coz M.L., Devarajan K., Wessels A., Soprano D.. Thymine DNA glycosylase is essential for active DNA demethylation by linked Deamination-Base excision repair. Cell. 2011; 146:67–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Cortázar D., Kunz C., Selfridge J., Lettieri T., Saito Y., Macdougall E., Wirz A., Schuermann D., Jacobs A.L., Siegrist F.. Embryonic lethal phenotype reveals a function of TDG in maintaining epigenetic stability. Nature. 2011; 470:419–423. [DOI] [PubMed] [Google Scholar]
- 19. Roberts R.J., Cheng X.. Base flipping. Annu. Rev. Biochem. 1998; 67:181–198. [DOI] [PubMed] [Google Scholar]
- 20. Klimasauskas S., Kumar S., Roberts R.J., Cheng X.. Hhal methyltransferase flips its target base out of the DNA helix. Cell. 1994; 76:357–369. [DOI] [PubMed] [Google Scholar]
- 21. Slupphaug G., Mol C.D., Kavli B., Arvai A.S., Krokan H.E., Tainer J.A.. A nucleotide-flipping mechanism from the structure of human uracil-DNA glycosylase bound to DNA. Nature. 1996; 384:87–92. [DOI] [PubMed] [Google Scholar]
- 22. Guéron M., Kochoyan M., Leroy J.L.. A single mode of DNA base-pair opening drives imino proton exchange. Nature. 1987; 328:89–92. [DOI] [PubMed] [Google Scholar]
- 23. Cao C., Jiang Y.L., Stivers J.T., Song F.. Dynamic opening of DNA during the enzymatic search for a damaged base. Nat. Struct. Mol. Biol. 2004; 11:1230–1236. [DOI] [PubMed] [Google Scholar]
- 24. Yin Y., Yang L., Zheng G., Gu C., Yi C., He C., Gao Y.Q., Zhao X.S.. Dynamics of spontaneous flipping of a mismatched base in DNA duplex. Proc. Natl. Acad. Sci. U.S.A. 2014; 111:8043–8048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wärmländer S., Anjana Sen A., Leijon M.. Imino proton exchange in DNA catalyzed by ammonia and trimethylamine: evidence for a secondary long-lived open state of the base pair. Biochemistry. 2000; 39:607–615. [DOI] [PubMed] [Google Scholar]
- 26. Dallmann A., Dehmel L., Peters T., Mügge C., Griesinger C., Tuma J., Ernsting N.P.. 2-Aminopurine incorporation perturbs the dynamics and structure of DNA. Angew. Chem. 2010; 49:5989–5992. [DOI] [PubMed] [Google Scholar]
- 27. Huang N., Banavali N.K., MacKerell A.D. Jr. Protein-facilitated base flipping in DNA by cytosine-5-methyltransferase. Proc. Natl. Acad. Sci. U.S.A. 2003; 100:68–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. And U.D.P., MacKerell A.D. Jr. Computational approaches for investigating base flipping in oligonucleotides. Chem. Rev. 2006; 37:489–505. [DOI] [PubMed] [Google Scholar]
- 29. Giudice E., Várnai P., Lavery R.. Base pair opening within B-DNA: free energy pathways for GC and AT pairs from umbrella sampling simulations. Nucleic Acids Res. 2003; 31:1434–1443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Várnai P., Canalia M., Leroy J.L.. Opening mechanism of G.T/U pairs in DNA and RNA duplexes: a combined study of imino proton exchange and molecular dynamics simulation. J. Am. Chem. Soc. 2004; 126:14659–14667. [DOI] [PubMed] [Google Scholar]
- 31. Folta-Stogniew E., Russu I.M.. Sequence dependence of base-pair opening in a DNA dodecamer containing the CACA/GTGT sequence motif. Biochemistry. 1994; 33:11016–11024. [DOI] [PubMed] [Google Scholar]
- 32. Leijon M., Gräslund A.. Effects of sequence and length on imino proton exchange and base pair opening kinetics in DNA oligonucleotide duplexes. Nucleic Acids Res. 1992; 20:5339–5343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Moe J.G., Russu I.M.. Kinetics and energetics of base-pair opening in 5′-d(CGCGAATTCGCG)-3′ and a substituted dodecamer containing G.T mismatches. Biochemistry. 1992; 31:8421–8428. [DOI] [PubMed] [Google Scholar]
- 34. Banavali N.K., MacKerell A.D. Jr. Free energy and structural pathways of base flipping in a DNA GCGC containing sequence. J. Mol. Biol. 2002; 319:141–160. [DOI] [PubMed] [Google Scholar]
- 35. Várnai P., Lavery R.. Base flipping in DNA: pathways and energetics studied with molecular dynamic simulations. J. Am. Chem. Soc. 2002; 124:7272–7273. [DOI] [PubMed] [Google Scholar]
- 36. Giudice E., Lavery R.. Nucleic acid base pair dynamics: the impact of sequence and structure using free-energy calculations. J. Am. Chem. Soc. 2003; 125:4998–4999. [DOI] [PubMed] [Google Scholar]
- 37. Fuxreiter M., Luo N., Jedlovszky P., Simon I., Osman R.. Role of base flipping in specific recognition of damaged DNA by repair enzymes. J. Mol. Biol. 2002; 323:823–834. [DOI] [PubMed] [Google Scholar]
- 38. Imhof P., Zahran M.. The effect of a G:T mispair on the dynamics of DNA. PLoS One. 2013; 8:e53305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Qi Y., Nam K., Spong M.C., Banerjee A., Sung R.J., Zhang M., Karplus M., Verdine G.L.. Strandwise translocation of a DNA glycosylase on undamaged DNA. Proc. Natl. Acad. Sci. U.S.A. 2012; 109:1086–1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Wang L., Chakravarthy S., Verdine G.L.. Structural basis for the Lesion-scanning mechanism of the bacterial MutY DNA glycosylase. J. Biol. Chem. 2017; 292:5007–5017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Wang L., Lee S.J., Verdine G.L.. Structural basis for avoidance of promutagenic DNA repair by MutY adenine DNA glycosylase. J. Biol. Chem. 2015; 290:17096–17105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Parker J.B., Bianchet M.A., Krosky D.J., Friedman J.I., Amzel L.M., Stivers J.T.. Enzymatic capture of a transient extrahelical thymine in the search for uracil in DNA. Nature. 2007; 449:433–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Schonhoft J.D., Stivers J.T.. Timing facilitated site transfer of an enzyme on DNA. Nat. Chem. Biol. 2012; 8:205–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Rowland M.M., Schonhoft J.D., Mckibbin P.L., David S.S., Stivers J.T.. Microscopic mechanism of DNA damage searching by hOGG1. Nucleic Acids Res. 2014; 42:9295–9303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Hendershot J.M., O’Brien P.J.. Search for DNA damage by human alkyladenine DNA glycosylase involves early intercalation by an aromatic residue. J. Biol. Chem. 2017; 292:16070–16080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Bellamy S.R., Krusong K., Baldwin G.S.. A rapid reaction analysis of uracil DNA glycosylase indicates an active mechanism of base flipping. Nucleic Acids Res. 2007; 35:1478–1487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Dunn A.R., Kad N.M., Nelson S.R., Warshaw D.M., Wallace S.S.. Single Qdot-labeled glycosylase molecules use a wedge amino acid to probe for lesions while scanning along DNA. Nucleic Acids Res. 2011; 39:7487–7498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Stivers J.T., And K.W.P., Watanabe K.A.. Kinetic mechanism of damage site recognition and uracil flipping by escherichia coli uracil DNA glycosylase. Biochemistry. 1999; 38:952–963. [DOI] [PubMed] [Google Scholar]
- 49. Friedman J.I., Stivers J.T.. Detection of damaged DNA bases by DNA glycosylase enzymes. Biochemistry. 2010; 49:4957–4967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Gorman J., Greene E.C.. Visualizing one-dimensional diffusion of proteins along DNA. Nat. Struct. Mol. Biol. 2008; 15:768–774. [DOI] [PubMed] [Google Scholar]
- 51. Nelson S.R., Dunn A.R., Kathe S.D., Warshaw D.M., Wallace S.S.. Two glycosylase families diffusively scan DNA using a wedge residue to probe for and identify oxidatively damaged bases. Proc. Natl. Acad. Sci. U.S.A. 2014; 111:2091–2099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Lee A.J., Wallace S.S.. Hide and seek: how do DNA glycosylases locate oxidatively damaged DNA bases amidst a sea of undamaged bases. Free Radic. Biol. Med. 2016; 107:170–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Chen L., Haushalter K.A., Lieber C.M., Verdine G.L.. Direct visualization of a DNA glycosylase searching for damage. Chem. Biol. 2002; 9:345–350. [DOI] [PubMed] [Google Scholar]
- 54. Blainey P.C., van Oijen A.M., Banerjee A., Verdine G.L., Xie X.S.. A base-excision DNA-repair protein finds intrahelical lesion bases by fast sliding in contact with DNA. Proc. Natl. Acad. Sci. U.S.A. 2006; 103:5752–5757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Blainey P.C., Luo G., Kou S.C., Mangel W.F., Verdine G.L., Bagchi B., Xie X.S.. Nonspecifically bound proteins spin while diffusing along DNA. Nat. Struct. Mol. Biol. 2009; 16:1224–1230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Schormann N., Ricciardi R., Chattopadhyay D.. Uracil-DNA glycosylases-structural and functional perspectives on an essential family of DNA repair enzymes. Protein Sci. 2014; 23:1667–1685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Zharkov D.O., Mechetin G.V., Nevinsky G.A.. Uracil-DNA glycosylase: Structural, thermodynamic and kinetic aspects of lesion search and recognition. Mutat. Res. 2010; 685:11–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Porecha R.H., Stivers J.T.. Uracil DNA glycosylase uses DNA hopping and short-range sliding to trap extrahelical uracils. Proc. Natl. Acad. Sci. U.S.A. 2008; 105:10791–10796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Qi Y., Spong M.C., Nam K., Banerjee A., Jiralerspong S., Karplus M., Verdine G.L.. Encounter and extrusion of an intrahelical lesion by a DNA repair enzyme. Nature. 2009; 462:762–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Banerjee A., Santos W.L., Verdine G.L.. Structure of a DNA glycosylase searching for lesions. Science. 2006; 311:1153–1157. [DOI] [PubMed] [Google Scholar]
- 61. Qi Y., Spong M.C., Nam K., Karplus M., Verdine G.L.. Entrapment and structure of an extrahelical guanine attempting to enter the active site of a bacterial DNA glycosylase, MutM. J. Biol. Chem. 2010; 285:1468–1478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Fromme J.C., Verdine G.L.. DNA lesion recognition by the bacterial repair enzyme MutM. J. Biol. Chem. 2003; 278:51543–51548. [DOI] [PubMed] [Google Scholar]
- 63. Bowman B.R., Lee S., Wang S., Verdine G.L.. Structure of Escherichia coli AlkA in complex with undamaged DNA. J. Biol. Chem. 2010; 285:35783–35791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Klimasauskas S., Szyperski T., Serva S., Wüthrich K.. Dynamic modes of the flipped-out cytosine during HhaI methyltransferase-DNA interactions in solution. EMBO J. 1998; 17:317–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Huang N., MacKerell A.D. Jr. Specificity in protein–DNA interactions: energetic recognition by the (cytosine-C5)-methyltransferase from HhaI. J. Mol. Biol. 2005; 345:265–274. [DOI] [PubMed] [Google Scholar]
- 66. Buechner C.N., Maiti A., Drohat A.C., Tessmer I.. Lesion search and recognition by thymine DNA glycosylase revealed by single molecule imaging. Nucleic Acids Res. 2015; 43:2716–2729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Drohat A.C., Coey C.T.. Role of base excision ‘Repair’ enzymes in erasing epigenetic marks from DNA. Chem. Rev. 2016; 116:12711–12729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Maiti A., Michelson A.Z., Armwood C.J., Lee J.K., Drohat A.C.. Divergent mechanisms for enzymatic excision of 5-formylcytosine and 5-carboxylcytosine from DNA. J. Am. Chem. Soc. 2013; 135:15813–15822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Maiti A., Noon M.S., MacKerell A.D. Jr, Pozharski E., Drohat A.C.. Lesion processing by a repair enzyme is severely curtailed by residues needed to prevent aberrant activity on undamaged DNA. Proc. Natl. Acad. Sci. U.S.A. 2012; 109:8091–8096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Maiti A., Morgan M.T., Drohat A.C.. Role of two strictly conserved residues in nucleotide flipping and N-glycosylic bond cleavage by human thymine DNA glycosylase. J. Biol. Chem. 2009; 284:36680–36688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Morgan M.T., Bennett M.T., Drohat A.C.. Excision of 5-Halogenated uracils by human thymine DNA glycosylase. J. Biol. Chem. 2007; 282:27578–27586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Bennett M.T., Rodgers M.T., Hebert A.S., Ruslander L.E., Eisele L., Drohat A.C.. Specificity of human thymine DNA glycosylase depends on N-glycosidic bond stability. J. Am. Chem. Soc. 2006; 128:12510–12519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Pidugu L.S., Flowers J.W., Coey C.T., Pozharski E., Greenberg M.M., Drohat A.C.. Structural basis for excision of 5-Formylcytosine by thymine DNA glycosylase. Biochemistry. 2016; 55:6205–6208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Coey C.T., Malik S.S., Pidugu L.S., Varney K.M., Pozharski E., Drohat A.C.. Structural basis of damage recognition by thymine DNA glycosylase: Key roles for N-terminal residues. Nucleic Acids Res. 2016; 44:10248–10258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Malik S.S., Coey C.T., Varney K.M., Pozharski E., Drohat A.C.. Thymine DNA glycosylase exhibits negligible affinity for nucleobases that it removes from DNA. Nucleic Acids Res. 2015; 43:9541–9552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Maiti A., Morgan M.T., Pozharski E., Drohat A.C.. Crystal structure of human thymine DNA glycosylase bound to DNA elucidates Sequence-Specific mismatch recognition. Proc. Natl. Acad. Sci. U.S.A. 2008; 105:8890–8895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Hashimoto H., Hong S., Bhagwat A.S., Zhang X., Cheng X.. Excision of 5-hydroxymethyluracil and 5-carboxylcytosine by the thymine DNA glycosylase domain: its structural basis and implications for active DNA demethylation. Nucleic Acids Res. 2012; 40:10203–10214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Case D.A., Cerutti D.S., Cheatham T.E. III, Darden T.A., Duke R.E., Giese T.J., Gohlke H., Goetz A.W., Greene D., Homeyer N. et al. . AMBER17. 2017; San Francisco: University of California; (April 2018, date last accessed)http://ambermd.org/. [Google Scholar]
- 79. Guy A.T., Piggot T.J., Khalid S.. Single-stranded DNA within nanopores: conformational dynamics and implications for sequencing; a molecular dynamics simulation study. Biophys. J. 2012; 103:1028–1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Hornak V., Abel R., Okur A., Strockbine B., Roitberg A., Simmerling C.. Comparison of multiple Amber force fields and development of improved protein backbone parameters. Proteins. 2006; 65:712–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Joung I.S., Cheatham T.E. III. Determination of alkali and halide monovalent ion parameters for use in explicitly solvated biomolecular simulations. J. Phys. Chem. B. 2008; 112:9020–9041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Joung I.S., Cheatham T.E.. Molecular dynamics simulations of the dynamic and energetic properties of alkali and halide ions using Water-Model-Specific ion parameters. J. Phys. Chem. B. 2009; 113:13279–13290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Hess B., Kutzner C., van der Spoel D., Lindahl E.. GROMACS 4: Algorithms for highly efficient, load-balanced, and scalable molecular simulation. J. Chem. Theory. Comput. 2008; 4:435–447. [DOI] [PubMed] [Google Scholar]
- 84. Van Der Spoel D., Lindahl E., Hess B., Groenhof G., Mark A.E., Berendsen H.J.. GROMACS: fast, flexible, and free. J. Comput. Chem. 2005; 26:1701–1718. [DOI] [PubMed] [Google Scholar]
- 85. Berendsen H.J., van der Spoel D., van Drunen R.. GROMACS: a message-passing parallel molecular dynamics implementation. Comput. Phys. Commun. 1995; 91:43–56. [Google Scholar]
- 86. Berendsen H.J.C., Postma J.P.M., van Gunsteren W.F., Hermans J.. Pullman B. Interaction Models for Water in Relation to Protein Hydration. Intermolecular Forces. 1981; Dordrecht: Reidel Publishing Company. [Google Scholar]
- 87. Essmann U., Perera L., Berkowitz M.L., Darden T., Lee H., Pedersen L.G.. A smooth particle mesh Ewald method. J. Chem. Phys. 1995; 103:8577–8593. [Google Scholar]
- 88. Hess B., Bekker H., Berendsen H.J.C., Fraaije J.G.E.M.. LINCS: a linear constraint solver for molecular simulations. J. Comp. Chem. 1997; 18:1463–1472. [Google Scholar]
- 89. Bussi G., Donadio D., Parrinello M.. Canonical sampling through velocity rescaling. J. Chem. Phys. 2007; 126:014101. [DOI] [PubMed] [Google Scholar]
- 90. Pérezhernández G., Paul F., Giorgino T., De F.G., Noé F.. Identification of slow molecular order parameters for Markov model construction. J. Chem. Phys. 2013; 139:015102. [DOI] [PubMed] [Google Scholar]
- 91. Pande C.R.S., Vijay S.. Improvements in markov state model construction reveal many non-native interactions in the folding of NTL9. J. Chem. Theory Comput. 2013; 9:2000–2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Naritomi Y., Fuchigami S.. Slow dynamics of a protein backbone in molecular dynamics simulation revealed by time-structure based independent component analysis. J. Chem. Phys. 2013; 139:215102. [DOI] [PubMed] [Google Scholar]
- 93. Naritomi Y., Fuchigami S.. Slow dynamics in protein fluctuations revealed by time-structure based independent component analysis. J. Chem. Phys. 2011; 134:065101. [DOI] [PubMed] [Google Scholar]
- 94. Harrigan M.P., Sultan M.M., Hernández C.X., Husic B.E., Eastman P., Schwantes C.R., Beauchamp K.A., Mcgibbon R.T., Pande V.S.. MSMBuilder: statistical models for biomolecular dynamics. Biophys. J. 2017; 112:10–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Da L.T., C E., Shuai Y., Wu S., Su X.D., Yu J.. T7 RNA polymerase translocation is facilitated by a helix opening on the fingers domain that may also prevent backtracking. Nucleic Acids Res. 2017; 45:7909–7921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Da L.T., Pardoavila F., Liang X., Silva D.A., Lu Z., Xin G., Dong W., Huang X.. Bridge helix bending promotes RNA polymerase II backtracking through a critical and conserved threonine residue. Nat. Commun. 2016; 7:11244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Shukla D., Hernández C.X., Weber J.K., Pande V.S.. Markov state models provide insights into dynamic modulation of protein function. Acc. Chem. Res. 2015; 48:414–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Da L.T., Shi Y., Ning G., Yu J.. Dynamics of the excised base release in thymine DNA glycosylase during DNA repair process. Nucleic Acids Res. 2017; 46:568–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Bowman G.R., Huang X., Pande V.S.. Using generalized ensemble simulations and Markov state models to identify conformational states. Methods. 2009; 49:197–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Zhu L., Fu K.S., Zeng X., Huang X.. Elucidating conformational dynamics of multi-body systems by constructing Markov State Models. PCCP. 2016; 18:30228–30235. [DOI] [PubMed] [Google Scholar]
- 101. Chodera J.D., Noé F.. Markov state models of biomolecular conformational dynamics. Curr. Opin. Struct. Biol. 2014; 25:135–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Prinz J.H., Wu H., Sarich M., Keller B., Senne M., Held M., Chodera J.D., Schütte C., Noé F.. Markov models of molecular kinetics: generation and validation. J. Chem. Phys. 2011; 134:174105. [DOI] [PubMed] [Google Scholar]
- 103. Noé F., Fischer S.. Transition networks for modeling the kinetics of conformational change in macromolecules. Curr. Opin. Struct. Biol. 2008; 18:154–162. [DOI] [PubMed] [Google Scholar]
- 104. And W.C.S., Pitera J.W., Suits F.. Describing protein folding kinetics by molecular dynamics simulations. 1. Theory†. J. Phys. Chem. B. 2004; 108:2084–2089. [Google Scholar]
- 105. Ghiani L., Denti P., Marcialis G.L.. Robust Perron cluster analysis in conformation dynamics. Linear Algebra Appl. 2005; 398:161–184. [Google Scholar]
- 106. Noé F., Schütte C., Vandeneijnden E., Reich L., Weikl T.R.. Constructing the equilibrium ensemble of folding pathways from short off-equilibrium simulations. Proc. Natl. Acad. Sci. U.S.A. 2009; 106:19011–19016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Weinan E., Vandeneijnden E.. Transition-Path theory and Path-Finding algorithms for the study of rare events. Annu. Rev. Phys. Chem. 2010; 61:391–420. [DOI] [PubMed] [Google Scholar]
- 108. Song K., Campbell A.J., Bergonzo C., Santos C.D.L., Grollman A.P., Simmerling C.. An Improved reaction coordinate for nucleic acid base flipping studies. J. Chem. Theory Comput. 2009; 5:3105–3113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Jiangyu L., Stivers J.T.. Mutational analysis of the Base-Flipping mechanism of uracil DNA glycosylase. Biochemistry. 2002; 41:11236–11247. [DOI] [PubMed] [Google Scholar]
- 110. Hu J., Ma A., Dinner A.R.. A Two-Step Nucleotide-Flipping mechanism enables kinetic discrimination of DNA lesions by AGT. Proc. Natl. Acad. Sci. U.S.A. 2008; 105:4615–4620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Law S.M., Feig M.. Base-flipping mechanism in postmismatch recognition by MutS. Biophys. J. 2011; 101:2223–2231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Bergonzo C., Campbell A.J., Santos C.D.L., Grollman A.P., Simmerling C.. Energetic preference of 8-oxoG eversion pathways in a DNA glycosylase. J. Am. Chem. Soc. 2011; 133:14504–14506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Zheng H., Cai Y., Ding S., Tang Y., Kropachev K., Zhou Y., Wang L., Wang S., Geacintov N.E., Zhang Y.. Base flipping free energy profiles for damaged and undamaged DNA. Chem. Res. Toxicol. 2010; 23:1868–1870. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.