


Abstract
Antisense oligonucleotides that are dependent on RNase H for cleavage and subsequent degradation of complementary RNA are being developed as therapeutics. Besides the intended RNA target, such oligonucleotides may also cause degradation of unintended RNA off-targets by binding to partially complementary target sites. Here, we characterized the global effects on the mouse liver transcriptome of four oligonucleotides designed as gapmers, two targeting Apob and two targeting Pcsk9, all in different regions on their respective intended targets. This study design allowed separation of intended- and off-target effects on the transcriptome for each gapmer. Next, we used sequence analysis to identify possible partially complementary binding sites among the potential off-targets, and validated these by measurements of melting temperature and RNase H-cleavage rates. Generally, our observations were as expected in that fewer mismatches or bulges in the gapmer/transcript duplexes resulted in a higher chance of those duplexes being effective substrates for RNase H. Follow-up experiments in mice and cells show, that off-target effects can be mitigated by ensuring that gapmers have minimal sequence complementarity to any RNA besides the intended target, and that they do not have exaggerated binding affinity to the intended target.
INTRODUCTION
RNase H is an endonuclease that specifically recognizes DNA/RNA heteroduplexes and cleaves the RNA strand (1). The two fragments of the cleaved RNA are not protected by a 5′-cap or poly-A tail at the cleaved ends, and are therefore rapidly degraded by endogenous exonucleases (2). Single-stranded DNA oligonucleotides can be designed to elicit RNase H-mediated cleavage and subsequent degradation of complementary RNA in cells (3). Such oligonucleotides usually have a gapmer design with chemically modified flanks conferring improved stability and binding, and a central DNA gap-region that supports RNase H binding. A gapmer targeting apolipoprotein B (APOB) was recently approved for the treatment of homozygous familial hypercholesterolemia (4), and many others are in clinical development (5).
It is well-known that gapmers may also cause degradation of unintended off-targets by binding to partially complementary target sites. Using Xenopus oocytes as a model system for investigating sequence-specificity of gapmers in vivo, it was demonstrated more than 20 years ago that gapmers can cause degradation of transcripts harboring only partially complementary target sites (6). More recently, this observation has also been confirmed for chemically modified gapmers (7). In fact, for gapmers with high-affinity modifications, such as locked nucleic acids (LNA) (8) or constrained ethyl (cEt) nucleosides (9), particular care must be taken to manage off-target effects (10,11). Generally, unintended targeting depends on whether the binding affinity between gapmer and RNA is sufficient to allow the formation of appreciable amounts of duplex, and whether RNase H tolerates the structural changes induced by duplex mismatches and bulges. Therefore, in general, as the number of mismatches and bulges in a partially complementary RNA target site increase, the probability of RNase H binding and cleavage decreases, as has been also shown in cells using library sequencing (12).
In a therapeutic context, unintended targeting may lead to unwanted or adverse effects. Indeed, recent experiments in mice suggest a direct relationship between the effective reduction in the number of unintended RNA targets in the liver following administration of high-affinity gapmers and the hepatotoxic potential of these oligonucleotides, as measured on the basis of clinical chemistry markers in the blood (13–16). The hepatotoxic potential of LNA-gapmers can be evaluated in primary hepatocytes by measurement of LDH and adenosine triphosphate levels, which allow some control of this type of toxicity prior to further investigations in animals (17). More recently, a cellular assay specifically focused on assessing the hybridization-dependent toxic potential of LNA-gapmers has been suggested. In this approach, a high concentration of gapmer is transfected into cells to drive binding and cleavage of all potential off-targets via mass-action kinetics (18). Potential adverse effects related to unintended targeting can therefore be managed both in cells and animals. Nevertheless, there is still a need for an improved understanding of how to design gapmers that avoid off-target effects, thereby reducing attrition in pre-clinical toxicity studies and consequently increasing the overall chance of developing gapmers with good drug-like properties.
In this study, we set out first to characterize the extent to which four different LNA-gapmers bind to and degrade unintended RNA targets in the livers of mice after intravenous injection. We evaluated the global effects on the liver transcriptomes by microarray analysis. To separate the effects of intended and unintended targeting, we designed two gapmers targeting the proprotein convertase subtilisin/kexin type 9 (Pcsk9) transcript in distinct, non-overlapping, regions and two targeting apolipoprotein B (Apob), also in non-overlapping regions. In this way, the downstream effects of reducing the intended target transcript levels will be the same for each pair of gapmers, while any effects of unintended targeting will be different because of their completely different sequences. In addition, it has been reported that Pcsk9 is involved in regulating Apob metabolism (19), and one of the downstream effects of reducing Apob mRNA levels is that Pcsk9 mRNA levels are also reduced, but not vice versa. This experimental design consequently allows separation of intended and off-target effects, and identification of sets of transcripts that are significantly and specifically reduced following treatment with each gapmer. Follow-up sequence analysis of each set of downregulated transcripts revealed, as expected, that target regions with no, or only one or two mismatches or bulges, were identified more often than expected by chance. Experimental evaluation of the binding affinity and RNase H-cleavage activity of the potential target regions identified in this manner confirmed that transcripts that are significantly and specifically reduced by gapmer treatment, and which harbor one- or two mismatches or bulges (but not more), can be substrates for RNase H.
These results suggest that off-target effects can be mitigated by ensuring that gapmers have no- or only a few off-targets, with less than three mismatches or bulges, and do not have exaggerated binding affinity beyond what is needed for optimal potency with respect to the intended target. This was directly demonstrated in two follow-up experiments. First, we evaluated the off-target effects in mice of five different gapmers targeting the same intended region and being of equal length, but having different binding affinities to the intended target. Second, we evaluated the off-target effects of four different gapmers in cells, targeting the same intended region and having similar binding affinities and potencies, but being of different lengths and therefore, having different numbers of mismatches and bulges to off-targets. Taken together, these results suggest a strategy for designing highly sequence-specific gapmers by optimizing length and LNA-modification patterns.
MATERIALS AND METHODS
Oligonucleotide synthesis and purification
LNA-modified gapmers were designed with fully modified phosphorothioate backbones and were synthesized on a MerMade 192X (Bioautomation, TX, USA) synthesizer following standard phosphoramidite protocols. The final 5′-dimethoxytrityl (DMT) group was left on the oligonucleotide. After synthesis, the oligonucleotides were cleaved from the solid support using aqueous ammonia and subsequently deprotected at 65°C for 5 h. The oligonucleotides were purified by solid phase extraction in TOP DNA cartridges (Agilent, Glostrup, Denmark) using the lipophilic DMT group as a chromatographic retention probe for purification purposes. After eluting impurities, the DMT group was removed by treatment with dichloroacetic acid. As the last step in the purification process, the oligonucleotides were eluted from the cartridge and the eluate was evaporated to dryness. The oligonucleotides were dissolved in phosphate-buffered saline (PBS) and the oligonucleotide concentration in solution determined using Beer–Lambert’s law by calculating the extinction coefficient and measuring UV-absorbance. Oligonucleotide identity and purity were determined by reversed-phase Ultra Performance Liquid Chromatography coupled to Mass Spectrometry (UPLC-MS).
Binding affinity of gapmers
Equal amounts of gapmer and RNA were dissolved in buffer (10 mM phosphate buffer, 100 mM NaCl, 0.1 nM ethylenediaminetetraacetic acid (EDTA), pH 7.0) to final concentrations of 1.5 mM for each. Samples were denatured at 95°C for 3 min and then allowed to anneal by slowly cooling to room temperature for 30 min. Thermal melting curves were recorded at 260 nm on a Lambda 40 UV/VIS Spectrophotometer equipped with a PTP6 Peltier Temperature Programmer (Perkin Elmer, Waltham, USA) using a temperature gradient that was increased by 1°C/min from 20°C to 95°C and then decreased to 58°C. First derivatives and the local maxima of both the melting and annealing were used to assess the duplex melting temperature (Tm), defined as the temperature at which half of the gapmers are duplexed with RNA.
Gapmer-mediated cleavage of RNA by human recombinant RNase H1
Gapmer (15 pmol) and 5′-FAM labeled RNA (45 pmol) was mixed with water to a total volume of 13 μl. Next, 6 μl annealing buffer (200 mM KCl, 2 mM EDTA, pH 7.5) was added, and the temperature raised to 90°C for 2 min. The sample was then allowed to reach room temperature. RNase H1 enzyme (LuBioScience, Zürich, Switzerland) was added (0.125 U in 3.125 μl of 750 mM KCl, 500 mM Tris–HCl, 30 mM MgCl2 and 100 mM dithiothreitol, pH 8.3) and the sample was incubated at 37°C for 30 min before the reaction was terminated by adding 4 μl EDTA solution (0.25 M). The following buffers were used: buffer A (10 mM NaClO4, 1 mM EDTA, 20 mM Tris–HCl, pH 7.8) and buffer B (1 mM NaClO4, 1 mM EDTA, 20 mM Tris–HCl, pH 7.8). Following RNase H1 treatment, 15 μl of each sample was mixed with 200 μl of buffer A, and 50 μl of the resulting mixture was subjected to Anion Exchange High Performance Liquid Chromatography (AIE-HPLC) on a DNAPac PA100 high-resolution column (2 × 250 mm). Using a flow rate of 0.25 ml/min, the gradient of buffer A to buffer B in the column was changed from 100%/0% at time t = 0 min, to 78%/22% at t = 22 min and then to 0%/100% at t = 25 min. This gradient was then maintained until t = 30 min. Subsequently, the flow rate was then increased to 0.5 ml/min, and the gradient of buffer A to buffer B was changed to 100%/0% at t = 31 min, and maintained until t = 35 min. The flow rate was then decreased again to 0.25 ml/min, and the gradient maintained at 100%/0% until t = 40 min. The FAM fluorophore on the RNA was excited at 494 nm, and the emitted light measured at 518 nm. Individual peaks in the resulting chromatograms corresponded to cleaved RNA fragments and remaining full-length RNA. The amount of each RNA fragment was quantified as the percentage area under the corresponding peak relative to the total area under all peaks measured. The percentage of full-length RNA cleaved represents the RNase H activity for the gapmer (referred to as % cleaved RNA).
In vivo studies
Inbred C57BL/6J female mice were obtained from Taconic (Ejby, Denmark) and fed with a standard diet ad libitum. At the start of the study, the mice weighed 20 ± 2 g (arithmetic mean ± standard deviation). The vivarium was maintained on a 12-hour light-dark cycle throughout each study. Two study schedules were used. For the short studies, mice were dosed intravenously on days 0, 1 and 2, and anesthetized (70% CO2/30% O2) before sacrifice by cervical dislocation on day 3. For the long studies mice, were dosed intravenously on days 0, 3, 7, 10 and 14, and sacrificed on day 16. The treatment groups (n = 5) received either 0.9% saline or saline-formulated gapmer administered by intravenous injection. At the end of the study, blood was sampled for serum analysis, and livers were snap-frozen in liquid nitrogen and stored at −80°C. All mouse protocols were approved by the Danish National Committee for Ethics in Animal Experiments.
Serum alanine-aminotransferase analysis
Serum was analyzed for alanine-aminotransferase (ALT) activity using an enzymatic assay (Horiba ABX Diagnostics) according to the manufacturer’s instructions adjusted to 96-well format. Measurements were correlated to a 2-fold diluted standard curve generated from an ABX Pentra MultiCal solution (Horiba ABX Diagnostics). The mean ALT activity in the saline treated group was calculated and all ALT levels presented as fold changes relative to that mean value.
RNA analyses from mouse liver: isolation, qRT-PCR and microarray
Total RNA from liver was isolated using Trizol (Invitrogen, Denmark), purified using mini RNeasy columns (Qiagen, Denmark), contaminating DNA removed by treating with DNase I (Sigma-Aldrich, Denmark) and the integrity and purity assessed using an Agilent Bioanalyzer (Agilent Technologies, Santa Clara, USA) and a NanoDrop RNA 6000 spectrophotometer (Thermo Fisher Scientific, Denmark). Quantification of mRNA by real-time quantitative reverse transcription polymerase chain reaction (qRT-PCR) was performed using TaqMan assays (Applied Biosystems, Foster City, USA) (Supplementary Table S1). The reverse transcription reaction was carried out with random decamers, 0.5 mg total RNA and the M-MLV RT enzyme (Ambion, Denmark) according to the protocol for first strand cDNA synthesis. Depending on expression levels, cDNA was subsequently diluted (5×) in nuclease-free water before addition to the RT-PCR reaction mixture. The Applied Biosystems 7500/7900/ViiA real-time PCR instrument was used for amplification. As a reference, mRNA levels were normalized to β-actin (Actb) and glyceraldehyde-3-phosphate dehydrogenase (Gapdh) and presented as fold-changes relative to average levels in saline controls. Quantification of mRNA by microarray was conducted using Affymetrix GeneChip Mouse Gene 1.0 ST Arrays according to manufacturer’s specifications (Affymetrix, Santa Clara, USA). Total RNA was amplified and labeled using the Whole Transcript Sense Target Labeling Assay (Affymetrix). Labeled ssDNA was hybridized to arrays in a GeneChip Hybridization Oven 640 (Affymetrix). The arrays were washed and stained in a GeneChip Fluidics Station 450 (Affymetrix), and subsequently scanned with a GeneChip Scanner 3000 (Affymetrix). The resulting images were analyzed using Affymetrix Expression Console Software (Affymetrix). Affymetrix probes were remapped to 22 361 different gene models using the Ensembl build 77 database (20). Using these redefined probesets, probe intensities were summarized and made comparable between arrays by quantile normalization using the Robust Multi-Array Average (RMA) expression measure (21). All data were submitted to the Gene Expression Omnibus with ID: GSE100699. Only the 13 959 genes with normalized and log2-transformed expression levels above 6 in at least one saline-treated sample were judged to be expressed in liver and analyzed further.
Isolation of mouse primary hepatocytes and RNA analysis by qRT-PCR
A C57BL/6J mouse (Taconic, Ejby, Denmark) was anesthetized with pentobarbital and the liver perfused at a flow rate of 7 ml per min through the vena cava using a two-step protocol: (i) pre-perfusion for 5 min with Hank’s balanced salt solution containing 15 mM HEPES and 0.38 mM EGTA and (ii) perfusion for 12 min with collagenase solution (Hank’s balanced salt solution containing 0.17 mg/ml Collagenase type 2 (Worthington 4176), 0.03% bovine serum albumin, 3.2 mM CaCl2 and 1.6 g/l NaHCO3). Following removal of the liver and opening of the liver capsule, the liver suspension was filtered through 70 μm cell strainer using William E medium (WME) followed by filtering through 40 μm cell strainer. Cells were washed with WME with 10% fetal bovine serum (WME w/FBS). The cell suspension was centrifuged for 5 min at 50 g at room temperature to pellet the hepatocytes. The cell pellet was re-suspended in 25 ml WME w/FBS, and 25 ml WME with 90% Percoll solution was added. The cell suspension was centrifuged for 10 min at 50 g at room temperature, and the resulting pellet re-suspended in 50 ml WME and again centrifuged for 3 min at 50 g. At last, the supernatant was removed and the cells re-suspended in 20 ml WME w/FCS. A volume of 100 μl of freshly isolated hepatocyte cells were seeded with 25 000 cells/ml in collagen-coated 96-well plates and incubated for 3–4 h at 37°C. After 3–4 h, the cells were washed with WME w/FBS and 95 μl/well WME w/FBS were added. To this was added a 5 μl dilution of oligonucleotide to reach the final concentration range for a 8-point concentration response curve starting at 50 μM with half-logarithmic dilution (reduced 3.16-fold per point). Cells incubated with oligonucleotide in this manner were harvested after 3 days by removal of media followed by addition of 125 μl PureLink©Pro 96 Lysis buffer (Invitrogen 12173.001A) and 125 μl 70% ethanol. RNA was purified according to the manufacture’s instruction and eluted in a final volume of 50 μl water resulting in an RNA concentration of 10–20 ng/μl. RNA was diluted 10-fold in water prior to the one-step qPCR reaction. For one-step qPCR reaction qPCR-mix (qScriptTMXLE 1-step RT-qPCR ToughMix®Low ROX from QauntaBio, cat.no 95134–500) was mixed with two Taqman probes in a ratio 10:1:1 (qPCR mix: probe 1: probe 2) to generate the mastermix. Taqman probes were acquired from LifeTechnologies (Supplementary Table S1). Mastermix (6 μl) and RNA (4 μl, 1–2 ng/μl) were mixed in a qPCR plate (MicroAmp®optical 384 well, 4309849). After sealing, the plate was given a quick spin, 1000 g for 1 min at room temperature and transferred to a ViiaTM 7 system (Applied Biosystems, Thermo), and the following PCR conditions used: 50°C for 15 min; 95°C for 3 min; 40 cycles of: 95°C for 5 s followed by a temperature decrease of 1.6°C/s followed by 60°C for 45 s. The data were analyzed using the QuantStudioTM Real_time PCR Software.
RNA isolation from human cells and analysis by qRT-PCR
The HeLa cell line was purchased from the European Collection of Authenticated Cell Cultures (through Sigma-Aldrich, Denmark) and maintained as recommended by the supplier in a humidified incubator at 37°C with 5% CO2. For assays, 2500 HeLa cells/well were seeded in a 96 multi-well plate in culture media. Cells were incubated for 30 min before addition of oligonucleotides dissolved in PBS and harvested 3 days later. RNA was extracted using the PureLink Pro 96 RNA Purification kit according to the manufacturer's instructions (Ambion). From this, cDNA was synthesized using M-MLT Reverse Transcriptase, random decamers RETROscript and RNase inhibitor (Ambion) with 100 mM dNTP set PCR Grade (Invitrogen) and DNase/RNase free water (Gibco) according the manufacturers’ instructions. For gene expression analysis, qPCR was performed using TaqMan Fast Advanced Master Mix (2×) (Ambion) in a doublex set-up. All primer sets were purchased from Life Technologies (Supplementary Table S1). The qPCR primer sets for the genes of interest were labeled with FAM-MGB dye, while the qPCR primer sets for the housekeeping PKG1 and PPIA were labeled with VIC-MGB dye.
Statistical analysis of concentration-response curves
Concentration-response curves (CRCs) of RNA levels after treatment with gapmer at eight different concentrations were analyzed by nonlinear least squares fitting of a four-parameter logistic function using the R software package drc (22). From each fitted curve the parameter and error estimates were tabulated. For each gapmer, selectivity ratios against each off-target were calculated as the EC50 (half-maximal effective concentration) value for that off-target relative to the EC50 value for the on-target. The standard deviation of the selectivity ratio was calculated by error propagation of the standard deviations of the individual EC50 estimates. Significance of differences between EC50 estimates are calculated from the corresponding standard errors of each EC50 using the delta method (22).
Statistical analysis of microarray data
For each transcript, the effects of gapmer treatment were contrasted with the effects of saline treatment by calculating log2-transformed expression fold-changes. The significance of this differential expression was evaluated by empirical Bayes-moderated F-statistics using the R software package LIMMA (23). Using the step-up procedure proposed by Benjamini and Hochberg in 1995 (24), transcripts with false discovery rates (FDRs) below 0.01 were considered significantly regulated or affected. Conversely, transcripts with FDRs > 0.1 were considered not significantly regulated or affected.
Sequence analysis
Hybridization between two complementary nucleic acid strands is mainly governed by hydrogen bonding between base pairs on opposite strands and base stacking. In addition, nucleic acid polymers are flexible and can form bulges or loops that also need to be considered when evaluating the thermodynamically optimal hybridization (25–27). Energy parameters for the binding of LNA-modified phosphorothioate oligonucleotides to RNA have not been published, although the LNA-modification is known to induce an RNA-like conformation of the DNA (28). In accord with previous reports of LNA-gapmers (12), we therefore made a rough estimate of the standard free energy of binding, ΔG, using parameters for RNA–RNA interactions (27). In addition, we calculated a simple binding score between oligonucleotide and RNA using the Needleman–Wunsch algorithm (29). The oligonucleotide sequences are listed in Table 1, and the pre-mRNA and mRNA sequences were retrieved from the Ensembl build 77 database (20). The Needleman–Wunsch alignment was performed without end gap and gap opening penalties, with a gap extension penalty of −1 and with substitution scores of 1 for each Watson–Crick base pair and zero otherwise. The maximal score using this scheme is equal to the length of the oligonucleotide.
Table 1. Sequence and LNA-modification patterns for gapmers in study.
| Name | Target | Sequence (5′ to 3′)a | Length (nt) | T m (°C) |
|---|---|---|---|---|
| P1 | Pcsk9 | GATacacctccACC | 14 | 69.0 |
| P2 | Pcsk9 | GTctgtggaaGCG | 13 | 63.8 |
| A1 | Abob | GTtgacactgTC | 12 | 54.1b |
| A2 | Apob | GCattggtatTCA | 13 | 59.5 |
| U1 | UBE3C | GTgtttctgcTGCT | 14 | 73.0 |
| U2 | UBE3C | GTGTttctgctgcTAT | 16 | 72.1 |
| U3 | UBE3C | GTGTttctgctgctATAA | 18 | 72.2 |
| U4 | UBE3C | TGgtgtttctgctgctATAA | 20 | 68.4 |
| T1 | Tradd | GctcatactcgtaggcCA | 18 | 66.8 |
| T2 | Tradd | GCtcatactcgtaggcCA | 18 | 69.7 |
| T3 | Tradd | GCtcatactcgtaggCCA | 18 | 72.1 |
| T4 | Tradd | GCTcatactcgtaggcCA | 18 | 73.3 |
| T5 | Tradd | GCTcatactcgtaggCCA | 18 | 76.3 |
aUpper case plus bold font indicates LNA and lower case indicates DNA. All nucleotides are phosphorothioate-linked.
bMelting curves did not have normal sigmoidal shape so Tm estimate may be biased.
RESULTS
Reproducible reduction of target mRNA in mouse liver after gapmer treatment
We designed and synthesized two different pairs of LNA phosphorothioate antisense oligonucleotides (gapmers). One pair, P1 and P2, targeted mouse Pcsk9 in non-overlapping but fairly close target regions, while the other pair, A1 and A2, targeted mouse Apob in widely distant target regions (Figure 1 and Table 1). Gapmers P2 (30), A1 (31) and A2 (32) have previously been investigated. To reduce the potential for mismatched binding of either P1 or P2 to Apob or for A1 or A2 to Pcsk9, the target regions of P1 and P2 in Pcsk9 were chosen so that there were at least three mismatches or bulges to any target region in Apob. Similarly A1 and A2 were designed with at least three mismatches or bulges to any target region in Pcsk9.
Figure 1.

Overview of gapmer designs and intended targets. Gene structures of mouse Pcsk9 with arrows indicating binding regions for gapmers P1 and P2, and of mouse Apob targeted by gapmers A1 and A2. Positions are relative to the transcription start site. For each gapmer sequence, uppercase bold indicates LNA and lower case indicates DNA.
C57BL/6J mice (n = 5 per group) were administered each of the four different gapmers by three intravenous injections on consecutive days. Knockdown of Pcsk9 and Apob mRNA in the liver at day 4 was measured by qRT-PCR (Figure 2). As seen in Figure 2A, the gapmers P1 and P2 significantly reduced Pcsk9 target mRNA levels to ∼50% and ∼30%, respectively, compared to those in saline control-treated mice. In Figure 2B, the gapmers A1 and A2 significantly reduced Apob target mRNA levels to <5% and ∼10%, respectively. To match activity on the intended target, based on the potency measured in primary mouse hepatocyes (data not shown), P1 and P2 were both dosed at 10 mg/kg, whereas A1 and A2 were dosed at 10 and 5 mg/kg, respectively. The Pcsk9 targeting gapmers, P1 and P2, did not affect Apob mRNA levels (Figure 2B), while conversely, the Apob targeting gapmers, A1 and A2, both reduced Pcsk9 mRNA levels to ∼60% (Figure 2A). This observation suggested that the reduction of Pcsk9 mRNA levels is a downstream effect of Apob mRNA knockdown, which is consistent with a previous study assessing the impact of human PCSK9 overexpression in mice (19).
Figure 2.
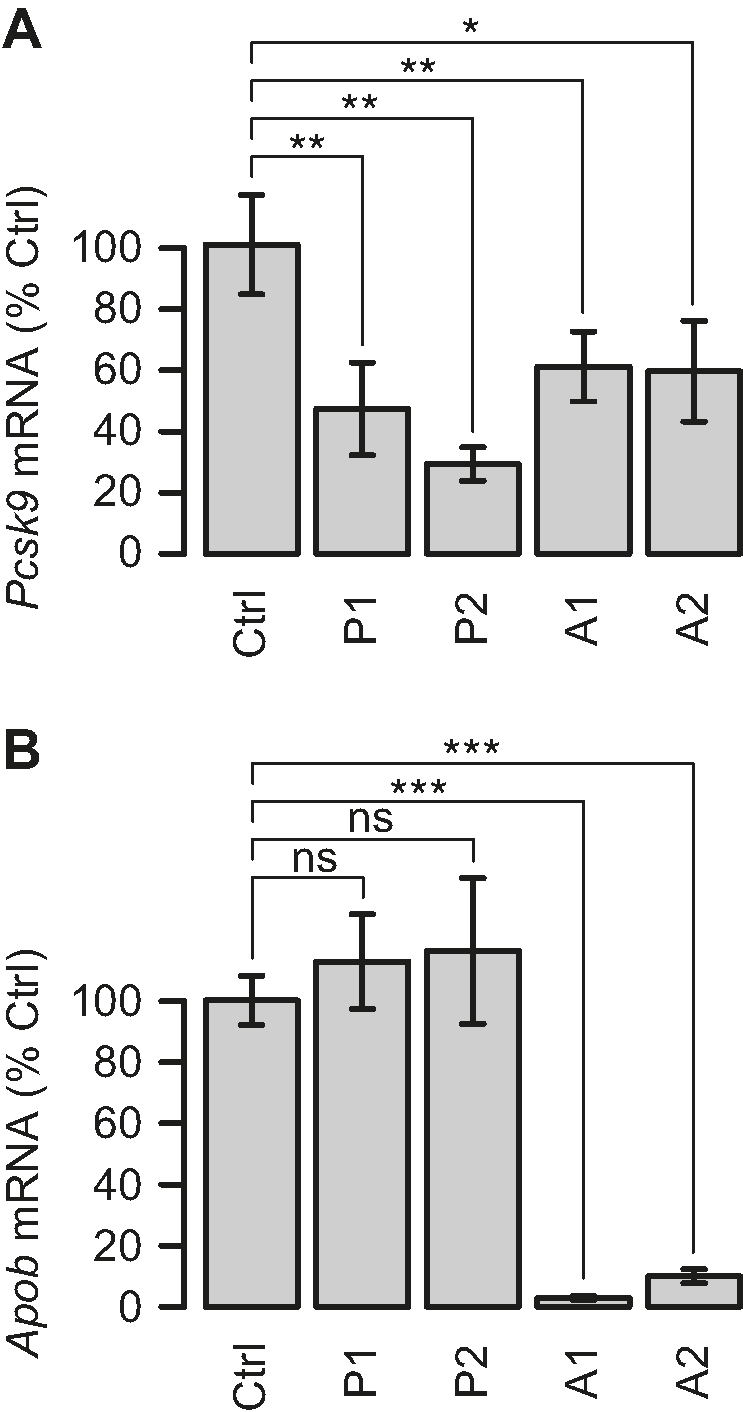
Confirmation of gapmer activity on intended targets. Knockdown of (A) Pcsk9 mRNA and (B) Apob mRNA in mouse liver as measured by qRT-PCR following three 10 mg/kg doses on consecutive days of gapmers P1 and P2 (targeted to Pcsk9), or 10 and 5 mg/kg, respectively, of A1 and A2 (targeted to Apob). Data represent the average ± one standard deviation relative to saline-treated controls (Ctrl) with n = 5 per treatment group. Significance of differences in average transcript levels between groups evaluated by Student's t-test. *P < 0.05, **P < 0.01, ***P < 0.001, ns: not significant.
These results were reproduced in an independent mouse experiment (Supplementary Figure S1). In summary, the gapmers P1, P2, A1 and A2 mediated significant knockdown of their intended targets in liver as expected.
Identification of effects from intended- and unintended targets in mice liver
To further characterize the effects of the four gapmers, P1, P2, A1 and A2, on the transcriptomes in mouse liver, we performed a microarray analysis of the RNA from the study presented in Figure 2. For each of the four gapmers, we found that levels of hundreds of transcripts were significantly lower or higher after treatment compared to those in the saline-treated control, when controlling for the FDR at 0.01 (Figure 3A and B).
Figure 3.

Overview of gapmer treatment effects on the transcriptome (A) Transcripts are divided into those significantly increased (up arrow), decreased (down arrow), or not affected (dot) after treatment with P1 (blue) or P2 (green). Similarly, for (B) significantly increased (up arrow), lowered (down arrow) or not affected (dot) transcript levels after treatment with A1 (orange) or A2 (purple) are shown. (C) The three overlapping sets of transcripts that are focused on in this analysis are outlined: those reduced by treatment with P1 but not affected by treatment with P2 (P1 only), those reduced by treatment with P2 but not affected by treatment with P1 (P2 only) and those significantly reduced by both P1 and P2. Similarly for (D) the overlapping sets of transcripts reduced by either A1 only, A2 only or by both A1 and A2.
At the significance cutoff-levels chosen, as for the qRT-PCR analysis (Figure 2), Apob mRNA was identified in the microarray analysis as significantly reduced by both A1 and A2, but not by P1 and P2. Furthermore, Pcsk9 mRNA was reduced by all four gapmers, although the P1 and P2 gapmers had the greatest effect (Supplementary Tables S2–5). Besides Pcsk9, the microarray analysis revealed 39 transcripts that were significantly reduced by both P1 and P2 (Figure 3C), which is a larger overlap than expected by chance (P < 10−16 by Fisher’s Exact test). Presumably, these transcripts are involved in downstream processes that are affected by reductions in Pcsk9 RNA levels. Moreover, 58 transcripts were identified that were significantly reduced by treatment with P1, but were not significantly affected (FDR > 0.1) by P2. In addition, 150 transcripts were significantly reduced by P2, but not by P1 (Figure 3C). This analysis was repeated for the Apob targeting gapmers, A1 and A2, to identify groups of transcripts that were significantly reduced by treatment with both gapmers (104 genes), or with only one of the gapmers (128 for A1 and 222 for A2) (Figure 3D). Furthermore, among the 104 transcripts significantly reduced by both A1 and A2, which again was a higher number than expected by chance (P < 10−16 by Fisher’s Exact test), 11 were found to also be significantly reduced by P1 and P2. This set of transcripts could be involved in Pcsk9-mediated downstream effects of Apob mRNA knockdown (Supplementary Table S6). Alternatively, the 11 transcripts could be reduced simply due to a general effect of treatment with any oligonucleotide. Re-analysis of previously published liver transcriptome data after treatment with a LNA-modified oligonucleotide (33) did not reveal such a general reduction relative to saline-treated controls for this set of genes (Supplementary Table S6), however these oligonucleotides were designed to sequester microRNAs, not activate RNase H. Further transcriptome studies with proper negative control oligonucleotides will be needed to confirm if these transcripts are indeed involved in Pcsk9-mediated downstream effects of Apob mRNA knockdown. With respect to identification of off-targets of A1, A2, P1 and P2, which is the focus in this study, the effects of gapmer A2-treatment on the transcriptome were largely reproduced in an independent mouse study, confirming the reproducibility of the microarray analysis and the identification of affected transcripts (Supplementary Figure S2).
Sequence analysis identifies partially complementary off-target sites
For each gapmer, we hypothesized that there was an enrichment of direct, RNase H-mediated, off-targets among the groups of transcripts identified in the microarray analysis as being reduced exclusively by that gapmer (Figure 3C and D). Such off-targets have, by definition, one or more target sites where the gapmer can bind and elicit cleavage via the RNase H-mechanism. We therefore analyzed all 13 959 RNA sequences identified as being expressed in mouse liver in the microarray analysis for potential binding sites. For each gapmer and RNA sequence, we identified the region in the RNA sequence with the computationally calculated lowest possible free energy of binding (ΔG) to the gapmer. For comparison of these calculated binding affinities for all possible unintended RNA targets (ΔGoff) with the calculated binding affinity to the intended RNA target (ΔGon), we calculated ΔΔG values as the difference between ΔGoff and ΔGon. That is, more positive ΔΔG values indicate weaker binding between the gapmer and unintended RNA, relative to the strength of the binding between the gapmer and the intended target RNA. In Figure 4A–D, for each of the four groups of transcripts identified as exclusively affected by each gapmer, P1, P2, A1 and A2, the corresponding ΔΔG values (colored boxes/lines) were compared to the ΔΔG values for the rest of the transcripts included in the microarray analysis (gray boxes/lines), as boxplots and cumulative fractions. For the transcripts in each of the four groups, those harboring strong binding regions to that gapmer in their RNA sequence (roughly ΔΔG-values < 5 kcal/mol) were significantly over-represented compared to all other transcripts (Figure 4A–D).
Figure 4.

Characterization of partially complementary off-target binding sites by calculation of free energy of binding. For each of the gapmers (A) P1, (B) P2, (C) A1 and (D) A2, transcripts were grouped into those specifically reduced by that gapmer (Figure 3C and D) and the rest, and the distribution of differences in free energy changes displayed as boxplots and cumulative fractions. Significance of differences between groups of transcripts were evaluated by Wilcoxon Rank Sum test for boxplots, and the Kolmogorov–Smirnov test for cumulative fractions. The test statisticD is the maximum vertical deviation between two cumulative fraction curves.
As an alternative approach to the identification of potential gapmer binding sites, which does not rely on estimating ΔG, we also counted the number of Watson–Crick base pairs between the gapmer and the RNA sequence in the region that allows optimal alignment. According to this method, a gapmer binding to a fully matched RNA target will have a binding score equal to the length of the gapmer, while a gapmer binding with one mismatch will have a binding score equal to its length minus 1, and so forth. As shown in Figure 5A–D, the results obtained using this method were consistent with those obtained when quantifying binding sites by estimating ΔG (Figure 4A–D), for each gapmer, P1, P2, A1 and A2. RNA sequences with high binding scores (≤2 mismatches or bulges) were significantly over-represented among the groups of transcripts exclusively affected by each gapmer, compared to all other transcripts.
Figure 5.

Characterization of partially complementary off-target binding sites by sequence score. For each of the gapmers (A) P1, (B) P2, (C) A1 and (D) A2, transcripts were grouped into those specifically reduced by that gapmer (Figure 3C and D) and the rest, and the distributions of binding scores displayed as histograms. Significance of differences between transcripts specifically reduced by a gapmer, and the rest of the transcripts, were evaluated for each binding score by Fisher’s Exact test. *P < 0.05, **P < 0.01, ***P < 0.001.
These results provide evidence for the over-representation of binding sites with characteristics as expected for direct, RNase H-mediated, off-targets among the transcripts significantly reduced by each of the gapmers, as identified either by thermodynamic evaluation of the change in free energy of binding (Figure 4A–D), or by the binding score of the number of Watson–Crick base pairs (Figure 5A–D).
Properties of potential off-targets as substrates for RNase H
To evaluate the capacity of potential off-target binding sites identified by sequence analysis as substrates for RNase H when duplexed with a gapmer, we selected 23 of the 222 transcripts reduced exclusively by A2 (Figure 3D). The criteria for the selection were: (i) transcript levels reduced by at least 2-fold following treatment with A2, and (ii) a binding score of at least 9; that is, no more than four mismatches or bulges. Of these 23 potential off-target binding sites, four were identical. Therefore, only 20 unique RNA oligonucleotides were synthesized (Supplementary Table S7). For each of the synthesized RNAs, we measured Tms and RNase H-cleavage activity toward A2 (Supplementary Table S7). The results are summarized in Figure 6A.
Figure 6.

Experimental characterization of potential off-target binding sites. (A) For the gapmer A2, Tm and average RNase H-cleavage activity (n = 3) toward the expected binding region sequence in 23 potential off-targets are shown. Sequences are divided into three groups based on their Tms and cleavage activities. Sequences represented by green dots had characteristics similar to the perfectly complementary Apob binding site (black dot). Sequences represented by blue dots had lower Tms, and sequences represented by orange dots had both lower Tm and lower cleavage activity, similar to the scrambled control characteristics (open dot). For the three groups of binding region sequences, differences in (B) ΔΔG values and (C) binding scores were evaluated by Wilcoxon rank sum test. *P < 0.05, **P < 0.01.
We identified three groups of RNA sequences. A group of 11 RNAs, represented by green dots in Figure 6A, had reasonably similar binding affinities to the fully matched Apob binding site for A2, represented by the black dot (Tm > 50°C), as well as RNase H-cleavage activity similar to or better than the that associated with the Apob binding site (more than 40% cleaved). Therefore, these data indicate that the 11 transcripts harboring these RNA target sites are A2 off-targets. A group of five RNA sequences, represented by orange dots in Figure 6A, had low binding affinity to A2 (Tm < 50°C) and only modest or no RNase H-cleavage activity when duplexed to A2 (<30% cleaved RNA). Based on these data, it is therefore not likely that these sites are functional. Alternatively, the reduced expression of these transcripts could be explained as secondary effects of engaging one or more of the off-targets of A2 identified in the green group (Figure 6A). For the group of four RNA sequences represented by blue dots, it was not possible to judge whether they are off-targets or not. On the one hand, the binding affinity was very low (Tm < 45°C), making it less likely that enough duplex can form in vivo to catalyze substantial degradation. On the other hand, the RNase H-cleavage activity is similar to, or better than, that for the Apob site (black dot), indicating that the cleavage reaction can be effective if enough duplex is formed in vivo. Stratification of the sequence analysis results for the set of 23 transcripts (Supplementary Table S7) into the three groups discussed above, revealed that the 11 transcripts with characteristic most similar to Apob binding site (green dots in Figure 6A), had significantly lower ΔΔG values (Figure 6B) and significantly higher binding scores (Figure 6C) than the rest in the selected set. We repeated the cleavage experiments with twice the concentration of RNase H enzyme and observed the same ranking of gapmers (data not shown).
Reducing off-target effects by lowering the binding affinity toward on- and off-targets
The binding affinity between gapmers and fully matched as well as mismatched target sites can be modulated by changing the number of LNA modifications in the gapmers. To investigate how modulating the binding affinity in this manner can affect knockdown activity of transcripts harboring fully matched or mismatched target sites, we designed five gapmers, T1–T5, targeting the same 18 nt site in the mouse tumor necrosis factor receptor type 1-associated DEATH domain protein (Tradd) transcript (Figure 7A). The gapmers were modified with three to six LNAs, with T1 having the lowest Tm value at 66.8°C and T5 having the highest at 76.3°C (Table 1 and Figure 7A). Gapmers designed as T2 and T5 have been evaluated previously in mice (34). Using sequence analysis, we identified 34 mouse transcripts with no more than two mismatches or bulges (binding score ≥ 16) to T1–T5. Of these, 11 were judged to be expressed in mouse liver based on the microarray data generated for the studies presented in Figures 2 and 3. Of these 11 transcripts, the two with the lowest ΔΔG values to their predicted binding sites, protein tyrosine phosphatase, receptor type D (Ptprd) and adiponectin receptor 1 (Adipor1), were selected as the most likely potential off-targets. We administered each of the five different gapmers by five intravenous injections of 15 mg/kg in C57BL/6J mice (n = 5 per group) over a 14-day period. The knockdown of Tradd, Ptprd and Adipor1 mRNA was measured in the liver at day 16 by qRT-PCR (Figure 7B).
Figure 7.

Evaluation of gapmer activity on intended and unintended targets for five gapmers with the same target regions but different binding affinities. (A) Expected binding regions in Tradd (intended target) and two unintended targets Ptprd and Adipor1. Mismatched bases in the five gapmers T1–T5 are indicated in red. Gray lines indicate gapmer binding region. For gapmer sequences, uppercase bold indicates LNA and lowercase indicates DNA. (B) Knockdown of the intended target, Tradd mRNA and two unintended targets, Ptprd and Adipor1 mRNA, in mouse liver following five 15 mg/kg doses of gapmers T1–T5 over 2 weeks as measured by qRT-PCR. (C) Levels of ALT in mouse serum following five 15 mg/kg doses of gapmers T1–T5 over 2 weeks. Data represent the average ± one standard deviation relative to saline-treated controls (Ctrl) with n = 5 per treatment group. Significance of differences in average transcript levels between groups were evaluated by Student’s t-test. *P < 0.05, ***P < 0.001. (D) Knockdown of Tradd mRNA at eight different concentrations in mouse primary hepatocytes for each of the gapmers T1–T5. CRCs found by least squares fitting of the four-parameter logistic function. Error bars indicate model-based standard errors (n = 4). Horizontal and vertical gray lines indicate estimated maximal efficacy and EC50, respectively.
For Tradd, all five gapmers significantly reduced mRNA levels to between 10 and 15%, with no significant statistical differences between the degrees of knockdown. In contrast, the greatest knockdown of Ptprd was observed with T3, which significantly reduced transcript levels to ∼40%. For Adipor1, only T3 significantly reduced transcript levels to ∼70% compared to the effects of the saline control.
For this dosing schedule in mice, the gapmer T1 significantly reduced Tradd mRNA levels without significantly affecting Ptprd or Adipor1. Increasing the binding affinity further with T2 to T5 did not significantly improve Tradd knockdown, whereas T2, T3 and T5 did mediate significant knockdown of Ptprd, as did T3 for Adipor1. Therefore, T1 was identified as the most sequence-specific gapmer in the set of T1–T5, achieving the best balance between knockdown of intended- and unintended targets.
The dosing schedule used in the mouse study is a standard schedule used for evaluating the hepatotoxic potential of LNA-modified gapmers (35). Measurements of ALT, a biomarker of hepatocellular injury, in serum at day 16 revealed that T2, T3 and T5 had significantly higher hepatotoxic potential compared to T1 (Figure 7C). In particular T3, which was found to be the least sequence-specific when comparing Tradd to Ptprd and Adipor1 (Figure 7B), had the highest hepatotoxic potential in the set of T1–T5, with ALT levels 60-fold above saline treated control.
To further evaluate efficacy and potency of the gapmers T1–T5, knockdown of Tradd was also evaluated by qRT-PCR in mouse primary hepatocytes (n = 4) at eight different concentrations between 0.016 and 50 μM. From the resulting CRCs, see Figure 7D, both maximal efficacy as well as EC50 values were estimated (Supplementary Table S9, horizontal and vertical dashed lines in Figure 7D). The maximal efficacies for T1–T5 were all around 15% mRNA relative to PBS control and not significantly different from each other. This efficacy is comparable to the knockdown observed in the mice studies, and suggests that the total administered dose of 75 mg/kg of each gapmer in mice resulted in very high intracellular concentrations of gapmers in liver, and in the maximally achievable knockdowns of the intended target (compare Figure 7B and D). The EC50 values for gapmers T1-T5 were between 1 and 3 μM, and also not significantly different from each other. For comparison, the two off-targets Ptprd and Adipor1 were also evaluated in mouse primary hepatocytes, but unfortunately even at the highest concentration of 50 μM, none of the gapmers T1–T5 reduced RNA levels significantly (data not shown). The fact that Ptprd and Adipor1 were reduced by some of the gapmers in the studies in mice supports the observation made above that the dosing schedule used in the mice studies resulted in very high intracellular concentrations of gapmers in the livers of mice.
Taken together, at very high dose levels in mice, the gapmer T1 was identified as the most sequence-specific gapmer in the set of T1–T5 (Figure 7B), and as the gapmer with the lowest hepatotoxic potential (Figure 7C). In primary hepatocytes, the gapmers T1–T5 were identified as having fairly similar efficacy and potency on the intended target Tradd, and with too weak potency on the off-targets Ptprd and Adipor1, to have measureable knockdown within the concentration range used.
Reducing off-target effects by lowering the binding affinity toward off-targets only
By increasing the length of a gapmer, the extended part may cover regions that are not fully matched to the unintended targets. The more mismatches or bulges between a gapmer and a RNA binding region, the lower the binding affinity relative to a fully matched RNA binding region, and presumably, the weaker the potency (11). To investigate the mechanism by which introduction of such differences in binding affinity between intended and unintended targets can affect off-target effects, we designed and synthesized four different gapmers, U1–U4. These gapmers targeted the same core region in the human ubiquitin protein ligase E3C (UBE3C) transcript, but ranged from 14–20 nucleotides in length (Table 1 and Figure 8A). The Tm values for each gapmer binding to the target region in UBE3C were in the range 72–73°C, except for U4, which was slightly lower at 68.4°C. We measured UBE3C knockdown by qRT-PCR at eight different concentrations in two different batches of HeLa cells, and estimated the concentration required to achieve half-maximal knockdown, EC50, from the resulting CRCs (Supplementary Table S8 and Figure S3).
Figure 8.

Evaluation of binding affinity and potency selectivity ratios for four gapmers of different length but the same core target region. (A) Expected binding regions in UBE3C (intended target) and four unintended targets. Mismatched bases in the four gapmers U1–U4 are indicated in red. Gray lines indicate nested gapmer binding regions. For gapmer sequences, uppercase bold indicates LNA and lowercase indicates DNA. (B) Tms for gapmers U1–U4 binding to UBE3C and each of the unintended targets. (C) For each of the unintended targets, the EC50 for each gapmer relative to the EC50 for UBE3C was estimated from an 8-point CRC measured by qRT-PCR (n = 2). Error bars represent one standard deviation as determined from the nonlinear regression using error propagation.
Based on sequence analysis, we identified 55 human transcripts with one mismatch or bulge (binding score 15) to the 16 nt gapmer, U2. Of these, we selected four that could be reliably measured by qRT-PCR in HeLa cells. Using the same approach as that used for UBE3C, we measured knockdown of each of the four potential unintended target transcripts and estimated EC50 values from the resulting CRCs (Supplementary Table S8 and Figure S3). We aligned the expected target regions in each transcript to the target region in UBE3C (Figure 8A). As shown in Figure 8A, for nudix hydrolase 19 (NUDT19), two potential target regions were identified, denoted A and B, respectively. A region identical to the NUDT19 (A) region was identified in rho guanine nucleotide exchange factor 7 (ARHGEF7) (Figure 8A).
Next, we synthesized the five RNA sequences (listed in Figure 8A), and measured the Tm to each of the gapmers, U1–U4 (Figure 8B). Comparison of Figure 8A and B showed that longer gapmers contained more mismatches (indicated in red), and had lower Tm toward the unintended target sites relative to the values for the intended target site in UBE3C.
At last, for each gapmer U1–U4, we calculated the selectivity ratio between the intended target on UBE3C and each of the four unintended targets as the ratio of estimated EC50 values (Figure 8C). Longer gapmers were associated with a larger selectivity ratio toward the unintended targets. This relationship corresponds to weaker potency of activity on unintended targets relative to that on the intended target, UBE3C.
DISCUSSION
It is a goal of drug discovery to ensure that the chemical compounds developed interact specifically with their intended biomolecular targets (36). For oligonucleotide therapeutics, improvements in potency arising from high-affinity chemical modifications and improved modes of cellular delivery are recognized in the field as factors that can potentially increase the risk of off-target-dependent toxicities (11,37). Although this type of adverse event has not been observed in clinical trials involving oligonucleotide therapeutics to date, recent studies in mice suggest that the number of off-targets that are effectively reduced in the liver after systemic administration of gapmers can be correlated with the hepatotoxic potential of the oligonucleotides, as measured using biochemical markers in the blood (13–16). This underlines the importance of understanding how to identify and avoid off-targets of gapmers.
To be able to conclude that phenotypic changes observed after administration of a gapmer are causally related to cleavage and degradation of the target RNA, it is necessary to test at least two gapmers complementary to the same RNA target, but with different nucleobase sequences. If treatment with both gapmers results in the same phenotype, whereas treatment with a control gapmer that is not complementary to the RNA target does not, it strongly suggests that the phenotype is mediated by target degradation (38).
In this study, we applied the same reasoning to the identification of off-target effects, and show for two different RNA targets, Apob and Pcsk9 in mice (Figures 1 and 2), that changes in the transcriptome resulting from engaging the intended target can be separated from those related to engaging unintended off-targets (Figure 3).
Both pairs of gapmers reduced the intended target to slightly different extents (Figure 2), and the separation of intended and unintended effects can therefore not be expected to be complete. Also, some transcripts might be reduced as downstream effects of a reduction in actual off-targets. As a result, not all transcripts in the sets of potential off-targets identified by the transcriptome analysis (Figure 3C and D) are expected to be off-targets themselves. Nevertheless, analysis of the binding site characteristics of the sets of potential off-targets revealed that binding sites with no- or only few mismatches or bulges to the gapmer, and consequently having a binding affinity close to that of the intended RNA target, were enriched in these sets (Figures 4 and 5).
For the transcripts that are not reduced by treatment and therefore not likely to be direct, RNase H-mediated, off-targets (gray lines and gray bars in Figures 4 and 5), some of them harbor sequence regions with no- or only few mismatches or bulges to the gapmer. This suggests that sequence regions with characteristics that allow gapmer binding are a necessary factor, but not the only factor, that influences gapmer activity. For example, kinetic modeling predicts that transcripts with high turnover rates are less likely to be reduced effectively with gapmers (39), and experiments in cells and in cell-free environments show that off-target regions are more likely to be found in unstructured and accessible parts of the RNA (40). This underscores the importance of combining sequence-analysis with transcriptomics to allow exclusion of false positives harboring binding sites that in principle allow gapmer binding, but where other factors negate possible gapmer activity, and as a consequence no appreciable reduction of RNA is seen.
Two hallmarks of binding sites in off-targets are: (i) sufficient binding affinity to the gapmer to form stable duplexes, and, (ii) sufficient RNase H-cleavage of the RNA in these duplexes to reduce overall RNA levels effectively. For a selected set of 23 potential off-target transcripts clearly reduced by treatment with gapmer A2, we therefore measured Tms and RNase H-cleavage activities for the most likely binding regions in each. We found that around half of these transcripts had Tms and cleavage activities similar to those measured for the duplex between the gapmer and intended target RNA (green dots in Figure 6). Sequence analysis revealed that this half had the fewest mismatches or bulges to their putative off-targets. Using the transcriptomics protocol presented here, combined with follow-up experimental validation of duplex stability and cleavage, off-target candidates can be identified with high probability. If needed, off-target potential could be further supported by characterizing cleavage products, using for example, 5′-RACE (41), and confirming that cleavage occurred at the predicted gapmer binding site. Interestingly, some of the off-targets of the gapmer A2 identified in this manner were found to be better substrates for RNase H than the intended target sequence in Apob, and were also reduced more effectively than the intended target (see Supplementary Table S7). This suggests that RNase H substrate preferences can in some cases contribute even more to the activity on off-targets than on the intended target. For other factors, such as RNA accessibility, a similar situation may exist. In some cases a structured region may negate potential binding and reduction, as discussed above, but in other cases, if the potential off-target region is even more unstructured and accessible than the region in the intended target, the off-target might be even more effectively reduced than the intended target because of this.
Given the ability to screen for likely off-targets then raises the question of how to best use this to avoid them. A simple trial and error strategy could be followed, where many gapmers are evaluated until one is found that is both sufficiently potent and specific. Instead of just discarding potent but unspecific gapmers, however, we suggest two approaches for optimizing the designs of such gapmers to generate versions that are more specific for the intended target. First, the overall binding affinity to the RNA could be reduced, for example, by decreasing the number of LNA modifications in the gapmer. We demonstrated this approach for five gapmers targeted to the same region in the Tradd transcript, and evaluated Tradd knockdown in mouse liver and primary mouse hepatocytes relative to the knockdown of two off-targets (Figure 7). The gapmer T1 with the lowest binding affinity of the five gapmers evaluated was identified as most sequence-specific and also the one with the lowest hepatotoxic potential. Second, the difference in binding affinities between intended and unintended RNA could be increased. This can be achieved by increasing the length of the gapmer in the cases where the off-target regions then become mismatched to the gapmer. For this approach to succeed, the binding affinity to the intended target region must remain as constant as possible, for example by reducing the number of LNA modifications as the length of the gapmer increases. We demonstrated this concept for four gapmers of different lengths targeted to the same core region in the UBE3C transcript, and evaluated UBE3C knockdown in human HeLa cells relative to the knockdown of four off-targets (Figure 8).
Recent studies relating gapmer binding affinity to potency (11,39,42) can be used to model and illustrate why sequence-specificity can be increased by the two approaches suggested here (Figure 9). The potency of a gapmer is related to the strength of the binding affinity between gapmer and RNA target (11). For gapmers with very low binding affinity, the propensity to duplex with RNA is low, and high concentrations of gapmer are needed to form enough duplex for effective RNase H-cleavage; therefore, the potency of these gapmers will be low. For gapmers with very high binding affinity, the gapmers will still bind strongly to the RNA also after cleavage, which stalls the catalytic cycle (39,42); this also weakens the potency of the gapmer. Consequently, there exists a region of optimal binding affinity, where the potency is highest (39). This parabolic, or U-shaped, relationship between binding affinity and potency exists for both intended and unintended targets (black and gray points in Figure 9, respectively). In this model, the first approach to reducing off-target effects can be seen as shifting both points to the right (Figure 9A), so that the intended target moves from having a higher-than-needed binding affinity, to having the optimal binding affinity for potency. In contrast, the reduced binding affinity to the off-target results in weaker potency, as also demonstrated experimentally (Figure 7). In the second approach, only the gray off-target point is shifted to the right (Figure 9B), reducing binding affinity to off-targets only. We demonstrated that this can be achieved by increasing the length of the gapmer and thereby, introducing mismatched base pairing to the off-targets (Figure 8). To maintain the same binding affinity to the intended target, the increased length of the gapmer is usually balanced by decreasing the number of LNA modifications. In our experience there is a limit to how long a gapmer can be without having weaker potency on the intended target compared to a shorter gapmer with similar affinity binding in the same target region. Such an effect can for example be explained by the gapmer being longer than the accessible region on the target, or by an increased propensity of the long gapmer to form self-structures. As a consequence, the gapmer length achieving optimal sequence-specificity varies between different target regions. A good starting point in most cases is probably a 17–18 nt long gapmer with a predicted Tm between 50°C and 55°C. We would rarely expect that gapmers more than 20 nt in length would be active enough to be taken into consideration.
Figure 9.

Affinity-potency relations suggesting how sequence-specificity can be optimized. Either (A) decrease binding affinity by reducing the number of high-affinity modifications, or (B) increase gapmer length and thereby, the number of mismatches to off-targets. For both suggestions, the extent of the optimization must be balanced against maintaining activity on the intended target.
A major strength of the antisense oligonucleotide technology is that it enables using sequence information and transcriptomics to predict potential hybridization-based off-target effects (11,37). The results presented here demonstrate how this can be achieved in practice, and suggest two approaches for optimizing potent gapmers for improved sequence-specificity. For RNA therapeutics, such optimization is an essential aspect of turning interesting leads into actual drugs, by allowing high potency to be combined with low toxicity for high-affinity RNase H-dependent antisense oligonucleotide drugs.
DATA AVAILABILITY
All microarray data were submitted to the Gene Expression Omnibus with ID: GSE100699
Supplementary Material
ACKNOWLEDGEMENTS
For their excellent technical assistance, we would like to thank Rikke Sølberg, Henriette L. Press, Heidii Mazur, Camilla T. Haugaard and Lisbeth Bang for handling of mice in animal facilities; Marianne B. Mogensen, Kirsten N. Petersen and Valesi Mukuka for synthesis and formulation of LNA-modified oligonucleotides; Charlotte Øverup for liver RNA and Tm measurements; and Bettina Nordbo for RNA measurements in primary mouse hepatocytes. Additionally, we are grateful to Art Levin and Andreas Petri for valuable discussions and to Bo R. Hansen for support in the preparation of the manuscript.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Danish Strategic Research Council [DSF-10-092320]. Funding for open access charge: Roche Innovation Center Copenhagen A/S.
Conflict of interest statement. All authors except M.P. are employees of Roche Pharma Research and Early Development, a part of F. Hoffmann-La Roche Ltd., a company that is developing LNA-modified oligonucleotides for therapeutic purposes.
REFERENCES
- 1. Stein H., Hausen P.. Enzyme from calf thymus degrading the RNA moiety of DNA-RNA Hybrids: Effect on DNA-Dependent RNA polymerase. Science. 1969; 166:393–395. [DOI] [PubMed] [Google Scholar]
- 2. Lima W.F., De Hoyos C.L., Liang X.-H., Crooke S.T.. RNA cleavage products generated by antisense oligonucleotides and siRNAs are processed by the RNA surveillance machinery. Nucleic Acids Res. 2016; 44:3351–3363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Zamecnik P.C., Stephenson M.L.. Inhibition of Rous sarcoma virus replication and cell transformation by a specific oligodeoxynucleotide. PNAS. 1978; 75:280–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Raal F.J., Santos R.D., Blom D.J., Marais A.D., Charng M.-J., Cromwell W.C., Lachmann R.H., Gaudet D., Tan J.L., Chasan-Taber S. et al. Mipomersen, an apolipoprotein B synthesis inhibitor, for lowering of LDL cholesterol concentrations in patients with homozygous familial hypercholesterolaemia: a randomised, double-blind, placebo-controlled trial. Lancet. 2010; 375:998–1006. [DOI] [PubMed] [Google Scholar]
- 5. Bennett C.F., Swayze E.E.. RNA targeting therapeutics: molecular mechanisms of antisense oligonucleotides as a therapeutic platform. Annu. Rev. Pharmacol. Toxicol. 2010; 50:259–293. [DOI] [PubMed] [Google Scholar]
- 6. Woolf T.M., Melton D.A., Jennings C.G.. Specificity of antisense oligonucleotides in vivo. Proc. Natl. Acad. Sci. U.S.A. 1992; 89:7305–7309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lennox K.A., Sabel J.L., Johnson M.J., Moreira B.G., Fletcher C.A., Rose S.D., Behlke M.A., Laikhter A.L., Walder J.A., Dagle J.M.. Characterization of modified antisense oligonucleotides in Xenopus laevis embryos. Oligonucleotides. 2006; 16:26–42. [DOI] [PubMed] [Google Scholar]
- 8. Koshkin A.A., Singh S.K., Nielsen P., Rajwanshi V.K., Kumar R., Meldgaard M., Olsen C.E., Wengel J.. LNA (Locked Nucleic Acids): Synthesis of the adenine, cytosine, guanine, 5-methylcytosine, thymine and uracil bicyclonucleoside monomers, oligomerisation, and unprecedented nucleic acid recognition. Tetrahedron. 1998; 54:3607–3630. [Google Scholar]
- 9. Seth P.P., Siwkowski A., Allerson C.R., Vasquez G., Lee S., Prakash T.P., Wancewicz E.V., Witchell D., Swayze E.E.. Short antisense oligonucleotides with novel 2′–4′ conformationaly restricted nucleoside analogues show improved potency without increased toxicity in animals. J. Med. Chem. 2009; 52:10–13. [DOI] [PubMed] [Google Scholar]
- 10. Kamola P.J., Kitson J.D.A., Turner G., Maratou K., Eriksson S., Panjwani A., Warnock L.C., Douillard Guilloux G.A., Moores K., Koppe E.L. et al. In silico and in vitro evaluation of exonic and intronic off-target effects form a critical element of therapeutic ASO gapmer optimization. Nucleic Acids Res. 2015; 43:8638–8650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hagedorn P.H., Hansen B.R., Koch T., Lindow M.. Managing the sequence-specificity of antisense oligonucleotides in drug discovery. Nucleic Acids Res. 2017; 45:2262–2282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Rukov J.L., Hagedorn P.H., Høy I.B., Feng Y., Lindow M., Vinther J.. Dissecting the target specificity of RNase H recruiting oligonucleotides using massively parallel reporter analysis of short RNA motifs. Nucleic Acids Res. 2015; 43:8476–8487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kakiuchi-Kiyota S., Koza-Taylor P.H., Mantena S.R., Nelms L.F., Enayetallah A.E., Hollingshead B.D., Burdick A.D., Reed L.A., Warneke J.A., Whiteley L.O. et al. Comparison of hepatic transcription profiles of locked ribonucleic acid antisense oligonucleotides: evidence of distinct pathways contributing to non-target mediated toxicity in mice. Toxicol. Sci. 2014; 138:234–248. [DOI] [PubMed] [Google Scholar]
- 14. Burel S.A., Hart C.E., Cauntay P., Hsiao J., Machemer T., Katz M., Watt A., Bui H.-H., Younis H., Sabripour M. et al. Hepatotoxicity of high affinity gapmer antisense oligonucleotides is mediated by RNase H1 dependent promiscuous reduction of very long pre-mRNA transcripts. Nucleic Acids Res. 2015; 44:2093–2109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kasuya T., Hori S.-I., Watanabe A., Nakajima M., Gahara Y., Rokushima M., Yanagimoto T., Kugimiya A.. Ribonuclease H1-dependent hepatotoxicity caused by locked nucleic acid-modified gapmer antisense oligonucleotides. Sci. Rep. 2016; 6:30377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kamola P.J., Maratou K., Wilson P.A., Rush K., Mullaney T., McKevitt T., Evans P., Ridings J., Chowdhury P., Roulois A. et al. Strategies for in vivo screening and mitigation of hepatotoxicity associated with antisense drugs. Mol. Ther. Nucleic Acids. 2017; 8:383–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sewing S., Boess F., Moisan A., Bertinetti-Lapatki C., Minz T., Hedtjaern M., Tessier Y., Schuler F., Singer T., Roth A.B.. Establishment of a predictive in vitro assay for assessment of the hepatotoxic potential of oligonucleotide drugs. PLoS One. 2016; 11:e0159431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Dieckmann A., Hagedorn P.H., Burki Y., Brügmann C., Berrera M., Ebeling M., Singer T., Schuler F.. A sensitive in vitro approach to assess the Hybridization-Dependent toxic potential of high affinity gapmer oligonucleotides. Mol. Ther. Nucleic Acids. 2018; 10:45–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sun H., Samarghandi A., Zhang N., Yao Z., Xiong M., Teng B.-B.. Proprotein convertase Subtilisin/Kexin type 9 interacts with apolipoprotein B and prevents its intracellular degradation, irrespective of the Low-Density lipoprotein receptor. Arterioscler. Thromb. Vasc. Biol. 2012; 32:1585–1595. [DOI] [PubMed] [Google Scholar]
- 20. Cunningham F., Amode M.R., Barrell D., Beal K., Billis K., Brent S., Carvalho-Silva D., Clapham P., Coates G., Fitzgerald S. et al. Ensembl 2015. Nucleic Acids Res. 2015; 43:D662–D669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Irizarry R.A., Hobbs B., Collin F., Beazer-Barclay Y.D., Antonellis K.J., Scherf U., Speed T.P.. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics. 2003; 4:249–264. [DOI] [PubMed] [Google Scholar]
- 22. Ritz C., Baty F., Streibig J.C., Gerhard D.. Dose-response analysis using R. PLoS One. 2015; 10:e0146021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Smyth G.K. Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat. Appl. Genet. Mol. Biol. 2004; 3:Article 3. [DOI] [PubMed] [Google Scholar]
- 24. Benjamini Y., Hochberg Y.. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Statist. Soc. B. 1995; 57:289–300. [Google Scholar]
- 25. Hofacker I.L., Fontana W., Stadler P.F., Bonhoeffer L.S., Tacker M., Schuster P.. Fast folding and comparison of RNA secondary structures. Monatsh. Chem. 1994; 125:167–188. [Google Scholar]
- 26. SantaLucia J., Hicks D.. The thermodynamics of DNA structural motifs. Annu. Rev. Biophys. Biomol. Struct. 2004; 33:415–440. [DOI] [PubMed] [Google Scholar]
- 27. Rehmsmeier M., Steffen P., Hochsmann M., Giegerich R.. Fast and effective prediction of microRNA/target duplexes. RNA. 2004; 10:1507–1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Bondensgaard K., Petersen M., Singh S.K., Rajwanshi V.K., Kumar R., Wengel J., Jacobsen J.P.. Structural studies of LNA:RNA duplexes by NMR: conformations and implications for RNase H activity. Chemistry. 2000; 6:2687–2695. [DOI] [PubMed] [Google Scholar]
- 29. Durbin R., Eddy S.R., Krogh A., Mitchison G.. Biological Sequence Analysis: Probabilistic Models of Proteins and Nucleic Acids. 1998; 1st ednCambridge: Cambridge University Press. [Google Scholar]
- 30. Lindholm M.W., Elmén J., Fisker N., Hansen H.F., Persson R., Møller M.R., Rosenbohm C., Ørum H., Straarup E.M., Koch T.. PCSK9 LNA antisense oligonucleotides induce sustained reduction of LDL cholesterol in nonhuman primates. Mol. Ther. 2012; 20:376–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Braendli-Baiocco A., Festag M., Dumong Erichsen K., Persson R., Mihatsch M.J., Fisker N., Funk J., Mohr S., Constien R., Ploix C. et al. From the cover: The minipig is a suitable Non-Rodent model in the safety assessment of single stranded oligonucleotides. Toxicol. Sci. 2017; 157:112–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Straarup E.M., Fisker N., Hedtjärn M., Lindholm M.W., Rosenbohm C., Aarup V., Hansen H.F., Ørum H., Hansen J.B.R., Koch T.. Short locked nucleic acid antisense oligonucleotides potently reduce apolipoprotein B mRNA and serum cholesterol in mice and non-human primates. Nucleic Acids Res. 2010; 38:7100–7111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Elmén J., Lindow M., Silahtaroglu A., Bak M., Christensen M., Lind-Thomsen A., Hedtjärn M., Hansen J.B., Hansen H.F., Straarup E.M. et al. Antagonism of microRNA-122 in mice by systemically administered LNA-antimiR leads to up-regulation of a large set of predicted target mRNAs in the liver. Nucleic Acids Res. 2008; 36:1153–1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Swayze E.E., Siwkowski A.M., Wancewicz E.V., Migawa M.T., Wyrzykiewicz T.K., Hung G., Monia B.P., Bennett C.F.. Antisense oligonucleotides containing locked nucleic acid improve potency but cause significant hepatotoxicity in animals. Nucleic Acids Res. 2007; 35:687–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hagedorn P.H., Yakimov V., Ottosen S., Kammler S., Nielsen N.F., Høg A.M., Hedtjärn M., Meldgaard M., Møller M.R., Ørum H. et al. Hepatotoxic potential of therapeutic oligonucleotides can be predicted from their sequence and modification pattern. Nucleic Acid Ther. 2013; 23:302–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Imming P., Sinning C., Meyer A.. Drugs, their targets and the nature and number of drug targets. Nat. Rev. Drug Discov. 2006; 5:821–834. [DOI] [PubMed] [Google Scholar]
- 37. Lindow M., Vornlocher H.-P., Riley D., Kornbrust D.J., Burchard J., Whiteley L.O., Kamens J., Thompson J.D., Nochur S., Younis H. et al. Assessing unintended hybridization-induced biological effects of oligonucleotides. Nat. Biotechnol. 2012; 30:920–923. [DOI] [PubMed] [Google Scholar]
- 38. Stein C.A. The experimental use of antisense oligonucleotides: a guide for the perplexed. J. Clin. Invest. 2001; 108:641–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Pedersen L., Hagedorn P.H., Lindholm M.W., Lindow M.. A kinetic model explains why shorter and less affine enzyme-recruiting oligonucleotides can be more potent. Mol. Ther. Nucleic Acids. 2014; 3:e149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Lima W.F., Vickers T.A., Nichols J., Li C., Crooke S.T.. Defining the factors that contribute to on-target specificity of antisense oligonucleotides. PLoS One. 2014; 9:e101752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Soutschek J., Akinc A., Bramlage B., Charisse K., Constien R., Donoghue M., Elbashir S., Geick A., Hadwiger P., Harborth J. et al. Therapeutic silencing of an endogenous gene by systemic administration of modified siRNAs. Nature. 2004; 432:173–178. [DOI] [PubMed] [Google Scholar]
- 42. Yamamoto T., Fujii N., Yasuhara H., Wada S., Wada F., Shigesada N., Harada-Shiba M., Obika S.. Evaluation of multiple-turnover capability of locked nucleic acid antisense oligonucleotides in cell-free RNase H-mediated antisense reaction and in mice. Nucleic Acid Ther. 2014; 24:283–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All microarray data were submitted to the Gene Expression Omnibus with ID: GSE100699