


Abstract
More than 140 post-transcriptional modifications (PTMs) are known to decorate cellular RNAs, but their incidence, identity and significance in viral RNA are still largely unknown. We have developed an agnostic analytical approach to comprehensively survey PTMs on viral and cellular RNAs. Specifically, we used mass spectrometry to analyze PTMs on total RNA isolated from cells infected with Zika virus, Dengue virus, hepatitis C virus (HCV), poliovirus and human immunodeficiency virus type 1. All five RNA viruses significantly altered global PTM landscapes. Examination of PTM profiles of individual viral genomes isolated by affinity capture revealed a plethora of PTMs on viral RNAs, which far exceeds the handful of well-characterized modifications. Direct comparison of viral epitranscriptomes identified common and virus-specific PTMs. In particular, specific dimethylcytosine modifications were only present in total RNA from virus-infected cells, and in intracellular HCV RNA, and viral RNA from Zika and HCV virions. Moreover, dimethylcytosine abundance during viral infection was modulated by the cellular DEAD-box RNA helicase DDX6. By opening the Pandora’s box on viral PTMs, this report presents numerous questions and hypotheses on PTM function and strongly supports PTMs as a new tier of regulation by which RNA viruses subvert the host and evade cellular surveillance systems.
INTRODUCTION
Enzymatic modification of RNA is rapidly emerging as a general mechanism for modulating the function of RNA (1–3). To date, ∼140 post-transcriptional modifications (PTMs) of the canonical ribonucleotides have been discovered (4,5), which contribute to the extraordinary structural diversity of natural RNA (6,7). At the molecular level, PTMs can affect the stability of individual base pairs and establish different hydrogen-bonding patterns to redefine the higher order structure of RNA (8,9). Moreover, PTMs influence intermolecular interactions involved in protein recruiting, DNA binding and recognition of other RNA. For instance, the absence of specific pseudouridines in rRNA of Escherichia coli and Saccharomyces cerevisiae undermines the stability of the entire fold, which leads to disruption of ribosome assembly (10,11). Unmodified tRNAs are not loaded efficiently with their respective aminoacyl groups, and some modifications prevent incorrect loading altogether (12). Hypermodified nucleotides in the tRNA anticodon loop can affect codon–anticodon interactions, which may impair the accuracy of RNA–RNA recognition and foster translational recoding (13). Last, methylation of the terminal nucleotide protects siRNAs and microRNAs from 3′-uridylation that promotes nuclease binding and degradation (14). The discovery of specific enzymes responsible for introducing, removing and interpreting PTMs (i.e. writers, erasers and readers, listed in the MODOMICS database) (15,16) accounts for the dynamic nature of PTMs and sensitivity to the general health, epigenetic, and metabolic state of the cell.
With the exception of well-known housekeeping classes, such as tRNA and rRNA, the incidence and distribution of PTMs across the different types of functional RNAs are still largely unexplored (1–3). Although the presence of PTMs on the genomes of RNA viruses has been known for decades, the elucidation of the biological significance is still in its infancy. In Dengue virus (DENV), the 5′ cap of the viral RNA is produced by methylation of the N7-guanosine and 2′-O-adenosine positions (17). These modifications are introduced by the viral NS5 methyltransferase, which can also methylate the 2′-O position of internal adenosines in viral and cellular RNAs (18,19). The presence of N6-methyladenosine (m6A) has been described in mRNAs of influenza and simian virus 40 (SV40) (20–22), as well as in the genome of Rous Sarcoma virus (RSV) (23). The function of this PTM in influenza and SV40 mRNAs is unknown, but is likely to involve modulation of RNA splicing in RSV (23). Recent groundbreaking studies have shown that m6A can affect replication and stability of HIV-1 RNA, which demonstrated for the first time the impact of PTMs on specific steps of a viral lifecycle (24–26). Similar m6A activities on viral RNA have been also described in human cells infected by Zika virus (ZIKV), DENV and hepatitis C virus (HCV) (27,28). Although different flaviviruses, coronaviruses and rhabdoviruses are known to encode enzymes with methyltransferase activity (19,29–30), cellular enzymes are largely responsible for installing and removing RNA modifications, thus suggesting that PTMs may play essential roles in determining virus–host interactions. Indeed, the addition of PTMs to disguise viral RNA as ‘self’ to elude host surveillance and promote structure within the viral RNA has already been described (31–33).
The groundbreaking functional studies of m6A in viruses were made possible by the development of an analytical approach that combines immunoprecipitation of modified RNA fragments with m6A-specific antibodies, followed by identification of modification sites by RNA-seq (34–36). The availability of specific antibodies or the susceptibility to ad hoc chemical reagents has aided the identification of a handful of other PTMs, including N1-methyladenosine (m1A) (37), cytosine-5-methylation (m5C) (38), guanosine methylation (m1G and m22G (39) (40)), 2-O’-methylation (41) and pseudouridylation (42). However, the investigation of the remaining known PTMs has been severely hampered by the chronic absence of suitable analytical tools. This obstacle could be overcome by the broader diffusion of mass spectrometric (MS) approaches, which analyze genuine RNA strands rather than the cDNA copies generated to complete RNA-seq analysis (43–45). This type of platform enables the unequivocal characterization of any PTMs present on RNA according to unique mass and fragmentation features (46–48). Indeed, MS analysis has been instrumental to the discovery of all known PTMs listed in the RNA Modification Database (4,5). Based on this platform, we have recently developed a robust approach for an agnostic analysis of PTMs at the whole transcriptome level (49,50). This comprehensive approach can reveal the effects of different environmental factors on the expression of the entire complement of modifications, including variations of distinct subsets contained in the cell, and thus enables the investigation of RNA modifications at a systems level (50).
Based on rapidly emerging evidence of the ubiquitous presence of modifications in cellular RNA (1–3), we hypothesized that the regulatory roles attributed to m6A are shared by many other PTMs. To test this hypothesis, we employed our analytical approach to investigate the impact of viral infection on the entire epitranscriptome of the host cell. The variations of composition and expression levels in virus-infected cells versus mock-infected controls revealed the elements of a concerted response mounted by the host against viral infection. In investigating changes across different virus–host systems, we dissected general and virus-specific responses. Moreover, examination of viral genomes isolated by antisense-probe affinity capture offered the first comprehensive view of the extraordinary dimensions and diversity of viral epitranscriptomes. By comparing the modification landscapes of viral genomes extracted from cells and virions, our analyses suggest possible functions of PTMs during the infectious cycle. These observations prompted RNAi depletion experiments aimed at evaluating the effects of a cellular RNA DEAD-box helicase on the expression of RNA modifications, which revealed possible links between PTM biogenetic enzymes and ribostasis. This work aimed at obtaining an initial but comprehensive evaluation of the effects of viral infection on RNA modifications, which would lead to the identification of common and differentiating features across viruses, stimulate hypotheses on the functional significance of viral epitranscriptomes and provide a valid framework for supporting full-fledged mechanistic studies of their possible roles in virus–host interactions.
MATERIALS AND METHODS
Cell culture
Human hepatocarcinoma (Huh7) cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM) (Life Technologies), 10% fetal bovine serum (FBS) (VWR Life Science Seradigm), 1% non-essential amino acids (Life Technologies) and 1% glutamine (Life Technologies). HeLa and 293T cells were cultured in DMEM, 10% FBS and 1% glutamine. All cell lines were maintained at 37°C and 5% CO2.
Virus infections/transfections
Infections were undertaken with the following single-stranded positive sense RNA [ss(+) RNA] viruses: ZIKV MR766 Uganda (gift from Brett Lindenbach, Yale University), DENV-2 strain 16681 (infectious clone from Richard Kinney and Claire Huang, CDC) (51), HCV JFH-1 (infectious clone from Takaji Wakita, National Institute of Infectious Diseases, Tokyo) (52) and poliovirus (PV) Mahonney strain (infectious clone from Peter Sarnow, Stanford University).
Huh7 cells were infected with ZIKV at a multiplicity of infection (MOI) of 1, and DENV at a MOI of 0.1. Huh7 cells were incubated with ZIKV and DENV for 1 h at 37°C. Hereafter, virus was removed, fresh medium was added to the cells and ZIKV and DENV infections were incubated at 37°C for 24 and 48 h, respectively. HeLa cells were similarly infected with ZIKV at a MOI of 1 for 24 h. Huh7 cells were also infected with HCV JFH-1 at a MOI of 0.01 for 6 h at 37°C. Hereafter, cells were trypsinized and re-plated, and JFH-1 infected cells were harvested 3 days post infection (p.i.). For PV infections, the medium was removed from Huh7 cells and cells were washed with cold PBS+ (PBS, 90 mM CaCl2, 100 mM MgCl2). Stock PV was resuspended in PBS+ and added to cells at a MOI of 0.01. Cells were incubated at 37°C for 30 min. Cells were then washed with PBS+, and cell culture medium was replaced onto the cells. HeLa and 293T cells were similarly infected with PV at a MOI of 0.01. PV-infected cells were harvested 5–7 h p.i.
Because HIV-1 cannot successfully replicate in Huh7 or HeLa cell lines, 293T cells were exclusively used for HIV-1 infections (53). Before transfection, 2 × 106 cells were seeded per 100 mm plate. After 24 h, the cells were given fresh medium and transfected with pNl4–3 (NIH AIDS Reagent Program) by using the ProFection® mammalian transfection system (Promega) following the manufacturer’s instructions. The cells were incubated overnight at 37°C. The following morning, the medium/transfection solution was replaced with fresh medium and incubated at 37°C for a further 60 h before harvesting.
Total RNA samples for mass spectrometric analysis
Total RNA samples were obtained from cell lysates and extracellular virions. Total intracellular RNA was obtained by washing cell pellets with cold PBS (Life Technologies) and then lysing them in TRIzol (Invitrogen). Viral RNA from ZIKV, DENV, HCV and HIV-1 virions was obtained from the medium of two 150 mm tissue cultures plates of either mock- or virus-infected cells. The medium was collected and submitted to ultrafiltration on 100K centrifugal filters (Amicon Ultra 15) per manufacturer’s protocol to harvest the desired virions. In contrast, PV virions were obtained by ultracentrifugation in a 10–60% sucrose gradient (54). The presence of virions in the various fractions was verified by re-infecting HeLa cells. The material was then extracted by using TRIzol LS (Invitrogen) according to the manufacturer’s specifications. In each case, the ensuing TRIzol extract was stored in 1/10 volume of 3 M ammonium acetate (Amresco) and 2.5 volumes of absolute ethanol (Fisher Bioreagents) at −20°C.
Each TRIzol extract, which may contain any type of nucleic acid component present in the initial material (e.g. intact RNA and DNA), as well as free mononucleotides and any other soluble component, was submitted to DNase 1 (New England Biolabs) digestion for 2 h at 37°C. The solution was then submitted to overnight ethanol precipitation to separate intact RNA from the deoxynucleotide monophosphate (dNMP) products of DNase 1 digestion and pre-existing mononucleotides, salts and soluble components. The pellets were reconstituted in 5–30 μl RNase free water (Sigma-Aldrich), depending on overall yields. RNA concentrations were determined by using UV 260 nm and adjusted to 40 ng/μl. The RNA was then treated with nuclease P1 and phosphodiesterase to obtain the desired NMP mixtures for mass spectrometric analysis, as previously described (49,50). It is important to note that this analytical platform is capable of correctly discriminating NMPs from dNMPs based on their masses and fragmentation properties. The fact that no significant amounts of dNMPs were ever observed in these samples confirmed the ability of the ethanol precipitation step to eliminate not only the products of DNase 1 digestion, but also any pre-existing mononucleotides in the TRIzol extracts. This observation is consistent with the fact that ethanol precipitation tends to be several fold more efficient with polymeric than monomeric nucleic acid components, thus enabling mutual separation under proper conditions.
Affinity capture for targeted isolation of viral RNA
Antisense oligodeoxynucleotides utilized to carry out the affinity capture of viral RNAs were designed by using sFold software (55). For each virus in the study, we targeted three unstructured regions that possessed the highest binding affinities for putative complementary probes (Supplementary Table S1). Streptavidin-coated paramagnetic beads (Qiagen) were first washed three times with 150 mM ammonium acetate (pH 6.8). A 500 μg aliquot of beads was treated with up to 15 μM of each biotinylated antisense oligodeoxynucleotide probe (Integrated DNA Technologies) in 150 mM ammonium acetate. After overnight incubation at 4°C under gentle agitation, the beads were washed three times with a solution of 150 mM ammonium acetate and 5 mM EDTA (pH 6.8) to remove any unbound probe.
Total RNA samples obtained by TRIzol extraction, DNase 1 digestion and overnight ethanol precipitation were redissolved in 150 mM ammonium acetate and 5 mM EDTA, and then added to the derivatized beads. The tube was heated at 95°C for 3 min followed by gentle agitation for 30 min at 37°C. The beads were then washed two times with the same solution, and finally once with RNase-free water (Sigma-Aldrich). The captured RNA was eluted off the beads in RNase-free water at 95°C for 2 min. The affinity-isolated RNA was immediately removed from the beads to prevent rebinding to the probe, and submitted to ethanol precipitation to eliminate salts and other soluble components. The pellet was redissolved and submitted to exonuclease digestion, as described above.
It should be noted, that the binding, washing, and elution conditions employed here were initially optimized by using synthetic oligonucleotides. The selected washing conditions proved to be sufficiently stringent to prevent the recovery of oligoes that were not completely complementary to the probe, as well as to significantly reduce that of oligoes that were fully complementary. Furthermore, the possible retention of weaker non-specific binders in the captured material was routinely assessed by performing RT-PCR amplification of rRNA—the most abundant RNA ‘contaminant’ in cell lysates.
Mass spectrometric analysis
Immediately before analysis, the mononucleotide mixtures obtained by exonuclease digestion of intact RNA were diluted to 4 ng/μl in 150 mM ammonium acetate and 10% isopropanol. All samples were analyzed on a Thermo Scientific LTQ-Orbitrap Velos instrument and a Waters Synapt G2 HDMS ion mobility spectrometry mass spectrometer (IMS-MS), as previously described (49,50). Analyses were accomplished by using direct infusion electrospray ionization (ESI) in negative ion mode.
The relative abundance of each PTM was expressed as Abundance versus Proxy (AvP), which was calculated according to the following equation:
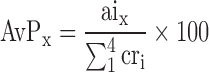 |
in which the signal intensity (aix) of each PTM was normalized against the sum of the intensities displayed in the same spectrum by the four canonical bases (cri).
For the sake of clarity, Table 1 translates relative abundances in AvP units to a hot-cold color gradient, whereas the actual numerical values are reported in full in Supplementary Table S2. The relative abundances displayed by the mock-infected samples were used as the baseline for comparisons with the corresponding infected samples. A different color was assigned only if the respective values were statistically different according to an unpaired Student t-test with a P-value < 0.05. Each data point was the result of at least three biological replicates, which were each separately analyzed five times (technical replicates). Therefore, each value represents the average and standard deviation of a total of 15 separate analyses.
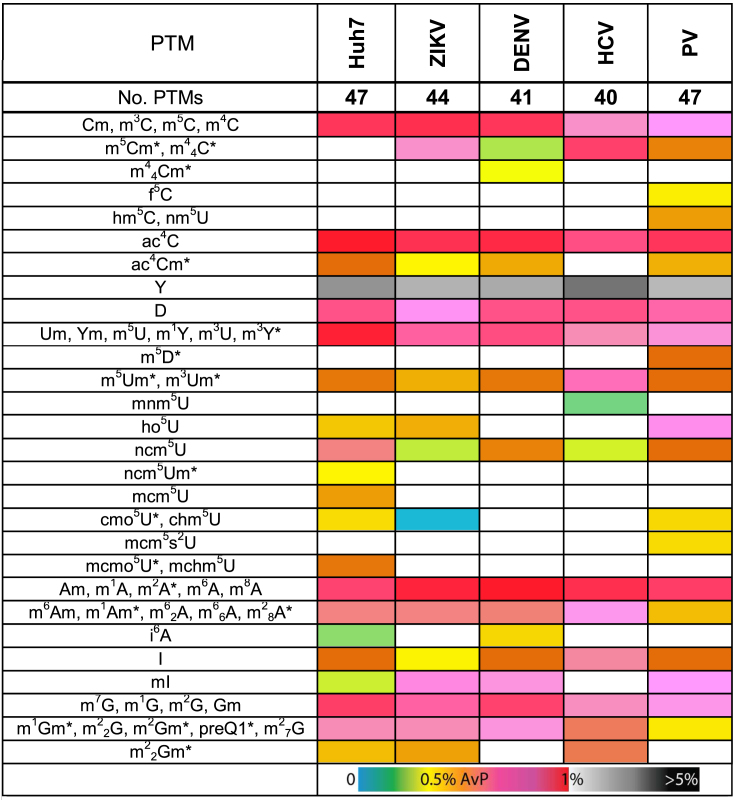
Table 1. Profiles of RNA post-transcriptional modifications obtained from total RNA extracts from mock-infected Huh7 cells (control); and Huh7 cells infected with ZIKV, DENV, HCV and PV (as described in the ‘Materials and Methods’ section). Supplementary Tables S2 and S3 provide respectively full names and actual numerical values of relative abundance, expressed in relation to those of the canonical ribonucleotides detected in the same sample (49). A cold-hot gradient covers relative abundances up to 1%, whereas shades of gray are assigned to values >1%. The different colors also express variations of relative abundance from the Huh7 column (mock-infected control) with a P < 0.05 statistical significance. An asterisk (*) indicates PTMs that to date have not been reported in mammalian systems.
|
Tandem mass spectrometry was carried out in both positive and negative mode to differentiate possible isobars detected during PTM analysis (49,50). This approach involves recognizing unique fragments produced by gas-phase activation, which reveal the position of modifying functional groups. In most cases, the contribution of each isobar to the initial signal can be estimated from the relative intensities of their unique fragments. The abbreviations and complete names of each PTM identified in this study are listed in Supplementary Table S3. This information is also available from the RNA Modifications (http://mods.rna.albany.edu/) and MODOMICS (http://modomics.genesilico.pl/) databases.
Determining virus infectivity
Coverslips were coated with poly-L-lysine hydrobromide (Sigma) and placed in a 48-well cell culture plate. Huh7 cells were seeded onto coverslips at 1 × 104 cells per well. After 24 h, the cells were infected with ZIKV (MOI of 1), DENV (MOI of 0.1), HCV (MOI of 0.01) and PV (MOI of 0.01). At specific p.i. intervals, the medium was removed and the cells were washed two times with PBS, and fixed with 4% paraformaldehyde/PBS and prepared for immunofluorescence analysis, as described previously (56). Virus-infected cells were detected with the J2 anti-double-stranded RNA monoclonal antibody (1:250; Scicons) and AlexaFluor 488 conjugated goat anti-mouse antibody (1:200; Invitrogen). Individual cells were detected by staining the nuclei with Hoechst 33258. Coverslips were mounted and uninfected and virus-infected cells were visualized with an EVOS FL Cell Imaging System (ThermoFisher Scientific). On each coverslip, two different fields of view and at least 100 cells/field were counted. This infectivity assay was repeated three times for each virus.
Determining viral titers
The previously described limiting dilution assay was used to determine the infectivity of DENV and HCV released into the extracellular media (57). This tissue culture infectious dose (TCID50) assay was performed by using the anti-HCV Core C7–50 monoclonal antibody (1:2000; Abcam), and anti-Flavivirus 4G2 monoclonal antibody (Millipore) incubated overnight at 4°C, and detected the following day with AlexaFluor 488 conjugated goat anti-mouse antibody (Invitrogen) incubated at 1:500 for 2 h at room temperature. TCID50 was calculated as previously described (58). Medium from PV-infected cells was collected and viral titers determined by plaque assay in HeLa cells (59). The titer of ZIKV virions in the extracellular medium was similarly determined by plaque assay in Vero cells by using a 10-fold serial dilution prepared in DMEM supplemented with 10% FBS and 10 mM HEPES. Vero cells in 6-well plates were incubated with 0.4 ml of diluted virus for 1 h at 37°C, and then overlayed with 3 ml DMEM (with 10% FBS and 10 mM HEPES) containing 0.6% Oxoid agar. The plates were incubated at 37°C and 5% CO2 for 4 days. Hereafter, the agar overlay was removed and the plates were stained with 1% crystal violet solution in 20% methanol.
siRNA and plasmid transfections
Control (Luciferase, GL2) and DDX6-specific siRNA oligonucleotides were synthesized by Integrated DNA Technologies. The siRNA sequences are as follows: siGL2 sense 5′-CGUACGCGGAAUACUUCGAUU-3′, siGL2 antisense 5′-UCGAAGUAUUCCGCGUACGUU-3′, siDDX6 sense 5′-GCAGAAACCCUAUGAGAUUUU-3′, and siDDX6 antisense 5′-AAUCUCAUAGGGUUUCUGCUU-3′. siRNA oligonucleotides were resuspended in RNase-free water and annealed as previously described (56). For plasmid transfection experiments, the 3xFlag-tagged Bacterial Alkaline Phosphatase (BAP; Sigma) plasmid was used as a control. DDX6 was subcloned from pRFP-DDX6 (a generous gift from Nancy Kedersha, Brigham and Women’s Hospital) into p3xFlag 3.1 cloning plasmid (Sigma) (54). Additional site-directed mutagenesis was completed to mutate the siRNA-targeting sequence within p3xFlag-DDX6 (p3xFlag-DDX6Δsi) (54). Following manufacturer’s protocol, Lipofectamine 2000 (Invitrogen) was used to co-transfect Huh7 cells with 100 nM siRNA and 1 μg plasmid. For mock-infections and infections with ZIKV, DENV and PV, transfected cells were trypsinized the following day, re-plated into 100 mm tissue culture plates and then infected 24 h later (i.e. 48 h post-transfection). In contrast, transfected cells were infected with HCV 24 h post-transfection, and then re-plated into 100 mm tissue culture plates following a 6-h infection.
Induction of a cellular stress response
Oxidative stress was induced by treatment with sodium arsenite. Specifically, Huh7 cells were seeded at 3 × 105 cells per 60 mm plate. The following day, cells were treated with 1 mM sodium arsenite (Sigma) in fresh culture medium for 30 min. After the treatment, TRIzol (Invitrogen) was added to the cells, and RNA was extracted as described. Cells were also treated with synthetic poly(I:C) (Sigma). Specifically, Huh7 cells were seeded in a 6-well plate. The following day, cells were transfected with of 3 μg IFN β-Firefly luciferase (generously provided by Michael Gale, University of Washington) and 1 μg TK-Renilla luciferase (pGL4.74, Promega) plasmids with Lipofectamine 2000 (Invitrogen). Twenty-four hours post-transfection of the plasmids, cells were transfected by using Transmessenger reagent (Qiagen) and 1 μg of poly(I:C) (Sigma) per well. Prior to transfection, the 10 μg/μl stock concentration of poly(I:C) was diluted to 1 μg/μl in PBS, heated at 65°C for 10 min and slow-cooled at room temperature for 1 h. The transfected cells were harvested the following day in 500 μl Passive Lysis Buffer (Promega). Activation of the IFN-β promoter as a result of the poly(I:C) was examined by luciferase activity in lysates that were analyzed in triplicate in a 96-well plate by using the Promega Dual-Luciferase Reporter Assay System on the BioTek Synergy H1 multi-mode microplate reader. RNA was extracted from the remaining fraction of the lysate (400 μl) and treated as described above for mass spectrometric analysis.
Western blotting
To confirm siRNA-depletion and 3xFlag-BAP or 3xFlag-DDX6Δsi plasmid overexpression (54), cells were harvested at specific times post-infection as described above. In brief, cells were washed twice in cold PBS. Hereafter, 1 ml PBS was added per plate and cells scraped off the plates using a cell lifter. One-third of cells were used for analysis by western blot, and two-thirds for northern blot. Cell pellets for protein analysis were lysed in RIPA buffer (100 mM Tris–HCl pH 7.4, 0.1% sodium dodecyl sulphate (SDS), 1% Triton X-100, 1% deoxycholic acid, 150 mM NaCl) with protease inhibitor (EDTA-free) on ice for 20 min and centrifuged at 4°C at 20 000 rpm. Protein concentrations in the clarified lysate were quantified by using BCA reagent (BioRad). Proteins (10 μg) were separated in a 10% SDS-polyacrylamide gel at 100 V for 2 h. Hereafter, proteins were transferred to a PVDF membrane at 100 V for 1 h at 4°C. Following transfer, the membrane was blocked in 5% skim milk powder in PBS and 1% Tween-20 (PBS-T/milk) for 1 h, and incubated in PBS-T/milk with in primary antibody at 4°C overnight. The following day, the blot was washed three times in PBS-T for 10 min. Secondary antibodies in PBS-T/milk were then added for 1 h at room temperature, and then washed an additional three times in PBS-T. Proteins were visualized by using Clarity Western ECL Substrate solution (BioRad) and imaged on a chemiluminescent BioRad imager. Western blot analysis of specific proteins was performed by using the following primary antibodies: rabbit anti-ZIKV Capsid (1:1,000; GeneTex GTX133317), rabbit anti-DENV-2 Capsid (1:1000; ThermoFisher Scientific PA5–34689), mouse anti-HCV Core 1b [C7–50] (1:2000; Abcam ab2740), rabbit anti-PV 3D (1:5000; generous gift from Karla Kirkegaard, Stanford University), rabbit anti-DDX6 (1:10 000; Bethyl Laboratories A300–460A), 3xFlag M2 monoclonal antibody conjugated horseradish peroxidase (1:10 000, Sigma F1804) and mouse anti-GAPDH (1:10,000; Millipore). Secondary antibodies (donkey anti-mouse-HRP (sc-2314) and donkey anti-rabbit-HRP (sc 2313)) were purchased from Santa Cruz Biotechnologies and used at 1:10 000 ratio.
Northern blotting
Viral RNA and actin mRNA from total cell extracts were detected by northern blotting. Specifically, TRIzol-extracted RNA was resuspended in 2× loading dye (22% formaldehyde (Fisher Bioreagents), 60% Formamide (Sigma), 18% 10× MOPS-EDTA-Sodium Acetate (MESA; Sigma), 0.004% Bromophenol blue), denatured at 65°C for 10 min and separated for 2 h at 100 V in a 1% agarose gel containing 6.7% formaldehyde and 3.7% MESA. RNA was transferred to a nitrocellulose membrane overnight. The following morning RNA was UV-crosslinked to the membranes. Membranes were pre-hybridized in ExpressHyb (Clontech) for 1 h at 55°C, followed by hybridization for 1 h at 65°C with α32P dATP RadPrime (Invitrogen) labeled probe. Hereafter, membranes were washed three times (30-min/wash) at 55°C in 0.1× saline-sodium citrate (SSC) buffer, 0.1% SDS buffer. Membranes were exposed to a phospho-screen overnight and imaged on a Typhoon 2400 imager. Viral genome and actin mRNA were detected using the following DNA probes targeting different regions within each RNA: ZIKV (nt 10 324–10 808), DENV (nt 10 507–10 526), HCV (nt 1–341) PV (nt 1–753) and actin (nt 685–1171).
Bioinformatics
A bioinformatics approach was employed to find possible relationships between PTM biogenetic enzymes and factors involved in prominent RNA processes. The MODOMICS database (15,16) was employed to identify putative enzymes responsible for each target PTM. In the absence of information on the human version of such enzymes, human homologues were identified by using the BLASTp package provided by the National Center for Biotechnology Information (https://blast.ncbi.nlm.nih.gov/Blast.cgi?PAGE=Proteins). The Cytoscape platform was employed to search numerous relational databases to generate the sought-after interactomics maps (60).
RESULTS AND DISCUSSION
Mining the epitranscriptome during viral infection
An essential first step toward understanding the role of RNA modifications in viral infection is to define the boundaries of the viral epitranscriptome. For this reason, we first examined the landscapes of PTMs of cell lines infected with five different single-stranded positive-sense [ss(+)] RNA viruses (see ‘Materials and Methods’ section). In particular, we determined the change in PTM composition and abundance upon infection with three viruses from the Flaviviridae family, representing two different genera: flavivirus (ZIKV and DENV) and hepacivirus (HCV). To determine whether the changes were unique to Flaviviridae or more ubiquitous to infection with ss(+) RNA viruses, we also examined the PTM profiles following infection with a picornavirus (PV), and a lentivirus (human immunodeficiency virus type 1, HIV-1). We performed a comprehensive investigation of PTMs on total RNA extracted directly from cell lysates, that is without fractionating the RNA into the individual classes of RNAs (see ‘Materials and Methods’ section). More specifically, total RNA was isolated by performing TRIzol extraction, DNase 1 digestion and then ethanol precipitation to eliminate unwanted DNA, as well as any pre-existing mononucleotides and soluble components, which may have been present in the initial extract (49,50). The intact RNA was then hydrolyzed into mononucleotide mixtures with specific exonucleases, and analyzed by direct-infusion nanoflow ESI (nanoESI, 61,62) mass spectrometry (see ‘Materials and Methods’ section). We first assigned PTMs by searching the observed masses against a non-redundant index of all known PTMs, which was generated in-house by combining data present in the RNA modifications (http://mods.rna.albany.edu/) and MODOMICS (http://modomics.genesilico.pl/) databases. We then confirmed the database hits by gas-phase dissociation techniques, such as multistage tandem MS (MSn) (63) and consecutive reaction monitoring (CRM) (64), which provided structure-specific fragmentation signatures (43,46–47 and references therein). We have previously shown that this two-step identification process is capable of identifying all PTMs contained in the database, including isomeric species that differ only by the position of the modification group. This workflow completes both absolute and relative quantifications with typical sensitivity in the low attomol (10−18 mol) range, quantification precision of ±7.1% RSD, and false discovery rate of 5.8% (49,50). The data presented in Table 1 were obtained by averaging the results of at least three biological replicates that were each analyzed five times (technical replicates; see ‘Materials and Methods’ section). The relative abundance of each PTM was calculated in relation to the total abundance of the canonical ribonucleotides (A, C, G and U) in the sample and expressed in percentage form (AvP; see ‘Materials and Methods’ section) (49). The utilization of relative rather than absolute abundances, enabled self-consistent comparisons of PTM expression levels across samples, regardless of possible variations of their total RNA contents (49). A color gradient was utilized to aid data interpretation and examination, whereas the corresponding numerical values are reported in full in Supplementary Table S2. In comparing the relative abundances of an individual PTM detected in matched mock- and virus-infected samples, a different color was assigned only when the variation was statistically significant with a P-value < 0.05 (see ‘Materials and Methods’ section) (49). The full names of the PTMs detected in these experiments were provided in Supplementary Table S3.
The global landscape of mock-infected Huh7 cells consisted of 47 unique PTMs (Table 1), which reflected not only the remarkable diversity of the cellular epitranscriptome, but also the comprehensive nature and sensitivity of the selected analytical platform. When examining the results against the information contained in the RNA Modification and MODOMICS databases, we immediately noted that 12 of the 47 PTMs had not been reported in human cell lines before (marked with * in Table 1). This observation highlights the chronic lack of data on the distribution of PTMs across different organisms and classes of RNAs (50). In most cases, the leading databases cite only the first discovery of the PTM, typically in a certain organism and class of RNA and fail to report subsequent detection in other systems. Moreover, the vast majority of the known PTMs were discovered in isolated tRNA and rRNA, which are abundant sources of modified ribonucleotides, thus overlooking PTMs present on other types of RNAs in the same organism. In contrast, the data displayed in Table 1 were obtained from total RNA samples, not from isolated classes of RNAs. Therefore, the detection of previously unreported PTMs should not be surprising. The most abundant PTM in the sample was pseudouridine, which has been dubbed the fifth nucleotide for its abundance in natural RNA (expressed with a separate grey scale) (65), followed by a variety of singly methylated variants of the four canonical ribonucleotides (Table 1). In our analyses, m6A was just one of the 21 singly-methylated ribonucleotides identified in the sample, indicating that the methylome is significantly broader than is currently appreciated. We also detected variants containing multiple methyl groups suggesting that monomethylated species are not necessarily an end product, but may also serve as intermediates in more complex regulatory pathways involving successive methylation steps.
The analysis of Huh7 cells infected with the various viruses revealed striking differences in the observed PTM landscapes compared not only to the mock-infected cells, but also to one another (Table 1). These differences were substantiated in both the overall number and identity of detected PTMs, with ZIKV, DENV, HCV and PV displaying respectively 44, 41, 40 and 47 total PTMs (Table 1). These differences could not be explained on the basis of possibly different infection rates afforded by the various viruses. In fact, infection with ZIKV, DENV, HCV and PV produced respectively 79, 51, 85 and 39% of infected cells under the selected infection conditions (see ‘Materials and Methods’ section and Supplementary Figure S1). In contrast, the common PTMs observed in such samples did not display equivalent proportions (Supplementary Table S2), thus downplaying possible dilution effects. Furthermore, the approach used to quantify the PTMs provides relative abundances that are self-consistent within each sample and, thus, are not significantly affected by sample-to-sample variations of total RNA. Therefore, the observed landscape variations could be safely ascribed to each virus inducing unique modulation of the cellular response.
A Venn diagram was generated from these data to differentiate unique versus common PTMs observed in mock- and virus-infected Huh7 cells (Figure 1). The diagram was based on the presence/absence of each PTM in the sample, while disregarding any observed variation of expression level. This analysis revealed 35 PTMs common to both mock- and virus-infected samples. Among these 35 common PTMs, a handful displayed similar expression levels, while the majority was either up- or downregulated in the virus-infected samples as compared to mock-infected (Table 1). For example, the relative abundance of monomethylated adenosines, such as m6A, showed either no change or an increase in virus-infected samples compared to the mock-infected control (Table 1). Conversely, 4 unique PTMs were detected in mock-infected cells, which were suppressed instead in all virus-infected samples (indicated in purple in Figure 1: mcm5U, ncm5Um, mcmo5U and mchm5U; full names provided in Supplementary Table S3). At the same time, m44Cm was exclusively detected in DENV; mnm5U was only in HCV; and m5D, hm5C, f5C, nm5U and mcm5s2U were only in PV-infected cells, while no unique PTM was identified in ZIKV-infected cells. Finally, the dimethylcytosine species m5Cm and m44C were consistently observed in ZIKV, DENV, HCV, and PV samples, but conspicuously absent in the mock-infected cells (indicated in light blue in Figure 1). Taken together, these data indicated that PTM landscapes are influenced by both virus-specific and non-specific factors. More significantly, the concerted appearance and disappearance of certain PTMs suggested that well-defined PTM regulatory pathways are operational in the host response to viral infection.
Figure 1.

Venn diagram showing unique and overlapping PTM profiles of total RNA extracts obtained from mock- and virus-infected Huh7 cells. Cultured cells were mock-infected with medium or infected with ZIKV, DENV, HCV and PV, and harvested at specific time points post-infection (see ‘Materials and Methods’ section). Because HIV-1 does not replicate in Huh7 cells, no corresponding data are included in this diagram. Instead, data obtained from HIV-1 grown in 293T cells are provided in Supplementary Table S4 and Figure S5. For the sake of simplicity, the diagram was based exclusively on the presence or absence of each PTM in the sample, whereas comprehensive quantitative data, which may help appreciate any up- and downregulation, are provided separately in Supplementary Table S2. Full PTM names are listed in Supplementary Table S3.
General stress response or virus-specific reaction
Although Huh7 cells infected with the various viruses afforded clearly different PTM profiles, the observation of common features raised the possibility that their expression may be the manifestation of a general cellular response to viral infection. We therefore compared viral infections in different cell lines to assess whether the same expression patterns would be replicated. Specifically, we compared ZIKV infections in Huh7 and HeLa cells; PV infections in Huh7, HeLa and 293T cells; and PV and HIV infections in 293T cells (Supplementary Tables S5-6 and Figures S2-3). Control experiments carried out on mock-infected Huh7, HeLa and 293T cells identified a common set of 31 PTMs, which may constitute a manifestation of the common regulatory/metabolic infrastructure possessed by human cell lines (Supplementary Table S7 and Figure S4). In ZIKV-infected Huh7 and HeLa cells, we identified 2 and 7 new PTMs, respectively (Supplementary Figure S2), whereas m5Cm and m44C were detected in both ZIKV-infected cell lines. Conversely, 5 and 4 PTMs present in mock-infected cells were absent or suppressed in ZIKV-infected Huh7 and HeLa cells respectively (Supplementary Table S5 and Figure S2). We similarly observed an increase or decrease of different sets of PTMs in 293T cells infected with PV or transfected with HIV-1 proviral DNA (Supplementary Figure S5). In the latter case, however, it is worth noting that transfection of HIV-1 proviral DNA does not represent a true infection, but rather the post-integration steps of viral replication, and it is therefore possible that the PTM profiles observed in these experiments may be incomplete. Regardless, these data showed that PTM landscapes were impacted by both the virus and cell line under consideration. The only exceptions were m5Cm and m44C, which were consistently detected in all virus-infected samples (i.e. ZIKV, DENV, HCV, PV and HIV-1), and absent in all mock-infected cell lines.
To discriminate which modifications were linked to a general stress response, rather than viral infection, we next assessed the PTM profiles induced by different stress conditions. Indeed, PTM of RNA is known to play an essential role in the cellular response to oxidative stress through tRNA-based translational recoding (66,67). Moreover, we recently showed that the PTM landscape of yeast is significantly affected by heat shock and osmotic stress through a mechanism that involves regulation of the mitogen-activated protein kinase (MAPK) pathway (50). In this study, however, we focused on conditions associated with the formation of stress granules, a cellular response common to ss(+) RNA virus infection (68). Stress granules are cytoplasmic protein-RNA aggregates that assemble in response to environmental stressors, such as heat, nutrient deprivation and a viral infection. Because replication of ss(+) RNA viruses proceeds via the synthesis of a negative (−) strand, a double-stranded (±) RNA replication intermediate forms and activates protein kinase R (PKR), a kinase that modulates the host innate immune response (69,70). Activated PKR phosphorylates eIF2α and stalls translation, and resulting stalled translation initiation–mRNA complexes, mRNAs and RNA binding proteins are sequestered in stress granules. Double-stranded RNA replication intermediates also initiate the innate immune response to induce synthesis of interferon (IFN) and subsequent expression of interferon stimulated genes (71). We induced stress granule assembly either by treating uninfected Huh7 cells with sodium arsenite, or by transfecting them with the RNA analog polyinosinic-polycytidylic acid [poly(I:C)], which mimics the host response to double-stranded replication intermediates (see ‘Materials and Methods’ section) (72). We monitored the cellular response to arsenite by visualizing the formation of stress granules (data not shown) (56), and evaluated the response to poly(I:C) by measuring the induction of firefly luciferase expression from an IFN-β promoter (Supplementary Figure S6) (73).
The data showed that induction of stress resulted in notable similarities and differences in the RNA modification landscapes from one another, as well as from the virus-infected samples (Table 1; Supplementary Tables S2 and 10). Mock- and ZIKV-infected Huh7 cells, and arsenite- and poly(I:C)-treated Huh7 cells shared 35 common PTMs (Figure 2). Conversely, 5 PTMs consistently appeared in mock-infected cells (i.e. ncm5Um, mcm5U, mcmo5U, mchm5U and i6A in Figure 2) and were absent from the poly(I:C)-treated and ZIKV- infected samples. In the case of arsenite, 12 additional PTMs were detected, which were absent in the poly(I:C) sample. Of these, m5Cm and m44C were in common with the ZIKV-infected sample, while mcm5s2U and f5Um were only identified in arsenite-treated cells. However, in comparing the PTM profiles after infection with the five ss(+) RNA viruses in the study, mcm5s2U was also identified in PV-infections, whereas f5Um was not identified in any viral-infected cells. (Figures 1 and 2). The partial overlap between the arsenite-treated and virus-infected cells is not particularly surprising considering that arsenite is a potent inducer of oxidative stress resulting in mitochondrial damage (74,75), and that viral infections alter mitochondrial structure and function. For example, DENV and ZIKV have been shown to dampen the innate immune response by altering the structure of the mitochondria during infection (76), whereas HCV infection induces oxidative stress and mitophagy (77–82) and HIV may re-localize and damage mitochondria (83,84). Therefore, the incomplete overlap may reflect a common response resulting from mitochondrial damage, as well as separate cellular response pathways that are uniquely activated by oxidative stress or viral infection.
Figure 2.

Venn diagram generated from PTM profiles of total RNA extracts from mock-infected, ZIKV-infected, arsenite-treated and poly(I:C)-transfected Huh7 cells (see ‘Materials and Methods’ section for details). The diagram was based exclusively on the presence or absence of each PTM in the samples, whereas comprehensive quantitative data, which may help appreciate any up- and downregulation, are provided separately in Supplementary Table S10. Full PTM names are listed in Supplementary Table S3.
The Huh7 cells transfected with poly(I:C) displayed 36 PTMs in common with mock- and ZIKV-infected samples, and 35 of these were also observed upon arsenite treatment (Figure 2). The absence of distinguishing PTMs suggests that poly(I:C) transfection may not be capable of accurately mimicking the virus-specific elements that modulate the host response, which lead to detectable variations of the PTM landscapes (Figure 2). The absence of characteristic features that distinguish cellular and viral RNAs from mere RNA homopolymers, such as terminal capping, highly-structured untranslated regions (UTRs) and polyadenylated tails, may contribute to explain the inability of poly(I:C) to elicit the same type of effects on the PTM profiles, as expressed in mock- and ZIKV-infected, and arsenite-treated Huh7 cells, where 7 PTMs were conspicuously absent in the poly(I:C) sample. These 7 PTMs may therefore depend on the presence of these characteristic features of cellular and viral RNAs.
The remarkable diversity of PTMs on viral RNA
The presence of m6A on HIV-1, HCV and ZIKV RNA was recently shown to affect the viral infectious cycles (24–28). To investigate whether other PTMs decorated viral RNAs, we established an affinity capture method to isolate viral RNAs from whole lysates of infected cells and from virions released into extracellular media, which employed biotinylated antisense oligonucleotides (see ‘Materials and Methods’ section). The captured RNA was similarly hydrolyzed into mononucleotides and analyzed according to the described workflow. The results showed that the genomes of all five viruses in the study were decorated with a plethora of different PTMs: 32 in ZIKV, 39 in DENV, 42 in HCV, 41 in PV and 36 in HIV-1 (see Supplementary Tables S8 and 9). A Venn diagram was generated to rationalize these data, which showed a set of 29 common PTMs present in all isolated genomes, as well as additional sets that were either unique for a specific virus, or shared only by select viruses (Figure 3, top panel). Consistent with these findings, the same 29 common PTMs were detected also on HIV-1 viral RNA affinity captured from 293T cultures (see Supplementary Table S8).
Figure 3.

Venn diagrams generated from PTM profiles of viral RNAs isolated from whole cell lysates of infected cells (top) and virions harvested from the culture media (bottom, see ‘Materials and Methods’ section for details). Only the ss(+) RNA viruses grown in Huh7 cells are included in the diagrams to preserve the comparison consistency. Data obtained from isolated HIV-1 in cell lysate and virions are provided separately in Supplementary Table S8. These diagrams were based exclusively on the presence/absence of each PTM, while comprehensive data displaying abundance variations are reported separately in Supplementary Table S9. Full PTM names are listed in Supplementary Table S3.
The corresponding viral RNAs captured from virions in extracellular media provided PTM landscapes that were comparable, but not identical to those obtained from those captured from cell lysates (see Supplementary Tables S8 and 10). In particular, 41 PTMs were detected on ZIKV genomes, 37 on DENV, 40 on HCV, 40 on PV and 33 on HIV-1. The ensuing Venn diagram showed 28 common PTMs on all virion RNAs (Figure 3, bottom panel). These PTMs were also detected in HIV-1 RNA captured from virions in extracellular media of transfected 293T cells (Supplementary Table S8). Smaller sets of PTMs were recognized, which were either partially shared between some of the viruses, or outright virus-specific (represented by dark and light blue cells in Table 2, respectively). For example, viral RNA from ZIKV virions displayed 1 PTM (f5Um) that was also identified on viral RNAs from PV and HIV-1 virions. However, 11 PTMs were specifically identified on the ZIKV RNA from virions, and absent on the viral RNA from cell lysates. That virus-specific PTMs were detected on either the intracellular or virion RNA, or both, but not in mock-infected samples, raises the possibility that such PTMs might facilitate common steps in the viral infectious cycle, such as translation, replication, and virus assembly. Such specificity could warrant the utilization of PTMs as possible diagnostic markers for the respective viral infections.
Table 2. Unique and common PTMs identified on viral RNAs isolated from virions. Dark color blocks show PTMs common to all ss(+) RNA viruses, and light color blocks highlight unique modifications on each respective viral RNA. The table does not show the unique and common PTMs on the viral RNA isolated from cell lysates, nor the abundance of each PTM. Comprehensive abundance data are reported separately in Supplementary Table S9. Full PTM names are listed in Supplementary Table S3.

|
Several modifications found on virion RNA matched those in the total RNA extracts from cells infected with the corresponding virus, but the abundance of the PTMs on the virion RNA was comparatively greater, likely due to the enrichment effects of the capture procedure. In considering these results, it should be noted that no RNA whatsoever was detected in capture samples obtained by exposing mock-infected lysates and corresponding culture media to the virus-specific antisense probes (Supplementary Figure S7). Explained by the absence of viral RNAs in non-infected material, this negative outcome confirmed the inability of the capture procedure to inadvertently pull down cellular RNA through non-specific interactions. In fact, the washing conditions described in ‘Materials and Methods’ section were sufficiently stringent to prevent the recovery of synthetic oligonucleotide standards that were not fully complementary to the probes, as well as limit that of oligonucleotides that were. This consideration is particularly important in light of the presence of abundant rRNA in cell lysates and the ability of virions to package different types of cellular RNAs (85,86).
The diversity and abundance of the PTMs detected in these experiments prompted numerous hypotheses on their functional significance, which could be immediately placed in the context of a steadily growing body of knowledge regarding their roles in RNA process. For example, both ZIKV and DENV encode a viral methyltransferase that adds a m7G cap to the 5′-end of the genome (17–19). However, this PTM was detected not only on ZIKV and DENV isolated genomes, but also on HCV and PV genomes that are known to lack 5′ caps (Supplementary Table S9). It should be noted that terminal m7G modifications are known to facilitate transport of mRNAs out of the nucleus and promote their translation. In contrast, the role of internal m7Gs is not understood, with the exception of guanosine 46 of yeast tRNA, which has been shown capable of modulating structure and promoting additional tRNA modifications (87). The current experimental workflow relies on digesting polymeric RNA into mononucleotide components and, thus, cannot differentiate between terminal and internal m7G modifications. Therefore, additional work aimed at locating the position of m7G on the viral RNA will be necessary to support a full-fledged functional elucidation.
When the abundances of the various 2′-O-methylated nucleotides were compared, we found that 2′-O-methyl-adenosine (Am) was significantly more abundant than the corresponding Gm, Um and Cm counterparts, not only in ZIKV and DENV, but also in HCV and PV genomes (Supplementary Figure S8). While DENV and ZIKV methyltransferase are capable of modifying the guanosine cap, as well as the 2′-O position of adenosine on both viral and cellular RNAs (18), HCV and PV are supposed to lack this methyltransferase activity. Nevertheless, PV genomes isolated from cells contained the highest amount of Am. Recently, the m6A eraser FTO was shown to preferentially convert m6Am to Am (88). While ZIKV, DENV, HCV, PV and HIV-1 RNA genomes contain m6A (Supplementary Table S9 and refs. (24–28)), it remains to be determined whether the presence of Am on viral genomes might be the result of the activity of FTO, or an unknown adenosine-specific 2′-O methyltransferase. To date, the only known bona fide 2′-O methyltransferases in humans methylate the 2′-O position of adenine, guanine, and uridine (88,89). Our data showed that Cm and Um on ZIKV, DENV, HCV and PV intracellular and virion genomes manifested with similar abundances, while Gm was significantly lower (Supplementary Figure S8).
The detection of m6A on all viral RNAs in our study was consistent with recent reports that this PTM decorates the genomes of HIV, HCV, ZIKV and DENV (24–25,27–28). Based on the relative abundance of this PTM in ZIKV RNAs (Supplementary Table S11), we estimated an average overall incidence of ∼11.4 m6A modifications per genome in viral RNAs captured from virions, which compares favorably with the 12 m6As per genome reported for virion-extracted RNA by Lichinchi et al. (28). Similarly, we estimated an incidence of ∼27.6 and ∼13.6 m6As per genome for HCV and DENV RNAs captured from intracellular versus 19 and 11 reported by Gokhale et al. (27). At the same time, we detected ∼10.8 and 5.4 m6A modifications per genome on PV RNA isolated from cell lysates and virions, respectively, for which there are no available data from alternative sources. The agreement between these statistics is particularly remarkable in light of the different types of cells and growth conditions employed between studies, as well as the fundamental difference between the selected analytical approaches: one involving the direct analysis of genuine RNA samples; the other relying on reverse transcription and amplification of RNA that was immunoprecipitated by m6A-specific antibodies (27,28).
A putative role for DDX6 in the expression of m5Cm and m44C during viral infection
In similar fashion, the consistent observation of m5Cm and m44C in cells infected with ZIKV, DENV, HCV and PV, or transfected with HIV-1 provirus, raised a series of questions that spurred the exploration of possible strategies for pursuing their functional investigation. These two PTMs were present on HCV intracellular RNA, and the RNA in ZIKV and HCV virions, but surprisingly absent from the viral genomes of DENV, PV and HIV-1 (Supplementary Tables S8 and 9). Moreover, arsenite treatment stimulated the production of m5Cm and m44C (Supplementary Table S10). We could therefore hypothesize that m5Cm and m44C may be installed on cellular RNAs that modulate the infectious cycles of these viruses. A possible strategy for testing this hypothesis would be to deplete or knock-out the writer enzymes responsible for the installation of m5Cm and m44C, and then examine the effects on the viral lifecycle. However, as is frequently the case in epitranscriptomics research, the identities of their writer, eraser and reader enzymes are still unknown in human cells; however they have been identified in E. coli (90). Based on the excellent homology between the E. coli RsmH and Homo sapiens METTL15 methylating enzymes (91), the latter was employed as input information in a comprehensive bioinformatics search aimed at identifying key nodes of RNA pathways, which could be manipulated to probe the role of dimethylcytosine modifications in viral infection (see ‘Materials and Methods’ section). Among the putative pathways returned by the search, the most promising one identified a possible link between key components of the mRNA decapping complex and m5Cm/m44C biogenesis. The most prominent of such components was the DEAD-box RNA helicase DDX6, a protein that is at the nexus of numerous RNA metabolism pathways. Furthermore, this protein can affect gene expression in different RNA viruses, including HCV and HIV-1 (56,92–93); is a suppressor of abnormal interferon-stimulated gene expression (94); and has been found, perhaps not coincidentally, to localize in the stress granules mentioned above (95).
The possible impact of DDX6 on m5Cm/m44C biogenesis during viral infection was initially explored by using target-specific siRNA to silence its production in Huh7 cells, which were subsequently mock- or virus infected. At specific times post-infection, we examined the effect of depleting DDX6 on viral titers and gene expression, as well as the expression of m5Cm and m44C in virus-infected cells (Figure 4). These experiments showed that m5Cm and m44C were not detected in cells treated with transfection reagent alone (data not shown), in mock-infected cells, in mock-infected cells depleted of DDX6 or following rescue with 3xFlag-DDX6Δsi (Figure 4). In contrast, siRNA-depletion of DDX6 significantly decreased HCV and ZIKV RNA, protein abundance and viral titers, but had little effect on DENV or PV RNA (Figure 4). Moreover, in all virus infections, depletion of DDX6 significantly decreased m5Cm and m44C levels to the point of being undetectable (Figure 4). At the same time, the levels of m5Cm and m44C were almost completely restored when a plasmid encoding a siRNA-resistant 3xFlag-tagged version of DDX6 was transfected into cells depleted of DDX6 (Figure 4). The data showed excellent correlation between the abundance of m5Cm/m44C and that of DDX6, thus suggesting a possible relationship between the biogenesis of m5Cm/m44C and DDX6. In this direction, it should be pointed out that DDX6 possesses intrinsic RNA-binding, ATPase and helicase activities (95), but lacks a methyltransferase domain and, thus, is not likely to play a direct enzymatic role in m5Cm/m44C biogenesis. It was recently shown, however, that the nuclear helicase DDX46 was capable of binding the m6A eraser enzyme ALKBH5 via the helicase domain, which induced demethylation of select interferon stimulated genes in response to vesicular stomatitis virus infection (96). Additionally, the ability of DDX6 to suppress anomalous expression of interferon genes supports an involvement in regulating the innate immune response of the host cell (94). Since DDX6 affects gene expression in both ZIKV and HCV, but not in DENV and PV (Figure 4, (54,56)), it is possible that DDX6 might similarly interact with writer, eraser or reader enzymes to modulate m5Cm/m44C modifications and expression of virus-specific interferon-stimulated genes. Therefore, understanding the relationship between DDX6 and m5Cm/m44C biogenesis could potentially help elucidate how viral RNA eludes innate immune response, which will be the object of future investigation.
Figure 4.

DDX6 modulates the presence of m5Cm and m44C during viral infection. (A) The abundance of m5Cm and m44C are presented above each western and northern blot. These levels show changes in m5Cm and m44C in viral infected cells without arsenite treatment. (B) Western blots show expression of viral protein (ZIKV and DENV capsid, HCV core and PV 3D proteins), DDX6, Flag MS2 and GAPDH in Huh7 cells transfected with control (siGL2) and DDX6-specific (siDDX6) siRNAs, and 3xFlag-Bacterial Alkaline Phosphatase (BAP) or 3xFlag-DDX6 resistant to siRNA targeting (DDX6Δsi). (*) indicates detection of 3xFlag- DDX6Δsi by using the anti-DDX6 antibody. (C) Northern blots show abundance of viral genomic RNA and actin mRNA. (D) Quantification of viral RNA on northern blot. Viral RNA was normalized to actin mRNA, and a percentage relative to infected Huh7 cells transfected with siGL2 is shown. The quantitative data were obtained from at least three independent experiments, and SD of mean is shown. (E) Titers of virions released into the extracellular media. The viral titers of ZIKV and PV were calculated by plaque assay (PFU/ml), and DENV and HCV by TCID50 assay (TCID50/ml). Titers are from at least three independent experiments, and SD of mean is shown. (F) The abundance of m5Cm and m44C in uninfected cells transfected with the respective siRNAs and 3xFlag plasmids, and treated with 1 mM sodium arsenite for 30 min at 37°C. The color gradient provides their abundances relative to the combined abundances of the canonical ribonucleotides, calculated as described in ‘Materials and Methods’ section. Any color change along the gradient carries a P < 0.05 statistical significance. Full PTM names are listed in Supplementary Table S3.
The data in Figure 4 showed a decrease in the abundance of HCV RNA, which was consistent with the recent observation that DDX6 modulates an essential interaction between HCV RNA and the liver-specific miR-122, with significant effects on the stability and replication of the viral genome (54). These data also showed for the first time that DDX6 similarly modulated ZIKV gene expression. In contrast to earlier reports (94,97), we found that depletion of DDX6 did not affect levels of DENV capsid, genomic RNA or viral RNA. Although these discrepancies could be due to different viral replication efficiencies, permissiveness between different cell passages and Huh7 lines (98,99), and efficiency of siRNA knockdown, the observed decrease of m5Cm/m44C in all virus-infected cells clearly supports the ability of DDX6 to modulate the expression of these PTMs’ biogenetic enzymes. The fact that m5Cm and m44C were detected on ZIKV and HCV RNAs isolated from virions, as well as intracellular HCV RNA, but were not observed at all on viral RNAs of DENV, PV and HIV-1 (either virion or intracellular), may be due to the timing of their capture from the initial samples. It is also possible that, at least in DENV and PV, these PTMs might decorate the negative-strand intermediates synthesized during replication. In the case of both ZIKV and HCV, the expression patterns observed for m5Cm and m44C are consistent with the presence of a unique, but common mode of regulation, which is operational during viral infection. Although the current data are not sufficient to draw firm conclusions, the range of possible hypotheses generated by these observations provides strong motivations for a full-fledged-mechanistic investigation.
CONCLUSION
This report capitalized on a very versatile and agnostic analytical approach to complete a comprehensive survey of RNA PTMs present on viral genomes and total cellular RNA obtained from virus-infected cells. The results exposed for the first time the remarkable dimensions and diversity of the viral epitranscriptome, which far exceed the handful of modifications reported thus far (100,101). Despite the well-established effects of some PTMs on RNA structure and their implication in maintaining cellular homeostasis (3,102), PTMs have been often perceived as mere passengers introduced in a stochastic fashion by the cellular machinery, or as non-specific components of general mechanisms of stress response. The fact that we observe considerable differences between PTM landscapes of mock- versus virus-infected cells, as well as between the landscapes associated with five different viruses, negates these perceptions. In determining the distribution of PTMs on cellular versus viral RNAs, as well as intracellular versus virion-packaged genomes, our study establishes the foundation to probe the boundaries of the viral epitranscriptome and traces possible roadmaps for elucidating PTM functions and regulation. Our study highlighted also the need to investigate not only the functions of individual PTMs, but also their uncharted synergies at the systems level.
The interpretation of the wealth of data generated by these experiments poses numerous challenges, as well as opportunities, which must be addressed in the context of the still limited but steadily growing knowledge of the functions of RNA modifications. The identification of common sets of PTMs, which are present not only in mock-, but also in virus-infected cells and isolated viral RNAs, suggests their possible implication in mechanisms by which viral genomes disguise as cellular RNAs to elude cellular surveillance. To this effect, 2′-O methylation of the penultimate nucleotide on cellular RNAs is known to prevent recognition by IFIT1 and to inhibit RNA degradation and turnover (103,104). ZIKV and DENV encode a methyltransferase responsible for introducing this modification on viral RNA (105,106), which helps ensure efficient translation of viral proteins and camouflages viral RNA from the cellular defense mechanisms against foreign RNA. In contrast, HCV and PV lack methyltransferase activity and are known to achieve similar goals through different means. In particular, these two viruses regulate translation via a cap-independent mechanism mediated by an internal ribosome entry site, which prevents IFIT1 detection and activation of the innate immune response pathway (107). In analogy with 2′-O methylation, some or all of the other 22 common PTMs found in virus-infected samples, isolated viral genomes and mock-infected controls could potentially contribute to camouflaging the viral RNA as ‘self’ to protect against the cell's innate immune response. Indeed, the presence of m6A and s2U, and to a lesser extent m5C, pseudouridine, and m5U, decreases the activation of Toll-like receptors (TLRs) by RNA, which leads to a decrease of the host innate immune response (31). Additionally, the presence of m6A and pseudouridine in viral RNA was recently shown to attenuate the activation of the RIG-I innate signaling pathway (33). With the sole exception of s2U, we observed all of these immunity-reducing PTMs during viral infection, thus supporting the possibility that ss(+) RNA viruses take advantage of this evasion mechanism. Because the response to infection by single-stranded positive-sense RNA viruses is largely regulated at the mRNA level (71), future studies will have to address the composition and dynamics of PTMs on cellular mRNAs, which will be expected to provide new insights into the activation and regulation of antiviral response pathways. The ability of viral genomes to acquire the same types of modifications present on cellular RNAs may also facilitate the recognition of the viral RNA by enzymatic complexes involved in RNA processing and trafficking. For example, the presence of m6A on HIV-1 RNA promotes export of the viral RNA out of the nucleus (24). The ability of viruses to mimic the PTM profiles on cellular RNAs may explain the extraordinary efficiency by which viruses usurp the host metabolic machinery. At the same time, however, the identification of virus-specific PTMs supports distinctive virus–host interactions, which do not parallel the regular interactions involving cellular RNAs. Further investigation of these sets of PTMs will be expected to provide new data on the mechanisms of viral adaptation to the host environment and subversion of cellular regulatory pathways.
The PTM landscapes induced by ss(+) RNA viruses and oxidative stress showed unique epitranscriptome fingerprints, as well as the appearance of m5Cm and m44C in both types of samples, thus suggesting specific relationships between the interactome and/or response pathways activated by viral infection or stress. The absence of direct knowledge on m5Cm/m44C biogenesis in humans gave us the opportunity to evaluate a general bioinformatics strategy for identifying possible intersections between viral processes and PTM regulatory pathways. Guided by this information, RNAi depletion of DDX6 revealed that interfering with decapping of cellular mRNA and microRNA gene regulation altered significantly the PTM landscapes, which may in turn affect the translation/stability of specific interferon-induced antiviral mRNAs (108). Considering the potential therapeutic implications, this preliminary examination of the putative DDX6/dimethylcytosine interaction provided a strong rationale for pursuing full-fledged mechanistic studies. At the same time, it also demonstrated the benefits of using the most comprehensive information possible to shape any divide-and-conquer strategy and to prioritize putative targets for follow-up investigations. It has become clear that the breadth of this systematic approach can significantly outpace the current knowledge of cellular factors regulating PTMs. Therefore, identifying the actual writers, erasers, and readers will be essential to sustain the advancement of the new and exciting field of viral epitranscriptomics.
It should be finally emphasized that, at the molecular level, modifications affect the normal binding characteristics of canonical nucleotides, which in turn influence the folding properties of the modified RNA and the ability to recruit cognate nucleic acids and protein factors. The RNA genome of ss(+) RNA viruses represents an essential component in each step of the virus lifecycle, in which each function may be modulated by RNA structure, cis-acting elements, and virus–host interactions (109). As such, RNA modifications have the ability to directly influence the complexion of these RNA structures and interactions. The remarkable diversity of PTMs that we have identified on both cellular and viral RNAs suggests very broad functional ramifications. Investigating the interplay and evaluating the significance of PTMs on viral infection will provide new exciting insights into unexplored epigenetic modes of regulation. Historically, viral systems have made significant contributions toward a greater understanding of fundamental biological functions. We similarly anticipate that the study of viral epitranscriptomics will lead to new far-reaching discoveries about essential cellular processes and disease states.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Brett Lindenbach (Yale University) for ZIKV MR766 Uganda, Richard Kinney and Claire Huang (CDC) for the DENV-2 strain 16681 infectious clone, Takaji Wakita (National Institute of Infectious Diseases, Tokyo) for the HCV JFH-1 infectious clone, Peter Sarnow (Stanford University) for PV Mahonney strain infectious clone, Karla Kirkegaard (Stanford University) for the rabbit anti-PV 3D antibody, Nancy Kedersha (Brigham and Women’s Hospital) for the pRFP-DDX6 plasmid, Michael Gale (University of Washington) for the pIFN β-Firefly luciferase reporter plasmid and Stacy Horner (Duke University) for helpful discussions on transfection of poly (I:C) and the IFN-β luciferase reporter. We also thank Marlene Belfort, Rebecca Dutch and Peter Sarnow for valuable comments and suggestions on the manuscript.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
University at Albany-SUNY and New York State Start-up Funds (to C.T.P.); Presidential Initiatives Fund for Research and Scholarship (PIFRS) (to C.T.P.); American Association for the Study of Liver Diseases (to C.T.P.); National Institute of Allergy and Infectious Diseases (NIAID) [R21 AI133617–01 to C.T.P., D.F.]; National Institute of General Medical Sciences [R01 GM121844–01 to D.F.]. Funding for open access charge: NIAID [R21 AI133617-01].
Conflict of interest statement. None declared.
REFERENCES
- 1. Shafik A., Schumann U., Evers M., Sibbritt T., Preiss T.. The emerging epitranscriptomics of long noncoding RNAs. Biochim. Biophys. Acta. 2015; 1859:59–70. [DOI] [PubMed] [Google Scholar]
- 2. Chen K., Zhao B.S., He C.. Nucleic acid modifications in regulation of gene expression. Cell Chem. Biol. 2016; 23:74–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Zhao B.S., Roundtree I.A., He C.. Post-transcriptional gene regulation by mRNA modifications. Nat. Rev. Mol. Cell Biol. 2017; 18:31–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Limbach P.A., Crain P.F., McCloskey J.A.. Summary: the modified nucleosides of RNA. Nucleic Acids Res. 1994; 22:2183–2196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Cantara W.A., Crain P.F., Rozenski J., McCloskey J.A., Harris K.A., Zhang X., Vendeix F.A.P., Fabris D., Agris P.F.. The RNA Modification Database: 2011 Update. Nucleic Acids Res. 2011; 39:D195–D201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Chang K.Y., Varani G.. Nucleic acids structure and recognition. Nat. Struct. Biol. 1997; 4(Suppl):854–858. [PubMed] [Google Scholar]
- 7. Carell T., Brandmayr C., Hienzsch A., Müller M., Pearson D., Reiter V., Thoma I., Thumbs P., Wagner M.. Structure and function of noncanonical nucleobases. Angew. Chem. Int. Ed Engl. 2012; 51:7110–7131. [DOI] [PubMed] [Google Scholar]
- 8. Kowalak J.A., Dalluge J.J., McCloskey J.A., Stetter K.O.. The role of posttranscriptional modification in stabilization of transfer RNA from hyperthermophiles. Biochemistry (Mosc.). 1994; 33:7869–7876. [DOI] [PubMed] [Google Scholar]
- 9. Helm M. Post-transcriptional nucleotide modification and alternative folding of RNA. Nucleic Acids Res. 2006; 34:721–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Gutgsell N.S., Del Campo M., Raychaudhuri S., Ofengand J.. A second function for pseudouridine synthases: a point mutant of RluD unable to form pseudouridines 1911, 1915, and 1917 in Escherichia coli 23S ribosomal RNA restores normal growth to an RluD-minus strain. RNA. 2001; 7:990–998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. King T.H., Liu B., McCully R.R., Fournier M.J.. Ribosome structure and activity are altered in cells lacking snoRNPs that form pseudouridines in the peptidyl transferase center. Mol. Cell. 2003; 11:425–435. [DOI] [PubMed] [Google Scholar]
- 12. Giegé R., Sissler M., Florentz C.. Universal rules and idiosyncratic features in tRNA identity. Nucleic Acids Res. 1998; 26:5017–5035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Grosjean H. Fine-Tuning of RNA Functions by Modification and Editing. Topics in Current Genetics. 2005; 12:Berlin: Springer. [Google Scholar]
- 14. Li J., Yang Z., Yu B., Liu J., Chen X.. Methylation protects miRNAs and siRNAs from a 3′-end uridylation activity in Arabidopsis. Curr. Biol. 2005; 15:1501–1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Dunin-Horkawicz S., Czerwoniec A., Gajda M.J., Feder M., Grosjean H., Bujnicki J.M.. MODOMICS: a database of RNA modification pathways. Nucleic Acids Res. 2006; 34:D145–D149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Machnicka M.A., Milanowska K., Osman Oglou O., Purta E., Kurkowska M., Olchowik A., Januszewski W., Kalinowski S., Dunin-Horkawicz S., Rother K.M. et al. MODOMICS: a database of RNA modification pathways–2013 update. Nucleic Acids Res. 2012; 41:D262–D267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Bisaillon M., Lemay G.. Viral and cellular enzymes involved in synthesis of mRNA cap structure. Virology. 1997; 236:1–7. [DOI] [PubMed] [Google Scholar]
- 18. Dong H., Chang D.C., Hua M.H.C., Lim S.P., Chionh Y.H., Hia F., Lee Y.H., Kukkaro P., Lok S.-M., Dedon P.C. et al. 2′-O methylation of internal adenosine by flavivirus NS5 methyltransferase. PLoS Pathog. 2012; 8:e1002642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Dong H., Fink K., Züst R., Lim S.P., Qin C.-F., Shi P.-Y.. Flavivirus RNA methylation. J. Gen. Virol. 2014; 95:763–778. [DOI] [PubMed] [Google Scholar]
- 20. Canaani D., Kahana C., Lavi S., Groner Y.. Identification and mapping of N6-methyladenosine containing sequences in simian virus 40 RNA. Nucleic Acids Res. 1979; 6:2879–2899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Krug R.M., Morgan M.A., Shatkin A.J.. Influenza viral mRNA contains internal N6-methyladenosine and 5′-terminal 7-methylguanosine in cap structures. J. Virol. 1976; 20:45–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Narayan P., Ayers D.F., Rottman F.M., Maroney P.A., Nilsen T.W.. Unequal distribution of N6-methyladenosine in influenza virus mRNAs. Mol. Cell. Biol. 1987; 7:1572–1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kane S.E., Beemon K.. Precise localization of m6A in Rous sarcoma virus RNA reveals clustering of methylation sites: implications for RNA processing. Mol. Cell. Biol. 1985; 5:2298–2306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lichinchi G., Gao S., Saletore Y., Gonzalez G.M., Bansal V., Wang Y., Mason C.E., Rana T.M.. Dynamics of the human and viral m6A RNA methylomes during HIV-1 infection of T cells. Nat. Microbiol. 2016; 1:16011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kennedy E.M., Bogerd H.P., Kornepati A.V.R., Kang D., Ghoshal D., Marshall J.B., Poling B.C., Tsai K., Gokhale N.S., Horner S.M. et al. Posttranscriptional m(6)A editing of HIV-1 mRNAs enhances viral gene expression. Cell Host Microbe. 2016; 19:675–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Tirumuru N., Zhao B.S., Lu W., Lu Z., He C., Wu L.. N(6)-methyladenosine of HIV-1 RNA regulates viral infection and HIV-1 Gag protein expression. Elife. 2016; 5:e15528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Gokhale N.S., McIntyre A.B.R., McFadden M.J., Roder A.E., Kennedy E.M., Gandara J.A., Hopcraft S.E., Quicke K.M., Vazquez C., Willer J. et al. N6-Methyladenosine in flaviviridae viral RNA genomes regulates infection. Cell Host Microbe. 2016; 20:654–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lichinchi G., Zhao B.S., Wu Y., Lu Z., Qin Y., He C., Rana T.M.. Dynamics of human and viral RNA methylation during Zika virus infection. Cell Host Microbe. 2016; 20:666–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Bouvet M., Debarnot C., Imbert I., Selisko B., Snijder E.J., Canard B., Decroly E.. In vitro reconstitution of SARS-coronavirus mRNA cap methylation. PLoS Pathog. 2010; 6:e1000863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ma Y., Wei Y., Zhang X., Zhang Y., Cai H., Zhu Y., Shilo K., Oglesbee M., Krakowka S., Whelan S.P.J. et al. mRNA cap methylation influences pathogenesis of vesicular stomatitis virus in vivo. J. Virol. 2014; 88:2913–2926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Karikó K., Buckstein M., Ni H., Weissman D.. Suppression of RNA recognition by Toll-like receptors: the impact of nucleoside modification and the evolutionary origin of RNA. Immunity. 2005; 23:165–175. [DOI] [PubMed] [Google Scholar]
- 32. Zhu J., Gopinath K., Murali A., Yi G., Hayward S.D., Zhu H., Kao C.. RNA-binding proteins that inhibit RNA virus infection. Proc. Natl. Acad. Sci. U.S.A. 2007; 104:3129–3134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Durbin A.F., Wang C., Marcotrigiano J., Gehrke L.. RNAs containing modified nucleotides fail to trigger RIG-I conformational changes for innate immune signaling. Mbio. 2016; 7:e00833–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Dominissini D., Moshitch-Moshkovitz S., Schwartz S., Salmon-Divon M., Ungar L., Osenberg S., Cesarkas K., Jacob-Hirsch J., Amariglio N., Kupiec M. et al. Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature. 2012; 485:201–206. [DOI] [PubMed] [Google Scholar]
- 35. Dominissini D., Moshitch-Moshkovitz S., Salmon-Divon M., Amariglio N., Rechavi G.. Transcriptome-wide mapping of N(6)-methyladenosine by m(6)A-seq based on immunocapturing and massively parallel sequencing. Nat. Protoc. 2013; 8:176–189. [DOI] [PubMed] [Google Scholar]
- 36. Meyer K.D., Saletore Y., Zumbo P., Elemento O., Mason C.E., Jaffrey S.R.. Comprehensive analysis of mRNA methylation reveals enrichment in 3′ UTRs and near stop codons. Cell. 2012; 149:1635–1646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Dominissini D., Nachtergaele S., Moshitch-Moshkovitz S., Peer E., Kol N., Ben-Haim M.S., Dai Q., Di Segni A., Salmon-Divon M., Clark W.C. et al. The dynamic N(1)-methyladenosine methylome in eukaryotic messenger RNA. Nature. 2016; 530:441–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Schaefer M., Pollex T., Hanna K., Lyko F.. RNA cytosine methylation analysis by bisulfite sequencing. Nucleic Acids Res. 2009; 37:e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Jackman J.E., Montange R.K., Malik H.S., Phizicky E.M.. Identification of the yeast gene encoding the tRNA m1G methyltransferase responsible for modification at position 9. RNA. 2003; 9:574–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ansmant I., Motorin Y., Massenet S., Grosjean H., Branlant C.. Identification and characterization of the tRNA:Psi 31-synthase (Pus6p) of Saccharomyces cerevisiae. J. Biol. Chem. 2001; 276:34934–34940. [DOI] [PubMed] [Google Scholar]
- 41. Dong Z.-W., Shao P., Diao L.-T., Zhou H., Yu C.-H., Qu L.-H.. RTL-P: a sensitive approach for detecting sites of 2′-O-methylation in RNA molecules. Nucleic Acids Res. 2012; 40:e157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Bakin A.V., Ofengand J.. Mapping of pseudouridine residues in RNA to nucleotide resolution. Methods Mol. Biol. 1998; 77:297–309. [DOI] [PubMed] [Google Scholar]
- 43. McCloskey J.A. Characterization of nucleosides by mass spectrometry. Nucleic Acids Symp. Ser. 1979; 6:s109–s113. [DOI] [PubMed] [Google Scholar]
- 44. Crain P.F. Preparation and enzymatic hydrolysis of DNA and RNA for mass spectrometry. Methods Enzymol. 1990; 193:782–790. [DOI] [PubMed] [Google Scholar]
- 45. Crain P.F., McCloskey J.A.. Applications of mass spectrometry to the characterization of oligonucleotides and nucleic acids. Curr. Opin. Biotechnol. 1998; 9:25–34. [DOI] [PubMed] [Google Scholar]
- 46. Limbach P.A. Indirect mass spectrometric methods for characterizing and sequencing oligonucleotides. Mass Spectrom. Rev. 1996; 15:297–336. [DOI] [PubMed] [Google Scholar]
- 47. Nordhoff E., Kirpekar F., Roepstorff P.. Mass spectrometry of nucleic acids. Mass Spectrom. Rev. 1996; 15:67–138. [DOI] [PubMed] [Google Scholar]
- 48. Fabris D. MS analysis of nucleic acids in the post-genomic era. Anal. Chem. 2011; 83:5810–5816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Rose R.E., Quinn R., Sayre J.L., Fabris D.. Profiling ribonucleotide modifications at full-transcriptome level by electrospray ionization mass spectrometry. RNA. 2015; 21:1361–1374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Rose R.E., Pazos M.A., Curcio M.J., Fabris D.. Global profiling of RNA post-transcriptional modifications as an effective tool for investigating the epitranscriptomics of stress response. Mol. Cell. Proteomics. 2016; 15:932–944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Kinney R.M., Butrapet S., Chang G.J., Tsuchiya K.R., Roehrig J.T., Bhamarapravati N., Gubler D.J.. Construction of infectious cDNA clones for dengue 2 virus: strain 16681 and its attenuated vaccine derivative, strain PDK-53. Virology. 1997; 230:300–308. [DOI] [PubMed] [Google Scholar]
- 52. Wakita T., Pietschmann T., Kato T., Date T., Miyamoto M., Zhao Z., Murthy K., Habermann A., Kräusslich H.-G., Mizokami M. et al. Production of infectious hepatitis C virus in tissue culture from a cloned viral genome. Nat. Med. 2005; 11:791–796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Park I.-W., Fan Y., Luo X., Ryou M.-G., Liu J., Green L., He J.J.. HIV-1 Nef is transferred from expressing T cells to hepatocytic cells through conduits and enhances HCV replication. PLoS One. 2014; 9:e99545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Biegel J.M., Henderson E., Cox E.M., Bonenfant G., Netzband R., Kahn S., Eager R., Pager C.T.. Cellular DEAD-box RNA helicase DDX6 modulates interaction of miR-122 with the 5′ untranslated region of hepatitis C virus RNA. Virology. 2017; 507:231–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Ding Y., Chan C.Y., Lawrence C.E.. Sfold web server for statistical folding and rational design of nucleic acids. Nucleic Acids Res. 2004; 32:W135–W141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Pager C.T., Schütz S., Abraham T.M., Luo G., Sarnow P.. Modulation of hepatitis C virus RNA abundance and virus release by dispersion of processing bodies and enrichment of stress granules. Virology. 2013; 435:472–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Lindenbach B.D., Evans M.J., Syder A.J., Wölk B., Tellinghuisen T.L., Liu C.C., Maruyama T., Hynes R.O., Burton D.R., McKeating J.A. et al. Complete replication of hepatitis C virus in cell culture. Science. 2005; 309:623–626. [DOI] [PubMed] [Google Scholar]
- 58. Lindenbach B.D. Measuring HCV infectivity produced in cell culture and in vivo. Methods Mol. Biol. Clifton N.J. 2009; 510:329–336. [DOI] [PubMed] [Google Scholar]
- 59. Kirkegaard K., Baltimore D.. The mechanism of RNA recombination in poliovirus. Cell. 1986; 47:433–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Cline M.S., Smoot M., Cerami E., Kuchinsky A., Landys N., Workman C., Christmas R., Avila-Campilo I., Creech M., Gross B. et al. Integration of biological networks and gene expression data using Cytoscape. Nat. Protoc. 2007; 2:2366–2382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Yamashita M., Fenn J.B.. Electrospray ion source. Another variation on the free-jet theme. J. Phys. Chem. 1984; 88:4671–4675. [Google Scholar]
- 62. Wilm M., Mann M.. Analytical properties of the nanoelectrospray ion source. Anal. Chem. 1996; 68:1–8. [DOI] [PubMed] [Google Scholar]
- 63. Collings B.A., Campbell J.M., Mao D., Douglas D.J.. A combined linear ion trap time-of-flight system with improved performance and MSn capabilities. Rapid Commun. Mass Spectrom. 2001; 15:1777–1795. [DOI] [PubMed] [Google Scholar]
- 64. Tomer K.B., Guenat C.R., Deterding L.J.. Consecutive reaction monitoring in a four-sector mass spectrometer: MS4 and one step beyond. Anal. Chem. 1988; 60:2232–2236. [DOI] [PubMed] [Google Scholar]
- 65. Davis F.F., Allen F.W.. Ribonucleic acids from yeast which contain a fifth nucleotide. J. Biol. Chem. 1957; 227:907–915. [PubMed] [Google Scholar]
- 66. Chan C.T.Y., Pang Y.L.J., Deng W., Babu I.R., Dyavaiah M., Begley T.J., Dedon P.C.. Reprogramming of tRNA modifications controls the oxidative stress response by codon-biased translation of proteins. Nat. Commun. 2012; 3:937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Endres L., Dedon P.C., Begley T.J.. Codon-biased translation can be regulated by wobble-base tRNA modification systems during cellular stress responses. RNA Biol. 2015; 12:603–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Reineke L.C., Lloyd R.E.. Diversion of stress granules and P-bodies during viral infection. Virology. 2013; 436:255–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Weber F., Wagner V., Rasmussen S.B., Hartmann R., Paludan S.R.. Double-stranded RNA is produced by positive-strand RNA viruses and DNA viruses but not in detectable amounts by negative-strand RNA viruses. J. Virol. 2006; 80:5059–5064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Williams B.R. PKR; a sentinel kinase for cellular stress. Oncogene. 1999; 18:6112–6120. [DOI] [PubMed] [Google Scholar]
- 71. Beachboard D.C., Horner S.M.. Innate immune evasion strategies of DNA and RNA viruses. Curr. Opin. Microbiol. 2016; 32:113–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Li K., Chen Z., Kato N., Gale M., Lemon S.M.. Distinct poly(I-C) and virus-activated signaling pathways leading to interferon-beta production in hepatocytes. J. Biol. Chem. 2005; 280:16739–16747. [DOI] [PubMed] [Google Scholar]
- 73. Foy E., Li K., Sumpter R., Loo Y.-M., Johnson C.L., Wang C., Fish P.M., Yoneyama M., Fujita T., Lemon S.M. et al. Control of antiviral defenses through hepatitis C virus disruption of retinoic acid-inducible gene-I signaling. Proc. Natl. Acad. Sci. U.S.A. 2005; 102:2986–2991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. McEwen E., Kedersha N., Song B., Scheuner D., Gilks N., Han A., Chen J.-J., Anderson P., Kaufman R.J.. Heme-regulated inhibitor kinase-mediated phosphorylation of eukaryotic translation initiation factor 2 inhibits translation, induces stress granule formation, and mediates survival upon arsenite exposure. J. Biol. Chem. 2005; 280:16925–16933. [DOI] [PubMed] [Google Scholar]
- 75. Yih L.H., Huang H.M., Jan K.Y., Lee T.C.. Sodium arsenite induces ATP depletion and mitochondrial damage in HeLa cells. Cell Biol. Int. Rep. 1991; 15:253–264. [DOI] [PubMed] [Google Scholar]
- 76. Chatel-Chaix L., Cortese M., Romero-Brey I., Bender S., Neufeldt C.J., Fischl W., Scaturro P., Schieber N., Schwab Y., Fischer B. et al. Dengue virus perturbs mitochondrial morphodynamics to dampen innate immune responses. Cell Host Microbe. 2016; 20:342–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Gong G., Waris G., Tanveer R., Siddiqui A.. Human hepatitis C virus NS5A protein alters intracellular calcium levels, induces oxidative stress, and activates STAT-3 and NF-kappa B. Proc. Natl. Acad. Sci. U.S.A. 2001; 98:9599–9604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Waris G., Siddiqui A.. Hepatitis C virus stimulates the expression of cyclooxygenase-2 via oxidative stress: role of prostaglandin E2 in RNA replication. J. Virol. 2005; 79:9725–9734. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 79. Waris G., Turkson J., Hassanein T., Siddiqui A.. Hepatitis C virus (HCV) constitutively activates STAT-3 via oxidative stress: role of STAT-3 in HCV replication. J. Virol. 2005; 79:1569–1580. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 80. Piccoli C., Scrima R., Quarato G., D’Aprile A., Ripoli M., Lecce L., Boffoli D., Moradpour D., Capitanio N.. Hepatitis C virus protein expression causes calcium-mediated mitochondrial bioenergetic dysfunction and nitro-oxidative stress. Hepatology. 2007; 46:58–65. [DOI] [PubMed] [Google Scholar]
- 81. Prensner J.R., Iyer M.K., Balbin O.A., Dhanasekaran S.M., Cao Q., Brenner J.C., Laxman B., Asangani I.A., Grasso C.S., Kominsky H.D. et al. Transcriptome sequencing across a prostate cancer cohort identifies PCAT-1, an unannotated lincRNA implicated in disease progression. Nat. Biotechnol. 2011; 29:742–749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Kim S.-J., Syed G.H., Siddiqui A.. Hepatitis C virus induces the mitochondrial translocation of Parkin and subsequent mitophagy. PLoS Pathog. 2013; 9:e1003285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Huang C.-Y., Chiang S.-F., Lin T.-Y., Chiou S.-H., Chow K.-C.. HIV-1 Vpr triggers mitochondrial destruction by impairing Mfn2-mediated ER-mitochondria interaction. PLoS One. 2012; 7:e33657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Groppelli E., Starling S., Jolly C.. Contact-induced mitochondrial polarization supports HIV-1 virological synapse formation. J. Virol. 2015; 89:14–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Kleiman L., Cen S.. The tRNALys packaging complex in HIV-1. Int. J. Biochem. Cell Biol. 2004; 36:1776–1786. [DOI] [PubMed] [Google Scholar]
- 86. Onafuwa-Nuga A.A., Telesnitsky A., King S.R.. 7SL RNA, but not the 54-kd signal recognition particle protein, is an abundant component of both infectious HIV-1 and minimal virus-like particles. RNA. 2006; 12:542–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Hori H. Methylated nucleosides in tRNA and tRNA methyltransferases. Front. Genet. 2014; 5:144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Mauer J., Luo X., Blanjoie A., Jiao X., Grozhik A.V., Patil D.P., Linder B., Pickering B.F., Vasseur J.-J., Chen Q. et al. Reversible methylation of m6Am in the 5′ cap controls mRNA stability. Nature. 2017; 541:371–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Lee K.-W., Bogenhagen D.F.. Assignment of 2′-O-methyltransferases to modification sites on the mammalian mitochondrial large subunit 16 S ribosomal RNA (rRNA). J. Biol. Chem. 2014; 289:24936–24942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Wei Y., Zhang H., Gao Z.-Q., Wang W.-J., Shtykova E. V., Xu J.-H., Liu Q.-S., Dong Y.-H.. Crystal and solution structures of methyltransferase RsmH provide basis for methylation of C1402 in 16S rRNA. J. Struct. Biol. 2012; 179:29–40. [DOI] [PubMed] [Google Scholar]
- 91. Kimura S., Suzuki T.. Fine-tuning of the ribosomal decoding center by conserved methyl-modifications in the Escherichia coli 16S rRNA. Nucleic Acids Res. 2010; 38:1341–1352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Reed J.C., Molter B., Geary C.D., McNevin J., McElrath J., Giri S., Klein K.C., Lingappa J.R.. HIV-1 Gag co-opts a cellular complex containing DDX6, a helicase that facilitates capsid assembly. J. Cell Biol. 2012; 198:439–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Yasuda-Inoue M., Kuroki M., Ariumi Y.. Distinct DDX DEAD-box RNA helicases cooperate to modulate the HIV-1 Rev function. Biochem. Biophys. Res. Commun. 2013; 434:803–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Lumb J.H., Li Q., Popov L.M., Ding S., Keith M.T., Merrill B.D., Greenberg H.B., Li J.B., Carette J.E.. DDX6 represses aberrant activation of interferon-stimulated genes. Cell Rep. 2017; 20:819–831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Presnyak V., Coller J.. The DHH1/RCKp54 family of helicases: an ancient family of proteins that promote translational silencing. Biochim. Biophys. Acta. 2013; 1829:817–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Zheng Q., Hou J., Zhou Y., Li Z., Cao X.. The RNA helicase DDX46 inhibits innate immunity by entrapping m6A-demethylated antiviral transcripts in the nucleus. Nat. Immunol. 2017; 18:1094–1103. [DOI] [PubMed] [Google Scholar]
- 97. Ward A.M., Bidet K., Yinglin A., Ler S.G., Hogue K., Blackstock W., Gunaratne J., Garcia-Blanco M.A.. Quantitative mass spectrometry of DENV-2 RNA-interacting proteins reveals that the DEAD-box RNA helicase DDX6 binds the DB1 and DB2 3′ UTR structures. RNA Biol. 2011; 8:1173–1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Lohmann V., Körner F., Dobierzewska A., Bartenschlager R.. Mutations in hepatitis C virus RNAs conferring cell culture adaptation. J. Virol. 2001; 75:1437–1449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Sainz B., Barretto N., Uprichard S.L.. Hepatitis C virus infection in phenotypically distinct Huh7 cell lines. PLoS One. 2009; 4:e6561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Gokhale N.S., Horner S.M.. RNA modifications go viral. PLoS Pathog. 2017; 13:e1006188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Gonzales-van Horn S.R., Sarnow P.. Making the mark: the role of adenosine modifications in the life cycle of RNA viruses. Cell Host Microbe. 2017; 21:661–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Gilbert W.V., Bell T.A., Schaening C.. Messenger RNA modifications: form, distribution, and function. Science. 2016; 352:1408–1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Daffis S., Szretter K.J., Schriewer J., Li J., Youn S., Errett J., Lin T.-Y., Schneller S., Zust R., Dong H. et al. 2′-O methylation of the viral mRNA cap evades host restriction by IFIT family members. Nature. 2010; 468:452–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Kimura T., Katoh H., Kayama H., Saiga H., Okuyama M., Okamoto T., Umemoto E., Matsuura Y., Yamamoto M., Takeda K.. Ifit1 inhibits Japanese encephalitis virus replication through binding to 5′ capped 2′-O unmethylated RNA. J. Virol. 2013; 87:9997–10003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Dong H., Ray D., Ren S., Zhang B., Puig-Basagoiti F., Takagi Y., Ho C.K., Li H., Shi P.-Y.. Distinct RNA elements confer specificity to flavivirus RNA cap methylation events. J. Virol. 2007; 81:4412–4421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Zhou Y., Ray D., Zhao Y., Dong H., Ren S., Li Z., Guo Y., Bernard K.A., Shi P.-Y., Li H.. Structure and function of flavivirus NS5 methyltransferase. J. Virol. 2007; 81:3891–3903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Hyde J.L., Diamond M.S.. Innate immune restriction and antagonism of viral RNA lacking 2′-O methylation. Virology. 2015; 479–480:66–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Li M.M.H., MacDonald M.R., Rice C.M.. To translate, or not to translate: viral and host mRNA regulation by interferon-stimulated genes. Trends Cell Biol. 2015; 25:320–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Iglesias N.G., Gamarnik A.V.. Dynamic RNA structures in the dengue virus genome. RNA Biol. 2011; 8:249–257. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.