


Abstract
Translocation of RNA polymerase (RNAP) along DNA may be rate-limiting for transcription elongation. The Brownian ratchet model posits that RNAP rapidly translocates back and forth until the post-translocated state is stabilized by NTP binding. An alternative model suggests that RNAP translocation is slow and poorly reversible. To distinguish between these two models, we take advantage of an observation that pyrophosphorolysis rates directly correlate with the abundance of the pre-translocated fraction. Pyrophosphorolysis by RNAP stabilized in the pre-translocated state by bacteriophage HK022 protein Nun was used as a reference point to determine the pre-translocated fraction in the absence of Nun. The stalled RNAP preferentially occupies the post-translocated state. The forward translocation rate depends, among other factors, on melting of the RNA–DNA base pair at the upstream edge of the transcription bubble. DNA–DNA base pairing immediately upstream from the RNA–DNA hybrid stabilizes the post-translocated state. This mechanism is conserved between E. coli RNAP and S. cerevisiae RNA polymerase II and is partially dependent on the lid domain of the catalytic subunit. Thus, the RNA–DNA hybrid and DNA reannealing at the upstream edge of the transcription bubble emerge as targets for regulation of the transcription elongation rate.
INTRODUCTION
Translocation is the one-base-pair (bp) movement of RNA polymerase (RNAP) along the DNA template between formation of two consecutive phosphodiester bonds (1). Translocation leaves a vacant active center at the RNA 3′ end to allow binding of the next NTP substrate (2). It has been suggested that faster translocation decreases transcription fidelity (3). Translocation is accompanied by conformational changes in the bridge helix and the trigger loop, the catalytic subunit domains located close to the RNAP active site (2,4,5). RNA–DNA hybridization at the upstream end of the RNA–DNA hybrid has been implicated in regulation of Thermus thermophilis RNAP translocation (6).
A ternary elongation complex (TEC) consists of RNAP core enzyme, nascent RNA and double-stranded DNA encompassing the transcription bubble. The TEC is stabilized by the RNA–DNA hybrid formed between template strand of the bubble and 3′-proximal 9-10 bases of the RNA (7). An extensive structural and biochemical analysis of TECs stalled on DNA by NTP deprivation or assembled on synthetic RNA–DNA scaffolds showed that the bubble starts 1–2 nucleotides beyond the 3′ end of the RNA with the two DNA strands reannealing immediately after the last base pair of the RNA–DNA hybrid (8). This structure is shared by bacterial and eukaryotic RNAPs. NMP incorporation by the post-translocated TEC extends the hybrid to 10 base pairs at the RNA 3′ end. Forward translocation restores the original 9 base pair hybrid length by melting one RNA–DNA base pair at the opposite end of the hybrid. In this scenario, the 9-bp and 10-bp RNA–DNA hybrids are characteristic for the post-translocated and pre-translocated TECs. Translocation also involves melting and reforming of a single DNA base pair at the downstream and upstream ends of the transcription bubble, respectively. Melting of the RNA–DNA hybrid and the downstream DNA occurs shortly after the phosphodiester bond formation, and has been proposed to be coordinated with RNAP translocation (9,10). The dynamics of the upstream dsDNA reannealing remains less clear.
Translocation represents a target for transcription regulation at the post-initiation level (1). Backward translocation is the key step in the development of the pauses caused by backtracking of RNAP (11). The strong hairpin-dependent regulatory pauses in the leader sequences of the his and trp biosynthetic operons in Escherichia coli (12) were long believed to be caused by the improper alignment of the 3′ RNA base with its complementary DNA base in the i+1 site. The misalignment prevents translocation of this RNA–DNA base pair to the more spatially constrained i site (reviewed in (10)). The most recent cryo-EM structures of the TECs paused in a hairpin-dependent manner suggest that pausing is caused by the asymmetric (13), partial (14), or asynchronous (15) translocation of RNAP, when the RNA 3′ end occupies a post-translocated state, but the i+1 DNA base fails to translocate and is not available for base pairing with the next incoming NTP.
Although some strong sequence-specific pauses of E. coli RNAP do not appear to involve a translocation block (16), slow translocation at intrinsic signals in DNA is often implicated in RNAP pausing (17). The sequence Logo identified for pause sites of E. coli and B. subtilis RNAPs determined by NET-seq reveals two separate sequence elements involved in transcription pausing in vivo. First, pausing appears to depend on the enhanced interaction of RNAP with a single DNA base in the non-transcribed DNA strand at the leading edge of transcription bubble. This base is located just beyond the active center of RNAP. Second, pausing appears to involve an interaction of RNAP with the RNA–DNA base pair (or the non-template DNA base) 10-bp from the RNA 3′ end. It has been proposed that interaction of RNAP with the RNA and/or DNA bases (typically with guanines) at both sites causes a problem with forward translocation of the enzyme along DNA (18,19). Although the role of these interactions has been firmly established for a transcription pause site, it is not clear if they also limit forward translocation during pause-free transcription where the elongation rate may reach up to a hundred nucleotides per second (17,18). Although slow translocation contributes to sequence-specific pauses over a broad range of dwell times, little is known about whether translocation also limits the >10 s−1 NMP incorporation rate characteristic for pause-free transcription (20,21).
The Brownian ratchet model of translocation suggests that, for the vast majority of template positions, translocation is rapid and reversible. This reversible translocation enables RNAP to equilibrate between the post-translocated and pre-translocated states prior to NTP binding (22,23). This model appears consistent with some, but not all in vitro observations (24). It seemed consistent in that multiple mutations in RNAP subunits induce changes in the RNAP footprints on the DNA, suggesting shifts in translocation equilibrium (25–27). Similar changes may be caused by specific sequences in the template near the active site or 9–10 bp upstream from the active site (16,28–30). Also, shifts in RNAP translocation equilibrium often correlate with the changes in NMP incorporation and pyrophosphorolysis, the catalytic activities uniquely specific to the post-translocated and the pre-translocated states, respectively (25,31,32). Pyrophosphorolysis was previously used to assess the translocation equilibrium of the TECs formed by different RNAPs (6,17,30,33).
However, the Brownian ratchet model of translocation did not explain well the results of the pre-steady-state analyses of transcription elongation, which were more consistent with a slow and poorly reversible translocation (10,24,26,34). It does not explain the properties of the rpb1-E1103G mutation in Saccharomyces cerevisiae RNA polymerase II (Pol II) that, according to footprinting results, stabilizes the pre-translocated state, but, paradoxically, promotes NTP binding, increases transcription rate, and decreases fidelity of NTP selection (25). More recent single-molecule analyses of transcription elongation also best fit a kinetic model with a slower translocation that is partially rate-limiting for the pause-free transcription by wild-type Pol II and fully rate-limiting for the rpb1-E1103G mutant Pol II (35). Finally, dynamics of the RNA–DNA base pairing at the rear edge of the transcription bubble and the DNA base pair melting at the downstream edge of the transcription bubble suggest that translocation is rate-limiting for transcription and poorly reversible (10).
A study of translocation equilibrium in elongation complexes deprived of NTPs is technically challenging due to the lack of conditions that would selectively inhibit translocation as opposed to NTP binding and catalysis. For instance, known translocation inhibitors α-amanitin (for Pol II) and tagetitoxin (for bacterial RNAPs) affect catalysis in addition to interfering with translocation (36–39). Thus, the basic characteristics of the pre-translocation state of RNAP such as the rate of pyrophosphorolysis and NTP binding are essentially unknown, and can only be indirectly determined in a mixture of the pre-translocated and post-translocated states of the same TEC. In this work, we solved this technical problem by placing RNAP in a pre-translocated state on a non-pausing sequence using Nun protein of HK022 phage to stabilize that state. The recent cryo-EM analysis of Nun bound to E. coli TEC showed that Nun inhibits translocation by binding to the upstream end of the RNA–DNA hybrid and to RNAP catalytic cleft, which anchors RNAP to DNA and prevents both forward or backward translocation of the enzyme along DNA (8). Nun appears to anchor RNAP to DNA by forming multiple contacts with the RNA–DNA hybrid, DNA template and β subunit of RNAP (8). Importantly, Nun binding does not alter the overall architecture of TEC including the active center, or the degree of DNA clamping and interaction of RNAP with the DNA-RNA hybrid. This property distinguishes Nun from all other known inhibitors of phosphodiester bond formation such as tagetitoxin, streptolydigin, and α-amanitin. Thus, Nun acts as a bona fide translocation inhibitor of RNAP with no direct impact on catalysis. This unique feature makes Nun an ideal tool for stabilization of a pre-translocated state of RNAP.
We generated TECs carrying the intact transcription bubble and RNA–DNA hybrids of altered length and sequence in the presence and the absence of Nun. A stably pre-translocated E. coli TEC obtained in the presence of Nun protein (28,40) was used as a reference point for determining the basal biochemical characteristics of the pre-translocated state as opposed to its post-translocated counterpart. We were able reconcile what appeared to be contradictory results of Exo III footprinting, pyrophosphorolysis, and NMP incorporation by demonstrating that translocation is poorly reversible, relatively slow [compared to the previously proposed Brownian ratchet model (22)], and rate-limiting during pause-free elongation. Furthermore, we show that translocation is regulated by the switching from RNA–DNA to DNA–DNA base pairing at the rear end of the transcription bubble.
MATERIALS AND METHODS
Protein purification
Purification of E. coli RNAP core enzymes carrying a hexahistidine-tagged β’ subunit as described in Supplemental Text. Nun was purified as described in (28). The 12-subunit core enzyme of S. cerevisiae Pol II was purified as described in (41).
TEC assembly and purification
The TECs were assembled from RNAP core enzyme or Pol II and synthetic RNA and DNA oligonucleotides as in (7) on the previously characterized sequence of cre gene of P1 bacteriophage (42). TECs C8 and C9 (the capital letter followed by the number indicate the 3′-terminal residue in the RNA and the length of RNA complementary to the template DNA) are assembled using RNA C8 and RNA C9, respectively, and walked 1 bp downstream by adding UMP. A typical assembly reaction was performed in 50 μl transcription buffer (TB) (20 mM Tris–HCl, pH 7.9, 5 mM MgCl2, 20 μM ZnSO4, 1 mM 2-mercaptoethanol) containing 40 mM KCl (TB40) with 10 pmol of RNA–DNA hybrid, 10 pmol RNAP, and 15 pmol non-template DNA strand. The 5′ to 3′ sequences of the RNA primers RNA C8 and RNA C9 are CCCCCCCCUCAUGAAC and CCCCCCCCGUCAUGAAC, respectively; of the non-template strand NDS57 – GGCTGGACCAATGTAAATATTGTCATGAACTGTATCCGTAACCTGGATAGTGAAACA, and of the template strand TDS57 – TGTTTCACTATCCAGGTTACGGATACAGTTCATGACAATATTTACATTGGTCCAGCC. NDS57C-10, (GGCTGGACCAATGTAAATATTCTCATGAACTGTATCCGTAACCTGGATAGTGAAACA, dC-10 is underlined) was used for the assembly of TECs carrying the dC–dC-10 mismatch in the DNA. The RNA oligonucleotides were labeled at the 5′ end with fluorescein. The resulting TECs C8 and C9 were purified by three rounds of dilution with 450 μl TB40 containing 0.2 mg/ml acetylated BSA (Sigma) and concentration to 25 μl in the Amicon Ultra centrifuge filters with a 100 kDa molecular weight cutoff (Millipore) (43).
The TECs U9 and U10 were derived from the C8 and C9 TECs by a 5 min incubation with 20 μM UTP (purified as described in (44)) and treated as described above, but a wash buffer (WB) containing 20 mM Tris–HCl, pH 7.9, 1 mM EDTA, and 1 mM 2-mercaptoethanol was used instead of TB for purification of E. coli RNAP TECs. MgCl2 was removed from the buffer used for purification of the TECs U9 and U10 because of a high susceptibility of TEC U10 to pyrophosphorolysis, which leads to partial cleavage of the RNA in the TEC U10 by trace amounts of the pyrophosphate in the buffer solutions. For some experiments, the TECs were immobilized on Ni-NTA agarose (Qiagen) using 20 μl of 50% Ni-NTA agarose suspension per 1 μg (2 pmol) of RNAP after the assembly. The immobilized TECs U9 and U10 were purified by five washes with 1 ml WB.
Exo III footprinting
The assay was performed as in (16), and the complete scaffolds used in the Exo III assay are in Supplementary Figure S1. The DNA products were analyzed by fluorescent imaging set up for Cy3 or Cy5 detection using the Typhoon Trio instrument (GE).
Transcription elongation assay
Equal volumes of TECs C8 and C9 in TB or TECs U9 and U10 in WB were mixed with 2 mM each GTP and UTP in TB containing 10 mM MgCl2 to achieve 1 mM GTP and UTP and 5 mM MgCl2 final concentrations. The reactions were stopped by addition of 1 M HCl or 0.5 M EDTA using the Rapid Quench Flow instrument RQF-3 (Kintek). The samples were processed as described in (43). The RNA products were analyzed by fluorescent imaging set up for fluorescein detection using the Typhoon Trio instrument (GE). The NMP incorporation rate constants were obtained by fitting the data with single or double exponential function.
Pyrophosphorolysis time course analysis
Pyrophosphorolysis time courses were taken at 5 s–12 min intervals by mixing TEC U9 or U10 in WB containing 2.5 units of apyrase (NEB) with inorganic pyrophosphate (PPi), and the reaction was started by addition of MgCl2. The final [PPi] was 2.5 mM, and the final [MgCl2] was 5 mM. For the Vmax and Km determination, the final [PPi] was varied from 10 μM to 2.5 mM. The time points shorter than 5 s were obtained using the RQF-3 instrument. Equal volumes of TEC U10 in WB containing 1 μM Nun (where indicated) and 5 mM PPi and TB containing 10 mM MgCl2 were mixed and the reaction was stopped with 1 M HCl. The samples were processed and the RNA products were analyzed as described for the elongation assay.
RESULTS AND DISCUSSION
Altering translocation equilibrium by changing the length of RNA–DNA hybrid in the TEC
The crystallographic data on bacterial RNA polymerases assembled on the RNA–DNA scaffolds strongly argue that the post-translocated and pre-translocated TECs contain 9-bp and 10-bp RNA–DNA hybrids, respectively (45). The precise coordination of NMP incorporation and melting of the –10th RNA–DNA base pair (9,10) suggests that RNA–DNA hybridization at the –10th position might regulate RNAP translocation (Figure 1A). To test if the change in the RNA–DNA hybrid length influences translocation in the regular non-paused TECs, we compared the elongation complexes with the 9- and 10-bp RNA–DNA hybrids obtained on the RNA–DNA scaffold within the otherwise identical sequence context. To obtain these complexes, we first assembled TECs C8 and C9 using RNA C8 and RNA C9, respectively, and walked them 1 bp downstream by adding UMP to obtain TECs U9 and U10 (the letters indicate the 3′ residue in the RNA and the numbers stand for the length of the RNA–DNA hybrid). The derivatives of TECs C8 and C9, TECs U9 and U10, contain the 9- and the 10-bp RNA–DNA hybrid, respectively. In TEC U10, the –10th rG base pairs with the template dC; the –10th rC in TEC U9 is not complementary to the template dC, limiting the RNA–DNA hybrid to 9-bp length (Figure 1 and Supplementary Figure S1). This approach produces two pairs of TECs, C8/C9 and U9/U10 stalled at the two adjacent template positions and each carrying 8 or 9-bp and 9 or 10-bp RNA–DNA hybrids, respectively. These two TECs have identical sequence context in the immediate proximity of the RNA 3′ end, suggesting that the active site rearrangements, including the trigger loop opening and closure (46), as well as phosphodiester bond formation or pyrophosphorolysis, should not be affected by this change in the RNA–DNA hybrid length.
Figure 1.

RNA–DNA-10 base pairing at the upstream end of the RNA–DNA hybrid in RNAP affects pyrophosphorolysis and translocation, but not NMP addition in a non-paused TEC. (A) Cryo-EM structure (PDB: 6ALG) of the rear end of transcription bubble in the post-translocated TEC by E. coli RNAP arrested by Nun protein (8). The non-transcribed DNA strand (NS, yellow), template strand (TS, cyan), and nascent RNA (R, green) in the RNA–DNA hybrid are shown. The lid domain in the β’ subunit of RNAP (249–265 aa) is shown as a tube model (gray). The cartoon on the right illustrates RNA–DNA rearrangement at the upstream edge of the bubble during forward 1-bp translocation of RNAP (Pre→Post transition). Melting of the –10th RNA–DNA base pair in the hybrid is coupled with re-annealing of the –10th DNA–DNA base pair at the rear end of the bubble are shown as shaded boxes with the arrows indicating movement of the corresponding bases. (B) Exonuclease mapping of the front boundary of RNAP. The C8 and C9 TECs were assembled with RNA C8 or RNA C9 hybridized to TDS57 carrying three phosphothioate bonds at the 3′ end, and NDS57 labeled at the 5′ end by Cy3 (Supplementary Figure S1). The RNAP footprint is shown by a light grey oval. Two possible outcomes, UMP incorporation followed by translocation and UMP incorporation without translocation, are illustrated. The TECs were incubated with Exo III for 10, 20, 40 and 90 s before (C8 and C9) or after (U9 and U10) addition of 10 μM UTP. The RNA was detected in the same gel as the DNA to confirm UMP incorporation. GMP incorporation (C) and pyrophosphorolysis (D) were analyzed in TECs U9 and U10. The cartoons depict the apparent relative abundance of the pre- and post-translocated states in the TECs U9 and U10.
First, we assessed the translocation state of these TECs using ExoIII footprinting (Figure 1B). ExoIII is broadly used to measure a dynamic of the front-end boundary of RNAP (translocation) in TECs deprived of NTPs (3,16,25). The footprint of TEC C8 shifts 1-bp downstream as a result of the 3′ RNA extension with UMP and formation of TEC U9. In contrast, extension of RNA C9 with UMP and formation of TEC U10 did not cause the characteristic 1-bp shift of the front-end boundary indicating that RNAP in TEC U10 has a problem with forward translocation after incorporation of UMP. Because both U9 and U10 complexes are identical except for a single RNA–DNA mismatch at the –10th position of the RNA–DNA hybrid, we conclude that the –10th base pair in the hybrid interferes to some extent with forward translocation. Note that footprinting by ExoIII provides a qualitative assessment of translocation equilibrium in TECs without providing information on the translocation rate and concentrations of the pre/post-translocated states [(3,28); Supplementary Figures S2 and S3]. Next, the catalytic rate for the next GMP incorporation by TECs U9 and U10 was determined (Figure 1C). Although ExoIII footprinting shows a significant difference in the translocation equilibrium in these complexes, they both represent the catalytically active state of RNAP, because the GMP incorporation at 1 mM GTP occurs at 60 s−1 in TECs U9 and U10 (Figure 1C). In contrast, these TECs are dramatically different in pyrophosphorolysis (Figure 1D). TECs U9 and U10 are identical except for a single –10th base in their nascent RNA located at a large distance from the active site (Figure 1A). Thus, the direct effect of this change on chemistry of pyrophosphorolysis or pyrophosphate binding was unlikely. Because pyrophosphorolysis occurs in the pre-, but not the post-translocated TEC (Figure 1D) we argue that the RNA/DNA mismatch at the –10th position in TEC U9 dramatically decreases the pre-translocated fraction, which is also consistent with the ExoIII data (Figure 1B). We further hypothesized that the pre-translocated state represents only a minor fraction in the mostly post-translocated TEC U10. This notion would explain why the effect of the –10th mismatch has no impact on elongation, but dramatically reduces pyrophosphorolysis. The limitations of the time-resolved ExoIII footprinting for analyses of RNAP translocation are discussed in Supplemental Materials.
Quantitative analysis of translocation equilibria in TECs U9 and U10 using Nun protein
To explore the translocation state of RNAP in TECs U9 and U10 quantitatively, we analyzed pyrophosphorolysis rates in these TECs relying on interesting conclusions published by Hein et al. (30). A simple kinetic model of pyrophosphorolysis reaction, suggesting that PPi binds exclusively to the pre-translocated TEC, predicts that the Km for PPi should be lower in the more pre-translocated TEC, and pyrophosphorolysis Vmax should not depend on the RNAP translocation state. However, Hein et al. (30) reported that, paradoxically, in the TECs formed by E. coli and T. thermophilis RNAPs, Vmax of pyrophosphorolysis, rather than Km for PPi, depends on the TEC translocation state. They explained their observations by proposing that PPi binds equally well to the post-translocated and pre-translocated TECs (30). In this case, Vmax of pyrophosphorolysis should be directly proportional to the size of the pre-translocated fraction in the TECs exposed to PPi. Therefore, pyrophosphorolysis rate at saturating [PPi] can be used as an indicator of the pre-translocated fraction abundance. However, to determine the absolute size of the pre-translocated fraction, a reference parameter—the pyrophosphorolysis rate of a stably pre-translocated TEC—should be determined.
To solve this problem, we used the translocation inhibitor Nun (28,40). Nun prevents exchange between the pre- and post-translocated states in the TEC deprived of NTPs (28). Nun inhibits transcription elongation by TEC U10 (Supplementary Figure S4), providing an excellent opportunity for kinetic analysis of the stably pre-translocated TEC U10. The apparent pyrophosphorolysis rate of TEC U10 increased ∼10-fold from 0.21 to 2.4 s−1 after pre-incubation with Nun (Figure 2A). Thus, the 2.4 s−1 rate represents the pyrophosphorolysis rate in the pre-translocated TEC U10. We assumed that pyrophosphorolysis rate is directly proportional to the abundance of the pre-translocated state (30) and calculated the pre-translocated fraction of TEC U10 in the absence of Nun by using the equation
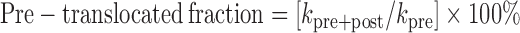 |
Figure 2.

RNA–DNA hybridization at the –10th position restricts translocation by a conserved, lid domain-dependent mechanism. Translocation inhibitor Nun stimulates pyrophosphorolysis by TEC U10 (A) and inhibits pyrophosphorolysis in a major fraction of TEC U9 (B). The TECs were pre-incubated with 1 μM Nun or the same volume of Nun storage buffer in the absence of Mg2+, and pyrophosphorolysis time courses were analyzed in the presence of 2.5 mM PPi and 5 mM MgCl2. The data points were fitted with a single exponential function. The translocation schematic at right in (A) and (B) show the pre-translocated fraction of TEC U10 and TEC U9 inferenced from the TEC U10 pyrophosphorolysis rate in the presence of Nun. The detailed description of the translocation schemes is in the text. (C) Pyrophosphorolysis rate constants at 2.5 mM PPi and 5 mM MgCl2 in TECs U9 and U10 formed by the WT RNAP, Δlid RNAP and yeast Pol II.
where kpre+post is pyrophosphorolysis rate in the absence of Nun, and kpre is pyrophosphorolysis rate in the presence of Nun.
For TEC U10, the pre-translocated fraction is (0.21/2.4) × 100% = 8.8%. Additional experiments to determine the pre-translocated fraction of TEC U10 are described in the Supplemental Text and Supplementary Figure S5.
It cannot be completely ruled out that Nun changes the equilibrium concentration of the pre- and post-translocation states by preferential binding and stabilization of one of them. This possibility is exemplified by the inhibitory effect of Nun on pyrophosphorolysis of TEC U9 (Figure 2B). The ExoIII footprint of TEC U9 (Figure 1B, lanes 5–8) and the slow rate of pyrophosphorolysis of TEC U9 (Figure 1D) indicate that this complex is predominantly post-translocated. Addition of Nun inhibits pyrophosphorolysis by TEC U9 (Figure 2B) indicating that Nun stabilizes the post-translocated state of TEC U9, which suggests that the pre-translocated state of TEC U9 is poorly accessible to Nun. The strong inhibition of pyrophosphorolysis suggests that Nun binding stabilized the post-translocated fraction of TEC U9. Therefore, the pyrophosphorolysis rate of the stably pre-translocated TEC U9 cannot be measured directly by using Nun. However, the pyrophosphorolysis rate strongly depends on the local sequence context around the RNA 3′ end (30). We prove that substitutions in the –10th position do not affect pyrophosphorolysis of the stably pre-translocated TEC by analyses of a pair of TECs with rG–dC-10 and rU–dA-10 formed in the otherwise identical sequence context (19). These TECs show identical susceptibility to PPi in the presence of Nun (Supplementary Figure S6), while in the absence of Nun the TEC with rU–dA-10 is significantly less susceptible to PPi than the TEC carrying rG–dC-10 [(19) and Supplementary Figure S6]. Therefore, the unknown pyrophosphorolysis rate of the stably pre-translocated TEC U9 should be the same as the pyrophosphorolysis rate of the stably pre-translocated TEC U10-Nun complex (2.4 s−1). Based on this conclusion, the pre-translocated fraction of TEC U9 can be determined by dividing pyrophosphorolysis rate of TEC U9 (8.4 × 10−3 s−1) by the pyrophosphorolysis rate in the pre-translocated TEC U10-Nun complex (2.4 s−1), which yields not more than 0.5% of TEC U9 being pre-translocated at any given moment.
Next, we measured pyrophosphorolysis rates in TEC U9, and TEC U10, and in the TEC U10 complex with Nun at different [PPi], and determined the apparent kinetic constants, Km and Vmax (Table 1). The apparent Kms for PPi are similar for all the TECs tested. The apparent maximum pyrophosphorolysis rate (Vmax) for TEC U10 is 30 times higher than that for TEC U9, and pre-incubation of TEC U10 with Nun results in an additional 10-fold increase in Vmax. The Km and Vmax measurements support the assumption that the pyrophosphorolysis rate at 2.5 mM PPi (the concentration significantly higher than Km for PPi) is directly proportional to the fraction of the pre-translocated state in the TEC and are fully consistent with the previously reported effects of RNAP translocation state on pyrophosphorolysis (30). The pyrophosphorolysis rates reported by Hein et al. (30) are the same or slower than those we observe for TEC U9 and TECs C8 and C9 (Supplementary Figure S2E and F). Taken together with our observations, those slow rates of pyrophosphorolysis might suggest that all the TECs characterized in (30) are predominantly post-translocated, and the differences in the pyrophosphorolysis rates and Exo III footprints reported in that work are explained by the change in the size of the minor pre-translocated fraction. Another report describes a TEC with an 8-bp RNA–DNA hybrid, which predominantly dwells in a pre-translocated conformation (6). However, that work employed TECs, which lacked the upstream part of the RNA–DNA hybrid. According to our findings, that region is essential for translocation control. The stable post-translocated state of TECs with 9-bp or shorter RNA–DNA hybrids is consistent with the conclusions that RNAP undergoes forward translocation immediately after NMP incorporation and remains in the post-translocated state, which is based on the Exo III footprinting (26) and the stop-flow analyses of RNA–DNA hybrid melting (10).
Table 1. Kinetic parameters of pyrophosphorolysis reaction. Susceptibility of TEC U10 to PPi was determined in the absence and in the presence of Nun. TECs U9 and U10 were incubated with [PPi] varying from 10 μM to 2.5 mM. For the TEC U10, the experiment was also performed in the presence of 1 μM Nun. The pyrophosphorolysis rate constants were obtained by single exponential fits of the averaged data points, plotted versus [PPi], and the Km and Vmax values were obtained by fitting the data to a Michaelis–Menten equation.
| TEC | Km, mM | V max, min−1 |
|---|---|---|
| U9 | 0.39 ± 0.09 | 0.5 ± 0.03 |
| U10 | 0.32 ± 0.14 | 15.7 ± 1.9 |
| U10 + Nun | 0.24 ± 0.02 | 160 ± 3 |
Lid domain in RNAP promotes forward translocation
In a cryo-EM structure, the lid domain of E. coli RNAP is positioned next to the –9th base pair of the RNA–DNA hybrid in the post-translocated TEC (Figure 1A). It was shown that this domain is involved in separation of the RNA and template DNA strands at the rear end of the transcription bubble (45). It is plausible that the lid may also stimulate forward translocation by interfering with formation of the RNA–DNA-10 base pair. To test this hypothesis, we compared pyrophosphorolysis rates at 2.5 mM PPi in TECs U9 and U10 assembled with the wild type (WT) and Δlid RNAP (Figure 2C and Supplementary Figure S2). While the pyrophosphorolysis by WT RNAP is ∼20-fold faster in the TEC U10 than in TEC U9, pyrophosphorolysis by Δlid RNAP is much less sensitive to the RNA–DNA hybrid length. Specifically, the lid deletion results in an 8-fold increase in pyrophosphorolysis rate for TEC U9. The effect of the lid deletion on pyrophosphorolysis for TEC U10 is much less pronounced (∼1.5-fold). This result indicates that the lid domain promotes forward translocation and/or inhibits the reverse translocation by interfering with RNA–DNA-10 base pairing. The much stronger negative effect of the lid deletion on translocation of TEC U9 than TEC U10 argues that the lid poorly competes with the stable rG–dC-10 base pair, but its effect on translocation might be stronger for the complexes lacking the –10th base pair (TEC U9) or carrying weak rU–dA-10 or rA-dT-10 base pairs. The lid may also have a stronger effect on forward translocation in TECs containing hairpin in nascent RNA immediately upstream from the 9-bp RNA–DNA hybrid. These hairpins have been shown to promote melting of the upstream part of the RNA–DNA hybrid (47), which may facilitate access of the lid to the –10th RNA–DNA base pair.
Interestingly, S. cerevisiae Pol II showed a significant decrease of pyrophosphorolysis rate in response to RNA–DNA hybrid shortening from 10 to 9 bp, similar to WT E. coli RNAP (Figure 2C and Supplementary Figure S7). This result indicates that the mechanism for regulation of forward translocation by the length of the RNA–DNA hybrid is shared by these two RNA polymerases.
Base pairing of the template and non-template DNA bases at the –10th register slows down reverse translocation
The majority of the TECs subjected to high resolution structural analyses were assembled on incomplete nucleic acid scaffolds. These scaffolds contained the RNA–DNA hybrid, but lacked the upstream DNA duplex (45,48,49) or carried the non-template DNA strand and the RNA upstream from the RNA–DNA hybrid that are not fully complementary to the template DNA strand (50,51). Recently published cryo-EM structures of the mammalian Pol II and E. coli (Figure 1A) TECs show that the two DNA strands form a duplex immediately upstream from the end of the RNA–DNA hybrid (51). Here we present functional evidence that DNA reanneals as close as 10 bp upstream from the RNA 3′ end in E. coli RNAP, consistent with recent the report by Turtola and Belogurov (34).
When dG-10 in the non-template DNA of TEC U10 is replaced with dC-10, creating a dC/dC-10 mismatch, the pyrophosphorolysis rate (and, by inference, the pre-translocated fraction) of TEC U10 increases about two-fold (Figure 3A). When the same dC/dC-10 mismatch is introduced to TEC U9 (a version of TEC U10 lacking rG–dC-10 base pair), it's very slow pyrophosphorolysis rate also increases about three-fold (Figure 3B). Therefore, it appears that the rG–dC-10 and dG–dC-10 hybridization affect the frequencies of forward and reverse translocation, respectively. Effects of all possible combinations of RNA, template DNA, and non-template DNA bases at –10 on pyrophosphorolysis are summarized in Supplementary Figure S8. In most sequence contexts, DNA–DNA base pairing at the rear edge (the –10th position) of the transcription bubble stabilizes the post-translocated state.
Figure 3.

Template DNA base pairing with the non-template DNA base at the –10th position promotes forward translocation. Pyrophosphorolysis rate constants were determined for TECs U10 (A) and U9 (B), and the fraction of the pre-translocated TEC was quantified assuming that pyrophosphorolysis in a stably pre-translocated TEC occurs at 2.4 ± 0.28 s−1. The TECs were formed with the non-template DNA strand (NDS) fully complementary to the template DNA strand (shaded bars) or with the NDS carrying dC-10, which creates a dC/dC mismatch in the DNA at the –10th position.
The clearly pronounced effect of the non-template DNA strand on translocation of the TEC with a weak rU–dA-10 base pair (Figure 4A, WT RNAP) might be important for the E. coli RNAP function in vivo. The previously proposed model for intrinsic transcription termination suggests that the upstream part of the bubble collapses to a DNA duplex as an essential step in termination (47). Our findings suggest that dT in the non-template DNA strand successfully competes with rU base pairing to the dA-10 template base (Figure 4A). dT–dA-10 reannealing might serve as a nucleation step for bubble collapse during intrinsic transcription termination.
Figure 4.
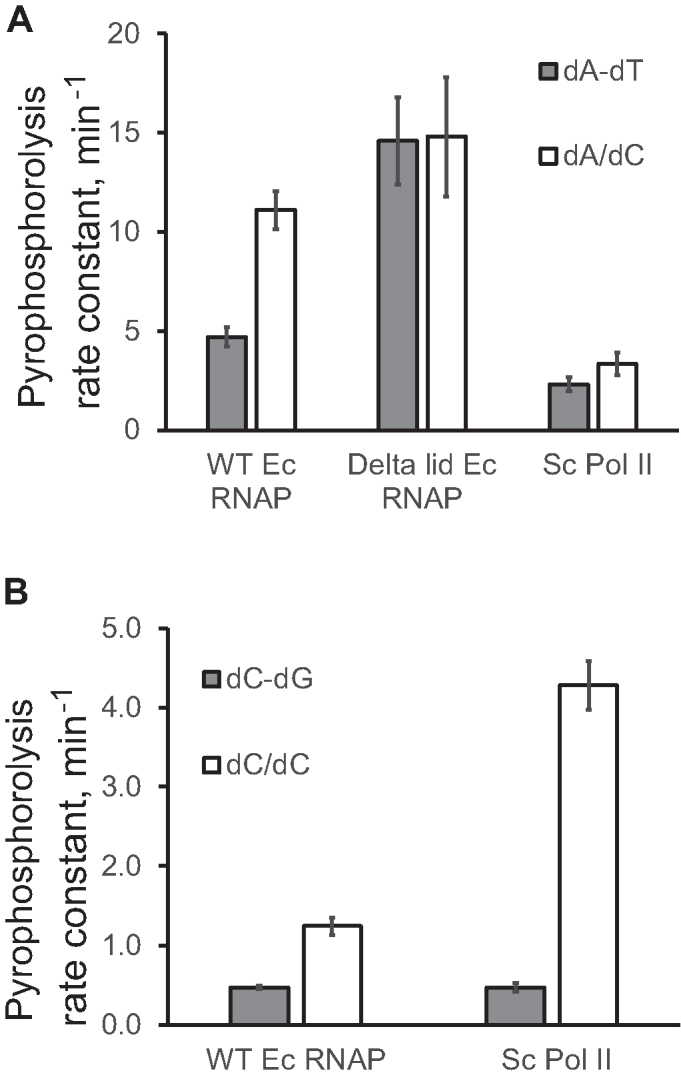
Translocation is promoted by the template DNA base pairing with the non-template DNA-10 base by a conserved mechanism shared by E. coli RNAP and S. cerevisiae Pol II. Pyrophosphorolysis rate constants obtained in 2.5 mM PPi and 5 mM MgCl2 are plotted for TEC U10 (A) carrying rU–dA-10 and the original TEC U9 (B). The nature of the mismatch in the DNA at the –10th position is specified in the chart legends.
We also observed some potentially important differences in the mechanisms of RNA–DNA hybridization and the effects of the DNA–DNA base pairing at –10 between E. coli RNAP and S. cerevisiae Pol II. Translocation of Pol II appears less sensitive to the RNA–DNA hybrid length. Specifically, pyrophosphorolysis of Pol II TEC U10 is slower than that of E. coli RNAP TEC U10 (Figure 2C). At the same time, Pol II TECs C9 and C8 pyrophosphorolize faster than their E. coli RNAP counterparts (compare Supplementary Figure S7 and Supplementary Figure S2E and F), suggesting that translocation of Pol II is less sensitive to RNA–DNA base pairing at the –10th position compared to E. coli RNAP. The opposite process, reannealing of the DNA duplex upstream from the RNA–DNA hybrid, appears to have a slightly different sequence specificity in Pol II compared to E. coli RNAP. For example, the dC-10 template base is more accessible to reannealing with the dG-10 non-template base (Figure 4B and Supplementary Figure S9) in TEC U9 formed by Pol II. At the same time, the dA/dC mismatch at the –10th position has a less pronounced effect in Pol II TEC than in the WT E. coli RNAP TEC (Figure 4B and Supplementary Figure S9). Interestingly, Pol II TECs, but not E. coli RNAP TECs are structurally unstable as they slowly decay in the absence of Mg2+. These Pol II TECs are significantly stabilized by a single DNA–DNA mismatch at the –10th position (Supplementary Figure S10). We concluded that displacement of the upstream end of the RNA–DNA hybrid by the non-template DNA strand may suggest a Pol II termination mechanism at the intrinsic pause sites localized downstream from polyadenylation signals (52).
Translocation during processive transition from one phosphodiester bond formation to another
The predominance of the post-translocated state in TECs U10 and U9 is fully consistent with the high GMP incorporation rates at 1 mM GTP (Figure 1C). The question arises whether the effect of the RNA–DNA-10 base pairing on the translocation equilibrium bears any physiological significance, if it hardly has any effect on transcript elongation by the TECs stalled by NTP deprivation outside the intrinsic pause sites in DNA. We note that when transcription is artificially stalled it is different from processive transcription when transition from one bond to another occurs without any interruption except a potential delay caused by slow forward translocation. To address this principal difference, we analyzed the properties of the TECs U9 and U10 during processive transcription – monitoring formation of two consecutive phosphodiester bonds accompanied by the translocation step between the bonds. We took advantage of the two-bond double-quench protocol originally developed by Burton and co-authors for characterization of transcription by human Pol II (53,54). Time courses of the first bond formation (escape from the stall) and the second bond formation (processive synthesis) are obtained using either hydrochloric acid or EDTA to stop the reactions. While the acid quench stops the reaction instantly, quenching with EDTA allows for completion of the bond formation in the TECs that have stably sequestered the substrate NTP but did not complete the bond formation when exposed to EDTA (25,43) (Figure 5A).
Figure 5.

Translocation in real time: the double-bond double-quench assay. (A) The cartoon shows the active site region of RNAP and the reaction intermediates detected by the two types of quenchers. (B) and (C) Dynamics of the first phosphodiester bond formation (HCl quench, black) and the NTP sequestration for the second bond formation (EDTA quench, red) by TEC C8 (B) and TEC C9 (C). Results of two independent experiments are combined in each panel (double dots). (D) The scheme depicts a ∼0.03 s delay detected in forward translocation of TEC U10 after UMP incorporation, which is caused by the rG–dC-10 hybridization. (E) The model shows that the RNA–DNA-10 and the DNA–DNA-10 base pairing decreases the frequency of forward and reverse translocation in TEC U10, respectively. The model is based on the coupling of disruption of the RNA–DNA-10 base pair and its replacement with the corresponding DNA–DNA-10 base during forward translocation. In this scenario, the RNA–DNA-10 base pair prevents forward translocation, whereas the subsequently formed DNA–DNA-10 base pair inhibits the reverse translocation.
In the experiment of Figure 5, TECs C8 and C9 were used to monitor the escape from the stall by UMP incorporation (TECs U9 and U10 formation) and the processive synthesis by the subsequent GMP incorporation to U9 and U10. TECs C8 and C9 were incubated with a mixture of 1 mM each UTP and GTP. The time courses of UMP incorporation (first bond) were obtained from the acid quench experiment and showed dynamics of the actual bond formation. The time courses of GMP incorporation (second bond) are from the EDTA quench parallels. Note that the time course of GTP sequestration by the newly formed TEC U9 coincides with the time course of UMP incorporation that precedes the GTP sequestration (Figure 5B). It means that, as soon as the first bond with UTP is formed, the new TEC binds and sequesters the next substrate NTP (GTP), making it inaccessible to quenching with EDTA. If we reasonably assume that only post-translocated TEC is capable of NTP sequestration, this result indicates that translocation of the TEC U9 is not rate-limiting for GMP incorporation. Remarkably, while the rates of UMP incorporation by the TECs C8 and C9 are also identical (54 s−1; Figure 5B and C), GTP sequestration by the newly formed TEC U10 (after UMP incorporation to TEC 9C) occurs slower than UMP incorporation (38 s−1; note the gap between the black and red traces in Figure 5C). This observation suggests that translocation of TEC U10 is at least partially rate-limiting for GTP sequestration and GMP incorporation during the processive synthesis (Figure 5D). Consistent with fast pyrophosphorolysis, translocation appears rate limiting in both TECs U9 and U10 formed by the Δlid RNAP (Supplementary Figure S11). The rate-limiting translocation is in agreement with the previously proposed kinetic models of the nucleotide addition cycle (35). The two-bond double-quench assay for translocation appears to have significant potential for understanding physiological aspects of translocation and its regulation.
CONCLUSIONS AND PERSPECTIVES
Here we provide a strong evidence that the RNA–DNA base pairing at the rear end of transcription bubble (the 10th, and the last, RNA–DNA base pair) inhibits forward translocation of RNAP, and causes translocation to become fully or partially rate-limiting for NMP incorporation during pause-free transcription. DNA–DNA base pairing at this end of the hybrid inhibits reverse translocation (Figure 5E). In the context of the two sequences that we characterized, RNAP does not undergo frequent forward and reverse translocations in sub-millisecond time-scale posited by the original Brownian ratchet model (22,55,56). Instead, forward translocation occurs in milliseconds and is poorly reversible. Our observations are consistent with the single molecule data (35) and with the stop-flow analysis of upstream RNA–DNA and downstream DNA–DNA base pair melting during the translocation step (10,34).
The two-bond double-quench assay further revealed that translocation is partially rate-limiting during pause-free transcription. The fully active (non-paused) TECs stalled on the template by substrate NTP deprivation exist in equilibrium having minor pre-translocated and major post-translocated conformations. The bona fide translocation inhibitor, Nun protein of bacteriophage HK022, was a valuable tool that enabled isolation of a stable pre-translocated fraction and its biochemical characterization (28). Using Nun protein, we successfully demonstrated that shortening of the RNA–DNA hybrid to 9 bp promotes forward translocation of RNAP, which is stabilized by the reannealing of the –10th template base and its non-template DNA base, leading to the disappearance of the pre-translocated fraction. We further demonstrated that different bases at the –10th position of the RNA have a capacity to stimulate or inhibit forward translocation. This regulatory signal is shared by bacterial and yeast RNA polymerases despite a significant difference in architecture of their respective DNA/RNA/protein interactions at the upstream edge of the transcription bubble. Analyses of the regulatory effect of transcription elongation factors (e.g. NusG for E. coli RNAP, and Spt4/5, and TFIIF for Pol II) on the RNA–DNA hybridization and DNA reannealing at the upstream edge of the transcription bubble warrant further investigation. Although the effect of base pairing at the –10th position on transcription pausing at hairpin-dependent and -independent intrinsic pause sites is beyond the scope of our work, these interactions at the upstream edge of the bubble may contribute to various types of transcription pausing, including those associated with the pre-translocated and the post-translocated forms of RNAP (11,12,16,57).
Supplementary Material
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Funding for open access charge: Intramural Research Program of the National Institutes of Health at National Cancer Institute’s Center for Cancer Research; National Cancer Institute, National Institutes of Health Grant [HHSN261200800001E to M.K.].
Conflict of interest statement. None declared.
REFERENCES
- 1. Kireeva M., Kashlev M., Burton Z.F.. Translocation by multi-subunit RNA polymerases. Biochim. Biophys. Acta. 2010; 1799:389–401. [DOI] [PubMed] [Google Scholar]
- 2. Brueckner F., Ortiz J., Cramer P.. A movie of the RNA polymerase nucleotide addition cycle. Curr. Opin. Struct. Biol. 2009; 19:294–299. [DOI] [PubMed] [Google Scholar]
- 3. Nedialkov Y.A., Burton Z.F.. Translocation and fidelity of Escherichia coli RNA polymerase. Transcription. 2013; 4:136–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Feig M., Burton Z.F.. RNA polymerase II with open and closed trigger loops: active site dynamics and nucleic acid translocation. Biophys. J. 2010; 99:2577–2586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Martinez-Rucobo F.W., Cramer P.. Structural basis of transcription elongation. Biochim. Biophys. Acta. 2013; 1829:9–19. [DOI] [PubMed] [Google Scholar]
- 6. Kashkina E., Anikin M., Tahirov T.H., Kochetkov S.N., Vassylyev D.G., Temiakov D.. Elongation complexes of Thermus thermophilus RNA polymerase that possess distinct translocation conformations. Nucleic. Acids. Res. 2006; 34:4036–4045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Sidorenkov I., Komissarova N., Kashlev M.. Crucial role of the RNA:DNA hybrid in the processivity of transcription. Mol. Cell. 1998; 2:55–64. [DOI] [PubMed] [Google Scholar]
- 8. Kang J.Y., Olinares P.D., Chen J., Campbell E.A., Mustaev A., Chait B.T., Gottesman M.E., Darst S.A.. Structural basis of transcription arrest by coliphage HK022 Nun in an Escherichia coli RNA polymerase elongation complex. eLife. 2017; 6:e25478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Malinen A.M., Turtola M., Belogurov G.A.. Monitoring translocation of multisubunit RNA polymerase along the DNA with fluorescent base analogues. Methods Mol. Biol. 2015; 1276:31–51. [DOI] [PubMed] [Google Scholar]
- 10. Malinen A.M., Turtola M., Parthiban M., Vainonen L., Johnson M.S., Belogurov G.A.. Active site opening and closure control translocation of multisubunit RNA polymerase. Nucleic Acids Res. 2012; 40:7442–7451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Artsimovitch I., Landick R.. Pausing by bacterial RNA polymerase is mediated by mechanistically distinct classes of signals. Proc. Natl Acad. Sci. U.S.A. 2000; 97:7090–7095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Toulokhonov I., Artsimovitch I., Landick R.. Allosteric control of RNA polymerase by a site that contacts nascent RNA hairpins. Science. 2001; 292:730–733. [DOI] [PubMed] [Google Scholar]
- 13. Guo X., Myasnikov A.G., Chen J., Crucifix C., Papai G., Takacs M., Schultz P., Weixlbaumer A.. Structural basis for NusA stabilized transcriptional pausing. Mol. Cell. 2018; 69:816–827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kang J.Y., Mishanina T.V., Bellecourt M.J., Mooney R.A., Darst S.A., Landick R.. RNA polymerase accommodates a pause RNA hairpin by global conformational rearrangements that prolong pausing. Mol. Cell. 2018; 69:802–815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Artsimovitch I., Belogurov G.A.. Uneven braking spins RNA polymerase into a pause. Mol. Cell. 2018; 69:723–725. [DOI] [PubMed] [Google Scholar]
- 16. Kireeva M.L., Kashlev M.. Mechanism of sequence-specific pausing of bacterial RNA polymerase. Proc. Natl Acad. Sci. U.S.A. 2009; 106:8900–8905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Imashimizu M., Kireeva M.L., Lubkowska L., Gotte D., Parks A.R., Strathern J.N., Kashlev M.. Intrinsic translocation barrier as an initial step in pausing by RNA polymerase II. J. Mol. Biol. 2013; 425:697–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Larson M.H., Mooney R.A., Peters J.M., Windgassen T., Nayak D., Gross C.A., Block S.M., Greenleaf W.J., Landick R., Weissman J.S.. A pause sequence enriched at translation start sites drives transcription dynamics in vivo. Science. 2014; 344:1042–1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Imashimizu M., Takahashi H., Oshima T., McIntosh C., Bubunenko M., Court D.L., Kashlev M.. Visualizing translocation dynamics and nascent transcript errors in paused RNA polymerases in vivo. Genome Biol. 2015; 16:98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Herbert K.M., Zhou J., Mooney R.A., Porta A.L., Landick R., Block S.M.. E. coli NusG inhibits backtracking and accelerates pause-free transcription by promoting forward translocation of RNA polymerase. J. Mol. Biol. 2010; 399:17–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Mejia Y.X., Nudler E., Bustamante C.. Trigger loop folding determines transcription rate of Escherichia coli's RNA polymerase. Proc. Natl Acad. Sci. U.S.A. 2015; 112:743–748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Bar-Nahum G., Epshtein V., Ruckenstein A.E., Rafikov R., Mustaev A., Nudler E.. A ratchet mechanism of transcription elongation and its control. Cell. 2005; 120:183–193. [DOI] [PubMed] [Google Scholar]
- 23. Abbondanzieri E.A., Greenleaf W.J., Shaevitz J.W., Landick R., Block S.M.. Direct observation of base-pair stepping by RNA polymerase. Nature. 2005; 438:460–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Dangkulwanich M., Ishibashi T., Bintu L., Bustamante C.. Molecular mechanisms of transcription through single-molecule experiments. Chem. Rev. 2014; 114:3203–3223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kireeva M.L., Nedialkov Y.A., Cremona G.H., Purtov Y.A., Lubkowska L., Malagon F., Burton Z.F., Strathern J.N., Kashlev M.. Transient reversal of RNA polymerase II active site closing controls fidelity of transcription elongation. Mol. Cell. 2008; 30:557–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Nedialkov Y.A., Nudler E., Burton Z.F.. RNA polymerase stalls in a post-translocated register and can hyper-translocate. Transcription. 2012; 3:260–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Nedialkov Y.A., Opron K., Caudill H.L., Assaf F., Anderson A.J., Cukier R.I., Wei G., Burton Z.F.. Hinge action versus grip in translocation by RNA polymerase. Transcription. 2018; 9:1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Vitiello C.L., Kireeva M.L., Lubkowska L., Kashlev M., Gottesman M.. Coliphage HK022 Nun protein inhibits RNA polymerase translocation. Proc. Natl Acad. Sci. U.S.A. 2014; 111:E2368–E2375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Bochkareva A., Yuzenkova Y., Tadigotla V.R., Zenkin N.. Factor-independent transcription pausing caused by recognition of the RNA–DNA hybrid sequence. EMBO J. 2012; 31:630–639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hein P.P., Palangat M., Landick R.. RNA transcript 3′-proximal sequence affects translocation bias of RNA polymerase. Biochemistry. 2011; 50:7002–7014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Nedialkov Y.A., Opron K., Assaf F., Artsimovitch I., Kireeva M.L., Kashlev M., Cukier R.I., Nudler E., Burton Z.F.. The RNA polymerase bridge helix YFI motif in catalysis, fidelity and translocation. Biochim. Biophys. Acta. 2013; 1829:187–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kireeva M.L., Opron K., Seibold S.A., Domecq C., Cukier R.I., Coulombe B., Kashlev M., Burton Z.F.. Molecular dynamics and mutational analysis of the catalytic and translocation cycle of RNA polymerase. BMC Biophys. 2012; 5:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Artsimovitch I., Landick R.. Interaction of a nascent RNA structure with RNA polymerase is required for hairpin-dependent transcriptional pausing but not for transcript release. Genes Dev. 1998; 12:3110–3122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Turtola M., Belogurov G.A.. NusG inhibits RNA polymerase backtracking by stabilizing the minimal transcription bubble. eLife. 2016; 5:e18096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Dangkulwanich M., Ishibashi T., Liu S., Kireeva M.L., Lubkowska L., Kashlev M., Bustamante C.J.. Complete dissection of transcription elongation reveals slow translocation of RNA polymerase II in a linear ratchet mechanism. eLife. 2013; 2:e00971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Yuzenkova Y., Roghanian M., Bochkareva A., Zenkin N.. Tagetitoxin inhibits transcription by stabilizing pre-translocated state of the elongation complex. Nucleic. Acids. Res. 2013; 41:9257–9265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Gong X.Q., Nedialkov Y.A., Burton Z.F.. Alpha-amanitin blocks translocation by human RNA polymerase II. J. Biol. Chem. 2004; 279:27422–27427. [DOI] [PubMed] [Google Scholar]
- 38. Brueckner F., Cramer P.. Structural basis of transcription inhibition by alpha-amanitin and implications for RNA polymerase II translocation. Nat. Struct. Mol. Biol. 2008; 15:811–818. [DOI] [PubMed] [Google Scholar]
- 39. Vassylyev D.G., Svetlov V., Vassylyeva M.N., Perederina A., Igarashi N., Matsugaki N., Wakatsuki S., Artsimovitch I.. Structural basis for transcription inhibition by tagetitoxin. Nat. Struct. Mol. Biol. 2005; 12:1086–1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Hung S.C., Gottesman M.E.. The Nun protein of bacteriophage HK022 inhibits translocation of Escherichia coli RNA polymerase without abolishing its catalytic activities. Genes Dev. 1997; 11:2670–2678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kireeva M.L., Hancock B., Cremona G.H., Walter W., Studitsky V.M., Kashlev M.. Nature of the nucleosomal barrier to RNA polymerase II. Mol. Cell. 2005; 18:97–108. [DOI] [PubMed] [Google Scholar]
- 42. Irvin J.D., Kireeva M.L., Gotte D.R., Shafer B.K., Huang I., Kashlev M., Strathern J.N.. A genetic assay for transcription errors reveals multilayer control of RNA polymerase II fidelity. PLoS Genet. 2014; 10:e1004532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kireeva M., Nedialkov Y.A., Gong X.Q., Zhang C., Xiong Y., Moon W., Burton Z.F., Kashlev M.. Millisecond phase kinetic analysis of elongation catalyzed by human, yeast, and Escherichia coli RNA polymerase. Methods. 2009; 48:333–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Lubkowska L., Kireeva M.L.. Direct competition assay for transcription fidelity. Methods Mol. Biol. 2015; 1276:153–164. [DOI] [PubMed] [Google Scholar]
- 45. Vassylyev D.G., Vassylyeva M.N., Perederina A., Tahirov T.H., Artsimovitch I.. Structural basis for transcription elongation by bacterial RNA polymerase. Nature. 2007; 448:157–162. [DOI] [PubMed] [Google Scholar]
- 46. Vassylyev D.G., Vassylyeva M.N., Zhang J., Palangat M., Artsimovitch I., Landick R.. Structural basis for substrate loading in bacterial RNA polymerase. Nature. 2007; 448:163–168. [DOI] [PubMed] [Google Scholar]
- 47. Komissarova N., Becker J., Solter S., Kireeva M., Kashlev M.. Shortening of RNA:DNA hybrid in the elongation complex of RNA polymerase is a prerequisite for transcription termination. Mol. Cell. 2002; 10:1151–1162. [DOI] [PubMed] [Google Scholar]
- 48. Weixlbaumer A., Leon K., Landick R., Darst S.A.. Structural basis of transcriptional pausing in bacteria. Cell. 2013; 152:431–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Westover K.D., Bushnell D.A., Kornberg R.D.. Structural basis of transcription: separation of RNA from DNA by RNA polymerase II. Science. 2004; 303:1014–1016. [DOI] [PubMed] [Google Scholar]
- 50. Barnes C.O., Calero M., Malik I., Graham B.W., Spahr H., Lin G., Cohen A.E., Brown I.S., Zhang Q., Pullara F. et al. . Crystal structure of a transcribing RNA polymerase II complex reveals a complete transcription bubble. Mol. Cell. 2015; 59:258–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Bernecky C., Herzog F., Baumeister W., Plitzko J.M., Cramer P.. Structure of transcribing mammalian RNA polymerase II. Nature. 2016; 529:551–554. [DOI] [PubMed] [Google Scholar]
- 52. Proudfoot N.J. Transcriptional termination in mammals: Stopping the RNA polymerase II juggernaut. Science. 2016; 352:aad9926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Nedialkov Y.A., Gong X.Q., Hovde S.L., Yamaguchi Y., Handa H., Geiger J.H., Yan H., Burton Z.F.. NTP-driven translocation by human RNA polymerase II. J. Biol. Chem. 2003; 278:18303–18312. [DOI] [PubMed] [Google Scholar]
- 54. Nedialkov Y.A., Gong X.Q., Yamaguchi Y., Handa H., Burton Z.F.. Assay of transient state kinetics of RNA polymerase II elongation. Methods Enzymol. 2003; 371:252–264. [DOI] [PubMed] [Google Scholar]
- 55. D O.M., Tadigotla V.R., Nudler E., Ruckenstein A.E.. A unified model of transcription elongation: what have we learned from single-molecule experiments. Biophys. J. 2011; 100:1157–1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Silva D.A., Weiss D.R., Pardo Avila F., Da L.T., Levitt M., Wang D., Huang X.. Millisecond dynamics of RNA polymerase II translocation at atomic resolution. Proc. Natl Acad. Sci. U.S.A. 2014; 111:7665–7670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Herbert K.M., La Porta A., Wong B.J., Mooney R.A., Neuman K.C., Landick R., Block S.M.. Sequence-resolved detection of pausing by single RNA polymerase molecules. Cell. 2006; 125:1083–1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.