


Abstract
In prokaryotes, the centromere is a specialized segment of DNA that promotes the assembly of the segrosome upon binding of the Centromere Binding Protein (CBP). The segrosome structure exposes a specific surface for the interaction of the CBP with the motor protein that mediates DNA movement during cell division. Additionally, the CBP usually controls the transcriptional regulation of the segregation system as a cell cycle checkpoint. Correct segrosome functioning is therefore indispensable for accurate DNA segregation. Here, we combine biochemical reconstruction and structural and biophysical analysis to bring light to the architecture of the segrosome complex in Type III partition systems. We present the particular features of the centromere site, tubC, of the model system encoded in Clostridium botulinum prophage c-st. We find that the split centromere site contains two different iterons involved in the binding and spreading of the CBP, TubR. The resulting nucleoprotein complex consists of a novel double-ring structure that covers part of the predicted promoter. Single molecule data provides a mechanism for the formation of the segrosome structure based on DNA bending and unwinding upon TubR binding.
INTRODUCTION
DNA segregation is a fundamental process that needs to be performed with high precision in order to ensure stable genome transmission during the cell cycle. In prokaryotes, plasmids have been used as tractable model systems to understand the minimal molecular requirements and the underlying mechanism during DNA segregation. Most of low-copy number plasmids use the so-called partition systems for their maintenance, which are compact genetic modules tightly auto-regulated by one or both of the gene products (1,2). These systems require only three elements: a centromere-like site, a centromere-binding protein (CBP), and a motor NTPase protein. The CBP binds to the centromere-like site forming a nucleoprotein complex known as segrosome (3,4), which is recognized by the motor protein that effectively moves the DNA within the cell. According to the nature of the motor protein, partition systems have been classified into: Type I (Walker-A ATPase), Type II (actin-like ATPase) and Type III (tubulin-like GTPase) (5).
Type III partition systems have been identified as part of the maintenance machinery of virulence plasmids in Bacillus and Clostridium species, where the CBP is a small protein (∼10 kDa) named TubR and, the motor protein, TubZ, is a prokaryotic tubulin homologue that assembles into helical cytomotive filaments (6,7). The most studied system is the one of B. thuriengensis plasmid pBtoxis, where the centromere-like site (tubC) has been characterized and there are high-resolution structures of TubZ, its related filament, and the adaptor protein TubR (6,8,9). Data from B. anthracis (pOX1), B. megaterium (pBM400), B. sphaericus (pBsph) plasmids and C. botulinum prophage (c-st) also contributed to the understanding of Type III partition systems. Common to all these systems, the centromere location is always upstream tubR, and contains several direct repeats that split into blocks forming a discontinuous tubC site (10,11). Upon TubR binding the resulting segrosome complex displays a dual function: one, repressing transcription from the operon promoter (7,11,12) and two, interacting with TubZ filaments for plasmid segregation (13). Studies focused on the nucleoprotein complex formation suggested that TubR interaction with the tubC site produces a flexible filamentous structure that eventually can close into a ring (10). However, the precise nature of the TubR-centromere interaction is not well defined due to the lack of a high-resolution structure. We are only certain about the different mechanism of interaction related to the classical binding through a Helix-Turn-Helix motif because in TubR, the recognition helix is part of the homo-dimerization interface, which forces different contacts with the DNA major groove (6,10). In addition, a structural study considering the whole operon is still missing. How TubR and the centromere region build a macromolecular partition complex within the plasmid environment and how such structure is able to control gene expression is key to understand the maintenance of virulence factors in those bacteria.
Here, we use the Type III partition model system encoded in C. botulinum prophage c-st to explore the segrosome complex formation and the mechanism involved in the transcriptional repression of the operon. We investigate the interaction of TubR with the centromere and the predicted promoter by a combination of biochemical and biophysical techniques including DNase I footprinting, analytical ultracentrifugation (AUC), atomic force microscopy (AFM), Magnetic Tweezers (MT), and electron microscopy (EM). We have found that tubC (previously defined as tubS, (14)) is a discrete region split into high and low affinity-binding sites. The interaction of TubR with the centromere results in a double-ring structure where each DNA loop is located on each of the binding sites of tubC. Furthermore, the ring containing the high affinity site also covers the predicted promoter region. We do not have experimental evidence of functionality of TubR promoter coverage, but this might repress the transcription of the partition operon as previously showed in pBtoxis and pBsph plasmids (7,11). Our data show that TubR not only bends but it also untwists the DNA upon binding, which has consequences in the supercoiling degree of the uncovered DNA in torsionally-constrained molecules. TubR bending and untwisting activities are independent and likely responsible for the assembly of the segrosome complex.
MATERIALS AND METHODS
Protein expression and purification
TubR was expressed in Escherichia coli C41 cells that were induced at OD600 ∼ 0.6 by the addition of 1 mM isopropyl-β-d-thiogalactopyranoside at 16°C during 15 h. Cells were pelleted and suspended in 50 mM Tris–HCl pH 8.0, 150 mM NaCl. DNase and lysozyme were added prior cell disruption by sonication. Cell debris was pelleted by centrifugation at 100 000 × g for 1 h. TubR was purified as described (14). Briefly, the protein was precipitated with an 80% of ammonium sulphate followed by a cation exchange chromatography and a gel filtration in 20 mM Tris–HCl pH 7.5, 150 mM KCl, 1 mM EDTA, 1 mM DTT. After concentration protein was kept at −80°C.
DNA substrates for EMSA and AFM experiments
Non-labeled DNAs tubC, SF1, SF2, SF3, CPRZ, NPDC, RCPR, CP1, PR1-9 and Random were obtained by PCR using the oligos of Supplementary Table S1 and pUC57 vector including the whole c-st partition operon (Shinegene). The NPDC fragment was made by ligation of a PCR product of tubC and the DNA related to the N-terminal domain of bacterial cell division protein FtsZ from B. subtilis. DNAs SF1b, SF3b and Shuffle (Supplementary Table S2) were obtained by annealing oligonucleotides (Metabion) in 10 mM Tris–HCl, pH 8.0, 50 mM NaCl, 1 mM EDTA. Hybridation of complementary strands was conducted in a thermocycler by heating the mixture to 95°C and cooling it down to 10°C at 1°C/min. 5′-fluorescein labeled SF1b and SF3b fragments were obtained by oligonucleotide annealing (Metabion), where the hybridization reaction contained a 10% excess of the unlabeled nucleotide in the mixture. The correct hybridation of these fragments was checked by polyacrylamide gel electrophoresis and Gel-Red (Biotium) staining. Two DNA molecules, AFM-C1 (508 bp) and AFM-C2 (2114 bp), not containing the centromere region were used as controls in AFM experiments. AFM-C1 was produced by PCR using appropriate oligos (Supplementary Table S1) and the plasmid pSP73-ParB (15). AFM-C2 was produced by enzymatic digestion of a plasmid derived from the pET28a-ParB (15) with BamHI and SphI.
DNA substrates for MT experiments
We fabricated different DNA constructs for magnetic tweezers experiments. The MT1 substrate consists of a large central insert of 5697 bp containing the c-st partition operon flanked by two smaller fragments, the handles (∼1 kb), labeled with biotins or digoxigenins. The labeled parts are used to specifically bind each DNA end to a glass surface covered by anti-digoxigenins and to streptavidin coated magnetic beads.
The large central part was generated by PCR amplification using the oligos F-NcoI-pUC57 and R-XhoI-pUC57 (Supplementary Table S1). After digestion, the PCR product was electrophoresed on a 1% agarose gel and extracted (QIAGEN Gel Extraction Kit). The handles labeled with biotins or digoxigenins were amplified by PCR from plasmid pSP73-JYo (16) using the oligos FMH-F2 and JOE-R1 (Supplementary Table S1) and biotin-dUTP or digoxigenin-dUTP (Roche). The labeled fragments were cut with NcoI (digoxigenin-labeled handle) or XhoI (biotinylated handle) and ligated with the central part. The final ligation included 4.4 nM of the central insert, 44 nM Bio-XhoI handle and 44 nM Dig-NcoI handle. The quality of ligated products was analyzed by electrophoresis and digestion with restriction enzymes. DNAs were never exposed to intercalant dyes or UV radiation during their production and were stored at 4°C. Details of other DNA substrates employed in MT experiments are described in supplementary information.
Electrophoretic mobility shift assays
For the binding reaction, variable amounts of protein were diluted in buffer BB (20 mM Tris–HCl, pH 8.0, 50 mM KCl, 1 mM EDTA) containing 0.5 mg/ml heparine and 200 nM of DNA. The mixture was incubated 5 min at RT, loaded into a 8% polyacrylamide gel (pre-run 30 min at 100 V), and run in 0.5× TBE (45 mM Tris borate, pH 8.2, 1 mM EDTA) at 100 V for 30–45 min. Gels were stained with a solution 1:10 000 of Gel-Red and visualized in a GelDoc (BioRad).
DNase I footprinting
Oligonucleotides were radioactively labeled at the 5′-end using [ γ-32P]-ATP (PerkinElmer) and T4 polynucleotide kinase (NEB). Reaction mixtures containing 25 pmol of oligonucleotide, 10× kinase buffer, 50 pmol of [ γ-32P]-ATP (3000 Ci/mmol) and 20 units of T4 polynucleotide kinase were incubated at 37°C for 30 min. Then, these reactions were incubated at 65°C for another 20 min to inactivate the kinase. The non-incorporated nucleotide was removed using Illustra MicroSpinTM G-25 columns (GE Healthcare). These oligonucleotides were used for PCR amplification to obtain dsDNA fragments labeled at either the coding or the non-coding strand. For the DNase I footprinting, the binding reactions contained 30 mM Tris–HCl (pH 7.6), 1% glycerol, 1.2 mM DTT, 0.2 mM EDTA, 50 mM NaCl, 1 mM CaCl2, 10 mM MgCl2, 500 mg/ml BSA, 4 nM of 32P-labeled DNA and increasing protein concentrations (126–960 nM). After 20 min at RT, 0.04 units of DNase I (Roche Applied Science) were added. Mixtures were further incubated for 5 min and the reactions stopped by the addition of Stop DNase I solution (2 M ammonium acetate, 0.8 M sodium acetate, 0.15 M EDTA). DNA was precipitated with ethanol, dried and dissolved in loading buffer (80% formamide, 1 mM EDTA, 10 mM NaOH, 0.1% bromophenol blue and 0.1% xylene cyanol). Samples were heated at 95°C for 5 min and loaded onto an 8 M urea–6% polyacrylamide gel. Dideoxy-mediated chain termination sequencing reactions were run in the same gel. Labeled products were visualized using a Fujifilm Image Analyzer FLA-3000 or by autoradiography. The intensity of the bands was quantified using Quantity One Software (BioRad).
Fluorescence anisotropy binding titrations
Binding reactions as described for EMSA experiments were repeated in the absence of heparine. We progressively increased the buffer ionic strength to avoid protein–DNA non-specific interactions and we found similar binding conditions when the binding buffer contained 150 mM KCl (buffer BB150). Anisotropy measurements were collected with Appliskan (Thermo Electro Corporation) using black U96 Nunc plates (Thermo Scientific) and polarized filters of excitation at 485 nm (band width 10 nm) and emission at 520 nm (band width 20 nm). The experiments were carried out by titrating TubR into a 150 μl of reaction buffer containing 20 nM of labeled DNA. Plates were shaken for 1 min (5 mm amplitude) and each well was measured for 1000 ms. The resulting anisotropy converged data were used to calculate the bound fraction and free protein concentration. These data were fit to a simple bimolecular binding model by non-linear regression (Equation 1) or to the Hill equation when cooperativity was present (Equation 2) to determine an apparent dissociation constant (Ka), and the degree of cooperativity (h).
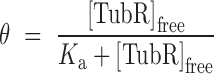 |
(1) |
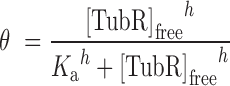 |
(2) |
where 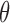 is the fraction bound, [TubR]free is the free concentration of protein, Ka is the apparent dissociation constant or ligand concentration at half occupancy and h is the Hill coefficient.
is the fraction bound, [TubR]free is the free concentration of protein, Ka is the apparent dissociation constant or ligand concentration at half occupancy and h is the Hill coefficient.
Analytical ultracentrifugation
Sedimentation velocity and equilibrium experiments were performed at 25°C in a Beckman Optima XLI analytical ultracentrifuge with absorption optics (260 and 233 nm), using an An50/Ti rotor with 12-mm double sector centrepieces. All experiments were done in buffer BB150 and samples contained 1.2 μM DNA, 1.2 μM DNA + 1.2 μM TubR, 1.2 μM DNA + 2.4 μM TubR, 1.2 μM DNA + 4.8 μM TubR and 1.2 μM DNA + 9.6 μM TubR. Speed experiments were conducted at 48 000 rpm and sedimentation coefficient distribution, c(s), was calculated using SEDFIT 12.51 (17). In equilibrium experiments the samples were centrifuged at speeds of 10 000, 13 000 and 15 000 rpm. Data were analyzed using Heteroanalysis 1.1.44 software (18). The binding of TubR to SF3b was measured from the increment over the buoyant molecular weight of the DNA at increasing protein concentrations (the protein contributed comparatively very little to the global absorbance at 260 nm), taking into account the free DNA measured in the speed experiments. The average buoyant molecular weight values were measured from the radial concentration gradient considering Equation (3),
 |
(3) |
where M is the monomer molar mass, ν is the partial specific volume, ρ is the solvent density, ω is the angular rotor speed, R is the molar gas constant and T is the temperature.
Rotary shadowing
Sample containing 5 nM DNA (tubC or RCPR) with or without 210 nM TubR in buffer BB was applied to a carbon-coated grid that was glow discharged and treated with 0.01% polylysine for 1 min, stained with 2% uranyl acetate, dried and shadowed. Rotary shadowing was performed with a BAF060 Freeze Etching System (Leica Microsystems) using a 3.5 nM Pt/C source at an angle of 3°.
Atomic force microscopy
TubR stock solution was first diluted in BB150 buffer to a final concentration of 1.71 μM. Different volumes of the solution were further diluted to the preferred concentration into a final volume of 20 μl containing 0.5, 1, or 2 nM DNA in 10 mM Tris–HCl pH 8.0, 75 mM KCl. The TubR–DNA mixture was incubated for 1 min and deposited onto freshly cleaved mica. The surface was thoroughly washed with 3 ml of Milli-Q water and dried under nitrogen air-flow (19).
Images were taken with an AFM from Nanotec (Nanotec Electrónica, Madrid, Spain) using PointProbePlus tips, PPP-NCH (Nanosensors, Neuchâtel, Switzerland). AFM was operated using tapping mode for imaging in air, at room conditions. Image processing and data extraction was done with WSxM software (20).
Correlation between DNA and protein localization in AFM images
Details of 240 × 240 nm2 were selected from images of 2.5 × 2.5 μm2 and processed (plane extraction, flattening, height selection and smooth filtering) using WSxM software (20). A threshold height was applied to every AFM image. Data over 0.75 nm represented only the location of DNAs, and data between 0.35 and 0.7 nm were considered as protein data. These two sets of heights were converted into plain text square matrixes of 49 points named as matrix A and matrix B. The correlation coefficient 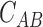 between matrixes was calculated following Equation (4),
between matrixes was calculated following Equation (4),
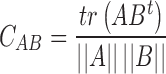 |
(4) |
Magnetic tweezers experiments
We used a custom-built MT apparatus similar to the one described previously (21–23). Optical images obtained with a CMOS camera operated at 120Hz is used to calculate beads’ three-dimensional position in real-time, using a custom made software programmed in LabVIEW. Forces are calculated using the Brownian motion method applied to DNA-tethered beads and corrected for low-pass filtering and aliasing (21,24,25). Alignment of magnets and force calibration of the instrument were done by performing force-extension curves of DNA molecules and fitting to the worm-like-chain model. The spatial resolution of our setup is 2–3 and 5 nm for in-plane and vertical positions, respectively (23). The nicked or torsionally-constrained character of each DNA tether was determined by performing rotation curves at different forces, as described (23). TubR protein flow was set to 20 μl/min.
RESULTS
TubR binds tubC on c-st Type III partition operon at multiple binding sites
In a previous work we showed the organization of the prophage c-st segregation locus (Figure 1A), but could not characterize the centromere region, tubC (14). To elucidate the molecular mechanism responsible for the formation of the partition complex we first investigated the extension and architecture of tubC by analyzing the interactions of recombinant TubR and the region upstream its locus. The non-coding DNA region between tubR and the following upstream gene (cst191) covers ∼500 bp enriched in direct and inverted repetitions, precluding the identification of TubR primary recognition site. Immediately upstream tubR there are a predicted Shine-Dalgarno sequence (at 26 bp) and a predicted promoter region, without proven functionality (here on assigned as ‘promoter’ for simplicity), with boxes −10 and −35 at 36 and 58 bp, respectively (phiSite, (26)). Hence, for the initial characterization of tubC and the study of the formation of the partition complex we choose the 352 bp DNA fragment between the promoter upstream tubR and the terminator region downstream cst191 (Figure 1A, Supplementary Table S2).
Figure 1.

tubC characterization and interaction with TubR. (A) Scheme of c-st encoded Type III partition systems including the number of bp of each gene and intergenic regions, and details related to the centromere-site and the promoter regions. The 352 bp tubC site was split into three overlapping fragments named SF1, SF2 and SF3. Fragments SF1 and SF3 show the localization of derived fragments SF1b and SF3b (related to Figure 2A). (B) EMSA experiments of 0.2 μM of each DNA fragment and increasing TubR concentrations in the presence of 0.5 mg/ml heparine as DNA competitor. EMSAs show the formation of multiple complexes that converge into a single one (red arrows) at protein saturating concentrations. Protein concentrations are 0.4–10 μM for all DNA fragments except for SF2 (0.6–7 μM). (C) DNase I footprinting experiments of 4 nM radiolabeled SF1 forward strand. Sequence of free DNA is shown by Maxam reaction (lane 1), free DNA (lane 2), 852 nM TubR (lane 3), 400 nM TubR (lane 4) and 252 nM TubR (lane 5) of TubR. Red and blue color lines represent the position of each iteron within the fragment (numbering is related to Figure 2A). Over-exposed bands (asterisks) and primary binding region in forward strand (dashed line) are marked on the right of the gel.
EMSA experiments demonstrated that purified TubR binds tubC specifically and showed the formation of multiple protein-DNA complexes, suggesting the presence of multiple binding sites (Figure 1B, upper panel). Further, at high protein concentrations the size of the complex or complexes prevented migration into the 8% native polyacrylamide EMSA gel. To specifically determine TubR recognition site and to perform DNase I footprinting experiments (see below) we decided to split the centromere-site into three overlapping subfragments of 146 bp (SF1), 141 bp (SF2) and 131 bp (SF3) (Figure 1A, Supplementary Table S2). EMSA experiments with these subfragments showed again the formation of multiple complexes, but importantly, in this case we could identify a single final complex at high protein concentration that could penetrate the gel (Figure 1B, lower panels). The only iteron in common between these three subfragments is A/TGAA with 6–7 repetitions, suggesting this might be the TubR primary binding site. Control experiments with random DNA of similar size (Supplementary Table S2) showed non-specific binding at much higher TubR concentration than that found for specific binding of TubR (Supplementary Figure S1D).
DNase I footprinting experiments of SF1 and SF3 showed that TubR protected most of the DNA from digestion when analysing both strands, denoting a high avidity of this protein for the centromere (Figure 1C, Supplementary Figure S1). Even more, we could identify the position of persistent and hypersensitive bands, always on the forward strand and at the lowest TubR concentrations, suggesting a directional DNA bending (Figure 1C and Supplementary Figure S1, asterisks), consistent with the ‘flexible’ and curved filaments found on pBtoxis and pBM400 tubCR complexes (10). However, our results support a centromere region covering more than 350 bp and therefore larger than those previously described in pBtoxis (∼130 bp) and pBsph (∼250 bp) (10,11).
The centromere tubC contains low and high affinity TubR binding sites
Sequence alignment of SF1 and SF3 revealed a very close pattern where the iteron A/TGAA (Figure 2A, red) mixes with another repetition, TTGAC (Figure 2A, blue), which appears three times spaced by 6 bp in SF1 and only once in SF3. Further, the densitometry of our footprinting assays suggested that this un-matching region supports the putative TubR primary binding site on both subfragments (Figure 1C, Supplementary Figure S1), denoting that iterons TTGAC might have an important function on the recognition of DNA by TubR. To shed light on this protein-DNA binding mechanism we further studied TubR binding to 51bp DNA subfragments containing these regions: SF1b includes three TTGAC iterons plus one A/TGAA and SF3b contains the mismatched site with one TTGAC repetition and three A/TGAA (Figure 2A, Supplementary Table S2). The initial protein-DNA binding interaction measured by the anisotropy change of fluorescein labeled subfragments with increasing protein concentrations revealed a clear difference on TubR binding mechanism (Figure 2B). The protein bound with similar apparent dissociation constant to both fragments (KaSF1b = 183.7 ± 4.04 nM and KaSF3b = 138.2 ± 7.7 nM), but only the interaction with SF1b displayed a cooperative binding mechanism, with a Hill coefficient of 3.1, denoting that the centromere contains stronger and weaker binding sites.
Figure 2.

tubC high and low affinity binding sites. (A) SF1 and SF3 DNA fragments alignment using Clustal Omega. Iterons TTGAC (in blue) and A/TGAA (in red) are numbered in each fragment from I-VIII. Derived subfragments SF1b and SF3b are marked on each fragment. (B) Anisotropy analysis of protein binding to fluorescein labeled SF1b and SF3b subfragments and fitting curves to cooperative (upper panel) and non-cooperative (lower panel) interactions for Ka and hill coefficient determinations. (C) EMSA experiments of 0.2 μM of non-labeled SF1b and SF3b fragments titrated with increasing TubR concentrations (0.3–7 μM) in the presence of 0.5 mg/ml heparine as DNA competitor. Red arrows indicate the identified complexes. (D–E) Analytical Ultracentrifugation Speed experiments of TubR interaction with SF1b (D) and SF3b (E) showing TubR (light blue), DNA (dark blue) and complexes formation (black) at increasing protein-DNA ratios (1:1, solid; 1:2, dotted; 1:4, dot-dot-dash and 1:8, dash lines). (E inset) Determination of the number of TubR molecules bound per SF3b fragment from the increment of mass at different protein concentrations in AUC equilibrium experiments.
Surprisingly, we found no correlation between the number of iterons in these sub-fragments and the complexes formed in EMSA experiments (Figure 2C). TubR binding to SF1b only produced two initial complexes that evolved into a single bigger complex at higher protein concentrations (Figure 2C, upper panel, see arrows), whereas the interaction with SF3b produced a single complex that barely changed in size at TubR increasing concentrations (Figure 2C, lower panel, see arrow). Importantly, TubR did not bind to a Shuffle 51bp fragment (Supplementary Figure S1E) denoting the importance of these repetitions on protein interaction. To further characterize those complexes we performed AUC speed experiments, where we could easily discern the sedimentation coefficients of TubR (1.2S), DNAs (∼4S) and complexes (>5S) (Figure 2D and E). Interestingly, TubR binding to any of these subfragments produced a single peak that rose on the sedimentation coefficients at increasing protein concentrations while the free DNA peak reduced. Therefore, we presume that the two complexes identified in the EMSA experiments when TubR interacts with SF1b must have a very close composition precluding their distinction in the AUC assays.
We further analyzed the single TubR-SF3b complex with the aim to estimate the number of TubR molecules bound per DNA. To this end, we used AUC speed experiments to determine the ratio of species (free DNA vs. protein bound DNA) at different TubR concentrations. Also, AUC equilibrium assays under the same conditions gave us the increment of mass of the complex due to TubR binding (methods). We further corrected these data by the fraction of DNA molecules that remains free to finally plot the fraction of protein bound related to the increment of TubR concentration (Figure 2E, inset). Interestingly, these data showed that despite TubR is a monomer in solution (14), as shown in current AUC speed experiments and AFM volumetric analysis (data not shown) (27), the lowest number of protein molecules bound to this fragment of DNA was two. This might represent a dimerization process on the DNA. We estimated up to 12 monomers at the highest protein concentration (Figure 2E, inset) suggesting that TubR not only binds to the four iterons present on SF3b sequence but also spreads along the whole DNA fragment, with an estimation of ∼ 2.5 TubR molecules per DNA turn.
The segrosome complex forms a double ring structure
Initial rotary shadowing EM images of tubC (352 bp) in the presence of TubR highlighted the formation of flexible filaments able to close into ring-like structures (> 70% of fragments, n > 500, Figure 3A and Supplementary Figure S2), resembling previous images showed with pBtoxis and pBM400 segrosome complexes (10). To avoid any potential alteration of the complexes due to the staining procedure we approached subsequent structural characterization by AFM. Interestingly, AFM images gave us a slightly different view of the nucleoprotein complex architecture since we observed a mixture of DNA configurations (Figure 3B), with single and double ring-like structures (40% and 50%, respectively, n > 1000), and about 10% of linear DNA molecules (presumably protein free DNAs). We observed a similar distribution of DNA configurations for different protein:DNA ratios up to 42:1 (Supplementary Figure S3A). Importantly, TubR in the presence of two control DNAs (without tubC) was unable to form loops (Supplementary Figure S4). This suggests that the nucleoprotein complex formation involves the assembly of two rings instead of one, where each loop could be related to the interaction of the protein with the stronger and the weaker binding sites we identified on the centromere sequence.
Figure 3.

Structural studies on the segrosome formation. (A) Electron micrographs of rotary shadowed TubR bound to tubC showing the bending of DNA-protein filaments from open loops to close rings (scale bar, 20 nm). (B) AFM images of TubR bound to tubC showing partially and fully closed loops at one or both ends (scale bar, 30 nm). (C, D) Top. Scheme of CPRZ/NPDC fragments composition. Medium. AFM images of TubR bound to fragment CPRZ (C) and NPDC (D), showing loops L1 (SF3) and L2 (SF1) and highlighting the presence of free DNA (black arrow) and DNAs with one (red arrow) or two (yellow arrow) loops (scale bar, 200 nm). Bottom. CPRZ/NPDC fragments scheme showing the localization of loops L1 and L2 and blue arrows following the distance measured to determine loop centre positions. (E) Scheme showing tubR upstream region highlighting the fitting of each loop with subfragments SF3 and SF1 and the promoter position. (F) AFM (scale bar, 50 nm) and (G) rotary shadowing images (scale bar, 100 nm) of TubR bound to RCPR fragment showing the formation of one or two partially or fully closed loops.
With the aim to correlate the position of the loops within the DNA sequence, we decided to build a longer DNA fragment of 866 bp (CPRZ) including tubC at the 5′- end, followed by the promoter region, tubR gene and the N-terminal region of tubZ gene (Figure 3C, Supplementary Table S2). Interestingly, AFM images considering different TubR:CPRZ ratios showed roughly the same DNA distribution as before, where again a low fraction of free DNA molecules (Figure 3C, black arrow) appeared together with DNAs containing one (red arrow) or two (yellow arrow) partially or completely closed loops (n > 1000, Supplementary Figure S3B). In this fragment, the L1 loop at the 5′-end (Figure 3C, lower cartoon) would fit with the weaker binding site (at SF3) and the second loop (L2) with the stronger binding site (at SF1). We then built a second fragment of 907 bp (NPDC) where the centromere region was at the 3′-end following a sequence of random non-phage DNA (Figure 3D and Supplementary Table S2). Similarly, AFM images displayed DNA molecules with one (red arrow) or two (yellow arrow) partially or fully closed loops with similar occupancies as in fragments tubC and CPRZ (n > 1000, Supplementary Figure S3C). However, in NPDC, the loop at the end of the fragment would fit with SF1 (L2) and the second loop with the SF3 region (L1) (Figure 3D, lower cartoon). To localize these loops on the centromere sequence, we measured the distance of the middle position of each loop from the free-loop DNA end (Figure 3C and D). In fragment CPRZ we found that L1 was at 270 ± 5 nm (794 ± 15 bp, Figure 3C cartoon and Supplementary Figure S5A) corresponding to the mismatch region between SF1 and SF3, whereas in NPDC it was at 196 ± 2 nm (576 ± 6 bp) corresponding to the 5′ end of SF3. In fragment CPRZ, L2 was at 180 ± 5 nm (529 ± 15 bp), very close to the promoter, and, in fragment NPDC was at 262 ± 3 nm (771 ± 9 bp), related to 5′end of SF1. Hence, we confirmed that the highly curved loop structures correlate with SF1 and SF3 regions, which contain the iterons described above (Figure 3E). The perimeter length, measured at the maximum height of the DNA, of partially closed rings was about 50 nm (Supplementary Figure S5C) whereas in fully closed rings this value expanded to up to 67 nm (Supplementary Figure S5D). This result supports the idea that the protein bound to the DNA induces a maximum bending angle and to fully close the ring, TubR needs to spread further on the DNA (see MT data below). Control AFM experiment of bare substrates did not show any feature similar to the loops reported above and contour lengths were in agreement with B-form DNA (Supplementary Figure S6).
It may be argued that the presence of a binding site at the very end of the DNA could favour protein binding and DNA bending, whereas in the plasmid, surrounding DNA flanks the centromere region. To discard any end-binding artefact in AFM imaging and to corroborate that TubR binds to two different sites and induce the formation of two separate rings, we built a new fragment of 977 bp (RCPR) where tubC (352 bp) sits between ∼300 bp of random DNA (upstream) and ∼320 bp of DNA including the promoter and tubR gene (downstream), (Figure 3F, G, Supplementary Figure S2, Supplementary Table S2). Similar to previous AFM images we identified one or two loops that were partially or totally closed at similar ratios as in other fragments (n > 600, Figure 3F), regardless of the amount of protein used (Supplementary Figure S3D). Interestingly, rotary shadowing EM images clearly showed double-ring structures assembled on a single DNA chain (Figure 3G, Supplementary Figure S2), denoting the importance of placing the centromere in the context of a longer DNA when analysing the segrosome complex formation (Figure 3A and G). Moreover, these EM ring-like structures showed diameters of 21.4 ± 2.8 nm (n > 300), which correlates very well with the diameter derived from the perimeter length measured in AFM images. We propose that the double-ring structure might be the typical segrosome complex architecture in Type III partition systems since all the centromeres characterized so far are split in at least two separated boxes (see Discussion).
The segrosome complex partially covers the predicted promoter region
Previous results suggested a direct interaction of TubR with the promoter region (11), which in our case is predicted (by a database of gene regulation in bacteriophages, phiSite) to be very close to the centromere's stronger binding site (downstream SF1) and therefore could be partially covered by the protein when this loop forms a complete ring-like structure (Figure 3E). Hence, to determine if TubR directly interacts with this promoter we prepared a set of nine DNA fragments (PR1-9, Supplementary Table S2) of 94bp long each. The first fragment (PR1) included the whole promoter region and subsequent fragments moved 10 bp downstream toward the tubR gene (Figure 4A). Then, we analyzed TubR ability to bind and form a complex with each of these fragments in 8% native polyacrylamide gels. First, we could confirm the direct binding of TubR to the promoter region. Second, we found that once the −10 and −35 predicted promoter boxes were absent within the fragment (PR5–PR9), there was a dramatic change of protein–DNA interaction characterized by the lost of binding affinity (free DNA molecules) and the formation of smaller size complexes and smeared DNA (usually due to unspecific or weak protein-DNA interactions). The formation of smaller size complexes is likely due to the presence of further A/TGAA repetitions appearing in the tubR gene at positions 15, 35, 62 and 80 bp from the ATG initial codon. This result supports the implication of A/TGAA on TubR spreading and suggests that the protein could spans over wider DNA regions. Unfortunately, AFM could not resolve individual protein-DNA complexes, and we could only infer the coverage of the protein by the length of the loop.
Figure 4.

Promoter implication on segrosome formation. (A) Schematic representation of the 94 bp fragments used to identify TubR binding to the predicted promoter region, indicating the positions of fragments PR1-9 with respect to the promoter and tubR gene. The lower part shows the migration of each fragment (0.2 μM) and the DNA band shift upon TubR addition (3.5 μM). (B) EMSA experiments of 0.2 μM of non-labeled fragments CP1, PR1 and PR9 titrated with increasing protein concentrations (0.3–7 μM), showing the earlier formation of complexes in CP1 than in PR1 (*). (C) Error bars graph representing the distances between both rings in all the fragments used in this work.
EMSA experiments using fragment PR1 and a control fragment (CP1) containing the centromere stronger binding site in a 94bp fragment length confirmed that despite TubR directly binds to the promoter region, the binding avidity is lower than to the region containing the three TTGAC repetitions (Figure 4B). Further, protein binding to PR9 gave smeared DNA at the highest protein concentrations used. Therefore, the covering of the promoter region by TubR might be a consequence of the segrosome complex formation rather than a primary protein-DNA interaction.
Finally, AFM images also allowed us to measure the distance between the centres of the two rings. Interestingly, we found that in the absence of the promoter region (tubC and NPDC) both rings were often closer than when the promoter was present in the DNA fragment (Figure 4C, CPRZ, RCPR), which supports the importance of the promoter region on the final segrosome complex conformation.
TubR untwists the DNA upon binding
MT experiments provide a measurement of the extension of individual DNA molecules as function of time for a given stretching force. In MT, a single DNA molecule is tethered between a glass surface coated with anti-digoxigenin and a streptavidin-coated magnetic bead. A set of permanent magnets is employed to stretch and twist the DNA by translation and rotation of the magnets, respectively (Figure 5A). Importantly, this technique is able to supercoil torsionally-constrained DNA (tcDNA) molecules, and this is reflected in a reduction of their extension due to the formation of plectonemic structures. Reciprocally, the action of proteins that twist a tcDNA molecule can be monitored by measuring changes in its extension in real time. We employed this unique capability of MT to investigate further the binding of TubR to the centromere region and hence, we assayed the protein-DNA interaction on a DNA molecule (MT1, Supplementary Table S2) of ∼5.7 kb that contained the Type III partition cassette (tubC, the promoter, tubR, tubZ and partial tubY, Figure 5A). This segregation locus was linked to two different DNA fragments (the handles) to attach the 5′-end of the molecule to the glass surface, and the 3′-end to the magnetic bead. Hence, in MT1, the centromere was located at ∼2.1 kb from the bead and ∼3.2 kb from the glass surface (Figure 5A).
Figure 5.

TubR bends and untwists the DNA upon binding. (A) Schematic representation of the MT1 molecule used in MT experiments where the DNA substrate contains the c-st partition operon, and the experimental configuration used to measure TubR binding with magnetic tweezers. (B) TubR binding assay. DNA molecules were first identified as tcDNA or nicked by doing a rotation curve (Supplementary Figure S7). At 1 pN stretching force, 100 nM TubR was injected in the fluid cell while the extension of the DNA molecules was recorded. Different amplitudes of extension reduction were observed for tcDNA and nicked DNA. The extension of tcDNA molecules increased upon negative (clockwise) rotations of the magnets (right panel). (C) Histogram of extension reduction for tcDNA and nicked DNA molecules upon TubR incubation. (D) Cartoons depicting the interpretation of extension reduction for tcDNA and nicked DNA. TubR untwist the DNA generating positive supercoiling in tcDNA molecules (left). Nicked molecules cannot be supercoiled and therefore the reduction in extension can only be explained by bending of the DNA by TubR (right).
We explored the behaviour of the system at an intermediate force of 1 pN. As expected, the DNA decreased its extension upon injection of 100 nM TubR, consistent with the formation of the rings observed in AFM images and EM micrographs (Figure 5B, left panel). The reduction of the extension of the DNA was progressive but self-limited, as the bead never reached the glass surface, and lasted for ∼40 s, well below the overall flowing time. Interestingly, we detected two populations of molecules, which reduction in extension correlated with DNA being torsionally-constrained or nicked (Supplementary Figure S7A, left panel). Nicked DNA molecules, which cannot be supercoiled, reduced its extension in 300 ± 49 nm (mean ± SD, n = 90), whereas tcDNA molecules reduced their total length in 579 ± 155 nm (mean ± SD, n = 110, Figure 5C).
We interpret the larger decrease of the extension observed for tcDNA molecules as a consequence of the generation of positive supercoils, due to DNA untwisting by TubR (Figure 5D). At 1 pN, a tcDNA molecule can only reduce its extension by over twisting its double-helix structure (Supplementary Figure S8A). This can be done by applying positive turns with the magnet, but also by the action of proteins that partially untwist a section of the DNA, which have to generate an excess of positive turns at the uncovered part of the molecule in order to maintain constant the Linking Number of the tether (Supplementary Methods). Indeed, we were able to recover the extension of these molecules by applying negative turns (Figure 5B, right panel). The fact that nicked DNA molecules also showed a decrease in their extension upon TubR binding indicates that TubR also bends the DNA and that the formation of rings might not require DNA (un)twisting (Figure 5D). Importantly, the rotation curve at 1 pN showed that the 279 nm length decrease (this is the difference in the reduction of extension between tcDNA and nicked DNA) is achieved upon the application of 18 positive turns (Supplementary Figure S8B). Hence, TubR should untwist the DNA by the same number of turns.
The DNA extension was partially recovered upon impairing the DNA-protein interaction by flowing buffer with an excess of salt (5 M NaCl solution, Supplementary Figure S7A, right panel), and fully recovered by further exchange with the reaction buffer (Supplementary Figure S7B and Supplementary Figure S7C). This confirmed that the DNA length reduction was protein-interaction dependent. Experiments with a DNA molecule devoid of partition cassette (MT2, Supplementary Table S2), showed no change of DNA extension upon TubR injection, supporting that TubR directly interacts with the segregation locus (Supplementary Figure S9A). However, an additional experiment employing a longer molecule (MT3, Supplementary Table S2) also devoid of partition cassette did show a decrease of extension (Supplementary Figure S9B). We note that the longer molecule contains many more repetitions of TTGAC and A/TGAA sequences, and we interpret the decrease of extension as interactions of TubR with these and/or other sequences. Importantly, we observed an identical differential behaviour for nicked and tcDNA molecules, and this confirms the mechanism proposed of DNA bending and untwisting by TubR (Figure 5D).
Experiments at higher forces (4 pN) reported a smaller decrease of tcDNA length and several nicked molecules did not show any reduction in extension, consistent with a force-dependent process (data not shown). A detailed study of the force dependence of TubR nucleoprotein filament formation will be the subject of a future work.
DISCUSSION
Common to all described centromeres in Type III partition systems, prophage c-st tubC is located in a single and discontinuous locus upstream of the operon (10,11), but it is larger and covers ∼350 bp, leaving ∼190 bp between the stronger and the weaker binding sites. Interestingly, we have found that this centromere harbours two different repetitions arranged in a slightly different pattern in each site and shaping the binding avidity of TubR. This is a rare feature but not unique since previous studies on the centromere site of prophage P1 also includes two different boxes (28). However important differences arise from both CBPs (ParB and TubR) that suggest a different mechanism on DNA recognition (see below).
We found that TubR independently binds to both sites likely by the recognition of the iteron TTGAC. Otherwise, the protein also binds to DNA fragments where there is no such sequence, like the inter-rings section corresponding to SF2 (Figure 1B) that contains the iteron A/TGAA. Our work also presents several evidences supporting TubR spreading. The estimation of TubR molecules per DNA fragment obtained from AUC experiments suggested the protein ability to bind and spread along the DNA (Figure 2E). Additionally, EMSA experiments proved TubR binding to non-centromeric DNAs like the promoter (PR1–4 fragments in Figure 4A), and AFM experiments also predicted the interaction with the promoter (Figures 3C and 4C). First, the length of the loops indicated a partial covering of the promoter (Figure 3C) and second, in the absence of the promoter region the distance between the two loops was smaller than in substrates with the promoter (Figure 4C), which could be explained by TubR covering this promoter region. Finally, MT experiments showed that the nucleoprotein complex spread along ∼1 kbp (the length corresponding to ∼300 nm at 1 pN force (Figure 5), although this technique does not allow to determine the exact region covered by TubR. All together, we propose that TubR primary recognition site might be the iteron TTGAC, whereas the repetition A/TGAA would help on protein spreading along the DNA. Control DNAs without these sequences did not produce defined complexes but a smear of DNA at very high TubR concentrations. Thus, TubR spreading might cover a region that involves the promoter but it also requires an initial nucleation site. Note, however, that our data only prove TubR covering of this region but not functionality. Evidences of spreading of CBPs beyond the specific binding sites have been reported for other types of partition systems (29). However, the mechanism of spreading of B.subtilis ParB is clearly different from CbTubR, as we do not see bridging (15,30), but unwinding and bending of DNA (see below).
Interestingly, TubR binding affinity to tubC in prophage c-st is lower than in pBtoxis (Ka of 138–184 nM versus 11 nM), which might be related to the monomeric versus dimeric states of each protein in solution. All known TubR proteins, apart from this one encoded in prophage c-st, arrange into strong dimers that could favour the formation of a larger protein-DNA interacting surface and so, promote a higher protein binding affinity. Otherwise, the monomeric state of TubR from prophage c-st could be key on the protein ability to interact with different iterons. Secondary structure prediction and sequence alignment highlight that prophage c-st TubR lacks the N-terminal dimerization helix and the C-terminal helical extension present in other TubR proteins, which could explain the absence of a dimeric state in solution. Dimeric DNA binding proteins interact with repetitions located at a particular distance, often imposed by the distance between the DNA binding motifs in those dimers. Prophage TubR only dimerizes once bound to the DNA (Figure 2E) and likely, this contributes to a higher flexibility of the protein to accommodate different bases during DNA binding, which could also facilitate spreading. Importantly, monomeric TubR also shows a higher cooperativity than dimeric TubR from pBtoxis (Hill coefficient of 3.1 versus 1.9), which may be explained by the higher chances of addition of a second monomer (rather than a dimer) after the formation of stable contacts within the DNA.
We have proved the topography of the complex by both AFM and EM. Images reveal a double-ring structure that covers the centromere-binding site and beyond. Importantly, all the centromeres described in Type III partition systems are split in several blocks, suggesting that this double-ring structure might be common among them. Measurements with MT showed coverage of TubR for over ∼1 kbp, larger than the length of the double-ring structure observed in AFM. On one hand, we cannot discard TubR binding to other sequences apart from to the partition cassette in MT experiments. It is also possible that the AFM ring structures might be incomplete (i.e. in 352bp tubC) or biased (in longer fragments NPRZ, NPDC) since these structures do not cover the whole fragment. Indeed, we have found a constant correlation on the presence of single and double rings when assayed different protein:DNA ratios in these experiments. Finally, in both AFM and EM we consider TubR covering by measuring the length of the bend structures lying on the surface, but here we miss the three dimensional component where lateral contacts could contribute on the segrosome complex stability (see model below).
The segrosome complex covers tubC, the promoter, and beyond. We have seen that TubR binds directly to the promoter region as previously shown in pBsph Type III partition operon (11). Substrates lacking the promoter showed lower distances between rings and we speculate that this might involve a conformational change on the double-ring structure. How this can affect subsequent steps on DNA mobilization is not clear, although it could be related to the correct tracking of the segrosome at TubZ filament/s minus ends (13). Importantly, the combination of our data with previous studies showing TubR implication on the operon transcriptional repression (7,11) suggests that the promoter is likely hijacked within the segrosome complex hampering the interaction of the transcriptional machinery. Hence, similar to Type II partition systems this nucleoprotein complex would be a molecular switch that turns off the expression of the partition genes, probably once the levels of the encoded proteins have reached a critical expression level for DNA mobilization. However, in Type II partition systems the promoter region is not part of the segrosome structure but forms a DNA loop that protrudes out (31,32).
Segrosome assembly via wrapping is a common mechanism in Type Ib and Type II partition systems, but the resulting segrosome complex is completely different. In Type Ib partition systems the CBP forms a left-handed helix that wraps only the centromere site (33,34), forming a rigid and filamentous nucleoprotein complex that does not bend the DNA. Then, in Type II partition systems the segrosome displays a continuous helical array, where the DNA wraps a CBP superhelix made of dimers-of-dimers (35,36). We have found that in Type III partition systems the segrosome complex might show a combination of both mechanisms. TubR binds and spreads on the centromere site and beyond with ∼3 monomers per turn on average according to our AUC estimations, which correlated with the number of molecules found in low-resolution structures (Protein Data Bank entries 4ASO and 4ASS (10)). Furthermore, these structures highlighted the formation of a TubR continuous right-handed helical arrangement wrapping one or both sides of the DNA. Importantly, we have seen strong changes on the DNA structure upon TubR binding. First, a DNA bending that was previously suggested (10), and we have proved that it is limited and involves the formation of a double-ring structure. Still, the underlying bending mechanism is unclear and will require further structural studies. Second, TubR also untwists the DNA in ∼6–7° by basepair according to our MT measurements (18 turns in a ∼1 kb covering). This value is far below the 15° per basepair untwisting measured on RecA (37), but essential for the generation of plectonemes on tcDNA molecules. Hence, it may have further implications on the phage genome packing to facilitate DNA translocation during segregation. Importantly, there is no correlation between both effects and so; DNA bending is independent from untwisting.
In the light of these results, we propose a new model where both protein wrapping and DNA bending combine on the formation of the final segrosome structure (Figure 6). Upon initial TubR binding (Figure 6A), the protein would dimerize and spread wrapping the DNA forming one or two right-handed helixes (Figure 6B). This binding bends the DNA but also generates a torque that untwists the DNA, inducing the formation of plectonemic structures in torsionally constrained molecules, the expected state in vivo. The combination of both, bending and untwisting the DNA, results in a helical ring-like structure that could display different three-dimensional arrangements (Figure 6C). In the previous single-ring segrosome model (10), the motor protein, TubZ, bound to the inner surface of the ring in a similar fashion as in Type II partition systems (38). In these systems, it is well known that the C-terminal domains of the CBPs (known to interact with the motor protein) cluster on the inside surface of the segrosome helical structure and hence, this is the place where they mediate the binding of the motor protein. In Type III partition systems the C-terminal region of TubR is also known to bind to the C-terminal tail of TubZ (6) and this interaction is only possible after the formation of the segrosome complex (10,13,14). However, we still have no clear information related to the arrangement of TubR binding motifs within the final nucleoprotein complex structure. In our model (Figure 6D), a TubZ filament (∼12 nm of diameter) could easily interact with the inside surface of each ring (∼20 nm of diameter). However, the final segrosome architecture would be critical on the tracking mechanism, allowing the interaction of one or two different TubZ filaments. This would have further implications on DNA segregation since in the case of two cytomotive filaments, these should work synchronized on pulling the DNA (13).
Figure 6.

Wrapping-Bending model of segrosome complex formation. TubR binds to DNA in a dimer conformation (A) and spreads wrapping the DNA by one or alternatively two sides (B), which induces certain DNA twisting (red arrow). Finally, DNA wrapped by TubR bends at the stronger and weaker binding sites (C) generating a two helical ring-like structures. Based on previous model (10), each ring could support one TubZ filament (D, left) for a synchronized DNA mobilization. Alternatively, a single TubZ filament could bind to the inter-ring region (D, right) favouring a simpler mechanism for DNA movement.
A key finding in this study is the implication of TubR on changing DNA structure by untwisting and bending. Both conformational changes are responsible of segrosome complex formation and presumably DNA mobilization upon segregation. Importantly, the segrosome complex structure is closely related to the architecture of the centromere site and covers a far wider extension of DNA, including the promoter. A direct consequence of the assembly of such nucleoprotein complex is the blockage of the transcription. Further structural and single molecule studies would help on understanding the molecular mechanisms involved in this protein-DNA interaction.
Supplementary Material
ACKNOWLEDGEMENTS
We would like to thank Dr JR Luque-Ortega (AUC facility at CSIC-CIB) for helpful discussion on AUC experiments.
Authors Contributions: M.A.O. and F.M.-H. designed the research. M.A.O., B.M.-G., A.M.-G., C.C., A.M.H.-A., R.R.-Q. and C.A.-R. performed research. M.A.O., F.M.-H., A.M.-G., A.M.H.-A. C.C. analyzed data. M.A.O. and F.M.-H. wrote the paper. All authors were involved in data interpretation and revising the manuscript.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Ministerio de Ciencia e Innovación [RYC-2011-07900 to M.A.O.]; Ministerio de Economía y Competitividad [BFU2013-47014-P to M.A.O. and BFU2016-75319-R, which were co-funded with an European Regional Development Fund (FEDER) and, FIS2014-58328-P to F.M.-H.]; European Research Council (ERC) under the European Union's Horizon 2020 Research and Innovation program [681299 to F.M.-H.]; Ministerio de Economía y Competitividad [BES-2015-071244 to A.M.-G.]. Funding for open access charge: Ministerio de Economía y Competitividad and FEDER.
Conflict of interest statement. None declared.
REFERENCES
- 1. Baxter J.C., Funnell B.E.. Plasmid partition mechanisms. Microbiol.Spectr. 2014; 2:1–20. [DOI] [PubMed] [Google Scholar]
- 2. Funnell B.E. ParB partition proteins: Complex formation and spreading at bacterial and plasmid centromeres. Front. Mol. Biosci. 2016; 3:44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Oliva M.A. Segrosome complex formation during DNA trafficking in bacterial cell division. Front. Mol. Biosci. 2016; 3:51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hayes F., Barilla D.. The bacterial segrosome: a dynamic nucleoprotein machine for DNA trafficking and segregation. Nat. Rev. Microbiol. 2006; 4:133–143. [DOI] [PubMed] [Google Scholar]
- 5. Schumacher M.A. Bacterial plasmid partition machinery: a minimalist approach to survival. Curr. Opin. Struct. Biol. 2012; 22:72–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ni L., Xu W., Kumaraswami M., Schumacher M.A.. Plasmid protein TubR uses a distinct mode of HTH-DNA binding and recruits the prokaryotic tubulin homolog TubZ to effect DNA partition. Proc. Natl. Acad. Sci. U.S.A. 2010; 107:11763–11768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Larsen R.A., Cusumano C., Fujioka A., Lim-Fong G., Patterson P., Pogliano J.. Treadmilling of a prokaryotic tubulin-like protein, TubZ, required for plasmid stability in Bacillus thuringiensis. Genes Dev. 2007; 21:1340–1352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Aylett C.H., Wang Q., Michie K.A., Amos L.A., Lowe J.. Filament structure of bacterial tubulin homologue TubZ. Proc. Natl. Acad. Sci. U.S.A. 2010; 107:19766–19771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Montabana E.A., Agard D.A.. Bacterial tubulin TubZ-Bt transitions between a two-stranded intermediate and a four-stranded filament upon GTP hydrolysis. Proc. Natl. Acad. Sci. U.S.A. 2014; 111:3407–3412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Aylett C.H., Lowe J.. Superstructure of the centromeric complex of TubZRC plasmid partitioning systems. Proc. Natl. Acad. Sci. U.S.A. 2012; 109:16522–16527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ge Y., Hu X., Zhao N., Shi T., Cai Q., Yuan Z.. A new tubRZ operon involved in the maintenance of the Bacillus sphaericus mosquitocidal plasmid pBsph. Microbiol. 2014; 160:1112–1124. [DOI] [PubMed] [Google Scholar]
- 12. Tang M., Bideshi D.K., Park H.W., Federici B.A.. Iteron-binding ORF157 and FtsZ-like ORF156 proteins encoded by pBtoxis play a role in its replication in Bacillus thuringiensis subsp. israelensis. J. Bacteriol. 2007; 189:8053–8058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Fink G., Lowe J.. Reconstitution of a prokaryotic minus end-tracking system using TubRC centromeric complexes and tubulin-like protein TubZ filaments. Proc. Natl. Acad. Sci. U.S.A. 2015; 112:E1845–E1850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Oliva M.A., Martin-Galiano A.J., Sakaguchi Y., Andreu J.M.. Tubulin homolog TubZ in a phage-encoded partition system. Proc. Natl. Acad. Sci. U.S.A. 2012; 109:7711–7716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Taylor J.A., Pastrana C.L., Butterer A., Pernstich C., Gwynn E.J., Sobott F., Moreno-Herrero F., Dillingham M.S.. Specific and non-specific interactions of ParB with DNA: implications for chromosome segregation. Nucleic Acids Res. 2015; 43:719–731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Fili N., Mashanov G.I., Toseland C.P., Batters C., Wallace M.I., Yeeles J.T., Dillingham M.S., Webb M.R., Molloy J.E.. Visualizing helicases unwinding DNA at the single molecule level. Nucleic Acids Res. 2010; 38:4448–4457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Schuck P., Perugini M.A., Gonzales N.R., Howlett G.J., Schubert D.. Size-distribution analysis of proteins by analytical ultracentrifugation: strategies and application to model systems. Biophys. J. 2002; 82:1096–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Cole J.L. Analysis of heterogeneous interactions. Methods Enzymol. 2004; 384:212–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lyubchenko Y.L., Shlyakhtenko L.S.. AFM for analysis of structure and dynamics of DNA and protein-DNA complexes. Methods. 2009; 47:206–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Horcas I., Fernandez R., Gomez-Rodriguez J.M., Colchero J., Gomez-Herrero J., Baro A.M.. WSXM: a software for scanning probe microscopy and a tool for nanotechnology. Rev. Sci. Instrum. 2007; 78:013705. [DOI] [PubMed] [Google Scholar]
- 21. Strick T.R., Allemand J.F., Bensimon D., Croquette V.. Behavior of supercoiled DNA. Biophys. J. 1998; 74:2016–2028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Seidel R.D. 3rd, Amor J.C., Kahn R.A., Prestegard J.H.. Conformational changes in human Arf1 on nucleotide exchange and deletion of membrane-binding elements. J. Biol. Chem. 2004; 279:48307–48318. [DOI] [PubMed] [Google Scholar]
- 23. Pastrana C.L., Carrasco C., Akhtar P., Leuba S.H., Khan S.A., Moreno-Herrero F.. Force and twist dependence of RepC nicking activity on torsionally-constrained DNA molecules. Nucleic Acids Res. 2016; 44:8885–8896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Daldrop P., Brutzer H., Huhle A., Kauert D.J., Seidel R.. Extending the range for force calibration in magnetic tweezers. Biophys. J. 2015; 108:2550–2561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. te Velthuis A.J., Kerssemakers J.W., Lipfert J., Dekker N.H.. Quantitative guidelines for force calibration through spectral analysis of magnetic tweezers data. Biophys. J. 2010; 99:1292–1302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Klucar L., Stano M., Hajduk M.. phiSITE: database of gene regulation in bacteriophages. Nucleic Acids Res. 2010; 38:D366–D370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Fuentes-Perez M.E., Gwynn E.J., Dillingham M.S., Moreno-Herrero F.. Using DNA as a fiducial marker to study SMC complex interactions with the atomic force microscope. Biophys. J. 2012; 102:839–848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Davis M.A., Austin S.J.. Recognition of the P1 plasmid centromere analog involves binding of the ParB protein and is modified by a specific host factor. EMBO J. 1988; 7:1881–1888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Breier A.M., Grossman A.D.. Whole-genome analysis of the chromosome partitioning and sporulation protein Spo0J (ParB) reveals spreading and origin-distal sites on the Bacillus subtilis chromosome. Mol. Microbiol. 2007; 64:703–718. [DOI] [PubMed] [Google Scholar]
- 30. Graham T.G., Wang X., Song D., Etson C.M., van Oijen A.M., Rudner D.Z., Loparo J.J.. ParB spreading requires DNA bridging. Genes Dev. 2014; 28:1228–1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hoischen C., Bussiek M., Langowski J., Diekmann S.. Escherichia coli low-copy-number plasmid R1 centromere parC forms a U-shaped complex with its binding protein ParR. Nucleic Acids Res. 2008; 36:607–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Salje J., Lowe J.. Bacterial actin: architecture of the ParMRC plasmid DNA partitioning complex. EMBO J. 2008; 27:2230–2238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Weihofen W.A., Cicek A., Pratto F., Alonso J.C., Saenger W.. Structures of omega repressors bound to direct and inverted DNA repeats explain modulation of transcription. Nucleic Acids Res. 2006; 34:1450–1458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Pratto F., Suzuki Y., Takeyasu K., Alonso J.C.. Single-molecule analysis of proteinxDNA complexes formed during partition of newly replicated plasmid molecules in Streptococcus pyogenes. J. Biol. Chem. 2009; 284:30298–30306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Schumacher M.A., Glover T.C., Brzoska A.J., Jensen S.O., Dunham T.D., Skurray R.A., Firth N.. Segrosome structure revealed by a complex of ParR with centromere DNA. Nature. 2007; 450:1268–1271. [DOI] [PubMed] [Google Scholar]
- 36. Moller-Jensen J., Ringgaard S., Mercogliano C.P., Gerdes K., Lowe J.. Structural analysis of the ParR/parC plasmid partition complex. EMBO J. 2007; 26:4413–4422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Stasiak A., Di Capua E.. The helicity of DNA in complexes with recA protein. Nature. 1982; 299:185–186. [DOI] [PubMed] [Google Scholar]
- 38. Gayathri P., Fujii T., Moller-Jensen J., van den Ent F., Namba K., Lowe J.. A bipolar spindle of antiparallel ParM filaments drives bacterial plasmid segregation. Science. 2012; 338:1334–1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.