

Abstract
Tissue-nonspecific alkaline phosphatase (TNAP) is an important isozyme of alkaline phosphatases, which plays different pivotal roles within the human body. Most importantly, it is responsible for maintaining the balanced ratio of phosphate and inorganic pyrophosphate, thus regulates the extracellular matrix calcification during bone formation and growth. The elevated level of TNAP has been linked to vascular calcification and end-stage renal diseases. Consequently, there is a need to search for highly potent and selective inhibitors of alkaline phosphatases (APs) for treatment of disorders associated with the over-expression of APs. Herein, a series of tricyclic coumarin sulphonate 1a-za with known antiproliferative activity, was evaluated for AP inhibition against human tissue nonspecific alkaline phosphatase (h-TNAP) and human intestinal alkaline phosphatase (h-IAP). The methylbenzenesulphonate derivative 1f (IC50 = 0.38 ± 0.01 μM) was found to be the most active h-TNAP inhibitor. Another 4-fluorobenzenesulphonate derivative 1i (IC50 = 0.45 ± 0.02 μM) was found as the strongest inhibitor of h-IAP. Some of the derivatives were also identified as highly selective inhibitors of APs. Detailed structure-activity relationship (SAR) was investigated to identify the functional groups responsible for the effective inhibition of AP isozymes. The study was also supported by the docking studies to rationalise the most possible binding site interactions of the identified inhibitors with the targeted enzymes.
Keywords: Alkaline phosphatase inhibitor, coumarin, molecular docking, structure-activity relationship, Tricyclic coumarin sulfonate
Introduction
Alkaline phosphatases (APs, E.C. 3.1.3.1) are membrane-bound ecto-enzymes with an extracellular-oriented active site1. They belong to the large family of ecto-enzyme known as ecto-nucleotidases, and are involved in various physiological functions within the human body, most importantly in phosphorylation and dephosphorylation reactions2. They are also responsible for the hydrolysis and breakdown of wide variety of nucleoside tri- and di-phosphates substrates to their respective monophosphates3,4. They also hydrolyse many other phosphate-containing substrates, such as inorganic pyrophosphate (PPi), glucose phosphate, polyphosphates, phosphomonoesters, and phosphatidates5. APs are further categorised into two types, the tissue-specific and tissue-nonspecific alkaline phosphatases (TNAPs). From the tissue-specific AP type, intestinal alkaline phosphatase (IAP) plays a pivotal role in the maintenance of physiological environment of the intestine6. Moreover, this isozyme is important for the regulation of bicarbonate secretion balance, duodenal luminal pH, dephosphorylation reaction, and helps in the maintenance of normal gut environment by detoxifying the bacterial toxins7. TNAP is widely distributed almost in each body part but is abundantly present in central nervous tissues, mineralising tissues and also in kidney8. As compared to the other body tissues, high concentration of TNAP is present in the mineralising tissues such as teeth and bones for the normal teeth and bone formation. But too high concentrations of TNAP in mineralising tissue results in abnormal calcification and mineral deposition9. The identification of effective inhibitors of AP isozymes becomes an emerging drug target for the disorders related to the hyper-activity of TNAP and IAP. To date, different inhibitors of APs based on triazole, pyrazoles, coumarin sulphonates, diaryl sulphonamides, and chromones have been identified as effective inhibitors of TNAP and IAP (Figure 1)10–13.
Figure 1.

APs inhibitors; (I) Levamisole (II) Theophylline (III) Aryl sulphonamide-based inhibitors (IV) trizole-based inhibitors (V) Pyrazole-based Inhibitors, (VI) coumarinyl sulphonates.
Recently, we have reported coumarinyl sulphonates derivatives as novel and potent inhibitors of h-TNAP and h-IAP13. In continuation of our efforts for searching the effective and selective inhibitors, herein we evaluated a series of 27 tricyclic-fused coumarin sulphonate derivatives 1a-za as inhibitors of APs which were already tested for their antiproliferative and anti-inflammatory activities14–17. We assumed that the fused cycloheptane or cyclooctane ring might contribute to stronger affinity than the previously reported bicyclic coumarin sulphonate derivatives through formation of additional hydrophobic interaction(s). Our assumption succeeded as shown in Figure 2. Except two derivatives 1w and 1 y, all the other compounds were found as effective AP inhibitors with sub-micromolar or very low micro-molar range IC50 values. Further, the most effective inhibitors of either h-TNAP or h-IAP were selected for the molecular docking studies to rationalise the possible binding interactions with the respective targeted enzymes.
Figure 2.

3D binding interactions of most potent inhibitor 1f within the active site of h-TNAP. Hydrogen bonds are shown in green dotted line while π–π interactions are shown in purple dotted line.
Experimental
Synthesis of the target molecules 1a-za
The synthetic procedures, purification methods, and the spectral analysis data have been reported14–17. The experimental details as well as representative analysis charts are provided in the supplementary file.
Bioactivity protocol
Alkaline phosphatase inhibition assay (h-TNAP and h-IAP)
The inhibition assay was performed by following the already reported method18 and the inhibitory activity was measured at the final concentration of 200 µM in diethanolamine (DEA) buffer as discussed in our published article19. The first step of the inhibition assay was the treatment of 10 µl of tested derivatives with 20 µl of h-TNAP (46 ng/well) or of h-IAP (57 ng/well). The plates were incubated at 37 °C for 5–10 min, and luminescence signals were recorded by microplate reader (BioTek FLx800, Instruments, Inc., Winooski, VT). Then, 20 µl of CDP-star® substrate was added to each reaction mixture and signals were again measured as after-read, after 15–20 min of incubation. The data were scrutinised by PRISM 5.0 (GraphPad, San Diego, CA) software and the inhibitory concentration values (IC50 values) were attained as discussed earlier.
Molecular docking studies
Molecular docking studies of compounds 1f and 1i, potent inhibitors of h-TNAP and h-IAP, respectively, were conducted to investigate the probable binding interactions of these compounds within the active site of the respective target enzyme. Our previously reported models of h-TNAP and h-IAP were used for docking calculation20. Before performing docking studies, protonation and energy minimisation of all enzyme structures were carried out using molecular operating environment (MOE) 2014, 09 software21. Structure generation as well as energy minimisation of the effective inhibitors were also carried out via MOE software. After preparation of all the prerequisite compounds and enzymes structures, total 50 independent docking runs for each ligand were executed by MOE docking program. The best scored pose of potent compounds in each target enzyme was selected for evaluation of binding interactions. Binding free energy for each pose was also determined and pose with the lowest free binding energy was selected for further visualisation studies. 3D visualisation of binding interaction between potent compound and respective target was carried out using Discovery studio visualizer v422.
Results and discussion
Chemistry
Scheme 1 illustrates the synthetic pathway via which the target compounds 1a-za were prepared. Cycloheptanone (2a) or cyclooctanone (2b) was reacted with diethyl carbonate in the presence of NaH in benzene at the reflux temperature to get the ester intermediates 3a,b that exist in keto-enol tautomers23. Subsequent reaction with (substituted) resorcinol in the presence of trifluoroacetic acid and concentrated sulphuric acid led to the production of the tricyclic phenolic intermediates 4a-f24. They were used in the next step as such with no prior purification. In the last step, those phenolic intermediates were treated with the appropriate sulphonyl chloride reagent in the presence of triethylamine as a catalyst to get the target sulphonate products 1a-za14–17. Table 1 illustrates the structure of each target compound.
Scheme 1.

Reagents and conditions: (i) diethyl carbonate, NaH, benzene, reflux, 90% (3a, n = 1), 85% (3 b, n = 2); (ii) (substituted) resorcinol, CF3COOH, conc. H2SO4, 0 °C; rt, 3 h; (iii) appropriate sulphonyl chloride derivative, triethylamine, CH2Cl2, 0 °C; rt, 1 h.
Table 1.
Structures of the target compounds 1a-za along with their yield percentages and in vitro inhibitory effects (IC50, μM) against h-TNAP and h-IAP.
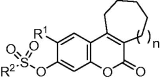 | ||||||
|---|---|---|---|---|---|---|
| IC50 (µM) ± SEMa |
||||||
| Compound No. | R1 | R2 | n | Yield% | h-TNAP | h-IAP |
| 1a | H | Me | 1 | 95 | 0.48 ± 0.01 | 1.98 ± 0.19 |
| 1b | H | Et | 1 | 92 | 2.13 ± 0.19 | 2.71 ± 0.23 |
| 1c | H | n-Pr | 1 | 93 | 1.52 ± 0.17 | 0.91 ± 0.06 |
| 1d | H | Cyclo-Pr | 1 | 85 | 0.98 ± 0.13 | 0.96 ± 0.08 |
| 1e | H | Ph | 1 | 93 | 8.57 ± 0.73 | 1.84 ± 0.19 |
| 1f | H | p-Tolyl | 1 | 95 | 0.38 ± 0.01 | 1.94 ± 0.11 |
| 1g | H | p-(CF3)C6H4 | 1 | 90 | 1.59 ± 0.12 | 2.64 ± 0.45 |
| 1h | H | p-(tert-butyl)C6H4 | 1 | 95 | 6.33 ± 0.99 | 3.92 ± 0.47 |
| 1i | H | p-(F)C6H4 | 1 | 82 | 1.06 ± 0.11 | 0.45 ± 0.09 |
| 1j | H | Et | 2 | 90 | 1.74 ± 0.14 | 0.84 ± 0.09 |
| 1k | H | n-Pr | 2 | 88 | 1.91 ± 0.11 | 3.37 ± 0.79 |
| 1l | H | Ph | 2 | 92 | 1.13 ± 0.21 | 2.82 ± 0.82 |
| 1m | H | p-Tolyl | 2 | 95 | 1.59 ± 0.16 | 2.12 ± 0.23 |
| 1n | H | p-(tert-butyl)C6H4 | 2 | 94 | 1.85 ± 0.13 | 1.28 ± 0.13 |
| 1o | OMe | Me | 1 | 85 | 1.21 ± 0.11 | 11.80 ± 1.91 |
| 1p | OMe | Me | 2 | 85 | 1.49 ± 0.13 | 1.51 ± 0.17 |
| 1q | OMe | n-Pr | 1 | 95 | 4.31 ± 0.99 | >100 |
| 1r | OMe | n-Pr | 2 | 91 | 2.60 ± 0.49 | 0.75 ± 0.07 |
| 1s | OMe | Ph | 1 | 92 | 1.26 ± 0.07 | 3.88 ± 0.32 |
| 1t | OMe | Ph | 2 | 87 | 2.68 ± 0.18 | 0.83 ± 0.06 |
| 1u | OMe | p-Tolyl | 1 | 96 | 1.65 ± 0.13 | 0.79 ± 0.05 |
| 1v | OMe | p-Tolyl | 2 | 90 | 0.64 ± 0.02 | 1.45 ± 0.04 |
| 1w | OMe | p-(CF3)C6H4 | 1 | 87 | >100 | >100 |
| 1x | OMe | p-(CF3)C6H4 | 2 | 85 | 1.29 ± 0.12 | 1.63 ± 0.19 |
| 1y | Cl | Me | 1 | 85 | >100 | >100 |
| 1z | Cl | Ph | 1 | 85 | 5.73 ± 1.06 | >100 |
| 1za | Cl | p-Tolyl | 1 | 86 | 2.22 ± 0.27 | 1.11 ± 0.01 |
| Levamisole | —— | —— | —— | —— | 20.2 ± 1.90 | —— |
| L-Phenylalanine | —— | —— | —— | —— | —— | 100 ± 3.15 |
IC50 is the concentration at which the 50% of the enzyme activity was inhibited. All the values were expressed as IC50 ± SEM (standard error mean), n = 3.
Alkaline phosphatase inhibition
All synthesised tricyclic coumarin sulphonate derivatives 1a-za were tested for their potential to inhibit h-TNAP and h-IAP. Except two compounds (1w and 1 y), all compounds were found to be active against both isozymes of AP with IC50 values in the lower micro-molar range. Compounds showed their h-TNAP inhibitory potential in the range of IC50 = 0.38 ± 0.01 to 8.57 ± 0.73 μM as compared to standard levamisole (IC50 = 20.21 ± 1.9 μM), and h-IAP inhibitory activity in the range of IC50 = 0.45 ± 0.09 to 11.8 ± 1.91 μM as compared to standard L-phenylalanine (IC50 = 100.1 ± 3.15 μM) (Table 1).
Structure-activity relationship (SAR)
In order to find out the comprehensive structure-activity relationship for AP inhibition, all tricyclic-fused coumarin sulphonates 1a-za were divided into two categories, on the basis of fused cycloheptane and cyclooctane ring. Moreover, the detailed structure-selectivity relationship was also rationalised on the basis of effect of unsubstituted, and chloro- and methoxy-substituted derivatives on the inhibition of both APs. Compounds 1q and 1z were found as selective inhibitors of h-TNAP while none of the derivative was found as a selective inhibitor of IAP.
Among the tested derivatives, compound 1f expressed maximum inhibitory effects on h-TNAP and compound 1i depicted the strongest potency against h-IAP. From the unsubstituted derivatives, except compound 1c, 1g, and 1m, the other target derivatives expressed varying inhibitory effects on h-TNAP. Compounds 1f, 1a, and 1d possessing p-toluene sulphonate, methane sulphonate, and cyclopropane substitution, respectively, depicted higher inhibition of h-TNAP than compounds 1c, 1g, and 1j-m. Among the cyclooctane-fused derivatives, compound 1l with benzenesulphonate substitution demonstrated the strongest inhibitory potential on h-TNAP (IC50 ± SEM = 1.13 ± 0.21 µM). Compounds 1c with the fused cycloheptane ring and compound 1j with the fused cyclooctane ring were found as stronger inhibitors of h-IAP with the inhibitory values of IC50 ± SEM = 0.91 ± 0.06 and 0.84 ± 0.09 µM, respectively. Among the cyclooctane-fused derivatives, compound 1j with ethanesulphonate substitution demonstrated the strongest inhibitory effect on h-IAP (IC50 ± SEM = 0.84 ± 0.09 µM). From the cycloheptane-fused derivatives, compound 1c with propanesulphonate substitution demonstrated the strongest inhibitory potential on h-IAP (IC50 ± SEM = 0.91 ± 0.06 µM). Although more lipophilic side chain was substituted in 1c, the inhibitory effect of this derivative was found less because of the presence of seven-membered fused ring. Moreover, from the obtained results this may be rationalised that the presence of more lipophilic side chain in compound 1c may form stronger hydrophobic interactions with the target enzyme.
The inhibitory effect by other derivatives; 1h and 1n with tert-butylbenzene suphonate moiety expressed the dual inhibition of both isozymes of APs. The derivative 1n depicted more inhibitory potential because of the presence of fused eight membered ring (h-TNAP; IC50±SEM = 1.85 ± 0.13 µM, h-IAP; IC50±SEM = 1.28 ± 0.13 µM). In comparison to 1h, the replacement of tert-butylbenzene with flouorobenzenesulphonate results in the most potent inhibitor of h-IAP i.e. 1i. Compound 1i was identified as the strongest inhibitor of h-IAP with and inhibitory value of IC50±SEM = 0.45 ± 0.09 µM. The increased potency might be due to the presence of most electronegative halogen.
On the other hand, the chloro- and methoxy-substituted derivatives were found more active against both APs than the unsubstituted analogues. With reference to h-TNAP, compound 1o (IC50±SEM = 1.21 ± 0.11 µM) was found more potent as compared to compound 1q (IC50±SEM = 4.31 ± 0.99 µM), 1p (IC50±SEM = 1.49 ± 0.13 µM), and 1r (IC50±SEM = 2.60 ± 0.49 µM) but with reference to h-IAP, 1p (IC50±SEM = 1.51 ± 0.17 µM) was found more better than 1o (IC50±SEM = 11.8 ± 1.91 µM) and 1r (IC50±SEM = 0.75 ± 0.07 µM) was found better than 1q (IC50 ± SEM = >100 µM). It means that the presence of functional group is playing more important role on the inhibitory effects. The derivatives with less carbon number were found more potent that the propane derivative. But, with detailed SAR it was found that the heptane ring was favourable for TNAP, while octanone derivative was found potent towards IAP. When the activity of 1o was compared with 1r and 1p, it was observed that the reduced inhibitory effect of both the derivatives on TNAP was due to the presence of n-propane substitution in case of 1r, and the presence of fused cyclooctane ring in case of 1p.
In comparison to 1o, compound 1s having benzenesulphonate substitution was found almost as an equipotent inhibitor of TNAP with the inhibitory value of 1.26 ± 0.07 µM. The other derivative i.e. 1t having the same substitution of benzenesulphonate as in compound 1s, but with the cyclooctane ring, a reduced inhibitory effect on TNAP was observed with an increased inhibitory effect on IAP isozymes (h-TNAP; (IC50±SEM = 2.68 ± 0.18 µM, h-IAP; IC50±SEM = 0.83 ± 0.06 µM). The derivatives 1w and 1x having the same substitution at R1 and R2 positions but with cycloheptane and cyclooctane ring in 1w and 1x, respectively. When the SAR of these two derivatives was considered, it was observed that cycloheptane derivative 1w was found less reactive than that of the cyclooctane derivative 1x which inhibited both isozymes effectively. This inhibitory effect confirms that like 1p, the presence of more carbon number in the derivative is responsible for the more inhibition.
An interesting inhibition of IAP was also exhibited by the aromatic derivative 1u and 1za processing cycloheptane ring along with p-tosylate substitution at R2 position but different substitution at R1 position. The derivative with electron-donating group i.e. OMe in case of 1u (IC50±SEM = 0.79 ± 0.05 µM) was found more active inhibitor as compared to the derivative 1za (IC50±SEM = 1.11 ± 0.01 µM) with electron-withdrawing substitution. These two derivative also exert some inhibitory potential on h-TNAP, but less (1u; IC50±SEM = 1.65 ± 0.13 µM, 1za; IC50±SEM = 2.22 ± 0.27 µM) in comparison to h-IAP. The other derivatives 1 y and 1z with methanesulphonate and benzenesulphonate substitution were found inactive towards h-IAP. Moreover, it was found that the aromatic sulphonate derivatives 1s, 1u, and 1za depicted stronger inhibitory effects on h-IAP than h-TNAP.
The results obtained here are in consistent with our previously reported work. The 2-methoxy- or 2-chloro- substituted derivatives possessing the fused cycloheptane ring were found stronger inhibitors of h-TNAP than the corresponding analogues having fused cyclooctane ring. Similarly, an inverse effect was observed towards h-IAP. It can be suggested from the obtained data that the derivatives with more lipophilic side chains led to the more inhibition of h-IAP and vice versa in case of h-TNAP. This lipophilic side chains might be involved in some hydrophobic interactions within the pocket of the receptor site and increase the affinity of such derivatives for the targeted enzyme and ultimately accounts for the enhanced h-IAP inhibition.
Molecular docking studies
Molecular docking studies of the most potent derivatives i.e. 1f and 1i were carried out within the respective target enzyme and their most probable binding interactions within the modelled enzymes are depicted in Figures 2 and 3. Detailed analysis of binding interaction of compound 1f within h-TNAP revealed that oxygen atom in both sulphonate group and carbonyl group are responsible for making three strong hydrogen bonds with amino acid residues. Two hydrogen bonds were formed by oxygen of sulphonate group with amino groups of Arg167 while the third hydrogen bond was formed by carbonyl oxygen in coumarin ring with amino acid Thr436. Additional to hydrogen bonding, second oxygen atom of the sulphonate group also formed direct interaction with the Zn+2 ion within the active site of h-TNAP. The excellent h-TNAP inhibition activity of compound 1f can be related with this direct binding interaction of sulphonate oxygen with the Zn+2 ion of the active site. Furthermore, two π–π interactions were formed by benzene ring adjacent to sulphonate group with amino acid residues His154 and His321. One π-alkyl interaction was also exhibited by the seven-membered ring fused with the coumarin ring and His434 as shown in Figure 2. This interaction may account for the stronger affinity and potency of this tricyclic compound compared to the previously reported bicyclic coumarin sulphonate derivatives13. Detailed binding interaction of 1i with h-IAP is shown in Figure 3. In comparison to docking interaction of compound 1f with h-TNAP, compound 1i formed four hydrogen bonds with different amino acids of h-IAP. Oxygen atom in sulphonate group of compound 1i also formed two hydrogen and one metal interaction. These two hydrogen bonds were formed by one oxygen atom in sulphonate group with amino acid residue of Arg166 and His143. While other oxygen of sulphonate group formed interaction with Zn+2 ion. Carbonyl oxygen in coumarin ring and fluorine atom in compound 1i also formed hydrogen bond with amino acid residues Arg150 and Gln108 as shown in Figure 3. Additional to hydrogen bonding, two π–π interaction were also formed by benzene ring of coumarin ring with amino acid residue of His317 and His320. Metal interaction of sulphonate group with metal ion is analogous to previously reported interaction of sulphonamides with different APs13. π-alkyl interaction between seven-membered ring adjacent to coumarin ring was not shown as in 1i. Docking of the inactive derivatives 1q and 1w showed weaker affinity and fewer bonds formed, which supports the SAR discussion. The figures are given in the supplementary file.
Figure 3.

3D binding interactions of most potent inhibitor 1i within the active site of h-IAP. Hydrogen bonds are shown in green dotted line while π–π interactions are shown in purple dotted line.
Conclusions
Tricyclic-fused coumarin sulphonates 1a-za were evaluated for their effective inhibitory potential against h-TNAP and h-IAP. Except 1w and 1 y derivatives, all the other compounds exhibited excellent inhibitory potential against both h-TNAP and h-IAP. Methylbenzenesulphonate derivative 1f (IC50 = 0.38 ± 0.01 μM) was found to be the most active h-TNAP inhibitor. Another 4-fluorobenzenesulphonate derivative 1i (IC50 = 0.45 ± 0.02 μM) was found as the strongest inhibitor of h-IAP. Detailed SAR studies and molecular docking studies were performed to identify the importance of functional groups within the structures and to explore the possible binding site interactions, respectively. Moreover, previously these derivatives have been reported for their antiproliferative effects against different cell lines including, leukaemia, CNS, prostate, renal, colon, lung cancer, melanoma, ovarian, and breast cancers. From that data we got 1i and 1 m as the strongest antiproliferative derivatives against colon cancer cell line along with the maximum anti-inflammatory effects. Here in this study, we have identified these compounds as stronger inhibitors of h-IAP isozymes. This can be rationalised that the IAP, responsible for the maintenance of intestinal homeostasis, might be over-expressed in the colon cancer cell lines. Moreover, the over-expression of h-TNAP can be associated with the lung cancer, melanoma, ovarian, breast cancers, and renal cancer. Therefore, the obtained results synergise with the previously reported data. It can be suggested that the potent inhibitors of both h-TNAP and h-IAP can be selected for further exploration at molecular level.
Supplementary Material
Funding Statement
J. Iqbal is thankful to the Organization for the Prohibition of Chemical Weapons (OPCW), The Hague, The Netherlands and Higher Education Commission, Pakistan for the financial support through Project No. 20–3733/NRPU/R&D/14/520. This work was also supported by Korea Institute of Science and Technology (KIST), KIST Project (E26900).
Disclosure statement
The authors have declared no conflict of interest.
References
- 1.Zimmermann H, Zebisch M, Sträter N.. Cellular function and molecular structure of ecto-nucleotidases. Purinergic Signal 2012;8:437–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Millán JL.Mammalian alkaline phosphatases: from biology to applications in medicine and biotechnology. Hoboken, NJ, USA: John Wiley & Sons; 2006. [Google Scholar]
- 3.Millán JL.Alkaline phosphatases: structure, substrate specificity and functional relatedness to other members of a large superfamily of enzymes. Purinergic Signal 2006;2:335–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Millán JL.The role of phosphatases in the initiation of skeletal mineralization. Calcif Tissue Int 2013;93:299–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Al-Rashida M, Iqbal J.. Inhibition of alkaline phosphatase: an emerging new drug target. Mini Rev Med Chem 2015;15:41–51. [DOI] [PubMed] [Google Scholar]
- 6.Kiffer-Moreira T, Sheen CR, da Silva Gasque KC, et al. Catalytic signature of a heat-stable, chimeric human alkaline phosphatase with therapeutic potential. PLoS One 2014;9:e89374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Guerville M, Boudry G.. Gastrointestinal and hepatic mechanisms limiting entry and dissemination of lipopolysaccharide into the systemic circulation. Am J Physiol Gastrointest Liver Physiol 2016;311:G1–G15. [DOI] [PubMed] [Google Scholar]
- 8.Buchet R, Millán JL, Magne D.. Multisystemic functions of alkaline phosphatases. Methods Mol Biol 2013;1053:27–51. [DOI] [PubMed] [Google Scholar]
- 9.Yadav MC, Simao AMS, Narisawa S, et al. Loss of skeletal mineralization by the simultaneous ablation of PHOSPHO1 and alkaline phosphatase function: a unified model of the mechanisms of initiation of skeletal calcification. J Bone Miner Res 2011;26:286–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chung TD, Sergienko E, Millán JL.. Assay format as a critical success factor for identification of novel inhibitor chemotypes of tissue-nonspecific alkaline phosphatase from high-throughput screening. Molecules 2010;15:3010–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dahl R, Sergienko EA, Su Y, et al. Discovery and validation of a series of aryl sulfonamides as selective inhibitors of tissue-nonspecific alkaline phosphatase (TNAP). J Med Chem 2009;52:6919–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Al-Rashida M, Raza R, Abbas G, et al. Identification of novel chromone based sulfonamides as highly potent and selective inhibitors of alkaline phosphatases. Eur J Med Chem 2013;66:438–49. [DOI] [PubMed] [Google Scholar]
- 13.Salar U, Khan KM, Iqbal J, et al. Coumarin sulfonates: new alkaline phosphatase inhibitors; in vitro and in silico studies. Eur J Med Chem 2017;131:29–47. [DOI] [PubMed] [Google Scholar]
- 14.Jang HL, El-Gamal MI, Choi HE, et al. Synthesis of tricyclic fused coumarin sulfonates and their inhibitory effects on LPS-induced nitric oxide and PGE2 productions in RAW 264.7 macrophages. Bioorg Med Chem Lett 2014;24:571–5. [DOI] [PubMed] [Google Scholar]
- 15.El-Gamal MI, Oh C-H.. Synthesis, in vitro antiproliferative activity, and in silico studies of fused tricyclic coumarin sulfonate derivatives. Eur J Med Chem 2014;84:68–76. [DOI] [PubMed] [Google Scholar]
- 16.El‐Gamal MI, Baek D, Oh C-H.. A new series of cycloalkane‐fused coumarin sulfonates: synthesis and in vitro antiproliferative screening. Bull Korean Chem Soc 2016;37:184–91. [Google Scholar]
- 17.El‐Gamal MI, Lee WS, Shin JS, et al. Synthesis of new tricyclic and tetracyclic fused coumarin sulfonate derivatives and their inhibitory effects on LPS-Induced Nitric Oxide and PGE2 Productions in RAW 264.7 Macrophages: part 2. Archiv Der Pharmazie 2016;349:853–63. [DOI] [PubMed] [Google Scholar]
- 18.Sergienko EA, Millán JL.. High-throughput screening of tissue-nonspecific alkaline phosphatase for identification of effectors with diverse modes of action. Nat Protoc 2010;5:1431–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Khan I, Shah SJA, Ejaz SA, et al. Investigation of quinoline-4-carboxylic acid as a highly potent scaffold for the development of alkaline phosphatase inhibitors: synthesis, SAR analysis and molecular modelling studies. RSC Advances 2015;5:64404–13. [Google Scholar]
- 20.Khan I, Ibrar A, Ejaz SA, et al. Influence of the diversified structural variations at the imine functionality of 4-bromophenylacetic acid derived hydrazones on alkaline phosphatase inhibition: synthesis and molecular modelling studies. RSC Advances 2015;5:90806–18. [Google Scholar]
- 21.Molecular Operating Environment (MOE) , 2014.09; Chemical Computing Group Inc., 1010 Sherbooke St. West, Suite #910, Montreal, QC, Canada, H3A 2R7, 2015. [Google Scholar]
- 22.Accelrys Software Inc. , Discovery Studio Modeling Environment, Release 4.0, Accelrys Software Inc., San Diego, CA; 2013. [Google Scholar]
- 23.Li C-J, Chen D-L, Lu Y-Q, et al. Metal-mediated two-atom carbocycle enlargement in aqueous medium. Tetrahedron 1998;54:2347–64. [Google Scholar]
- 24.Woo LWL, Purohit A, Malini B, et al. Potent active site-directed inhibition of steroid sulphatase by tricyclic coumarin-based sulphamates. Chem Biol 2000;7:773–91. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.