

Abstract
The essential biological function of phosphodiesterase (PDE) type enzymes is to regulate the cytoplasmic levels of intracellular second messengers, 3′,5′-cyclic guanosine monophosphate (cGMP) and/or 3′,5′-cyclic adenosine monophosphate (cAMP). PDE targets have 11 isoenzymes. Of these enzymes, PDE5 has attracted a special attention over the years after its recognition as being the target enzyme in treating erectile dysfunction. Due to the amino acid sequence and the secondary structural similarity of PDE6 and PDE11 with the catalytic domain of PDE5, first-generation PDE5 inhibitors (i.e. sildenafil and vardenafil) are also competitive inhibitors of PDE6 and PDE11. Since the major challenge of designing novel PDE5 inhibitors is to decrease their cross-reactivity with PDE6 and PDE11, in this study, we attempt to identify potent tadalafil-like PDE5 inhibitors that have PDE5/PDE6 and PDE5/PDE11 selectivity. For this aim, the similarity-based virtual screening protocol is applied for the “clean drug-like subset of ZINC database” that contains more than 20 million small compounds. Moreover, molecular dynamics (MD) simulations of selected hits complexed with PDE5 and off-targets were performed in order to get insights for structural and dynamical behaviors of the selected molecules as selective PDE5 inhibitors. Since tadalafil blocks hERG1 K channels in concentration dependent manner, the cardiotoxicity prediction of the hit molecules was also tested. Results of this study can be useful for designing of novel, safe and selective PDE5 inhibitors.
Keywords: E-pharmacophore modeling, molecular docking, molecular dynamics (MD) simulations, phosphodiesterase (PDE) type enzymes, virtual screening
Introduction
Phosphodiesterase (PDE) type enzymes are located in multiple tissues and organs of vertebrate systems in mammalian organisms1. Their essential biological function is to regulate the cytoplasmic levels of intracellular second messengers, 3′,5′-cyclic guanosine monophosphate (cGMP) and/or 3′,5′-cyclic adenosine monophosphate (cAMP)2–4. Twenty one different genes promote 11 isoenzymes of the PDE family. Of these enzymes, PDE5 has attracted a special attention over the years after its recognition as being the target enzyme in treating erectile dysfunction (ED) where millions of men suffer from this disease, worldwide. Since the revolutionary discovery of Sildenafil (ViagraTM) by Pfizer in 19985, academic studies as well as clinical and pre-clinical industrial drug discovery programs have been widely carried out for more potent and selective PDE5 inhibitors. Although successful examples exist in the market, such as Tadalafil-CialisTM, Vardenafil-LevitraTM and Avanafil-StendraTM, main hurdle about PDE5 inhibitors has been focused on the circumvention of the undesired cross-reactivity with other PDE enzymes, especially towards PDE6 and PDE11 in the discovery and design studies6–28. PDE5 enzymes can be found in diverse tissues in human body in addition to the corpus cavernosum; such as lung, brain, platelets, kidney and liver which raise the physiological importance of this enzyme and in turn leads PDE5 inhibitors as drugs for treating other diseases such as pulmonary hypertension (RevatioTM) and Raynaud’s disease (ViagraTM), as well. This convenient situation marks the possibility of evaluating PDE5 inhibitors in drug repositioning applications as sildenafil is presumably the most important example at this field.
Structural assembly of PDE5 is a homodimer that consists of two regulatory GAF domains (GAFA and GAFB) which are the allosteric binding regions for the enzyme substrate (cGMP), phosphorylation site (at Ser92 position) which takes role in the activation mechanism of the enzyme and catalytic site located at the C-terminal end of the protein (amino acid residues: 535–860) which contains the divalent metal (Zn2+ and possibly, Mg2+) binding domain. The mechanism of “PDE5 drug activity” in the corpus cavernosum starts with the competitive binding with cGMP at the active site of the enzyme, intervening the neurotransmitter nitric oxide (NO) mediated sexual stimulation which eventually reverts the smooth muscle contraction via depression of the intracellular Ca2+ concentration. On the other hand, PDE6 enzyme is the key effector enzyme for the phototransduction cascade in the rod and cone segments of the retina in the mammalian eyes. It has a function in visual transduction and respond to light via shifting mechanism from its inactivated to the activated states, regulated by its unique “γ-subunit” which is absent among other PDEs29–33. Due to the amino acid sequence and the secondary structural similarity of its catalytic domain with PDE5, first-generation PDE5 inhibitors (i.e. sildenafil and vardenafil) are also competitive inhibitors of PDE6. In addition, tadalafil has a better PDE5/PDE6 selectivity as compared to sildenafil and vardenafil21,34,35. Visual disorders such as functional blindness, blue (cynopsia) and blurred vision and enhanced light sensitivity have been attributed to the cross-reactivity with the PDE6 catalytic site, upon intake of PDE5 inhibitors in patients with ED. Apart from this reasonable basis for the foundation of vision-related side effects by consequence of direct inhibition of PDE6 which is in line with the location/function of PDE6; another hypothesis ascribed these side effects to the reactivity of these drugs targeting to PDE5 isoenzymes that are distributed in tissues other than the corpus cavernosum36,37. In addition, tadalafil is a dual inhibitor of PDE5 and PDE11 enzymes which is thought to be the reason of back and muscle pain (myalgia) during the treatment of men with tadalafil since PDE11 enzymes are abundantly found in skeletal muscle cells38,39. Although the catalytic site of PDE11 is the most similar one with PDE5, the absence of crystal structure together with the inadequate knowledge about the physiological role of this enzyme in human body restrict the understanding of the mechanism of the inhibitory activity of this target.
Numerous scientific approaches appearing in medicinal chemistry area cover different experimental techniques along with computer-aided molecular modeling methods in the course of discovering more potent and selective PDE5 inhibitors. Especially, structure–activity relationship (SAR) studies are very helpful where distinct chemical synthesis routes guided by oral bioavailability, solubility, membrane permeability, toxicity, lipophilicity and other pharmacokinetic and physiochemical tests have given rise to novel potent compounds to emerge as PDE5 inhibitors8,13–16,18–23,25–28,40,41. Scaffold hopping strategies have been implemented in designing structurally different – in terms of core architecture – novel compounds9. An unusual study was conducted by Pfizer in 2008, introducing chirality concept on sildenafil in order to improve PDE5/PDE6 selectivity42. On the other hand, in silico strategies including quantitative structure–activity relationship (QSAR) studies, pharmacophore hypothesis generations, virtual screening along with molecular docking and molecular dynamics (MD) simulations have been emerged as popular techniques in identifying new leads and understanding the key concepts in protein–drug interactions6,17,43–54.
Since the major challenge in designing novel PDE5 inhibitors is to decrease their cross-reactivity with PDE6 and PDE11, we attempt to identify potent tadalafil-like PDE5 inhibitors that have PDE5/PDE6 and PDE5/PDE11 selectivity as well as preserved principal target activity (i.e. comparable or higher binding affinity for PDE5 enzyme as compared to tadalafil). For this aim, similarity-based virtual screening protocol is applied for the “clean drug-like subset of ZINC database” that contains more than 20 million small compounds. Another outcome of the present work is to illuminate the structural background for high/low binding tendencies to PDE5, PDE6 and PDE11 targets from molecular perspective. In this respect, we implemented step by step procedure in finding active hits as tadalafil-like compounds by combining ligand-based virtual screening (i.e. molecular fingerprint-based protocol) with structure-based modeling techniques (homology modeling, molecular docking and MD simulations). The flowchart of the current study is briefly illustrated in Scheme 1. Since it is known that current PDE5 inhibitors block hERG1 K channels in concentration dependent manner, the cardiotoxicity prediction of the hit molecules was also tested. Moreover, a novel approach for deriving structure-based pharmacophores (E-pharmacophore) was also applied for the selected hit compounds.
Scheme 1.

Flowchart of the current study in the effort for identifying novel and selective PDE5 inhibitors.
Methods
Ligand and protein preparations
The crystal structure of PDE5 enzyme (PDB ID, 2H42)55 was used as template protein for homology modeling and protein engineering procedures of catalytic domains of PDE6 and PDE11 targets. Protein preparations were handled with MOE molecular modeling package56 after retrieving the bound-sildenafil and water molecules from PDE5 enzyme. Energy minimizations and conformational search (CS) of the ligands were realized with MMFF94X force field by means of MOE molecular modeling package. Low MD method was utilized in the CS step by generating 50 conformers for each molecule.
Virtual library screening
“Clean drug-like compound library” in the ZINC database (>20 million compounds)57 was screened via molecular fingerprint and similarity search tools implemented in MOE. Molecular fingerprints of the molecules were computed with MACCS structural keys scheme. Subsequently, Tanimoto coefficient of each molecule in the database was calculated based on the characteristic MACCS structural keys of tadalafil. Tanimoto coefficient is defined as; TC=NAB/(NA+NB−NAB) where NAB represents the number of common MACCS structural key elements between molecule A and molecule B whereas NA and NB are the total number of MACCS structural key elements in molecule A and molecule B, respectively. MACCS structural keys include 166 structural elements that match the corresponding smart patterns in a compound. The applied similarity search strategy lies on an expectation that structurally similar molecules may also show similar biological activities58,59. A coefficient value of 0.8 (Tanimoto coefficient) was chosen in the filtering step of the database which enabled us to obtain 1309 hit candidates that are further subjected to flexible molecular docking simulations.
Flexible molecular docking simulations
GOLD (Genetic Optimization for Ligand Docking, v.5.3.0) program60 was utilized for the docking simulations. Chemscore scoring function was used for generating the predicted binding energies of the ligands within the targets. Ten amino acid residues at the drug binding region of PDE5, PDE6 and PDE11 targets were handled with flexibility by utilizing the rotamer library implemented in the GOLD docking program. Default settings were used for population and genetic operations steps. Early termination was switched-off and 20 docking poses were generated for each molecule.
Molecular dynamics simulations
Gromacs v.4.6.5 package61 was used for the MD simulations. Topology parameters for the ligands were prepared with PRODRG Server62. Partial charges of ligands were calculated with density functional theory (DFT) by B3LYP/G(d,p) basis set. Gaussian 09 program package63 was used for this calculation. Production stages of the simulations were conducted in isothermal-isobaric ensemble (NPT) at 310 K and 1 atm with periodic boundary conditions. GROMOS96 43A1 force field was used with leap-frog integrator. 2 fs time-step was used in simulations. SPC water model was used to solvate the systems in a cubic box and Na+ and Cl− ions were added to the systems as counter ions. Long-range electrostatic interactions were handled with Particle Mesh Ewald (PME) algorithm. Energy minimizations with 5000 steps of steepest descent (SD) method followed by 5000 steps using conjugate gradient (CG) algorithm were performed for apo and holo state systems, using a threshold value of 100 kJ/mol.nm. Subsequently, two-steps restrained dynamics were applied in order to equilibrate the systems to the desired temperature and density: (i) systems were heated in isochoric-isothermal ensemble (NVT) with a total of 0.1 ns simulations until the temperature reaches 310 K; (ii) then 2 ns NPT simulations were applied. Finally, 50 ns MD production runs were carried out without any restraints on atoms in an NPT ensemble. V-rescale temperature coupling scheme and Parrinello–Rahman pressure coupling scheme were used in order to control the temperature and pressure during the simulations, respectively. 5000 frames were collected through the 50 ns production runs for the post-processing MD analyses.
Molecular mechanics generalized born solvation (MM-GBSA) calculations
Protein–ligand binding free energies of the selected hits as well as tadalafil were estimated using MM-GBSA method, implemented in Prime module of Schrodinger’s molecular modeling package64, based on the MD trajectory frames. Prime uses the VSGB 2.0 solvation model and the OPLS2005 force field to simulate the interactions.
Results and discussion
A molecular finger print-based virtual screening is performed for ZINC small molecules database in order to identify novel and potent PDE5 inhibitors. Molecular docking simulations were realized under “Protein H-bonding Constraint” (oxygen atom of the invariant Glutamine residues – Gln817, Gln773 and Gln869 in PDE5, PDE6 and PDE11, respectively – were enforced to participate in H-bonding interaction with any H-bond donor atom of the ligands) which augment the elimination of mis-docked outcomes. Thus, integration of ligand-based similarity screening protocol with constraint docking method was able to yield reasonable ligand orientations in the active site of the proteins and to analyze crucial active site residues–ligand interactions. Figure 1 points out that the dominant interaction to fulfill the receptor complimentary is the positively charged region (shown with blue meshes) around Gln817, Gln773 and Gln869 residues for PDE5, PDE6 and PDE11, respectively.
Figure 1.

Electrostatic maps of the active sites of the enzymes (left panel). Blue, red and white colours represent positively charged, negatively charged and hydrophobic preferences built at the drug-binding cavity site (near 5 Å distance from the ligand). Tadalafil fulfils the positively charged electrostatic requirement created by the acceptor atom (Oɛ) of the invariant Glutamine side chain in each active sites via carrying a hydrogen-bond donating moiety (–NH) at the amide fragment (namely, Glutamine Switch). On the other hand, hydrophobic residues, Phe820, Phe776 and Trp 820, sandwich the ligand (namely, Hydrophobic Clamp). 2D ligand–protein interaction diagrams (right panel). Green arrows indicate hydrogen bonding interactions. Green and purple discs show hydrophobic and polar residues, respectively. Representations are created with MOE molecular modeling package.
Validation of the docking methodology
Tadalafil is retrieved from its crystal-bound enzyme (PDB ID, 1XOZ)65 and docked into the drug-binding region of PDE5, PDE6 and PDE11 targets by GOLD docking program. The ligand RMSD value yielded a value of <1.5 Å deviation (for PDE5) from its bioactive conformation at the crystal structure which supports the binding pose prediction power of docking methodology employed for the current study. Tadalafil exhibits monodentate hydrogen bonding interaction with invariant Glutamine residues of these enzymes as can be seen in Figure 2.
Figure 2.

Top docking poses of tadalafil with PDE enzymes. Only polar hydrogens are shown for clarity. Protein residues within 2.5 Å distance around tadalafil are depicted in the figures. Hydrogen bonds between amide hydrogen of tadalafil and Oɛ atom of invariant Glutamine amino acid residue are represented with red dashed lines.
Constructing the homology models of the catalytic domains of PDE6 (amino acid residues: 482–816) and PDE11 (amino acid residues: 587–910)
In the absence of crystal structures of PDE6 and PDE11, homology models of the catalytic domains of these enzymes were constructed based on the available crystal structure of PDE555 as a template. This crystal structure (PDB ID, 2H42) was chosen as a template because it is not chimerically hybridized or mutated like the other PDE5 crystal structures65,66 in the literature and also it does not have any missing elements67,68. The amino acid sequence of PDE6 (P16499) which belongs to the α-subunit of a human rod cell and PDE11 (Q9HCR9) were downloaded from Uniprot69, respectively and further used in the sequence alignment procedure. MOE software was used in building the homology models with Amber99 forcefield. BLOSUM62 matrix was used in the sequence alignment step. 10 different homology models were built; the best homology model was selected among the intermediate models according to the Generalized Born/Volume Integral (GB/VI) methodology and further subjected to refinement and energy minimization procedures. The RMSD values between PDE5 and the generated PDE6 and PDE11 are 0.46 Å and 0.90 Å, respectively, based on the Cα atoms. Sequence alignments of PDE5 with PDE6 and PDE11 are provided in the Supplementary Materials, Figure S1. PDE5 enzyme has a sequence similarity and identity of 64% and 42%; 68% and 47% for PDE6 and PDE11, respectively. 3D structures of PDE5 and PDE6 resemble each other except a clear difference in the β-hairpin domain of PDE6 (amino acid residues: Q687–M701) and its corresponding residues in PDE5 (amino acid residues: R739–L746) which is also observed in other homology model of PDE6 in the literature47. The reliability of the homology models is checked by Ramachandran’s plot (Figure S2, Supplementary Materials). All torsional angles of amino acid residues of derived protein models are in either favored or allowed regions except one and three outliers that their torsional angles are slightly away from allowed regions for PDE6 and PDE11, respectively. In addition, the stereochemical qualities of the homology models are checked by protein geometry report module in MOE program, carefully, by computing the atom clashes, backbone bond length and angle violations and side chain rotamer outliers (rotamer strain energy cutoff value of 5 kcal/mol). No atom clashes and backbone bond length violations from the expected values (atom–atom pair repulsion energy cutoff value of 0.5 kcal/mol and Z-score value <5, respectively) were reported for the models. Besides, one reported bond angle outlier (D609 for PDE11) and rotamer deviation (M702 for PDE6) are far away from the critical drug-binding cavity which in turn do not effect the docking experiments in the current work. Since the models are well aligned onto PDE5 (Figure 3), finally Zn2+ and Mg2+ counterions were embedded to the metal binding sites of PDE6 and PDE11 after overlaying the homology models on the PDE5 structure. Also, the comparison of the contact energy profiles (Figure S3, Supplementary Materials) are plotted for the models and compared with PDE5 which show correlation with each other.
Figure 3.

Superposition of PDE5, PDE6 and PDE11, illustrated with blue, cyan and red colors, respectively. The counterions, Zn2+ and Mg2+, are shown with blue circles at the metal binding side.
Binding affinity and binding pattern analysis of the hit compounds and tadalafil with PDE5, PDE6 and PDE11
More than 20 million compounds were downloaded from ZINC database and these ligands were prepared at physiological conditions. Similarity analysis of these compounds with tadalafil were performed with molecular fingerprint and similarity search tools. At the end of the similarity search, we were able to filter out 1309 tadalafil-like molecules. These molecules were docked into the drug-binding cavities of PDE5, PDE6 and PDE11 in order to check their predicted binding energies at these targets. After completion of docking calculations, 27 molecules were presented, herein, based on their high docking score against the principal target-PDE5 and also their selectivity over PDE6 and PDE11 (Table 1). The common feature of these compounds is that all of them form strong hydrogen bonding interactions with Gln817 in PDE5 except for ZINC23055991, ZINC23183710 and ZINC32995890 compounds. This is due to the absence of polar hydrogens in ZINC23055991 and ZINC23183710. However, they still fitted very well to the substrate pocket in PDE5 (dG.Chemscore = −43.24 and −38.01 kJ/mol for ZINC23055991 and ZINC23183710, respectively) via mostly van der Waals interactions (for ZINC23055991) whereas via H-bonding interactions and π–H interaction (i.e. His613, Tyr612 and Phe786, respectively, for ZINC23183710). On the other hand, ZINC32995890 compound is oriented in such a way that its 1–4 benzodioxine fragment makes a π–H stacking interaction with Leu804 and two π–π interactions by its 1,3-dioxo isoindolin-2-yl fragment with aromatic Phe820 residue. Detailed protein–ligand interaction diagrams of each compound with three PDEs can be found in the Supplementary Materials, Figure S4. All of the molecules occupy the narrow and deep hydrophobic catalytic pockets of the enzymes (volume of around 300 Å3); the superpositions of the compounds can be seen in Figure 4. None of the compounds directly interacts with the metal atoms at the M site which is in line with the identified crystal structures of the PDE5, so far. We have especially focused on two ligands, namely ZINC02120502 and ZINC16031243, due to their high docking scores for PDE5 (−48.79 kJ/mol and −46.40 kJ/mol, respectively) and relatively low predicted binding affinities at the binding cavities of PDE6 and PDE11 targets (−37.16 kJ/mol and −39.43 kJ/mol for ZINC02120502; and −34.96 kJ/mol and −38.49 kJ/mol for ZINC16031243 compounds) as compared to other hits. The key interactions of these two ligands within the target enzyme (PDE5) are summarized as follows: ZINC02120502 makes a π–π and π–H interaction with Phe820 and Val782 residues, respectively and a H-bonding interaction with Gln817 with its NH group within a distance of 2.07 Å (Figure 5). It is in close contact with Met816, Phe786, Ile813, Leu804, Leu725, Ala779 residues and; Ile665 and Ser 663 on the H-loop site residues via van der Waals interactions. The orientation of this compound at the catalytic pocket of PDE5 resembles to the crystal orientation of tadalafil in many aspects, whereas the top-scored docked poses within the PDE6 and PDE11 differ significantly compared to its orientation in PDE5 and share common conformations and interaction patterns within each other in PDE6 and PDE11. ZINC02120502, is mainly sandwiched by the aromatic ring of Phe776 residue and Val738 via π–H bonding interaction at the binding pocket of PDE6. The other dominant interactions that stabilize the ligand in the substrate binding pocket are the hydrophobic interactions with Leu721, Ile724, Leu671, Val734, Ala723 and also ligand is surrounded by His559, Glu628, Asp720 and Gln731. ZINC02120502 is oriented at the binding cavity of PDE11 as in PDE6, i.e. its terminal alkyl chain, –CH2–CH2–CH–(CH3)2– is pointed towards the metal site and its 4-ethoxy-3-methoxy-phenyl ring is clamped between the bulkier aromatic residue Trp872 (replaced with Phe776 in PDE6) and Val834 (replaced with Val738 in PDE6). The two ligands (ZINC02120502 and ZINC16031243) are mainly stabilized at the active sites of PDE6 and PDE11 by van der Waals interactions where they do not form any hydrogen bonding interactions with the active site residues (Figure 5). As Table 1 clearly demonstrates, all these 27 molecules show either higher or similar predicted binding affinities towards PDE5 as compared to tadalafil; also most of them show some selectivity against PDE6 and PDE11. (Binding scores were calculated by Chemscore fitness function implemented in GOLD Docking Program and Chemscore.dG values were converted to predicted IC50 values according to the following formula; ΔGbinding = RTlnIC50 (T, 300 K) for the purpose of selectivity comparison.) ZINC16031243 is another compound that has a predicted IC50-ratio (PDE6/PDE5) value of 98.16 and predicted IC50-ratio (PDE11/PDE5) value of 23.84 with dG.Chemscore (PDE5) value of −46.40 kJ/mol. A close look at the superimpositions of this compound (ZINC16031243) with the proteins (Figure 5) shows that the crucial residues that stabilize these two compounds (ZINC16031243 and ZINC02120502) at the PDE6 and PDE11 drug pockets are located on the flexible loops of the proteins (namely, H-loop and M-loop) besides the Q pocket residues. The position of each molecule is dramatically shifted from its PDE5 docked pose. The importance of these two loops have also been pointed out by Cahill et al., earlier34. They proposed that M-loop residues together with its linked α-14 helix (Figure 5) are mainly responsible for the selectivity of tadalafil (PDE6/PDE5) in a comparative study with vardenafil; whereas Huang et al.47 pointed out that the Q pocket residues, Val782(PDE5)-Val738(PDE6) and Leu804(PDE5)-Met760(PDE6) play a critical role in binding affinity reduction of tadalafil (towards PDE6) as compared to sildenafil and vardenafil. However, it should be emphasized that the docked orientation of tadalafil differs in their generated homology models of PDE6 in these two studies. Moreover, an analysis of the dynamical behaviour of these loops by Zagrovic and Van Gunsteren53 showed a pattern of which they called “loop clamp” in order to explain the approaching of M and H loops to each other upon ligand binding to PDE5. Hence, by the motivation of the understanding of the structural aspects of the PDEs and ligand interactions, we further derived relatively long (50 ns) classical MD simulations trajectories.
Table 1.
Predicted binding free energies (Chemscore.dG) and 2D structures of ZINC compounds against the principal target, PDE5 and off-target enzymes, PDE6 and PDE11 (binding scores are expressed in kJ/mol and calculated by Chemscore fitness function implemented in GOLD Docking Program). Chemscore.dG values were converted to IC50 values – for the purpose of selectivity comparison – according to the formula; ΔGbinding= RTlnIC50, where T is taken as 300 K.
| ZINC Compounds | Chemscore.dG(PDE5) | Chemscore.dG(PDE6) | Chemscore.dG(PDE11) | Predicted IC50 ratio(PDE6/PDE5) | Predicted IC50 ratio(PDE11/PDE5) | |
|---|---|---|---|---|---|---|
| 1 | 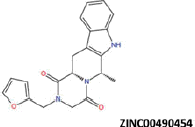 |
−37.83 | −31.30 | −34.92 | 13.71 | 3.21 |
| 2 | 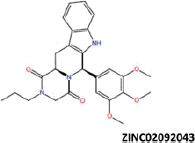 |
−43.96 | −36.83 | −37.63 | 17.44 | 12.65 |
| 3 | 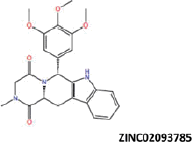 |
−42.55 | −33.56 | −35.76 | 36.76 | 15.22 |
| 4 | 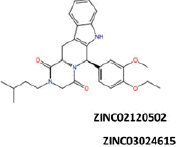 |
−48.79 | −37.16 | −39.43 | 105.93 | 42.64 |
| 5 | 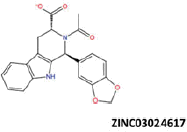 |
−38.99 | −31.70 | −36.99 | 18.59 | 2.23 |
| 6 | 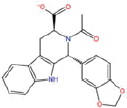 |
−37.61 | −30.59 | −39.00 | 16.69 | 0.57 |
| 7 | 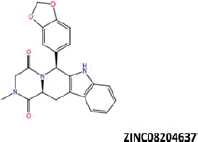 |
−45.24 | −38.76 | −36.49 | 13.44 | 33.39 |
| 8 | 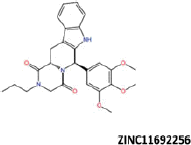 |
−43.12 | −38.58 | −37.54 | 6.17 | 9.37 |
| 9 | 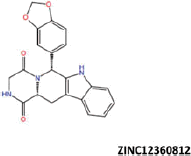 |
−38.01 | −29.55 | −37.96 | 29.72 | 1.02 |
| 10 | 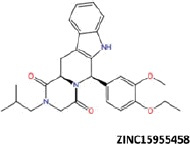 |
−47.98 | −39.31 | −39.24 | 32.33 | 33.25 |
| 11 | 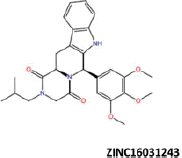 |
−46.40 | −34.96 | −38.49 | 98.16 | 23.84 |
| 12 | 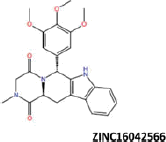 |
−43.21 | −36.87 | −36.05 | 12.70 | 17.65 |
| 13 | 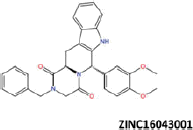 |
−45.29 | −38.80 | −40.26 | 13.49 | 7.51 |
| 14 | 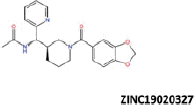 |
−39.22 | −32.47 | −36.60 | 14.97 | 2.86 |
| 15 | 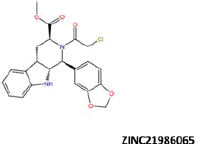 |
−36.84 | −27.13 | −39.64 | 49.06 | 0.33 |
| 16 | 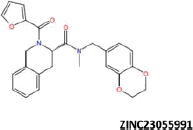 |
−43.24 | −36.50 | −38.60 | 14.91 | 6.43 |
| 17 | 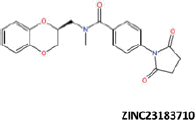 |
−38.01 | −30.89 | −36.09 | 17.37 | 2.16 |
| 18 | 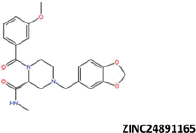 |
−37.69 | −30.66 | −37.68 | 16.75 | 1.00 |
| 19 | 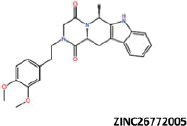 |
−43.03 | −36.01 | −38.09 | 16.69 | 7.25 |
| 20 | 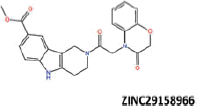 |
−39.30 | −32.80 | −35.77 | 13.55 | 4.12 |
| 21 | 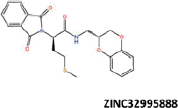 |
−36.85 | −30.20 | −36.81 | 14.38 | 1.02 |
| 22 | 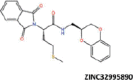 |
−38.00 | −29.48 | −34.92 | 30.45 | 3.44 |
| 23 | 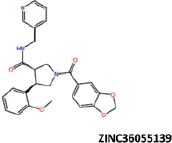 |
−41.49 | −36.02 | −37.69 | 8.96 | 4.59 |
| 24 | 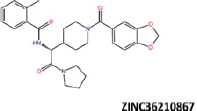 |
−39.87 | −33.30 | −35.83 | 13.93 | 5.05 |
| 25 | 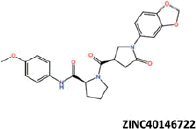 |
−39.05 | −29.85 | −37.44 | 39.99 | 1.91 |
| 26 | 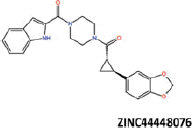 |
−39.56 | −33.03 | −38.33 | 13.71 | 1.64 |
| 27 | 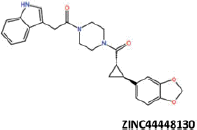 |
−43.16 | −35.41 | −37.4 | 22.36 | 10.07 |
Figure 4.

Superposition of 27 selected compounds at the end of the docking simulations.
Figure 5.
(A) Docked pose of ZINC02120502 at the active site of PDE6. (B) Docked pose of ZINC02120502 at the active site of PDE11. (C) Overlay of ZINC02120502 (tan) onto the crystal orientation65 of tadalafil (blue). (D) Overlay of ZINC16031243 docked poses at the active site of PDE5 (blue), PDE6 (pink) and PDE11 (tan). Protein?ligand interaction diagrams of ZINC02120502 and ZINC16031243 with PDEs (shown in the bottom of the figure).


MD simulations of apo and holo states of PDE5, PDE6 and PDE11 bound with the selected hit compounds (ZINC02120502 and ZINC16031243) and tadalafil
Relatively long MD simulations (50 ns) were carried out in order to understand the structural and dynamical behaviour of the selected hits within the binding pockets of the enzymes. Backbone RMSD values during the whole simulations time show that studied systems did undergo in metastable states mostly after about 10 ns (Figure 6). Both apo and holo states of PDE5 systems have reasonably converged trajectories at around 3 Å which are slightly lower as compared to PDE6 and PDE11 systems (around 4 Å) which can be expected owing to the construction of the models based on the PDE5 coordinates. It should be noted that, in the case of the PDE11 + tadalafil system, the relative late convergence (around 30 ns) also corresponds to a sudden jump in the ligand RMSD trace due to a ligand conformational change around its one rotatable bond (Figure 7). The H-bonds that were established during the docking simulations between the oxygens atom of Gln 817, Gln773, Gln869 in PDE5, PDE6 and PDE11, respectively and –NH hydrogen of the common indole rings of the ligands were generally stable during the MD simulations (Figure 8), except for the ZINC16031243 in its complex with PDE5. In this case, this H-bond was broken at the beginning of the simulation due to the dramatic shifts in the dihedral position of the residue, Gln817. This residue is pointed towards the ligand according to the position of Gln775 and moreover, depends on its orientation whether Gln775 forms an intramolecular hydrogen bond with the donor or acceptor atoms of Gln817, as well. Simulation trajectories were clustered based on the clustering algorithm implemented in UCSF Chimera program (version 1.10.2)70. Representative frames for the most populated cluster were chosen for each simulation and shown in Figure 9. Obviously, there is an energetic complimentary between the hydrogen bond network of the protein and the ligand–protein interactions. On the other hand, there is also considerable effect of configurations of water molecules – surrounded around the ligands – on the crucial residue–ligand interactions; e.g. the water molecule pulls the oxygen of Gln817 to itself causing a loss of a H-bond between ZINC02120502 and PDE5 during the simulation time (Figure 9). Besides, another H-bond is formed between Asn661 located on the H-loop and the oxygen of tetracyclic ring of ZINC02120502 in PDE5. It should be noted that the position of Phe820 in PDE5 kept its horizontal position according to the tetracyclic rings of each ligand, during the simulation time. Actually, the mentioned importance of rigid tetracyclic fragments in terms of high inhibition potency towards PDE5 was pointed out earlier71. Moreover, Asn607 makes a strong hydrogen bonding interaction within a distance of 1.84 Å with the same fragment of ZINC02120502 in PDE6, as well. Although the top docking poses of this compound overlay well within PDE6 and PDE11, the representative frame of ZINC02120502 in the catalytic pocket of PDE11 indicates an orientational change through the simulation time course due to a considerable conformational change in overall protein structure. Specifically, H-loop migrates towards the Q-pocket residues and adopts a one-turn helix and the bend in the α-14 helix also lets the emergence of a H-bond with its Gln869 and indole hydrogen of the ZINC02120505 (Figure 10). Additionally, the indole ring is sandwiched by Phe838. In the case of PDE6 + ZINC16031243 system, these motile loops are connected each other via an efficient H-bonding interaction via Tyr610 (H-loop) and Tyr816 (M-loop) within a 1.80 Å distance. Also a water molecule constructs a H-bond with Tyr816 which further gives rise to a unique, nearly overlapping conformational state of this part of the protein. These results point out that critical consideration should also be given to the residues on this highly mobile (which was confirmed by RMSF calculations, Figure 11) structural parts of the proteins (H and M loops) along with the Q pocket residues in terms of designing potent and selective PDE5 inhibitors.
Figure 6.

Traces of protein backbone RMSD (root-mean-squared-deviation) evaluation during the whole production stages of the MD Simulations.
Figure 7.

The RMSD evaluation of the ligands during the simulation time.
Figure 8.

Traces of hydrogen bonding interactions throughout simulation time (x and y axis represents the simulation time and distances between O? atoms of Gln817 (PDE5), Gln773 (PDE6), Gln869 (PDE11) and indole fragments hydrogen in the ligands, respectively). Color codes: orange: PDE5 + ZINC16031243; purple: PDE11 + tadalafil; blue: PDE5 + ZINC02120502; red: PDE5 + tadalafil; green: PDE6 + tadalafil.
Figure 9.

Simulated structures of ZINC0210502 and ZINC16031243 at the substrate pockets of PDE5, PDE6 and PDE11.
Figure 10.

Overlay of docking pose (blue) and representative structure of ZINC02120502 (white) in the catalytic pocket of PDE11.
Figure 11.

Root-mean-square fluctuation (RMSF) values per residue during the MD simulations.
MM-GBSA calculations
Predicted binding energies of the selected compounds as well as tadalafil are calculated by MM/GBSA calculations. Table 2 summarizes the derived results. Results verify higher predicted binding affinities of the selected hits from screening compared to tadalafil. Moreover, PDE5/PDE6 and PDE5/PDE11 selectivity profiles of these compounds are similar or higher than tadalafil.
Table 2.
Comparison of protein–ligand free energy results of tadalafil with selected hit compounds using MM/GBSA calculations.
| Targets/compounds | ΔGMM/GBSA (kcal/mol) | Selectivity ratio |
|---|---|---|
| PDE5 + tadalafil | −89.23 | |
| PDE6 + tadalafil | −75.63 | 1.18 |
| PDE11 + tadalafil | −88.31 | 1.01 |
| PDE5 + ZINC02120502 | −130.48 | |
| PDE6 + ZINC02120502 | −116.41 | 1.12 |
| PDE11 + ZINC02120502 | −110.20 | 1.18 |
| PDE5 + ZINC16031243 | −113.29 | |
| PDE6 + ZINC16031243 | −102.52 | 1.11 |
| PDE11 + ZINC16031243 | −101.05 | 1.12 |
hERG K+ ion channel activity of the compounds
hERG (KCNH2 or Kv11.1) is the name of a gene that encodes the α-subunit of a voltage-gated potassium channel. hERG channels are expressed in various types of tissue and cell types such as heart muscles, brain and retina. Its topology consists of six transmembrane α-helices (S1–S6) where S5 and S6 helices form the channel inner cavity and S1–S4 helices constitute the voltage sensing domain. The function of the channel is to permeate potassium cations across the cell membrane via its unique selectivity filter (SVGFG). Since the inhibition of the potassium current by direct binding of the drugs at the central cavity causes abnormalities in the cardiac action potential72 which may further lead long QT prolongation (LQTS), many drugs have been withdrawn from the drug market over the years or their usage has been restricted73. It was shown that tadalafil also inhibits the hERG channel by concentration dependent manner with an IC50 value of 100 μM74. In this study, for docking calculations, we used the refined structural models of hERG1 in different conformational states that were generated previously by our group and have been extensively validated in experimental and theoretical studies75–85. Accordingly, we report the in silico activities of the compounds with the channel (Table 3). ZINC02120502, ZINC16031243 and tadalafil show similar predicted binding affinities at the central cavities of the hERG K+ channel model.
Table 3.
Predicted binding affinities of the selected compounds within the hERG K+ channel. Each compound was docked into the central cavities of the channel by GOLD docking software with Chemscore fitness function. Dockings were realized by considering the two known conformational states of the channel. OS and OIS states stand for the open and open-inactivated states. dG.Chemscore values are expressed in kJ/mol. Tadalafil and the two selected potent and selective PDE5 inhibitor compounds, ZINC02120502 and ZINC16031243 are shown in bold in the table.
| Compounds | dG.Chemscore | dC.Chemscore |
|---|---|---|
| (hERG-OS state) | (hERG-OIS state) | |
| Tadalafil | −32.22 | −48.72 |
| ZINC00490454 | −34.97 | −39.74 |
| ZINC02092043 | −35.76 | −47.82 |
| ZINC02093785 | −32.8 | −46.75 |
| ZINC02120502 | −35.04 | −47.32 |
| ZINC03024615 | −29.46 | −38.31 |
| ZINC03024617 | −33.34 | −46.48 |
| ZINC08204637 | −32 | −47.9 |
| ZINC11692256 | −34.06 | −43.99 |
| ZINC12360812 | −32.71 | −41.98 |
| ZINC15955458 | −36.54 | −53.43 |
| ZINC16031243 | −35.12 | −47.06 |
| ZINC16042566 | −32.95 | −40.86 |
| ZINC16043001 | −40.52 | −53.84 |
| ZINC19020327 | −30.57 | −42.01 |
| ZINC21986065 | −29.05 | −42.83 |
| ZINC23055991 | −35.17 | −45.54 |
| ZINC23183710 | −33.42 | −38.81 |
| ZINC24891165 | −30.73 | −39.23 |
| ZINC26772005 | −37.63 | −41.57 |
| ZINC29158966 | −34.38 | −41.1 |
| ZINC32995888 | −33.84 | −43.22 |
| ZINC32995890 | −32.06 | −43.31 |
| ZINC36055139 | −34.8 | −53.11 |
| ZINC36210867 | −35.18 | −49.36 |
| ZINC40146722 | −32.34 | −44.02 |
| ZINC44448076 | −37.69 | −44.14 |
| ZINC44448130 | −36.76 | −45.99 |
E-pharmacophore studies
The structure-based pharmacophore modeling (E-pharmacophore) uses advantages of both ligand – and structure-based approaches by deriving energetically optimized structure-based pharmacophore models. For this aim, representative conformer from MD simulations of one of the selected hit compounds (ZINC02120502) is used for E-pharmacophore studies. Six-sited (RRRHHH) hypothesis was found as top-scored pharmacophore model. These main interactions were aromatic rings (labeled as “R”) and hydrophobic interactions (labeled as “H”, Figure 12). The RRRHHH hypothesis is then used for ligand screening of Otava Drug-like Green Collection (around 176000 compounds) using Glide/SP docking protocol from Schrodinger’s Maestro molecular modeling package86 and top-1000 scored compounds were collected. Compounds that show high docking scores and high fitness scores are also shown in Figure 12.
Figure 12.

(Top) Derived top-scored six-sited (RRRHHH) E-pharmacophore model; (bottom) 176 000 compounds from Otava small-molecules database are screened against derived pharmacophore model and top-1000 compounds that have high Fitness scores with these sites are then docked at the PDE5 binding pocket using Glide/SP (standard precision). Compounds that show high docking scores as well as high fitness scores are shown in the figure. 2D ligand interaction diagram of selected Otava compound (1094821) is also represented in the figure.
Conclusions
In this study, the similarity-based virtual screening protocol is applied for the ZINC small molecules database that contains more than 20 million small compounds. Based on Tanimato coefficient values, 1309 molecules from this database showed 80% or more structural similarity with the PDE5 inhibitor tadalafil. These compounds are then docked in PDE5 as well as structurally similar other isoforms PDE6 and PDE11. Results showed that 27 compounds have high predicted binding affinities towards the principle target, PDE5. Especially two hits (ZINC02120502 and ZINC16031243) from 27 compounds represented some selectivities against PDE6 and PDE11. Thus, these two compounds as well as tadalafil are used in classical MD simulations and post-processing MD analyses which showed some insights about their structural and dynamical behaviors at the studied targets. Finally these hits are also tested at the hERG K+ channel models in order to predict their possible cardiotoxicity side effects. Selected two hits showed similar predicted binding affinities at the hERG channels with tadalafil. Moreover, a structure-based pharmacophores (E-pharmacophore) study was also applied for the selected hit compounds. Results of this study can be useful for designing of novel, safe and selective PDE5 inhibitors.
Disclosure statement
The authors report no declarations of interest.
Funding
We acknowledge the National Center for High Performance Computing of Turkey (UHEM) under Grant 10982010, Istanbul Technical University Research Fund (BAP 30492) and The Scientific and Technological Research Council of Turkey (TUBITAK) under 2214-A Research Grant, for supporting this study.
Supplementary Material
References
- 1.Wang R, Burnett AL, Heller WH, et al. Selectivity of Avanafil, a PDE5 inhibitor for the treatment of erectile dysfunction: implications for clinical safety and improved tolerability. J Sex Med 2012;9:2122–9. [DOI] [PubMed] [Google Scholar]
- 2.Corbin JD, Francis SH, Webb DJ.. Phosphodiesterase Type 5 as a pharmacologic target in erectile dysfunction. Urology 2002;60:4–11. [DOI] [PubMed] [Google Scholar]
- 3.Rotella D.Phosphodiesterase 5 Inhibitors: Current Status and Potential Applications. Nat Rev Drug Discov 2002;1:674–82. [DOI] [PubMed] [Google Scholar]
- 4.Rybalkin SD, Yan C, Bornfeldt KE, Beavo JA.. Cyclic GMP phosphodiesterases and regulation of smooth muscle function. Circ Res 2003;93:280–91. [DOI] [PubMed] [Google Scholar]
- 5.Terrett NK, Bell AS, Brown D, Ellis P.. Sildenafil (Viagra(TM)), a potent and selective inhibitor of Type 5 CGMP phosphodiesterase with utility for the treatment of male erectile dysfunction. Bioorg Med Chem Lett 1996;6:1819–24. [Google Scholar]
- 6.Srivani P, Srinivas E, Raghu R, Sastry GN.. Molecular modeling studies of pyridopurinone derivatives-potential phosphodiesterase 5 inhibitors. J Mol Graph Model 2007;26:378–90. [DOI] [PubMed] [Google Scholar]
- 7.Sakamoto T, Koga Y, Hikota M, et al. 8-(3-Chloro-4-Methoxybenzyl)-8H-pyrido[2,3-d]pyrimidin-7-one derivatives as potent and selective phosphodiesterase 5 inhibitors. Bioorg Med Chem Lett 2015;25:1431–5. [DOI] [PubMed] [Google Scholar]
- 8.Sakamoto T, Koga Y, Hikota M, et al. The discovery of avanafil for the treatment of erectile dysfunction: a novel pyrimidine-5-carboxamide derivative as a potent and highly selective phosphodiesterase 5 inhibitor. Bioorg Med Chem Lett 2014;24:5460–5. [DOI] [PubMed] [Google Scholar]
- 9.Sakamoto T, Koga Y, Hikota M, et al. Design and synthesis of novel 5-(3,4,5-trimethoxybenzoyl)-4-aminopyrimidine derivatives as potent and selective phosphodiesterase 5 inhibitors: scaffold hopping using a pseudo-ring by intramolecular hydrogen bond formation. Bioorg Med Chem Lett 2014;24:5175–80. [DOI] [PubMed] [Google Scholar]
- 10.Wang Z, Zhu D, Yang X, et al. The selectivity and potency of the new PDE5 inhibitor TPN729MA. J Sex Med 2013;10:2790–7. [DOI] [PubMed] [Google Scholar]
- 11.Duan H, Zheng J, Lai Q, et al. 2-Phenylquinazolin-4(3H)-One, a class of potent PDE5 inhibitors with high selectivity versus PDE6 . Bioorg Med Chem Lett 2009;19:2777–9. [DOI] [PubMed] [Google Scholar]
- 12.Choi H, Lee J, Kim YH, et al. Discovery of potent, selective, and orally bioavailable PDE5 inhibitor: methyl-4-(3-chloro-4-methoxybenzylamino)-8-(2-hydroxyethyl)-7-methoxyquinazolin-6-ylmethylcarbamate (CKD 533). Bioorg Med Chem Lett 2010;20:383–6. [DOI] [PubMed] [Google Scholar]
- 13.Owen DR, Walker JK, Jon Jacobsen E, et al. Identification, synthesis and SAR of amino substituted pyrido[3,2b]pyrazinones as potent and selective PDE5 inhibitors. Bioorg Med Chem Lett 2009;19:4088–91. [DOI] [PubMed] [Google Scholar]
- 14.Kim YH, Choi H, Lee J, et al. Quinazolines as potent and highly selective PDE5 inhibitors as potential therapeutics for male erectile dysfunction . Bioorg Med Chem Lett 2008;18:6279–82. [DOI] [PubMed] [Google Scholar]
- 15.Arnold NJ, Arnold R, Beer D, et al. Potent and selective xanthine-based inhibitors of phosphodiesterase 5. Bioorg Med Chem Lett 2007;17:2376–9. [DOI] [PubMed] [Google Scholar]
- 16.Giovannoni MP, Vergelli C, Biancalani C, et al. Novel pyrazolopyrimidopyridazinones with potent and selective phosphodiesterase 5 (PDE5) inhibitory activity as potential agents for treatment of erectile dysfunction. J Med Chem 2006;49:5363–71. [DOI] [PubMed] [Google Scholar]
- 17.Yang GF, Lu HT, Xiong Y, Zhan CG.. Understanding the structure–activity and structure–selectivity correlation of cyclic guanine derivatives as phosphodiesterase-5 inhibitors by molecular docking, CoMFA and CoMSIA analyses. Bioorg Med Chem 2006;14:1462–73. [DOI] [PubMed] [Google Scholar]
- 18.Xia G, Li J, Peng A, et al. Synthesis and phosphodiesterase 5 inhibitory activity of novel pyrido[1,2-e]purin-4(3H)-one derivatives . Bioorg Med Chem Lett 2005;15:2790–4. [DOI] [PubMed] [Google Scholar]
- 19.Jiang W, Alford VC, Qiu Y, et al. Synthesis and SAR of tetracyclic pyrroloquinolones as phosphodiesterase 5 inhibitors. Bioorg Med Chem 2004;12:1505–15. [DOI] [PubMed] [Google Scholar]
- 20.Pissarnitski DA, Asberom T, Boyle CD, et al. SAR development of polycyclic guanine derivatives targeted to the discovery of a selective PDE5 inhibitor for treatment of erectile dysfunction. Bioorg Med Chem Lett 2004;14:1291–4. [DOI] [PubMed] [Google Scholar]
- 21.Daugan A, Grondin P, Ruault C, et al. The discovery of tadalafil: a novel and highly selective PDE5 inhibitor. 2: 2,3,6,7,12,12a-hexahydropyrazino[1′,2′:1,6]pyrido[3,4-b]indole-1,4-dione analogues. J Med Chem 2003;46:4533–42. [DOI] [PubMed] [Google Scholar]
- 22.Ukita T, Nakamura Y, Kubo A, et al. 1,7- and 2,7-naphthyridine derivatives as potent and highly specific PDE5 inhibitors . Bioorg Med Chem Lett 2003;13:2341–5. [DOI] [PubMed] [Google Scholar]
- 23.Maw GN, Allerton CM, Gbekor E, Million WA.. Design, synthesis and biological activity of beta-carboline-based type-5 phosphodiesterase inhibitors. Bioorg Med Chem Lett 2003;13:1425–8. [DOI] [PubMed] [Google Scholar]
- 24.Jiang W, Sui Z, Macielag MJ, et al. Furoyl and benzofuroyl pyrroloquinolones as potent and selective PDE5 inhibitors for treatment of erectile dysfunction. J Med Chem 2003;46:441–4. [DOI] [PubMed] [Google Scholar]
- 25.Wang Y, Chackalamannil S, Hu Z, et al. Design and synthesis of xanthine analogues as potent and selective PDE5 inhibitors. Bioorg Med Chem Lett 2002;12:3149–52. [DOI] [PubMed] [Google Scholar]
- 26.Bi Y, Stoy P, Adam L, et al. The discovery of novel, potent and selective PDE5 inhibitors. Bioorg Med Chem Lett 2001;11:2461–4. [DOI] [PubMed] [Google Scholar]
- 27.Wang G, Liu Z, Chen T, et al. Design, synthesis, and pharmacological evaluation of monocyclic pyrimidinones as novel inhibitors of PDE5. J Med Chem 2012;55:10540–50. [DOI] [PubMed] [Google Scholar]
- 28.Bi Y, Stoy P, Adam L, et al. Quinolines as extremely potent and selective PDE5 inhibitors as potential agents for treatment of erectile dysfunction . Bioorg Med Chem Lett 2004;14:1577–80. [DOI] [PubMed] [Google Scholar]
- 29.Zhang Z, Artemyev NO. Determinants for phosphodiesterase 6 inhibition by its gamma-subunit. Biochemistry 2010;49:3862–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Barren B, Gakhar L, Muradov H, et al. Structural basis of phosphodiesterase 6 inhibition by the C-terminal region of the gamma-subunit. EMBO J 2009;28:3613–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liu YT, Matte SL, Corbin JD, et al. Probing the catalytic sites and activation mechanism of photoreceptor phosphodiesterase using radiolabeled phosphodiesterase inhibitors. J Biol Chem 2009;284:31541–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang X, Feng Q, Cote RH.. Efficacy and selectivity of phosphodiesterase-targeted drugs in inhibiting photoreceptor phosphodiesterase (PDE6) in retinal photoreceptors. Investig Ophthalmol Vis Sci 2005;46:3060–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Granovsky AE, Artemyev NOA.. Conformational switch in the inhibitory γ-subunit of PDE6 upon enzyme activation by transducin. Biochemistry 2001;40:13209–15. [DOI] [PubMed] [Google Scholar]
- 34.Cahill KB, Quade JH, Carleton KL, Cote RH.. Identification of amino acid residues responsible for the selectivity of tadalafil binding to two closely related phosphodiesterases, PDE5 and PDE6. J Biol Chem 2012;287:41406–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pissarnitski D.Phosphodiesterase 5 (PDE 5) inhibitors for the treatment of male erectile disorder: attaining selectivity versus PDE6. Med Res Rev 2006;26:369–95. [DOI] [PubMed] [Google Scholar]
- 36.Kerr NM, Danesh-Meyer HV.. Phosphodiesterase inhibitors and the eye . Clin Exp Ophthalmol 2009;37:514–23. [DOI] [PubMed] [Google Scholar]
- 37.Foresta C, Caretta N, Zuccarello D, et al. Expression of the PDE5 enzyme on human retinal tissue: new aspects of PDE5 inhibitors ocular side effects. Eye (Lond) 2008;22:144–9. [DOI] [PubMed] [Google Scholar]
- 38.Makhlouf A, Kshirsagar A, Niederberger C.. Phosphodiesterase 11: a brief review of structure, expression and function. Int J Impot Res 2006;18:501–9. [DOI] [PubMed] [Google Scholar]
- 39.Bischoff E.Potency, selectivity, and consequences of nonselectivity of PDE inhibition. Int J Impot Res 2004;16:S11–S14. [DOI] [PubMed] [Google Scholar]
- 40.Ukita T, Nakamura Y, Kubo A, et al. Novel, potent, and selective phosphodiesterase 5 inhibitors: synthesis and biological activities of a series of 4-aryl-1-isoquinolinone derivatives. J Med Chem 2001;44:2204–18. [DOI] [PubMed] [Google Scholar]
- 41.Mohamed HA, Girgis NMR, Wilcken R, et al. Synthesis and molecular modeling of novel tetrahydro-β-carboline derivatives with phosphodiesterase 5 inhibitory and anticancer properties. J Med Chem 2011;54:495–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bunnage ME, Mathias JP, Wood A, et al. Highly potent and selective chiral inhibitors of PDE5: an illustration of Pfeiffer's rule . Bioorg Med Chem Lett 2008;18:6033–6. [DOI] [PubMed] [Google Scholar]
- 43.Simon A, Barabas P, Kardos J.. Structural determinants of phosphodiesterase 6 response on binding catalytic site inhibitors. Neurochem Int 2006;49:215–22. [DOI] [PubMed] [Google Scholar]
- 44.Chen G, Wang H, Robinson H, et al. An insight into the pharmacophores of phosphodiesterase-5 inhibitors from synthetic and crystal structural studies. Biochem Pharmacol 2008;75:1717–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cichero E, D’Ursi P, Moscatelli M, et al. Homology modeling, docking studies and molecular dynamic simulations using graphical processing unit architecture to probe the type-11 phosphodiesterase catalytic site: a computational approach for the rational design of selective inhibitors. Chem Biol Drug Des 2013;82:718–31. [DOI] [PubMed] [Google Scholar]
- 46.Xiong Y, Lu H, Zhan C.. Dynamic structures of phosphodiesterase-5 active site by combined molecular dynamics simulations and hybrid quantum mechanical/molecular mechanical calculations. J Comput Chem 2008;29:1259–67. [DOI] [PubMed] [Google Scholar]
- 47.Huang YY, Li Z, Cai YH, et al. The molecular basis for the selectivity of tadalafil toward phosphodiesterase 5 and 6: a modeling study. J Chem Inf Model 2013;53:3044–53. [DOI] [PubMed] [Google Scholar]
- 48.Mittal A, Paliwal S, Sharma M, et al. Pharmacophore based virtual screening, molecular docking and biological evaluation to identify novel PDE5 inhibitors with vasodilatory activity. Bioorg Med Chem Lett 2014;24:3137–41. [DOI] [PubMed] [Google Scholar]
- 49.Li Y, Wu W, Ren H, et al. Exploring the structure determinants of pyrazinone derivatives as PDE5 3HC8 inhibitors: an in silico analysis. J Mol Graph Model 2012;38:112–22. [DOI] [PubMed] [Google Scholar]
- 50.Tömöri T, Hajdú I, Barna L, et al. Combining 2D and 3D in silico methods for rapid selection of potential PDE5 inhibitors from multimillion compounds’ repositories: biological evaluation. Mol Divers 2012;16:59–72. [DOI] [PubMed] [Google Scholar]
- 51.Antunes JE, Freitas MP, da Cunha EFF, et al. In silico prediction of novel phosphodiesterase type-5 inhibitors derived from sildenafil, vardenafil and tadalafil. Bioorg Med Chem 2008;16:7599–606. [DOI] [PubMed] [Google Scholar]
- 52.Yoo J, Thai KM, Kim DK, et al. 3D-QSAR studies on sildenafil analogues, selective phosphodiesterase 5 inhibitors . Bioorg Med Chem Lett 2007;17:4271–4. [DOI] [PubMed] [Google Scholar]
- 53.Zagrovic B, Van Gunsteren WF.. Computational analysis of the mechanism and thermodynamics of inhibition of phosphodiesterase 5A by synthetic ligands. J Chem Theory Comput 2007;3:301–11. [DOI] [PubMed] [Google Scholar]
- 54.Weeks JL II, Corbin JD, Francis SH.. Interactions between cyclic nucleotide phosphodiesterase 11 catalytic site and substrates or tadalafil and role of a critical Gln-869 hydrogen bond. J Pharmacol Exp Ther 2009;331:133–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wang H, Liu Y, Huai Q, et al. Multiple conformations of phosphodiesterase-5: implications for enzyme function and drug development. J Biol Chem 2006;281:21469–79. [DOI] [PubMed] [Google Scholar]
- 56.Molecular Operating Environment (MOE), 2013.08. Canada: Chemical Computing Group Inc; 2015. [Google Scholar]
- 57.Irwin JJ, Sterling T, Mysinger MM, et al. ZINC: a free tool to discover chemistry for biology. J Chem Inf Model 2012;52:1757–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sheridan RP, Kearsley SK.. Why do we need so many chemical similarity search methods? Drug Discov Today 2002;7:903–11. [DOI] [PubMed] [Google Scholar]
- 59.Willett P.Similarity-based virtual screening using 2D fingerprints. Drug Discov Today 2006;11:1046–53. [DOI] [PubMed] [Google Scholar]
- 60.Jones G, Willett P, Glen RC, et al. Development and validation of a genetic algorithm for flexible docking. J Mol Biol 1997;267:727–48. [DOI] [PubMed] [Google Scholar]
- 61.Berendsen HJC, Spoel van der D, Drunen van R. GROMACS: a message-passing parallel molecular dynamics implementation. Comp Phys Commun 1995;91:43–56. [Google Scholar]
- 62.Schüttelkopf AW, van Aalten DM.. PRODRG: a tool for high-throughput crystallography of protein–ligand complexes . Acta Crystallogr D Biol Crystallogr 2004;60:1355–63. [DOI] [PubMed] [Google Scholar]
- 63.Frisch MJ, Trucks GW, Schlegel HB, et al. Wallingford, CT: Gaussian Inc; 2009. [Google Scholar]
- 64.Rastelli G, Rio del A, Degliesposti G, Sgobba M. Fast and accurate predictions of binding free energies using MM-PBSA and MM-GBSA. J Comput Chem 2010;31:797–810. [DOI] [PubMed] [Google Scholar]
- 65.Card GL, England BP, Suzuki Y, et al. Structural basis for the activity of drugs that inhibit phosphodiesterases. Structure 2004;12:2233–47. [DOI] [PubMed] [Google Scholar]
- 66.Zhang KYJ, Card GL, Suzuki Y, et al. A glutamine switch mechanism for nucleotide selectivity by phosphodiesterases. Mol Cell 2004;15:279–86. [DOI] [PubMed] [Google Scholar]
- 67.Sung B, Hwang K, Jeon Y, et al. Structure of the catalytic domain of human phosphodiesterase 5 with bound drug molecules. Nature 2003;425:98–102. [DOI] [PubMed] [Google Scholar]
- 68.Wang H, Ye M, Robinson H, et al. Conformational variations of both phosphodiesterase-5 and inhibitors provide the structural basis for the physiological effects of vardenafil and sildenafil. Mol Pharmacol 2008;73:104–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.The UniProt Consortium. UniProt: a hub for protein information. Nucleic Acids Res 2015;43:D204–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Pettersen EF, Goddard TD, Huang CC, et al. UCSF Chimera – a visualization system for exploratory research and analysis. J Comput Chem 2004;25:1605–12. [DOI] [PubMed] [Google Scholar]
- 71.El-Gamil DS, Ahmed NS, Gary BD, et al. Design of novel β-carboline derivatives with pendant 5-bromothienyl and their evaluation as phosphodiesterase-5 inhibitors. Arch Pharm (Weinheim) 2013;346:23–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Witchel HJ.Drug-induced hERG block and long QT syndrome. Cardiovasc Ther 2011;29:251–9. [DOI] [PubMed] [Google Scholar]
- 73.Sanguinetti MC, Tristani-Firouzi M.. hERG potassium channels and cardiac arrhythmia. Nature 2006;440:463–9. [DOI] [PubMed] [Google Scholar]
- 74.Dustan Sarazan R, Crumb WJ, Beasley CM, et al. Absence of clinically important HERG channel blockade by three compounds that inhibit phosphodiesterase 5 – sildenafil, tadalafil, and vardenafil. Eur J Pharmacol 2004;502:163–7. [DOI] [PubMed] [Google Scholar]
- 75.Lees-Miller JP, Subbotina JO, Guo J, et al. Interactions of H562 in the S5 helix with T618 and S621 in the pore helix are important determinants of hERG1 potassium channel structure and function. Biophys J 2009;J96:3600–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Durdagi S, Subbotina J, Lees-Miller J, et al. Insights into the molecular mechanism of hERG1 channel activation and blockade by drugs. Curr Med Chem 2010;17:3514–32. [DOI] [PubMed] [Google Scholar]
- 77.Subbotina J, Yarov-Yarovoy V, Lees-Miller J, et al. Structural refinement of the hERG1 pore and voltage-sensing domains with ROSETTA-membrane and molecular dynamics simulations. Proteins 2010;78:2922–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Durdagi S, Duff HJ, Noskov SY.. Combined receptor and ligand-based approach to the universal pharmacophore model development for studies of drug blockade to the hERG1 pore domain. J Chem Inf Model 2011;51:463–74. [DOI] [PubMed] [Google Scholar]
- 79.Durdagi S, Deshpande S, Duff HJ, Noskov SY.. Modeling of open, closed, and open-inactivated states of the hERG1 channel: structural mechanisms of the state-dependent drug binding. J Chem Inf Model 2012;52:2760–74. [DOI] [PubMed] [Google Scholar]
- 80.Guo J, Durdagi S, Changalov M, et al. Structure driven design of novel human ether-a-go-go-related-gene channel (hERG1) activators. PLoS One 2014;9:e105553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Durdagi S, Randall T, Duff HJ, et al. Rehabilitating drug-induced long-QT promoters: in-silico design of hERG-neutral cisapride analogues with retained pharmacological activity. BMC Pharmacol Toxicol 2014;15:14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Guo J, Cheng YM, Lees-Miller JP, et al. NS1643 interacts around L529 of hERG to alter voltage sensor movement on the path to activation. Biophys J 2015;108:1400–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Durdagi S, Guo J, Lees-Miller J, et al. Structure-guided topographic mapping and mutagenesis to elucidate binding sites for the Herg1 potassium channel (KCNH2) activator-NS1643. J Pharmacol Exp Ther 2012;342:441–52. [DOI] [PubMed] [Google Scholar]
- 84.Anwar-Mohamed A, Barakat KH, Bhat R, et al. A human ether-a-go-go-related (hERG) ion channel atomistic model generated by long supercomputer molecular dynamics simulations and its use in predicting drug cardiotoxicity. Toxicol Lett 2014;230:382–92. [DOI] [PubMed] [Google Scholar]
- 85.Dempsey CE, Wright D, Colenso CK, et al. Assessing hERG pore models as templates for drug docking using published experimental constraints: the inactivated state in the context of drug block. J Chem Inf Model 2014;54:601–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Sastry GM, Adzhigirey Day T, Annabhimoju R, Sherman W.. Protein and ligand preparation: parameters, protocols, and influence on virtual screening enrichments. J Comput Aided Mol Des 2013;27:221–34. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.