

Abstract
A series of symmetric molecules incorporating aryl or pyridyl moieties as central core and 1,4-substituted triazoles as a side bridge was synthesised. The new compounds were investigated as lactate dehydro-genase (LDH, EC 1.1.1.27) inhibitors. The cancer associated LDHA isoform was inhibited with IC50 = 117–174 µM. Seven compounds exhibited better LDHA inhibition (IC50 117–136 µM) compared to known LDH inhibitor – galloflavin (IC50 157 µM).
Keywords: Lactate dehydrogenase, triazole, inhibitors
Introduction
The lactate dehydrogenase (LDH, EC 1.1.1.27) is one of the most abundant proteins and it is expressed in all tissues1. The main function of LDH is interconversion of lactate and pyruvate with accompanying interconversion of NAD+ and NADH.
Three LDH isoforms are present in humans – LDHA, LDHB and LDHC. LDHA and LDHB are expressed in all cells, whereas LDHC is produced only in testis1. In the active form, LDH is a tetramer formed of LDHA and LDHB in various ratios making five tetramers: LDH1 (4 × LDHB), LDH2 (1 × LDHA/3 × LDHB), LDH3 (2 × LDHA/2 × LDHB), LDH4 (1 × LDHA/3 × LDHB) and LDH5 (4 × LDHA)2.
In normal cells, predominant is LDHB where it converts lactate to pyruvate with interconversion of NAD+ into NADH, which allows cells to use lactate as a nutrient source for oxidative metabolism, and/or for gluconeogenesis2. LDHA is the predominant isoform found in skeletal muscle and other highly glycolytic tissues. In contrast to LDHB, LDHA has a higher affinity for pyruvate, that is, LDHA and LDH5 tetramer in particular predominantly converts pyruvate to lactate with consumption of one NADH molecule to produce NAD+ which in turn is essential in glycolysis3. Cancer cells mainly generate energy through glycolysis even in the presence of normal oxygen pressure4. Since the LDHA is the final enzyme in glycolysis pathway where generated NAD+ is necessary for continued high glycolysis rate in cancer cells (Warburg effect)4, LDHA is an important supporter of glucose metabolism in cancer cells and can affect tumourigenesis and metastasis5. Additionally, elevated levels of LDHA are markers of many tumours, the majority of them are highly glycolytic, and high LDHA levels are related with poor prognosis, for instance in several human malignancies6. Therefore, LDHA is defined as anticancer drug target6,7. Notably, a limited number of LDHA inhibitors is reported in the literature so far8. In several papers, very promising LDHA inhibition results have been reported. As a one of the first inhibitors with low-micromolar inhibition activity (Ki = 13.5 μM) compound FX11 (Figure 1) along with two similar compounds was reported in 20109. Utilising NMR and SPR (surface plasmon resonance) fragment based hit identification technique and further optimisation of structure a series with low- and submicromolar LDHA inhibition was obtained with best lead A (IC50 = 0.27 μM)10. In the other study, also using fragment-based hit identification, series of new LDHA inhibitors was obtained. In this series, IC50 values ranged from 59 to 0.12 μM, where the best inhibition was observed for compound B.11 It was noted that in this series carbohydrate moiety in the middle of the molecule and its stereochemistry had significant influence on the inhibition activity. In very recent work through docking-based virtual screening, several potential LDHA inhibitors were identified. One of the compounds identified (C) showed very good LDHA inhibition potency in vitro with an IC50 value of 0.33 μM.12
Figure 1.

Examples of chemical structures of known LDHA inhibitors.
Results and discussions
Here, we report the synthesis of symmetric molecules incorporating aryl or pyridyl moieties as central core and 1,4-substituted triazoles as a side bridge and they evaluation as LDHA inhibitors.
At the beginning of our study based on literature data11, we assumed that V-shape structures are beneficiary for good LDHA inhibition. Our assumption was based on published X-ray structures, for instance for B–LDHA complex (PDB code 4I9H). It was also obvious that besides V-shape of the molecule and appropriate length of V-“arms” the terminal groups have to be able to make hydrogen bonding, therefore we chose carboxylic, sulphonamide and nitro groups as terminal ones for this study.
Chemistry
The synthesis of desired inhibitors 7a-c was started from commercially available 1,3-dibromobenzene (1) which was reacted with trimethylsilylacetylene in Sonogashira reaction to provide bis-TMS protected derivative 2 (Scheme 1). Following deprotection with KF afforded building block 3 in good yield over two steps13. Azides 6a-c necessary for Cu-mediated click reaction were prepared in two steps from commercially available anilines 4a-c. Acylation of anilines 4a-c with chloroacetyl chloride afforded chlorides 5a-c14–16 in good yields and following treatment with NaN3 provided azide building blocks 6a-c also in good yields. Reaction of building block 3 with 6a-c under acidic click reaction condition17 provided inhibitors 7a-c.
Scheme 1.
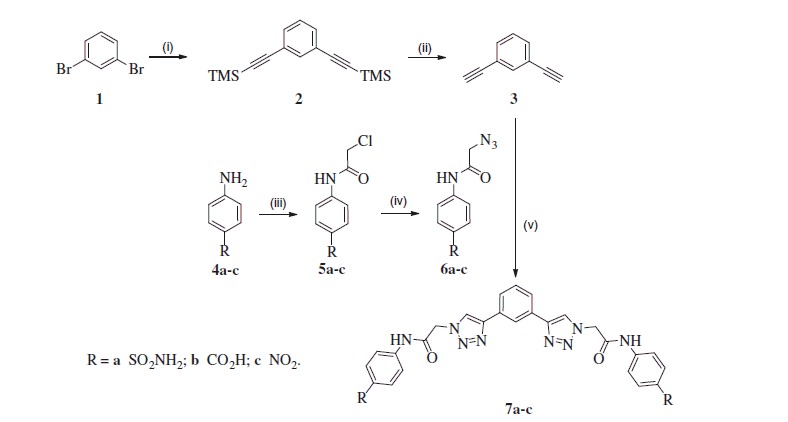
Reagents and conditions: (i) trimethylsilylacetylene, Pd2(PPh3)2, CuI, i-Pr2NH, THF, 70 °C; (ii) KF, MeOH/THF, rt, 69% for two steps; (iii) chloroacetyl chloride, K2CO3, THF, 0 °C, 5a (98%), 5b (84%), 5c (89%); (iv) NaN3, DMF, rt, 6a (97%), 6b (68%), 6c (95%); (v) CuSO4.5H2O, sodium ascorbate, AcOH, DMF/H2O, rt, 7a (32%), 7b (71%), 7c (41%). All detailed experimental procedures are provided in the Supplemental data.
The same strategy as described earlier was utilised for the synthesis of inhibitors 11a and 11b. First, aminomethylphenyl derivatives 8a,b were acylated with chloroacelyl chloride to obtain intermediates 9a and 9b, which were converted into corresponding azides 10a,b by treatment with NaN3 (Scheme 2). Following reaction of azides 10a,b with building block 3 under acidic click reaction conditions provided desired inhibitors 11a and 11b.
Scheme 2.
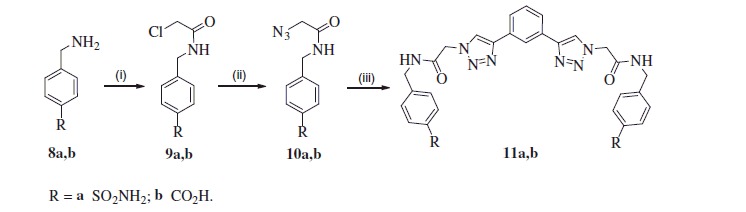
Reagents and conditions: (i) chloroacetyl chloride, K2CO3, THF, 0 °C to rt, 9a (33%), 9b (69%); (ii) NaN3, DMF, rt, 10a (78%), 10b (77); (iii) 3, CuSO4.5H2O, sodium ascorbate, AcOH, DMF/H2O, rt, 11a (17%), 11b (59%).
Bis-acetylenylpyridine building block 14 was prepared in similar way to compound 3, where 3,5-dibromopyridine 12 was first reacted with TMS-acetylene under Sonogashira reaction conditions and then TMS were eliminated in obtained intermediate 13 by treatment with K2CO3 proving building block 14 in 67% yield over two steps (Scheme 3)18.
Scheme 3.
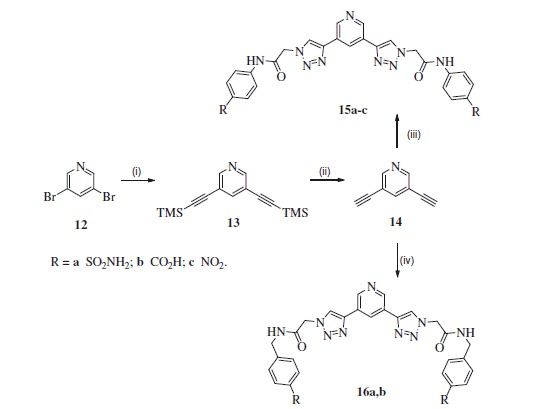
Reagents and conditions: (i) trimethylsilylacetylene, Pd2(PPh3)2, CuI, Et3N, 75 °C; (ii) K2CO3, MeOH/THF, rt, 67% for two steps; (iii) 6a-c, CuSO4.5H2O, sodium ascorbate, AcOH, DMF/H2O, rt, 15a (85%), 15b (39%), 15c (73%); (iv) 10a,b, CuSO4.5H2O, sodium ascorbate, AcOH, DMF/H2O, rt, 16a (63%), 16b (46%).
The reaction between building blocks 13 and 6a-c under acidic click reaction conditions provided inhibitors 15a-c (Scheme 3). In turn, treatment of 13 by azides 10a,b under the same condition provided inhibitors 16a,b with extended bridge.
And finally, for the synthesis of inhibitors 20a-c and 21a,b, anisole building block 19 was synthesised in analogy to 3 and 14 as above, starting synthesis from dibromoanisole 17. Bis-TMS protected intermediate 18 was obtained under Sonogashira reaction conditions and following deprotection by potassium hydroxide provided building block 19 in 61% yield over two steps (Scheme 4)19.
Scheme 4.
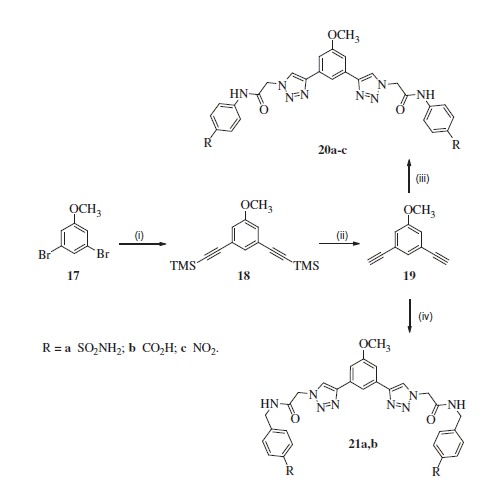
Reagents and conditions: (i) trimethylsilylacetylene, Pd2(PPh3)2, CuI, Et3N, THF 65 °C; (ii) KOH, MeOH/THF, rt, 61% for two steps; (iii) 6a-c, CuSO4.5H2O, sodium ascorbate, AcOH, DMF/H2O, rt, 20a (64%), 20b (56%), 20c (47%); (iv) 10a,b, CuSO4.5H2O, sodium ascorbate, AcOH, DMF/H2O, rt, 21a (31%), 21b (85%).
Treatment of 19 with azides 6a-c under the same condition as described above provided inhibitors 20a-c (Scheme 4). In turn, treatment of 19 with azides 10a,b afforded desired inhibitors 21a,b.
The structures of all new compounds synthesised were fully approved by 1H and 13C NMR, IR and HRMS data (see Supplemental data).
Inhibition studies
All 15 symmetrical compounds were evaluated for their ability to inhibit LDHA, for the comparison galloflavin as a known isoform nonselective LDHA inhibitor20 was used. Our choice of galloflavin was based on the fact that it is extensively studied as potent anticancer agents in recent years21–25; additionally, galloflavin is already commercially available.
Even though all compounds exhibited similar LDHA inhibition and it is difficult to perform structure-activity-relationship (SAR) analysis, we can divide this compound into two groups. First one, the most active compounds (7a, 7b, 15a, 15b, 16b, 20b and 21b) showed better LDHA inhibition compared to galloflalvin, ranging IC50 values from 117 to 136 µM for compounds in first group, whereas galloflavin has IC50 = 157 µM (Table 1). The most active compound in this group was 16b (IC50 = 117 µM), which incorporated pyridyl moiety as a central core and carboxylic groups as a terminal ones. Second group of compounds (7c, 11a, 11b, 15c, 16a, 20a, 20c and 21a) exhibited equal or weaker inhibition compared to galloflavin, ranging IC50 values from 156 to 174 µM. The most active compounds in this group were 11a, 11b and 15c (IC50 158, 156 and 156 µM, respectively). Similar activity of this three compounds is hard to explain, where compounds 11a and 11b have phenyl ring as a central core and sulphonamide and carboxyl groups as terminal ones on aminomethylphenyl moieties, but compounds 15c has pyridyl central moiety and nitro groups as terminal one on shorter, aniline containing, bridge.
Table 1.
Inhibition data of human LDHA.
| Compound | Inhibition, IC50 (μM)* |
|---|---|
| 7a | 128 |
| 7b | 120 |
| 7c | 172 |
| 11a | 158 |
| 11b | 156 |
| 15a | 136 |
| 15b | 125 |
| 15c | 156 |
| 16a | 174 |
| 16b | 117 |
| 20a | 165 |
| 20b | 127 |
| 20c | 163 |
| 21a | 173 |
| 21b | 128 |
| Galloflavin | 157 |
| Blank | 0 |
Mean from three different assays, by colorimetric assay measuring the absorbance at 450 nm (errors were in the range of ±5–10% of the reported values).
We hypothesise that the putative interaction of the inhibitors described in this work with LDHA is similar to the published one, that is, inhibitors bound in the active centre close to conserved residues involved in the catalytic processing of LDHA substrates26. We also assume that at the same time the NADH cofactor is bound in the active centre near the inhibitor molecule. This binding region of the protein is known to undergo considerable conformational changes during the catalytic cycle, that is, it has rather high flexibility. Therefore, inhibitor binding might be unstable and this reflects in IC50 values obtained.
Conclusions
In conclusion, a series of new symmetric molecules have been designed and synthesises as potent LDHA inhibitors. The compounds synthesised exhibited promising in vitro LDHA inhibition activity, where seven compounds were better inhibitors (IC50 117–136 µM) as known LDH inhibitor – galloflavin (IC50 157 µM), and other eight showed equal or slightly lower inhibitory activity (IC50 156–174 µM) as galloflavin. The results obtained are promising base for further development of novel LDH inhibitors.
Supplementary Material
Funding Statement
This project was supported by the National Plan of Science, Technology and Innovation [Grant No. 12-MED2980–54], Prince Sattam bin Abdulaziz University, Alkharj, PO Box 173, 11942.
Disclosure statement
The authors declare no conflict of interest. The authors alone are responsible for the content and writing of this article.
References
- 1.Di Stefano G, Manerba M, Di Ianni L, et al. . Lactate dehydrogenase inhibition: exploring possible applications beyond cancer treatment. Future Med Chem 2016;8:713–25. [DOI] [PubMed] [Google Scholar]
- 2.Doherty JR, Cleveland JL.. Targeting lactate metabolism for cancer therapeutics. J Clin Invest 2013;123:3685–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dawson DM, Goodfriend TL, Kaplan NO. Lactic dehydrogenases: functions of the two types rates of synthesis of the two major forms can be correlated with metabolic differentiation. Science 1964;143:929–33. [DOI] [PubMed] [Google Scholar]
- 4.Warburg O.On the origin of cancer cells. Science 1956;123:309–14. [DOI] [PubMed] [Google Scholar]
- 5.Talaiezadeh A, Shahriari A, Tabandeh MR, et al. . Kinetic characterization of lactate dehydrogenase in normal and malignant human breast tissues. Cancer Cell Int 2015;15:19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fiume L, Manerba M, Vettraino M, et al. . Inhibition of lactate dehydrogenase activity as an approach to cancer therapy. Future Med Chem 2014;6:429–45. [DOI] [PubMed] [Google Scholar]
- 7.Zhao Y, Butler EB, Tan M.. Targeting cellular metabolism to improve cancer therapeutics. Cell Death Dis 2013;4:e532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rani R, Kumar V.. Recent update on human lactate dehydrogenase enzyme 5 (hLDH5) inhibitors: a promising approach for cancer chemotherapy. J Med Chem 2016;59:487–96. [DOI] [PubMed] [Google Scholar]
- 9.Le A, Cooper CR, Gouw AM, et al. . Inhibition of lactate dehydrogenase A induces oxidative stress and inhibits tumor progression. PNAS 2010;107:2037–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ward RA, Brassington C, Breeze AL, et al. . Design and synthesis of novel lactate dehydrogenase A inhibitors by fragment-based lead generation. J Med Chem 2012; 55:3285–306. [DOI] [PubMed] [Google Scholar]
- 11.Kohlmann A, Zech SG, Li F, et al. . Fragment growing and linking lead to novel nanomolar lactate dehydrogenase inhibitors. J Med Chem 2013;56:1023–40. [DOI] [PubMed] [Google Scholar]
- 12.Fang A, Zhang Q, Fan H, et al. . Discovery of human lactate dehydrogenase A (LDHA) inhibitors as anticancer agents to inhibit the proliferation of MG-63 osteosarcoma cells. Med Chem Commun 2017;8:1720–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Neenan TX, Whitesides GM.. Synthesis of high carbon materials from acetylenic precursors. Preparation of aromatic monomers bearing multiple ethynyl groups. J Org Chem 1988;53:2489–96. [Google Scholar]
- 14.Addy PS, Saha B, Singh PND, et al. . 1,3,5-Trisubstituted benzenes as fluorescent photoaffinity probes for human carbonic anhydrase II capture. Chem Commun 2013;49:1930–2. [DOI] [PubMed] [Google Scholar]
- 15.Harte AJ, Gunnlaugsson T.. Synthesis of α-chloroamides in water. Tetrahedron Lett 2006;47:6321–4. [Google Scholar]
- 16.Deepkumar J, Kalpesh P.. Synthesis and evaluation of novel benzimidazole derivatives as antimicrobial agents. Med Chem Res 2014;23:1290–9. [Google Scholar]
- 17.Shao C, Wang X, Xu J, et al. . Carboxylic acid-promoted copper(I)-catalyzed azide-alkyne cycloaddition. J Org Chem. 2010;75:7002–5. [DOI] [PubMed] [Google Scholar]
- 18.Goto H, Heemstra JM, Hill DJ, et al. . Single-site modifcation and their effect on the folding stability of m-phenylene ethynylene oligomers. Org Lett 2004;6:889–92. [DOI] [PubMed] [Google Scholar]
- 19.Beves JE, Blanco V, Blight BA, et al. . Towards metal complexes that can directionally walk along tracks: controlled stepping of a molecular biped with a palladium(II) foot. J Am Chem Soc 2014;136:2094–100. [DOI] [PubMed] [Google Scholar]
- 20.Manerba M, Vettraino M, Fiume L, et al. . Galloflavin (CAS 568-80-9): a novel inhibitor of lactate dehydrogenase. ChemMedChem 2012;7:311–17. [DOI] [PubMed] [Google Scholar]
- 21.Farabegoli F, Vettraino M, Manerba M, et al. . Galloflavin, a new lactate dehydrogenase inhibitor, induces the death of human breast cancer cells with different glycolytic attitude by affecting distinct signaling pathways. Eur J Pharm Sci 2012;47:729–38. [DOI] [PubMed] [Google Scholar]
- 22.Vettraino M, Manerba M, Govoni M, et al. . Galloflavin suppresses lactate dehydrogenase activity and causes MYC downregulation in Burkitt lymphoma cells through NAD/NADH-dependent inhibition of sirtuin-1. Anticancer Drugs 2013;24:862–70. [DOI] [PubMed] [Google Scholar]
- 23.Manerba M, Di Ianni L, Fiume L, et al. . Lactate dehydrogenase inhibitors sensitize lymphoma cells to cisplatin without enhancing the drug effects on immortalized normal lymphocytes. Eur J Pharm Sci 2015;74:95–102. [DOI] [PubMed] [Google Scholar]
- 24.Han X, Sheng X, Jones HM, et al. . Evaluation of the anti-tumor effects of lactate dehydrogenase inhibitor galloflavin in endometrial cancer cells. J Hematol Oncol 2015;8:2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Manerba M, Di Ianni L, Govoni M, et al. . Lactate dehydrogenase inhibitors can reverse inflammation induced hanges in colon cancer cells. Eur J Pharm Sci 2017;96:37–44. [DOI] [PubMed] [Google Scholar]
- 26.Dragovich PS, Fauber BP, Corson LB, et al. . Identification of substituted 2-thio-6-oxo-1,6-dihydropyrimidines as inhibitors of human lactate dehydrogenase. Bioorg Med Chem Lett 2013;23:3186–94. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.