

Abstract
Alzheimer’s disease (AD) is a neurodegenerative disorder, which is complex and progressive; it has not only threatened the health of elderly people, but also burdened the whole social medical and health system. The available therapy for AD is limited and the efficacy remains unsatisfactory. In view of the prevalence and expected increase in the incidence of AD, the design and development of efficacious and safe anti-AD agents has become a hotspot in the field of pharmaceutical research. Due to the multifactorial etiology of AD, the multitarget-directed ligands (MTDLs) approach is promising in search for new drugs for AD. Tacrine, which is the first acetylcholinesterase (AChE) inhibitor, has been selected as the ideal active fragment because of its simple structure, clear activity, and its superiority in the structural modification, thus it could be introduced into the overall molecular skeletons of the multi-target-directed anti-AD agents. In this review, we have summarized the recent advances (2012 to the present) in the chemical modification of tacrine, which could provide the reference for the further study of novel multi-target-directed tacrine derivatives to treat AD.
Keywords: Alzheimer’s disease, tacrine, multitarget-directed, AChE inhibitors
Introduction
Alzheimer’s disease (AD), which is also called senile dementia of the Alzheimer type (SDAT), was firstly identified by the German psychiatrist and neuropathologist Alois Alzheimer in 1906. The most prominent form of AD is an age-related progressive neurodegenerative brain disorder, resulting in decline in language skills, loss of memory and cognitive functions, and accompanied by behavioral disturbances like aggression and depression1. Besides, AD is the fourth leading cause of death in people over 65 years old in the world. According to the data from the European Prevention of Alzheimer's Dementia (EPAD), AD affected more than 40 million people worldwide and its prevalence is projected to double over the next 20 years2. Therefore, AD is considered as the most common neurodegenerative disorder and a major health concern to the societies worldwide.
At present, there is no efficacious treatment available that allows the recovery or even slows the progression of AD and clinical treatments have only palliative effects. Such treatments include acetylcholinesterase inhibitors (AChEIs): tacrine (1), donepezil (2), rivastigmine (3), galantamine (4) and N-methyl-D-aspartic acid (NMDA) receptor antagonist memantine (5) (Figure 1), these inhibitors restore neurotransmitter deficits that are responsible for the symptomatic phase of the disease (cognitive, functional and neuropsychiatric deficits) that appear a decade or more after the onset of the neurodegenerative process3.
Figure 1.

Drugs approved for the treatment of AD.
However, due to the pathogenesis of AD is complex, these drugs that modulate such a single target can only enable a palliative treatment instead of curing or preventing the neurodegeneration4. At the same time, a large number of biological targets for potential therapeutics have been identified5 included amyloid β-peptide (Aβ) protein, tau protein, receptors (cholinergic, glutamatergic, serotoninergic, dopaminergic, noradrenergic, histaminergic) and enzymes (AChE, BuChE, α-, β-, and γ-secretase, monoamine oxidase A, monoamine oxidase B). Furthermore, a number of processes such as excitotoxicity, oxidative stress, calcium, and metal dys homeostasis, neuroinflammation and mitochondrial damage have been involved in the pathomechanism of AD, which are considered as promising directions in the search for AD treatment. Thus, drug discovery in AD is gradually moving from the development of molecules with “one-target, one-disease” to the “multi-target-directed ligands” (MTDLs)6, which is capable to simultaneously address several key pathophysiological processes as described earlier, with the aim of enhancing efficacy or improving safety relative to drugs that address only a single target7–10.
Tacrine was the first approved potent and clinically effective AChE inhibitor by the FDA in 1993 for the treatment of AD. However, due to its hepatotoxicity, this drug was soon withdrawn from the pharmaceutical market in 1998. Because of the good anti-AChE activity, much low molecular weight (MW: 198, lower than other approved AChEIs), potential attenuating Aβ-induced neurotoxicity and synthetic accessibility, tacrine once again became a hot research topic and many efforts have been concentrated on the synthesis of tacrine derivatives as MTDLs for AD treatment11–13. Most importantly, the liver toxicity induced by tacrine is closely related to the free primary amine group14–16, so in the multitarget-directed tacrine derivatives design, the amino could be modified by binding other functional fragment, thus greatly reduced the hepatotoxicity of tacrine. In addition, recent studies also demonstrated that homo- and hetero-dimers could improve the biological profile of tacrine and even overcame some of its side effects17. So more and more researches have focused on the combination of the potent AChE inhibitory property of tacrine with additional biological properties for the design of new MTDLs. The most common strategy to design MTDLs is to connect two distinct classes of compounds in one molecule by choosing a proper spacer, and the general structure of multi-target-directed tacrine derivatives (Figure 2) could be represented as follows.
Figure 2.
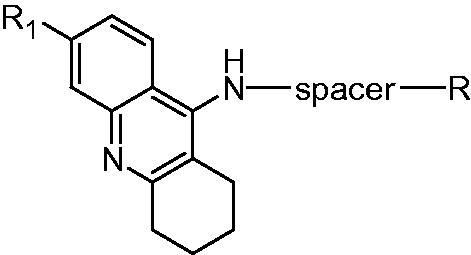
The chemical structure of multitarget-directed tacrine derivatives.
This review mainly summarizes the recent advances (2012 to the present) in the design of multi-target-directed anti-AD agents based on the chemical structure of tacrine, which could provide the reference for the further study of novel multi-target-directed tacrine derivatives for the treatment of AD, the previous research could be seen in the related reviews11–13.
Structural biology of acetylcholinesterase
Brain atrophy is the most obvious clinical finding in AD in which the levels of acetylcholine (ACh), a neurotransmitter responsible for the conduction of electrical impulses from one nerve cell to another nerve cell, are decreased due to its rapid hydrolysis by cholinesterases (ChE)18. That is the “cholinergic hypothesis” which was initially presented about 30 years ago and provided the first rational approach in the treatment of AD. We now know that ACh could be degraded by two types of cholinesterases including AChE and butyrylcholinesterase (BuChE). Indeed, AD therapy is bolstered mainly by AChEI nowadays.
AChE, which is one of the most crucial enzymes for nerve response and function, catalyzes the hydrolysis of acylcholinesters with a relative specificity for ACh. AChE is widely distributed in a variety of nerve muscle tissue in the brain, such as the substantia nigra, hippocampus, caudate nucleus and cerebellum. In 1991, the three-dimensional structure of Torpedo californica AChE (TcAChE) was analysised19. Subsequently, the crystal structure of mouse AChE (mAChE) and human AChE (hAChE) have also been reported20,21. Knowledge of the three-dimensional structure of AChE is essential for understanding its remarkable catalytic efficacy, for rational drug design and for developing new therapeutic approaches. The structures of the catalytic domains of the AChE from such species as Torpedo californica, mouse, and human are quite similar. In all cases, the active site is found at the bottom of a 20 Å long channel leading from the surface of the enzyme, and a group of aromatic side chains forms a bottleneck partway down the channel19–21.
Most crystallographic studies of AChE involve the electric ray (Torpedo californica) or the mouse homologs22. Of the over 100 AChE structures deposited in the Protein Data Bank, only four human AChE structures of the catalytic core have been solved to date, of which none is in complex with therapeutic drugs used for the treatment of disease. TcAChE contains 537 amino acids and possesses an ellipsoidal shape with dimensions of about 45 Å by 60 Å by 65 Å. The molecule consists of a 12-stranded central mixed β-sheet surrounded by 14α-helices and bears a striking resemblance to several hydrolases23,24. All current kinetic models for TcAChE proposed the existence of at least two substrate-binding sites: the active site, near the bottom of the active-site gorge and the peripheral anionic site (PAS), near its entrance25. The active site composes two subsites, and in the catalytic anionic subsite, it has been proposed that the choline moiety of TcAChE is stabilized principally via a cation-π interaction with Trp84 and also interacts with Glu199 and Phe3304. The esteratic subsite in TcAChE contains a typical serine-hydrolase catalytic triad, Ser200-His440-Glu327. A substantial contribution to ACh binding within the active site also arises from stabilization of the carbonyl oxygen within the oxyanion hole composed of Gly118, Gly119, and Ala201, and of the acetyl group in the “acyl-pocket” composed of Trp233, Phe288, Phe290, and Phe33126. The PAS contains three principal amino acids including Trp279, Tyr70, and Asp72, and binding the ligands at the PAS affects catalytic activity, this is established by site-directed mutagenesis and by the binding of inhibitors27. Szegletes et al.28 demonstrated that the primary physiologic role of the PAS was to accelerate the hydrolysis of ACh. The gorge of the active site was so deep and broad that it could bind many different substrates and inhibitors. The initial step in the catalytic pathway was substrate bond to the PAS; therefore, the design of new type AChEIs should be based on the hypothesis of dual binding mode. The PAS could act as an uncompetitive inhibitor-binding site, the strong binding of inhibitors at the PAS might hinder the entry of ACh to the enzyme gorge, and such types of inhibitors, which were able to bind to PAS could act as a steric blocker. Mutagenesis experiments showed that PAS mutations cause AChE inhibitor activity decreased significantly. In addition, PAS was confirmed to have relation with Aβ accumulation induced by AChE; therefore, blocking PAS could help to reduce the formation of amyloid plaques29.
hAChE and TcAChE have a high structural identity (∼53%) with a very low root-mean-square deviation (∼1 Å). The catalytic triad (Ser203-His447-Glu334) of hAChE is surrounded by three subsites that are important for catalytic activity30: (1) the esteratic subsite or choline-binding site (ES) consists of aromatic residues (Trp86, Tyr133, Tyr337, and Phe338), which bind to the quaternary trimethylammonium moiety of the choline group in the substrate for optimal positioning of the carbonyl carbon in the ester at the acylation site. The catalytic process is facilitated by orientation of the substrate’s carbonyl oxygen toward the oxyanion hole formed by the hydrogens from Gly120, Gly121, and Ala204; (2) the anionic subsite or acyl pocket (AS) of AChE formed by the side chains of Phe295 and Phe297 is the binding pocket for the acyl group of the substrate or the methyl group of methylphosphonate; (3) the PAS, which plays a significant role in excess substrate concentration kinetics, is located at the rim of the gorge and consists of residues including Tyr72, Asp74, Tyr124, Trp286, and Tyr341.
AChE was reported to colocalize with Aβ in neuritic plaques and could enhance the rate of formation of Aβ fibrils, forming stable complexes with them. Moreover, AChE was suggested to be a pathological chaperone, which induced a conformational transition in Aβ leading to aggregation and fibril formation31,32. Recent studies have also shown that selective AChE/BuChE inhibitors could reduce Aβ-induced inflammatory processes33. On the other hand, a number of new evidence have suggested that inhibitors of both cholinesterase enzymes might have beneficial functions in AD. Indeed, in a healthy human brain, BuChE mainly plays a supportive role in the hydrolysis of ACh, while in an AD brain, its role becomes predominant with AD progression as AChE levels decline and BuChE levels rise34. More importantly, in vivo experiments showed that brain-targeted BuChE inhibitors not only improved the cognitive performance of aged rats, without the classic adverse effects associated with AChE inhibition, but they also lowered Aβ brain levels in transgenic mice over expressing human mutant amyloid precursor protein (APP) and ameliorated the Aβ-induced cognitive dysfunction in mice35,36. Furthermore, it has been also demonstrated that both AChE and BuChE played an important role in Aβ-aggregation during the early stages of senile plaque formation. Therefore, AChE and BuChE inhibition have been documented as critical targets for the effective management of AD by an increase in the availability of acetylcholine in the brain regions and decrease in the Aβ deposition37.
In the tacrine–TcAChE complex, the tacrine moiety is stacked against Trp84, with the nitrogen in the ring forming a hydrogen bond with the main chain carbonyl oxygen of His440, its amino nitrogen binds to a water molecule. The Phe330 ring rotates to lie parallel to tacrine, which is sandwiched between the Phe330 and Trp84 rings, this binding mode clearly explains the reason why tacrine has the good inhibitory activity to AChE at the atomic level, and tacrine has been used as a reference to compare the other AChEIs for both clinical efficacy and side effects in the clinical development38.
Design of multitarget-directed tacrine derivatives
In recent years, the treatment of AD by multitarget-directed strategy has gradually become the consensus. The design of tacrine inhibitors with dual binding mode from previous studies39–46 lays the foundation for designing multitarget-directed tacrine derivatives at the molecular level. Selecting tacrine as AChE-binding fragment and introducing different types of functional fragments to regulate other important therapeutic target of AD could obtain multifunctional anti AD drugs, because these multi targeting derivatives have stronger anti-AD activity and less liver toxicity compared with tacrine17.
Tacrine derivatives with cholinesteraseinhibition and β-amyloid antiaggregation properties
The progressive deposition of Aβ in the brain of AD patients is generally considered to be fundamental to the development of neurodegenerative pathology. The cell toxicity associated with Aβ fibril aggregation provides an explanation for the neuronal cell loss found in AD patients47. Therefore, Aβ fibril aggregation in the brain is currently another potential target for the treatment of AD48. Aβ is a 39- to 43-residue peptides generated by the sequential cleaving of the APP by β- and γ-secretases. Aβ (1–40) and Aβ (1–42) are the main isoforms of Aβ peptides. Though the amount of Aβ (1–42) is only 10% of Aβ (1–40), Aβ (1–42) tends to aggregate more rapidly and displays stronger neuronal toxicity than Aβ (1–40). Therefore, the prevention of Aβ (1–42) aggregation attracts much attention. Recent study showed that AChE could also play a key role in accelerating senile Aβ plaques deposition49,50. It was likely that AChE interacted with Aβ and promoted amyloid fibril formation through a pool of amino acids located in the proximity of PAS29. Taking into account that the AChE and Aβ aggregation were particularly important targets for inhibition, the structure of tacrine was thus used as a pharmacophoric moiety in the development of MTDLs endowed with an inhibitory activity against cholinesterases and Aβ fibril formation11.
β-carboline alkaloids possesses a wide range of pharmacological properties relate to a variety of neurological disorders, studies indicated that naturally occurring as well as the chemically synthesized β-carboline analogs exhibited potent AChE inhibitory activity, especially bivalent β-carbolines with IC50 values were in the nanomolar range for AChE inhibition51,52. For tetrahydro-β-carbolines, these alkaloids occur and accumulate in mammalian tissues, fluids, brain and are able to directly scavenge a variety of reactive oxygen species53–55, making the β-carboline another useful scaffold for AD drug design. Lan et al.56 selected β-carboline to hybridize with tacrine by alkylene linkers to design a series of new hybrids: tacrine was used to inhibit AChE through its binding to the CAS of AChE, while β-carboline was used to interact potentially with the PAS due to its aromatic character. In vitro studies showed compound 6 (Figure 3) presented the greatest ability to inhibit cholinesterase (IC50 = 21.6 nM for eeAChE, 63.2 nM for hAChE, and 39.8 nM for BuChE), more potent than the reference compound tacrine (IC50 = 260 nM for eeAChE). Furthermore, 6 also showed good inhibition against Aβ aggregation (65.8% at 20 μM) relative to that of curcumin (42.4% at 20 μM), and good antioxidant activity (1.57 Trolox eq.). In addition, compound 6 could inhibit Cu2+-induced Aβ aggregation effectively by chelating Cu2+, reduce PC12 cells death induced by oxidative stress and penetrate the blood–brain barrier (BBB).
Figure 3.

The chemical structures of tacrine derivatives as cholinesterase and Aβ aggregation inhibitors.
Based on that benzothiazole derivatives could interact with Aβ peptides and have been used as Alzheimer’s brain imaging agents57, in 2013, Keri et al. synthesized a series of novel multifunctional compounds bearing two pharmacophoric groups: tacrine and a benzothiazole moiety connected by different linkers, containing an amide bond, an alkyl or arylalkyl chain58. All these new tacrine–benzothiazole hybrids displayed high in vitro activities with IC50 values were in the low micromolar to submicromolar concentration range (0.34–1.84 μM) toward the inhibition of AChE. In addition, the compounds also showed high inhibitory activity against self-induced Aβ1–42 aggregation at 50 μM ranging from 22.3% to 61.3%. Among them, compound 7 (Figure 3) presented the moderate inhibitory activity against AChE (IC50 = 0.57 μM) compared with the commercial drug tacrine (0.19 μM). Interestingly, it exhibited the highest activity as anti-Aβ42 self-aggregation (61.3% at 50 μM) due to the biaryl heterocyclic scaffold of BTA-moiety could recognize and interact with the abnormal β-sheet conformation of the Aβ peptide.
Because of the extensive biological actions, such as protecting neurons from ischemic damage and anti-inflammatory activity of phenylthiazole derivatives59, Wang et al.8 designed and synthesized a series of novel multifunctional compounds by conjugating tacrine with 4-phenyl-2-aminothiazole group, through different middle linkers including an alkyl chain and succinamide or glycinamide fragments. The inhibitory activities against AChE, BuChE, and Aβ-aggregation as well as a calcium overload blockade effect were studied for these phenylthiazole–tacrine hybrids, and the results showed that these hybrids were potent inhibitors of cholinesterase with pIC50 values ranged from 5.78 to 7.14 nM for AChE and from 5.75 to 10.35 nM for BuChE. Most compounds showed good inhibitory potency on Aβ1–42 self-aggregation and their inhibitory effects depended on the type of middle linker and substitutions at 4′-position of 4-phenyl-2-aminothiazole. In addition, some hybrids also displayed the Ca2+ overload blockade effect in the primary cultured cortical neurons. Among them, compound 8 (72.0% at 20 μM) (Figure 3) exhibited the highest inhibitory potency against Aβ1–42 self-aggregation, higher than the propidium iodide (57.1% at 20 μM) and moderate AChE inhibition (pIC50 = 6.80 nM) compared with tacrine (pIC50 = 7.19 nM).
Lately, some researches have suggested that electron-rich aromatics would bind to the peripheral binding site, which has been confirmed in a comparison of aromatics with differing electron density60,61. In order to further explore the anti-Alzheimer potential of the electron-rich aromatics based on MTDLs strategy, Zhang’s group62 synthesized a series of novel multifunctional compounds by conjugating a tacrine and electron-rich aromatics including benzoates, phenylacetates, cinnamates, and acetylindole. The screening results showed that most of them exhibited a significant ability to inhibit ChEs, certain selectivity for AChE over BuChE and strong potency against self-induced Aβ aggregation. All IC50 values of biological activity were at the nanomolar range. Especially, compound 9 (Figure 3) with a trimethoxyl phenylacetate group, showed the most potent inhibition for AChE with its IC50 value was 5.63 nM, which was 13 times stronger than tacrine and the highest selectivity (BuChE/AChE = 64.6). Moreover, it also exhibited a potent inhibitory against Aβ aggregation with an IC50 value was 51.81 nM. A Lineweaver–Burk plot and molecular modeling analysis showed that compound 9 targeted both the CAS and PAS of ChEs, in addition, the electron density of aromatic ring, which was linked with tacrine through acetyl group, played a significant role in determining the inhibitory activity.
Recent studies indicated that a naphthoquinone linked to a tertiary benzylamine through a proper spacer showed optimal anticholinesterase and antiaggregating features63. Nepovimova et al. proposed quinone as an antiamyloid privileged motif and they demonstrated that it might interfere with several amyloid proteins, due to its possibility to form favorable hydrogen bond and π-stacking interactions64,65. These results were also in agreement with the work of Scherzer-Attaliet et al., who described that naphthoquinone-tryptophan hybrids could effectively inhibit Aβ oligomerization both in vitro and in vivo66. Motivated by these considerations, Bolognesi’s group67 rationally designed a novel series of hybrids by linking the naphthoquinone moiety to a tacrine scaffold via a methylene spacer. All compounds displayed excellent in vitro AChE inhibitory potencies with IC50 values ranged from micromolar to subnanomolar concentrations, and interesting capabilities to block Aβ aggregation. Among them, compound 10 (Figure 3) exhibited outstanding inhibitory activity (IC50 = 0.72 nM for hAChE) in complex with AChE by the X-ray analysis. More meaningful was that the compound 10 showed negligible toxicity in immortalized mouse cortical neurons Neuro2A and primary rat cerebellar granule neurons, as well as was able to completely revert the decrease in viability induced by Aβ in Neuro2A.
Tacrine derivatives with cholinesterase and BACE1 inhibition properties
At current, the most popular hypothesis is that AD is caused by aggregates of the Aβ, which is generated by cleavage of APP by β-secretase1 (BACE1) follows by γ-secretase, making BACE1 a particularly good drug target for the generation of inhibitors that lower Aβ. The identification and cloning of this enzyme led to better understanding of its physiological function and to the development of BACE1 inhibitors68. Recently, numerous BACE1 inhibitors have been discovered, and a few of them have been tested in the early stage of clinical trials69,70, so the identification of BACE1 as a novel class of type-I transmembrane aspartic protease has been a land-mark discovery in AD71.
Galdeano et al.72 designed a series of tacrine–huperzine A hybrids, by combining the 4-aminoquinoline moiety of tacrine with huprine fragments through an alkyl or alkylamine linker, as potential inhibitors for AChE, BuChE, and BACE1. All the tested compounds were potent hAChE inhibitors with IC50 values were in the subnanomolar to low nanomolar range (0.31–9.09 nM). The strongest inhibitory activity towards hAChE was displayed by hybrid with a six carbon atom linker. The elongation of the tether led to a decreased activity, while the insertion of a methylamine group in the linker had a positive effect on the hAChE inhibitory activity. The new compounds were moderately potent inhibitors against hBuChE with IC50 values ranged from 24.6 to 139 nM. The 6-chlorotacrine–huprine hybrids showed a good inhibitory potency against BACE1 with the most active compound was 11 (Figure 4) (IC50 = 4.9 μM for hBACE1), these data indicated that the huprine–tacrine heterodimers could be considered as promising lead compounds for AD.
Figure 4.

The chemical structures of tacrine derivatives as cholinesterase and BACE1 inhibitors.
Flavonoids, which are ubiquitously present in fruits and vegetables, have attracted much attention in recent years because they could limit the neurodegeneration associated with a variety of neurological disorders73. Flavonoids mediate their effects by several routes including their capacity to scavenge neurotoxic species, such as free radicals, and their interactions with important neuronal enzymes, such as BACE174,75. In 2012, Fernandez-Bachiller et al. choose a flavonoid scaffold derive from 4-oxo-4H-chromene for its antioxidant, BACE1 inhibitory activities76 as well as its potential interaction with the AChE-PAS due to the aromatic character to connect with tacrine by linkers of different lengths (from 7 to 12 carbon atoms), and modified by an introduction of various substituents for the treatment of AD. The developed compounds showed a stronger inhibitory activity against human AChE and BuChE, with IC50 values were in the nano- and picomolar ranges, being more potent than the parent inhibitor tacrine. Compound 12 (Figure 4) showed potent inhibition activities against human BACE1 (IC50 = 2.8 μM) and cholinesterases (IC50 = 8.0 nM for hACHE, IC50 = 1.5 nM for hBCHE), as well as good antioxidant properties (1.3 Trolox eq.). Thus, it was expected that this hybrid could augment patient cognition (by increasing levels of acetylcholine), protect neurons from oxidative stress (by capturing free radicals), and reduce the formation of senile plaques (from inhibition of BACE-1).
Considering the widened biological profile toward amyloid aggregation from previous research77–81, Zha et al. selected the benzofuran nucleus (and the modification thereof) as the framework to connect with tacrine by an amido or amino linkage to build new MTDLs for AD82. Based on the previous studies have shown that the presence of a properly spaced protonable amino group and aromatic residues could facilitate the interaction with the PAS, and the introduction of an amido group coupled with aromatic planar moieties might enhance the binding properties toward BACE-183, their synthetic hybrids exhibited good inhibitory activities on CHEs and Aβ self-aggregation. In particular, some compounds displayed significant inhibition of hBACE1. Among the hybrids, 13 (Figure 4) showed the most interesting profile as a subnanomolar selective inhibitor of hAChE (IC50 = 0.86 nM for hAChE) and a good inhibitor of both Aβ aggregation (hAChE- and self-induced, 61.3% at 50 μM and 58.4% at 100 μM, respectively) and hBACE-1 activity (IC50 = 1.35 μM). Kinetic studies indicated that 13 acted as a slow, tight-binding, mixed-type inhibitor, while X-ray crystallographic studies highlighted the ability of 13 to induce large-scale structural changes in the active-site gorge of Torpedo californica AChE. In vivo studies confirmed that 13 significantly ameliorated performances of scopolamine-treated ICR mice and did not exhibit significant hepatotoxicity. Thus, 13 could be used for further studies to help understand the structural requirements for an optimal MTDL for AD treatment.
Tacrine derivatives with cholinesterase inhibition and antioxidant properties
Oxidative stress is characterized by an imbalance between the production of reactive oxygen species (ROS) and their removal by antioxidative mechanisms. Recent researches have demonstrated that the antioxidant defense system in elderly people lost its capacity to neutralize oxidative species. In the early stage of the AD, the oxidative stress plays a key role in initiating the aggregation of Aβ and tau protein hyperphosphorylation. So the oxidative stress might be a risk factor for the initiation and progression of AD. Extensive evidences suggested that free radicals might be involved in the pathogenesis of AD, because the brain tissues in patients with AD were exposed to oxidative stress during the development of the disease. ROS production is due to a variety of sources including mitochondrial abnormalities, disturbances in the level of transition metals and amyloid peptides themselves. Thus, antioxidant therapy in dementia might bring benefits, particularly in the early stage of AD. Many evidences proved that antioxidants could attenuate the syndrome of AD and prevent the progression of the disease84. Searching for cholinesterase inhibitors with additional antioxidant properties is one of the trends in the development of an effective therapy for AD.
Trolox is a powerful antioxidant agent widely used in biological or biochemical applications to reduce oxidative stress. Numerous reports indicated that trolox could protect liver from the damage induced by chemical insults both in vitro and in vivo85. In addition, it showed neuroprotective effect through scavenging ROS and attenuated the neurotoxicity mediated by Aβ and H2O2 on hippocampal neurons86. Furthermore, as a lipid-soluble analog of trolox, vitamin E has been already used to prevent the tacrine-induced hepatotoxicity in clinic and delayed the AD progression in patients with moderately severe AD87. Xie et al.88 connected the trolox with tacrine moiety via various methylene chain and obtained a novel series of compounds. Among them, compound 14 (Figure 5) was the most potent inhibitor against AChE (IC50 = 9.8 nM for eeAChE and 23.5 nM for hAChE), it was also a strong inhibitor against BuChE (IC50 = 22.2 nM for eqBuChE and 20.5 nM for hBuChE). Fortunately, in vivo hepatotoxicity assays indicated that compound 14 did not show signs of hepatotoxicity, which was much safer than tacrine.
Figure 5.

The chemical structures of tacrine derivatives with cholinesterase inhibition and antioxidant properties.
Carbazoles, naturally occurring phytochemicals, are widely present in many plant species and possess a wide range of biological activities associated with AD. It has been reported that naturally occurring carbazole derivatives were able to directly scavenge a variety of ROS and possessed strong antioxidant activities89. Thiratmatrakul et al.90 studied a series of carbazole derivatives extracted from root bark of Clausena Harmandiana as an antioxidant and found that 7-methoxyheptaphylline and heptaphylline possessed a strong antioxidant effect in vitro. So they combined the tacrine to the active functions of natural antioxidants, 7-methoxyhepta phylline and heptaphylline, as potential multifunctional agents for the treatment of AD. The synthesized compounds showed high inhibitory activity on AChE with IC50 values ranged from 0.48 to 1.03 nM and exhibited good inhibition selectivity against AChE over BuChE. The compounds showed higher radical-scavenging activity (IC50 = 8.34–11.24 μM) than trolox. Furthermore, they displayed a neuroprotective effect against the oxidative stress induced by H2O2 in the neuroblastoma cells and against the toxicity induced by Aβ1–42 peptide in C6 astroglioma cells at 100 μM. Compound 15 (Figure 5), bearing 7-methoxyheptaphylline and a five carbon atom spacer, exhibited the highest potency for both radical scavenging and AChE inhibitory activity and could improve both short- and long-term memory deficits through the enhancement of cholinergic signaling in behavioral studies91.
Based on that tacrine–lipoic acid92, tacrine–ferulic acid93, and tacrine–caffeic acid94 hybrids showed a good multiple biological profile, displaying AChE inhibition property and antioxidant activity. Digiacomo et al.95 synthesized a new class of tacrine derivatives by inserting 1,3-diamino-2-propanol to widen the pharmacological profile of the hybrids, which was applied successfully to a number of aspartyl proteases including BACE1. For example, compound 16 (Figure 5) possessed a good ability to inhibit Aβ self-aggregation (53.2% at 50 μM), submicromole AChE (IC50 = 0.15 μM)/BuChE (IC50 = 0.36 μM) inhibition and modest BACE1 inhibition (13.07% at 10 μM). Importantly, compound 16 was also a DPPH radical scavenger (60.87% at 10 μM) and copper chelator, as well as a potent neuroprotective agent against glutamate-induced cell death with low toxicity in HT22 cells. Similarly, Quintanova et al.96 reported a series of novel tacrine–cinnamate and tacrine–cinnamylidene acetate hybrids in 2015. These hybrid compounds all presented high activity against AChE, with IC50 values were mostly in the sub-micromolar range, and the best inhibitor was 17 (Figure 5) (IC50 = 0.09 μM) with 3,4-dimetoxy substitutions in the cinnamate unit. Moreover, the compounds with free catechol moieties showed very high antioxidant activity (EC50 in the low micromolar range), eventually due to their iron-chelating contribution.
5,6,7-Trimethoxyflavone, methylations of the hydroxyl groups of oroxylin A or baicalein,97 possesses a wide range of pharmacological properties such as free radical scavenging effect, Aβ fibril inhibition effect, anticancer activity, anti-inflammatory activity, and neuroprotective effect98. Based on these, Liao et al.99 created a series of novel hybrids that combined 5,6,7-trimethoxyflavone with 6-chlorotacrine, although the molecular weight of hybrids were more than 500, the hybrids possessed more complementary biological activities and could cross the BBB. The results showed that the target compounds exhibited good AChE inhibitory potency, high selectivity toward AChE over BuChE, potential antioxidant activity and significant inhibitory potency against self-induced Aβ-aggregation. Among them, compound 18 (Figure 5) showed the strongest inhibitory activity against AChE with its IC50 value of 12.8 nM was higher than the reference compound 6-chlorotacrine (IC50 = 78.5) and indicated remarkable inhibition on self-induced Aβ1–42 aggregation with inhibition ratio of 33.8% at 25 μM. Furthermore, compound 18 acted as an antioxidant, as well as a good neuroprotective agent against H2O2-induced PC12 cell injury determined by the ORAC-FL method (Oxygen Radicals Absorbance Capacity by Fluorescence)100.
Ebselen, an organoselenium found in 1996, is known as a classic glutathione peroxidase mimic, reacted in a rapid reaction with peroxynitrite101, possessed antioxidant activity and anti-inflammatory property and exhibited neuroprotective effect in several preclinical studies102,103. Mao et al.104 synthesized a series of tacrine–ebselen hybrids as multifunctional anti-AD agents with cholinesterase and antioxidant activities. Most of these compounds were potent inhibitors of AChE and BuChE (with IC50 values ranged from 2.55 to 657 nM for AChE and from 2.80 to 38.88 nM for BuChE), they also demonstrated similar hydrogen peroxide and peroxynitrite-scavenging activity as ebselen by horseradish peroxidase and peroxynitrite scavenging activity assay. Compound 19 (Figure 5) exhibited superior activity and a better balance of AChE and BuChE activities than tacrine (IC50 = 2.55 nM for EeAChE, IC50 = 2.80 nM for EqBuChE) and showed effective hydrogen peroxide and peroxynitrite scavenge property (neuroprot. vs. H2O2 = 83.7% at 25 μM), indicating that this hybrid was a good multifunctional drug candidate for the treatment of AD.
Tacrine derivatives with cholinesterase inhibition andNO-releasing properties
Nitric oxide (NO) is an essential signaling molecule involved in various physiological functions in humans, and the over and under production of NO are responsible for a number of pathological conditions. The biosynthesis of NO by brain neuronal NOS is associated with stroke and chronic neurodegenerative diseases, such as Alzheimer’s, Parkinson’s, and Huntington’s diseases105. Increasing evidences suggested that NO, a free radical gas, might be beneficial for the treatment of AD by increasing blood supply and regulating the cerebral circulation. Being involved in a variety of physiological and pathophysiological processes, NO is regarded as a potential tool in the pharmacotherapy of AD, therefore NO-releasing drugs might be especially beneficial for the treatment of AD106.
Chen et al.107 conjugated a nitrate moiety to the tacrine–ferulic acid hybrid compounds via alkylene side chain in order to obtain new multifunctional AChEIs, which endowed with additional properties at least such as vessel relaxation activity on the basis of previous study105,108. All the synthetic NO-donating tacrine–ferulic acid hybrids were potent cholinesterase inhibitors with IC50 values ranged from 3.6 nM to 44.3 nM against eeAChE and 1.0 nM to 24.9 nM against eqBuChE. Compound 20 (Figure 6) was one of the most potent inhibitors toward AChE (IC50 = 10.9 nM) and BuChE (IC50 = 17.1 nM), respectively, and possessed an activity correlated with the NO production (0.314 μg/ml) compared with the reference isosorbide dinitrate in the vascular relaxation assay. In addition, compound 20 was confirmed to have beneficial effects for the short-term learning ability, and could improve memory impairment in the passive avoidance test in the scopolamine-induced cognition impairment animal model, the hepatotoxicity study also proved that 20 was much safer than tacrine. These tests demonstrated that the strategy of NO-donating drugs was rational and beneficial.
Figure 6.

The chemical structures of tacrine derivatives with cholinesterase inhibition and NO-releasing properties.
To search for multifunctional anti-AD agents with good safety, the previously synthesized tacrine–flurbiprofen hybrids were modified into tacrine–flurbiprofen–nitrate trihybrids by Liao team109. These compounds displayed higher cholinesterase inhibitory activity relative to the bivalent hybrids with IC50 values were in the nanomolar ranges. The results revealed that the length of the alkyl, which was connected to nitrate moiety of the target compounds, could significantly influence the AChE inhibitory activity. When the length increased, the activity decreased and the optimal spacer length was two carbon atoms. Compound 21 (Figure 6) was the most potent trihybrid compound, which released moderate NO (0.348 μg/ml), exerted blood vessel relaxative activity and showed significant Aβ inhibitory effects whereas tacrine and flurbiprofen did not exhibit any Aβ inhibitory activity at the same dose. In addition, compound 21 was active in improving memory impairment in vivo and was much safer than tacrine in the hepatotoxicity study, and the higher safety of 21 qualified that it was more beneficial for the treatment of AD.
Nitroxides are stable free radicals that rapidly cross cell membranes, preempt free-radical formation by oxidizing redoxactive metal ions, and function both as intra- and as extracellular SOD mimics. Several studies indicated that modification of cardioprotective agents110, PARP-inhibitors111, or neuroprotective ebselen112 with nitroxides had a beneficial influence on their activity, being supplemented by the nitroxide’s “in status nascent acting” antioxidants and radical scavengers. With these concepts in mind, Kalai et al.113 combined nitroxide and/or nitroxide precursors (amines and hydroxylamines) with tacrine via methylene or piperazine space. The synthetic compounds were tested for their hydroxyl radical and peroxyl radical-scavenging ability, acetylcholinesterase inhibitor activity, and protection against Aβ-induced cytotoxicity. Compounds 22 and 23 (Figure 6) were found to be the most efficient AChEIs and radical scavengers, and they exhibited remarkable cell protection toward Aβ-induced toxicity, providing direction for further development of additional candidates with dual functionality (anti Alzheimer’s and antioxidant).
Tacrine derivatives with cholinesterase inhibition and neuroprotective properties
Neurodegeneration is characterized by a progressive loss of the structure and function of neurons. The purpose of neuroprotection is to counteract this process by targeting the most common mechanisms leading to it, like oxidative stress, NMDA receptor, synaptic plasticity, mitochondrial dysfunction, excitotoxicity, inflammatory changes, iron accumulation, and protein aggregation.
In 2012, Minarini et al. designed a novel multipotent anti-AD agent by linking two tacrine moieties via cystamine (2,2′-disulfanediyldiethanamine) for its important activities as antioxidant, cyto-, and neuroprotective agent114. In particular, systemic administration of cystamine was reported to diminish neural toxicity associated with different toxins and to protect against neurodegeneration. The obtained cystamine–tacrine compound 24 (Figure 7) displayed an inhibitory activity against both AChEs with its IC50 values were in the nanomolar range (IC50 = 5.04 nM for hAChE, IC50 = 4.23 nM for hBuChE), and inhibitory properties against the self-induced Aβ aggregation assay with its IC50 value was 24.2 μM. It also exerted a neuroprotective activity on SH-SY5Y cell line against H2O2-induced oxidative injury. Fortunately, 24 endowed with a lower toxicity in comparison to bis(7)tacrine.
Figure 7.

The chemical structures of tacrine derivatives as cholinesterase inhibitors with neuroprotective properties.
The long-term potentiation (LTP) in hippocampus has been widely considered as a key cellular mechanism underlying learning and memory115, and chemical entities containing mercapto group, such as dithiothreitol (DTT), glutathione (GSH) and N-acetyl cysteine (NAC) were confirmed to facilitate the induction of LTP in the normal rats and even reversed the LTP impairment in aged rats116–118. Therefore, Wang et al.119 choose the mercapto groups to cooperate with tacrine in a single molecular for novel multifunctional compounds as anti-AD potential drugs. These mercapto–tacrine derivatives displayed a synergistic pharmacological profile of LTP enhancement, cholinesterase inhibition, neuroprotection and less hepatotoxicity. Moreover, they could improve the memory and cognitive impairment efficiently with fewer side effects and diminish the oxidative damage caused by free radicals. Compound 25 (Figure 7), in which the mercapto group was covered with the acetyl group, exhibited the neuroprotective effect mostly in a concentration-dependent manner, with the maximal effect at 30 μM (56.5%), emerging as promising molecule for the therapy of age-related neurodegenerative diseases. Soon after that, Santos’s group120 also synthesized a set of mercaptotacrine derivatives by conjugation of a tacrine moiety with an S-allylcysteine (garlic constituent) or S-propargylcysteine moiety, aimed at improving the cholinergic system and neuroprotective capacity, and the most promising result was achieved by compounds 26 (Figure 7) for the AChE inhibition (IC50 = 0.3 μM) and 27 (Figure 7) for the remarkable prevention of superoxide production and Aβ-induced cellular toxicity.
A codrug of the natural product silibinin and the AChEI tacrine was synthesized by Chen et al.,121 these compounds showed potent AChE and BuChE inhibitory activities, while the toxicities of the co-drugs were markedly reduced in comparison to that of tacrine. Moreover, using a neuronal cell line (HT-22), a neuroprotective effect against glutamate-induced toxicity could be observed that the co-drugs showed no hepatotoxicity and no induction of the cytochrome P450 system. In a scopolamine-induced cognitive impairment model, the codrugs were as potent as tacrine in reversing memory dysfunction, being superior to the physical mixture of tacrine and silibinin. The obtained silibinin–tacrine codrug 28 (Figure 7) exhibited pronounced pro-cognitive effects just like tacrine, but lacked tacrine’s therapy-limiting hepatotoxic effects completely both in vitro and in vivo.
Tacrine derivatives with cholinesterase inhibition andmetal-chelating properties
Recent studies indicated another hypothesis, called metal hypothesis, might also contribute to AD pathologyn. It was observed that the level of metal ions such as iron (Fe), copper (Cu), and zinc (Zn) in AD patients was 3–7-folds higher than that of healthy individuals. The abnormal accumulation of metals was able to accelerate the formation of Aβ aggregates and neurofibrillary tangles (NFTs), which promoted inflammation and activated neurotoxic pathway, leading to dysfunction and death of brain cells122. In addition, the redox-active ions like Cu2+ and Fe2+ were involved in the production of ROS, and oxidative stress was also critical for Aβ neurotoxicity. Thus, the modulation of the level of these biometals in the brain is a potential therapeutic strategy for treating AD123,124, and multifunctional metal chelators might block metal-related oxidative stress as well as modulate Aβ aggregation.
In 2014, Li et al. combined tacrine and a rhein scaffold for its metal-chelating ability as novel multifunctional potent AD drug125. Most compounds inhibited AChE in the nanomolar range in vitro effectively. Among them, compound 29 (Figure 8) was one of the most potent inhibitors and was 5-fold more active than tacrine toward AChE, it also showed a moderate BuChE inhibition with IC50 value was 200 nM. In the inhibition of the AChE-induced Aβ aggregation assay, compound 29 (70.2% at 100 μM) showed the greatest inhibitory activity. Moreover, in metal-chelating effect assay, compound 29 could effectively chelate Cu2+ and Fe2+, and thereby could serve as a metal chelator in treating AD.
Figure 8.

The chemical structures of tacrine derivatives as cholinesterase inhibitors with metal-chelating properties.
PBT2 (an 8-hydroxyquinoline derivative) was selected by Fernandezbachiller et al.126 to combine with tacrine for its metal-chelating, neuroprotective, and antioxidant properties to synthesize tacrine-PBT2 hybrids as potential candidates for the treatment of AD. These new tacrine-PBT2 hybrids displayed potent inhibitory activities against human AChE and BuChE with their IC50 values were in the nano- and subnanomolar ranges, which were more potent than the parent fragment tacrine. They could diminish Aβ aggregation promoted by AChE as well as show better antioxidant properties than trolox due to the interaction with the PAS of AChE and Cu2+ complexing properties. In human neuroblastoma cells, these hybrids also showed protective properties against damage caused by mitochondrial free radicals. Compound 30 (Figure 8) as a representative hybrid was further confirmed that it controlled several pathological processes at the cellular and neuronal level and might have significant beneficial effects in neurodegenerative diseases127.
Presently, several natural compounds bearing pyridin-2(1H)-ones (2-pyridones) structure have emerged with numerous pharmacological activities, such as AChEI, antioxidant and antitumour128–130, while many synthetic or semisynthetic clinical drugs also enclose those nontoxic moieties131. Therefore, Chand et al.132 conjugated 2-hydroxybenzoyl-2-pyridone (HBP) with tacrine through different alkyl spacers and evaluated them as AChEIs, antioxidants and biometal chelators to combat AD. The results showed that all of them were dual-binding site AChE inhibitors with activity ranged in submicromolar (IC50 = 0.57–0.78 μM), which were comparable to the parent tacrine, and had good 2,2-diphenyl-1-picrylhydrazyl (DPPH) radical scavenging capacity (EC50 = 204–249 μM) conferred by the HBP moiety. The higher anti-AChE activity of compound 31 (IC50= 0.57 μM) (Figure 8) confirmed the possible better fitting of this compound between CAS and PAS as well as its capacity for H-bonding by molecular-modeling studies. The chelating capacity evaluation proved that the HBP moiety of this biometal acted as a moderate/good chelator, being able to form complexes with β-phenol-keto coordination mode.
Recent studies have demonstrated that hydroxyl-substituted coumarins could act as effective redox-active metal chelators, free radical scavengers and powerful chain-breaking antioxidants133,134, and coumarin derivatives in view of their marked implication in the improvement of cognitive functions of patients with neurodegenerative disease attracted people’s attention135. Hrabinova et al.136 synthesized a series of hybrids consist of a tacrine unit and a 7-hydroxycoumarin moiety substitutes at the 4-position, linked by alkylenediamine or alkylene polyamine tethers of different lengths via an amide functionality. Most compounds inhibited hAChE/hBuChE at nanomolar concentrations, being more potent than tacrine. The thioflavin T assay showed that these tacrine–coumarin hybrids were able to interfere with Aβ1–40 aggregation in a concentration-dependent manner. The ABTS assay and EPR spectroscopy indicated that they could suppress the formation of reactive hydroxyl radicals, by the chelation of free copper (II) ions due to the carbonyl oxygen and imino nitrogen atoms of the spacer were coordinated to the copper (II) ion, and confirmed the reduction of Cu(II) to Cu(I), which were further proved by in vitro DNA damage protection experiments. Among them, compound 32 (Figure 8) containing along with the AChE inhibitory segment, also an antioxidant moiety capable of alleviating metal (copper)-induced oxidative stress, might be of importance in the treatment of Alzheimer’s disease.
Other types of multitarget-directed tacrine derivatives
Tacrine derivatives with cholinesterase and MAO inhibition properties
Monoamine oxidases (MAOs) receive increasing attention in recent years due to their roles in the treatment of AD. MAOs are flavin adenine dinucleotide (FAD)-containing enzymes and localize in the outer mitochondrial membrane in various cells including nerve terminals, liver, intestinal mucosa and other tissues137, which are responsible for the oxidative deamination of neurotransmitters. Based on their substrate and inhibitor specificities, MAOs exist in two distinct enzymatic isoforms, MAO-A and MAO-B. Selective inhibitors of MAO-A have been shown to be effective antidepressants and anti-anxiety agents in the clinic, whereas MAO-B inhibitors are useful in several neurodegenerative disorders such as Parkinson’s disease, AD, Huntington chorea and amyotrophic lateral sclerosis138. Evidence showed that high expression levels of MAO-B in neuronal tissue could result in an increase in the level of H2O2 and oxidative free radicals, which played a major role in the etiology of AD139. Therefore, MAO-B inhibitors are considered to be potential candidates for anti-Alzheimer drugs.
Among the MAO inhibitors, selegiline, which is an irreversible and selective MAO-B inhibitor, acts as a neuroprotective agent in cellular and animal models of AD140, so Lu et al.141 designed tacrine hybrids that were connected by carbon spacers of different lengths with selegiline. Results showed that all the compounds were potent AChE and BuChE inhibitors (IC50 = 14.2–456 nM for eeAChE, IC50 = 2.03–66.0 nM for eqBuChE) and were effective inhibitors of both hMAO-A and hMAO-B enzymes in the micromolar range. Compound 33 (Figure 9) with a nine carbon atom tether, turned out to be a potent inhibitor of eeAChE (IC50 = 22.6 nM) and eqBuChE (IC50 = 9.37 nM) and a balanced inhibitor of both monoamine oxidases (IC50 = 0.372 μM for hMAO-A, IC50= 0.181 μM for hMAO-B). Additional experiments on 33 indicated that it was a mixed-type inhibitor of AChE and an irreversible inhibitor of hMAO-B, which could enhance patient cognition by increasing levels of acetylcholine and protect neurons by keeping the activities of selegiline.
Figure 9.

The chemical structures of tacrine derivatives with cholinesterase and MAO inhibitory activities.
Figure 10.
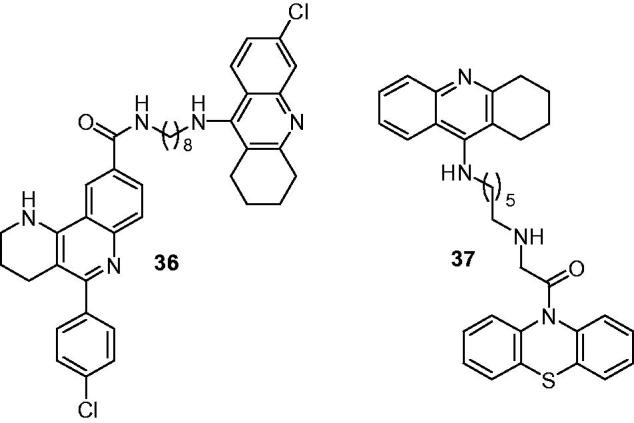
The chemical structures of tacrine derivatives as cholinesterase and tau-aggregation inhibitors.
Sun et al. chose homoisoflavonoid as the pharmacophore for its known MAO-B inhibitory activity to design the novel multi-target-directed tacrine derivatives142,143. Evaluated as inhibitors of cholinesterase and MAOs, all the compounds were potent cholinesterase inhibitors with activities in the nanomolar range and were selective MAO-B inhibitors. Compound 34 (Figure 9) with a six carbon linker between tacrine and (E)-7-hydroxy-3–(4-methoxybenzylidene) chroman-4-one, provided the best results for cholinesterase inhibition (IC50 = 67.9 nM for eeAChE, IC50 = 33.0 nM for eqBuChE) better than tacrine (IC50 = 111.0 nM for eeAChE) and preferable hMAO-B inhibition (IC50 = 0.401 μM). The PAMPA-BBB assay indicated that compound 34 could cross the BBB to target the enzymes in the CNS.
Xie et al.144 selected piperazine side-armed alkane spacers of different lengths to connect tacrine and coumarin for that the piperazine side-armed alkane spacers could be lodged in the narrow enzymatic cavity, which allowed the compounds simultaneously binding to the PAS, CAS and mid-gorge sites of AChE. The coumarin scaffold was used to inhibit the MAO-B and interacted with the PAS of AChE due to its aromatic character. Most of these tacrine–coumarin hybrids showed potent inhibitory activity toward AChE and BuChE and clearly selective inhibition for MAO-B. Among these compounds, 35 (Figure 9) exhibited strong inhibitory activity for AChE (IC50 = 33.63 nM for eeAChE, IC50 = 16.11 nM for hAChE) and BuChE (IC50 = 80.72 nM for eqBuChE, IC50 = 112.72 nM for hBuChE), and the competitive and highest inhibitory activity against hMAO-B (IC50 = 0.24 μM) from covering the substrate and entrance cavities of MAO-B. Moreover, 35 could penetrate the CNS and showed low cell toxicity.
Tacrine derivatives with cholinesterase inhibition and tauantiaggregating properties
Tau proteins are microtubule-associated proteins that are abundant in neurons in the central nervous system, the main physiological function of tau proteins is to interact with tubulin to stabilize microtubules and promote tubulin assembly into microtubules145. Evidences showed that hyperphosphorylation of tau appeared to be an early and central event in tau related neurodegeneration including AD, which was involved in the pathogenesis of AD146. Phosphorylation of tau is regulated by a host of kinases, for example, PKN, a serine/threonine kinase. When PKN is activated, it phosphorylates tau, resulting in the self-assembly of tangles of paired helical filaments and straight filaments, and eventually disrupting the microtubule organization, leading to collapse of the neuron’s transport system. Thus, inhibition of abnormal hyperphosphorylation of tau offers a promising therapeutic target for AD and related tauopathies147.
Di Pietro et al. obtained eight methylene-linked 1,2,3,4-tetrahydrobenzo[h][1,6]naphthyridine-6-chlorotacrine hybrids, by optimization of an essentially inactive 3,4-dihydro-2H-pyrano[3,2-c] quinoline carboxylic ester derivative as PAS-binding motif by double O→NH bio-isosteric replacement, molecular hybridization with the CAS inhibitor 6-chlorotacrine and molecular dynamics-driven optimization of the length of the linker148. Further biological profiling of hybrids have shown that these eight hybrids were potent inhibitors of hAChE/hBuChE and moderately potent Aβ42 and tau antiaggregating agents, with IC50 values were in the submicromolar and low micromolar range, respectively. The in vitro studies using an artificial membrane model showed these hybrids had good brain permeability. Compound 36 (Figure 10), with the most potent inhibition against hAChE, exhibited IC50 of 2.06 nM which was better than the reference compound 6-chlorotacrine (IC50 = 5.90 nM), as well as possessed the most potent and balance with self-ind Aβ1–42 antiaggregating (77.5% at 10 μM) and tau antiaggregating (68.7% at 10 μM) activities.
Phenothiazine, the key pharmacophore of rember (methylene blue, MB), could prevent tau filament formation and regenerate cognition, which is considered to be a bright and promising AD drug structure targeting tau protein149. A series of tacrine–phenothiazine hybrids via an alkylenediamine-type spacer were designed, according to the principle of multitarget drugs by Hui et al.,150 with the help of computational chemistry software and molegro virtual docker. Among them, 37 (Figure 10) was found to be the most potent compound with its IC50 was 89 nM. Meanwhile, 37 markedly prevented tau hyperphosphorylation induced by okadaic acid in N2α cell with 39.5% inhibition when tested at 10−5 M and could interact with Aβ fibril (fAβ) using surface plasmon resonance, and the data of KD was 5.51 × 10−8 M.
Conclusions
The more recent insights about the pathophysiology of AD have proven a complex interconnected variety of deleterious events, with multiple pathways involving in its etiology and a variety of factors that are associated with the installation, progress and severity of AD. Therefore, the development of effective drugs for AD becomes more and more difficult. Even though there is a great number of very interesting compounds with diverse pharmacological profile in preclinical studies, the majority of them fail in the clinical trials. The results of the analysis published by Cumming et al.151 is advanced to the FDA and approved for marketing. Currently, 108 clinical trials for AD therapies are being conducted, and only 14 agents reach phase 3. Ladostigil, the multifunctional agent, which fails phase-3 clinical trials as AD drug, is currently investigated as potential agent for mild cognitive impairment. The statistics indicate that there is no simple way of searching for AD therapy, and the presented MTDLs approach that enabled researchers to obtain compounds endowed with advantageous properties, resulting from possible interactions with more than one target involved in the pathogenesis of AD, gives hope for further development of new and effective therapy for AD.
Last decades, efforts have been employed in the design and search for new biological chemical entities capable to direct simultaneously with different molecular targets, which were involved in the pathogenesis of AD, and many new significantly active and promise molecules have been discovered. Tacrine, which is the first AChE inhibitor, has been selected as the ideal active fragment because of its simple structure, clear activity and its superiority in the structural modification, and thus, it could be introduced into the overall molecular skeletons of the multitarget-directed anti-AD agents. This review presents a series of novel multitarget-directed tacrine derivatives up to now, which are classified by the biological targets and chemical structure. New drug prototype candidates have been identified and they are the starting point of a revolutionary new way of thinking drug design, and some drug candidates are under preclinical evaluation. Despite none of these MTDL drug candidates have reached clinical phase of development, we hope that in the next few years the system biology approach could effectively contribute to the medicinal chemists in the discovery of innovative chemical entities, which are able to understand and effectively modify the course of this devastating neurodegenerative disorder.
Funding Statement
This work was supported by National Natural Science Foundation of China (No. 81274058, 21302225), Natural Science Foundation of Jiangsu Province (BK20151563), the Program for New Century Excellent Talents by the Ministry of Education (NCET-12?0741), 333 High-level Talents Training Project Funded by Jiangsu Province, Six Talents Project Funded by Jiangsu Province (2013-YY-010), Technology Innovation Venture Fund by Nanjing University of Chinese Medicine (CX201301), Program for Excellent Talents in School of Pharmacy of Nanjing University of Chinese Medicine (15ZYXET-1), Jiangsu Collaborative Innovation Center of Chinese Medicinal Resources Industrialization (ZDXMHT-1?13), Project Funded by the Priority Academic Program Development of Jiangsu Higher Education Institutions and Project Funded by the Flagship Major Development of Jiangsu Higher Education Institutions (PPZY2015A070), and Key Laboratory of Therapeutic Material of Chinese Medicine, Jiangsu Province.
Acknowledgements
This work was supported by National Natural Science Foundation of China (No. 81274058, 21302225), Natural Science Foundation of Jiangsu Province (BK20151563), the Program for New Century Excellent Talents by the Ministry of Education (NCET-12–0741), 333 High-level Talents Training Project Funded by Jiangsu Province, Six Talents Project Funded by Jiangsu Province (2013-YY-010), Technology Innovation Venture Fund by Nanjing University of Chinese Medicine (CX201301), Program for Excellent Talents in School of Pharmacy of Nanjing University of Chinese Medicine (15ZYXET-1), Jiangsu Collaborative Innovation Center of Chinese Medicinal Resources Industrialization (ZDXMHT-1–13), Project Funded by the Priority Academic Program Development of Jiangsu Higher Education Institutions and Project Funded by the Flagship Major Development of Jiangsu Higher Education Institutions (PPZY2015A070), and Key Laboratory of Therapeutic Material of Chinese Medicine, Jiangsu Province.
Disclosure statement
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of this article.
References
- 1.Querfurth HW, Laferla FM.. Alzheimer's disease. N Engl J Med 2010;362:329–44. [DOI] [PubMed] [Google Scholar]
- 2.Ritchie CW, Molinuevo JL, Truyen L, et al. . Development of interventions for the secondary prevention of Alzheimer's dementia: the European Prevention of Alzheimer's Dementia (EPAD) project. Lancet Psychiatry 2016; 3:179–86. [DOI] [PubMed] [Google Scholar]
- 3.Guzior N, Wieckowska A, Panek D, Malawska B. Recent development of multifunctional agents as potential drug candidates for the treatment of Alzheimer's disease. Curr Med Chem 2015;22:373–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Leon R, Garcia AG, Marco-Contelles J.. Recent advances in the multitarget-directed ligands approach for the treatment of Alzheimer's disease. Med Res Rev 2013;33:139–89. [DOI] [PubMed] [Google Scholar]
- 5.Martinez A, Emerging drugs and targets for Alzheimer’s disease: Volume 1: Beta-amyloid, Tau protein and Glucose metabolism. Cambridge: Royal Society of Chemistry; 2010:938–939. [Google Scholar]
- 6.Cavalli A, Bolognesi ML, Minarini A, et al. . Multi-target-directed ligands to combat neurodegenerative diseases. J Med Chem 2008;51:347–72. [DOI] [PubMed] [Google Scholar]
- 7.Chen X, Decker M.. Multi-target compounds acting in the central nervous system designed from natural products. Curr Med Chem 2013;20:1673–85. [DOI] [PubMed] [Google Scholar]
- 8.Wang Y, Wang F, Yu JP, et al. . Novel multipotent phenylthiazole-tacrine hybrids for the inhibition of cholinesterase activity, β-amyloid aggregation and Ca2+ overload. Bioorg Med Chem 2012;20:6513–22. [DOI] [PubMed] [Google Scholar]
- 9.Decker M, Kraus B, Heilmann J.. Design, synthesis and pharmacological evaluation of hybrid molecules out of quinazolinimines and lipoic acid lead to highly potent and selective butyrylcholinesterase inhibitors with antioxidant properties. Bioorg Med Chem 2008;16:4252–61. [DOI] [PubMed] [Google Scholar]
- 10.Mohamed T, Rao PP.. Alzheimer's disease: emerging trends in small molecule therapies. Curr Med Chem 2011;18:4299–320. [DOI] [PubMed] [Google Scholar]
- 11.Romero A, Cacabelos R, Oset-Gasque MJ, et al. . Novel tacrine- related drugs as potential candidates for the treatment of Alzheimer's disease. Bioorg Med Chem Lett 2013;23:1916–22. [DOI] [PubMed] [Google Scholar]
- 12.Minarini A, Milelli A, Simoni E, et al. . Multifunctional tacrine derivatives in Alzheimer's disease. Curr Top Med Chem 2013;13:1771–86. [DOI] [PubMed] [Google Scholar]
- 13.Musial A, Bajda M, Malawska B.. Recent developments in cholinesterases inhibitors for Alzheimer's disease treatment. Curr Med Chem 2007;14:2654–79. [DOI] [PubMed] [Google Scholar]
- 14.Gupta RC.Tacrine. Encyclopedia of Toxicology 2005; 332:130–1. [Google Scholar]
- 15.Meng Q, Ru J, Zhang GL, et al. . Re-evaluation of tacrine hepatotoxicity using gel entrapped hepatocytes. Toxicol Lett 2007;168:140–7. [DOI] [PubMed] [Google Scholar]
- 16.Chen Y, Sun HP, Li W.. Progress in novel multi-target-directed tacrine derivatives. Prog Pharm Sci 2014;38:656–64. [Google Scholar]
- 17.Tumiatti V, Minarini A, Bolognesi ML, et al. . Tacrine derivatives and Alzheimer's disease. Curr Med Chem 2010;17:1825–38. [DOI] [PubMed] [Google Scholar]
- 18.Ladner CJ, Lee JM.. Pharmacological drug treatment of Alzheimer disease: the cholinergic hypothesis revisited. J Neuropathol Exp Neurol 1998;57:719–31. [DOI] [PubMed] [Google Scholar]
- 19.Sussman JL, Harel M, Frolow F, et al. . Atomic structure of acetylcholinesterase from Torpedo californica: a protopic acetylcholine-binding protein. Science 1991;253:827–79. [DOI] [PubMed] [Google Scholar]
- 20.Bourne Y, Grassi J, Bougis PE, Marchot P.. Conformational flexibility of the acetylcholinesterase tetramer suggested by x-ray crystallography. J Biol Chem 1999;274:30370–6. [DOI] [PubMed] [Google Scholar]
- 21.Kryger G, Harel M, Giles K, et al. . Structures of recombinant native and E202Q mutant human acetylcholinesterase complexed with the snake-venom toxin fasciculin-II. Acta Crystallogr D Biol Crystallogr 2000;56:1385–94. [DOI] [PubMed] [Google Scholar]
- 22.Cheung J, Rudolph MJ, Burshteyn F, et al. . Structures of human acetylcholinesterase in complex with pharmacologically important ligands. J Med Chem 2012;55:10282–6. [DOI] [PubMed] [Google Scholar]
- 23.Dvir H, Silman I, Harel M, et al. . Acetylcholinesterase: from 3D structure to function. Chem Biol Interact 2010;187:10–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Harel M, Sonoda LK, Silman I, et al. . Crystal structure of thioflavin T bound to the peripheral site of Torpedo californica acetylcholinesterase reveals how thioflavin T acts as a sensitive flu. J Am Chem Soc 2008;130:7856–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bourne Y, Taylor P, Radic Z, Marchot P.. Structural insights into ligand interactions at the acetylcholinesterase peripheral anionic site. EMBO J 2003;22:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Harel M, Quinn DM, Nair HK, et al. . The X-ray structure of a transition state analog complex reveals the molecular origins of the catalytic power and substrate specificity of acetylcholinesterase. J Am Chem Soc 1996;118:2340–6. [Google Scholar]
- 27.Colletier JP, Fournier D, Greenblatt HM, et al. . Structural insights into substrate traffic and inhibition in acetylcholinesterase. EMBO J 2006;25:2746–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Szegletes T, Mallender WD, Rosenberry TL.. Nonequilibrium analysis alters the mechanistic interpretation of inhibition of acetylcholinesterase by peripheral site ligands. Biochemistry 1998;37:4206–16. [DOI] [PubMed] [Google Scholar]
- 29.De Ferrari GV, Canales MA, Shin I, et al. . A structural motif of acetylcholinesterase that promotes amyloid beta-peptide fibril formation. Biochemistry 2001;40:10447–57. [DOI] [PubMed] [Google Scholar]
- 30.Chambers C, Luo C, Tong M, et al. . Probing the role of amino acids in oxime-mediated reactivation of nerve agent-inhibited human acetylcholinesterase. Toxicol In Vitro 2015;29:408–14. [DOI] [PubMed] [Google Scholar]
- 31.Bartolini M, Bertucci C, Cavrini V, Andrisano V.. beta-Amyloid aggregation induced by human acetylcholinesterase: inhibition studies. Biochem Pharmacol 2003;65:407–16. [DOI] [PubMed] [Google Scholar]
- 32.Dinamarca MC, Sagal JP, Quintanilla RA, et al. . Amyloid-beta-acetylcholinesterase complexes potentiate neurodegenerative changes induced by the Abeta peptide. Implications for the pathogenesis of Alzheimer's disease. Mol Neurodegener 2010;5:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Reale M, Di Nicola M, Velluto L, et al. . Selective acetyl- and butyrylcholinesterase inhibitors reduce amyloid-beta ex vivo activation of peripheral chemo-cytokines from Alzheimer’s disease subjects: exploring the cholinergic anti-inflammatory pathway. Curr Alzheimer Res 2014;11:608–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ballard CG, Greig NH, Guillozet-Bongaarts AL, et al. . Cholinesterases: roles in the brain during health and disease. Curr Alzheimer Res 2005;2:307–18. [DOI] [PubMed] [Google Scholar]
- 35.Greig NH, Utsuki T, Ingram DK, et al. . Selective butyrylcholinesterase inhibition elevates brain acetylcholine, augments learning and lowers Alzheimer beta-amyloid peptide in rodent. Proc Natl Acad Sci USA 2005;102:17213–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Furukawahibi Y, Alkam T, Nitta A, et al. . Butyrylcholinesterase inhibitors ameliorate cognitive dysfunction induced by amyloid-beta peptide in mice. Behav Brain Res 2011;225:222–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Anand P, Singh B.. A review on cholinesterase inhibitors for Alzheimer's disease. Arch Pharm Res 2013;36:375–99. [DOI] [PubMed] [Google Scholar]
- 38.Harel M, Schalk I, Ehret-Sabatier L, et al. . Quaternary ligand binding to aromatic residues in the active-site gorge of acetylcholinesterase. Proc Natl Acad Sci USA 1993;90:9031–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pang YP, Quiram P, Jelacic T, et al. . Highly potent, selective, and low cost bis-tetrahydroaminacrine inhibitors of acetylcholinesterase. Steps toward novel drugs for treating Alzheimer's disease. J Biol Chem 1996;271:23646–9. [DOI] [PubMed] [Google Scholar]
- 40.Cappelli A, Gallelli A, Manini M, et al. . Further studies on the interaction of the 5-hydroxytryptamine3 (5-HT3) receptor with arylpiperazine ligands. Development of a new 5-HT3 receptor ligand showing potent acetylcholinesterase inhibitory properties. J Med Chem 2005;48:3564–75. [DOI] [PubMed] [Google Scholar]
- 41.Hu MK, Wu LJ, Hsiao G, Yen MH.. Homodimeric tacrine congeners as acetylcholinesterase inhibitors. J Med Chem 2002;45:2277–82. [DOI] [PubMed] [Google Scholar]
- 42.Shao D, Zou C, Luo C, et al. . Synthesis and evaluation of tacrine-E2020 hybrids as acetylcholinesterase inhibitors for the treatment of Alzheimer's disease. Bioorg Med Chem Lett 2004;14:4639–42. [DOI] [PubMed] [Google Scholar]
- 43.Camps P, Formosa X, Galdeano C, et al. . Novel donepezil-based inhibitors of acetyl- and butyrylcholinesterase and acetylcholinesterase-induced beta-amyloid aggregation. J Med Chem 2008;51:3588–98. [DOI] [PubMed] [Google Scholar]
- 44.Alonso D, Dorronsoro I, Rubio L, et al. . Donepezil-tacrine hybrid related derivatives as new dual binding site inhibitors of AChE. Bioorg Med Chem 2005;13:6588–97. [DOI] [PubMed] [Google Scholar]
- 45.Munoz-Ruiz P, Rubio L, Garcia-Palomero E, et al. . Design, synthesis, and biological evaluation of dual binding site acetylcholinesterase inhibitors: new disease-modifying agents for Alzheimer’s disease. J Med Chem 2005;48:7223–33. [DOI] [PubMed] [Google Scholar]
- 46.Krasinski A, Radic Z, Manetsch R, et al. . In situ selection of lead compounds by click chemistry: target-guided optimization of acetylcholinesterase inhibitors. J Am Chem Soc 2005;127:6686–92. [DOI] [PubMed] [Google Scholar]
- 47.Lorenzo A, Yankner BA.. Beta-amyloid neurotoxicity requires fibril formation and is inhibited by congo red. Proc Natl Acad Sci USA 1994;91:12243–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Thorsett ED, Latimer LH.. Therapeutic approaches to Alzheimer's disease. Curr Opin Chem Biol 2000;4:377–82. [DOI] [PubMed] [Google Scholar]
- 49.Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science 2002;297:353–6. [DOI] [PubMed] [Google Scholar]
- 50.Paul S, Planque S, Nishiyama Y.. Beneficial catalytic immunity to Aβ peptide. Rejuvenation Res 2010;13:179–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Schott Y, Decker M, Rommelspacher H, Lehmann J. 6-Hydroxy- and 6-methoxy-beta-carbolines as acetyl- and butyrylcholinesterase inhibitors. Bioorg Med Chem Lett 2006;16:5840–3. [DOI] [PubMed] [Google Scholar]
- 52.Rook Y, Schmidtke KU, Gaube F, et al. . Bivalent beta-carbolines as potential multitarget anti-Alzheimer agents. J Med Chem 2010;53:3611–17. [DOI] [PubMed] [Google Scholar]
- 53.Herraiz T, Galisteo J.. Tetrahydro-β-carboline alkaloids that occur in foods and biological systems act as radical scavengers and antioxidants in the ABTS assay. Free Radic Res 2002;36:923–8. [DOI] [PubMed] [Google Scholar]
- 54.Herraiz T, Galisteo J. Tetrahydro-beta-carboline alkaloids occur in fruits and fruit juices. Activity as antioxidants and radical scavengers. J Agric Food Chem 2003;51:7156–61. [DOI] [PubMed] [Google Scholar]
- 55.Matsutomo T, Stark TD, Hofmann T.. In vitro activity-guided identification of antioxidants in aged garlic extract. J Agric Food Chem 2013;61:3059–67. [DOI] [PubMed] [Google Scholar]
- 56.Lan JS, Xie SS, Li SY, et al. . Design, synthesis and evaluation of novel tacrine-(β-carboline) hybrids as multifunctional agents for the treatment of Alzheimer's disease. Bioorg Med Chem 2014;22:6089–104. [DOI] [PubMed] [Google Scholar]
- 57.Choi MM, Kim EA, Hahn HG, et al. . Protective effect of benzothiazole derivative KHG21834 on amyloid beta-induced neurotoxicity in PC12 cells and cortical and mesencephalic neurons. Toxicology 2007;239:156–66. [DOI] [PubMed] [Google Scholar]
- 58.Keri RS, Quintanova C, Marques SM, et al. . Design, synthesis and neuroprotective evaluation of novel tacrine-benzothiazole hybrids as multi-targeted compounds against Alzheimer's disease. Bioorg Med Chem 2013;21:4559–69. [DOI] [PubMed] [Google Scholar]
- 59.Zhang WT, Ruan JL, Wu PF, et al. . Design, synthesis, and cytoprotective effect of 2-aminothiazole analogues as potent poly (ADP-ribose) polymerase-1 inhibitors. J Med Chem 2009;52:718–25. [DOI] [PubMed] [Google Scholar]
- 60.Elsinghorst PW, Tanarro CMG, Gutschow M.. Novel heterobivalent tacrine derivatives as cholinesterase inhibitors with no table selectivity toward butyrylcholinesterase. J Med Chem 2006;49:7540–4. [DOI] [PubMed] [Google Scholar]
- 61.Ucar G, Gokhan N, Yesilada A, Bilgin AA.. 1-N-substituted thiocarbomoyl-3-phenyl-5-thienyl-2-prozolines: a novel cholinesterase and selected monoamine oxidase B inhibitors for the treatment of Parkinson’s and Alzheimer’s diseases. Neurosci Lett 2005;382:327–31. [DOI] [PubMed] [Google Scholar]
- 62.Zhang C, Du QY, Chen LD, et al. . Design, synthesis and evaluation of novel tacrine-multialkoxybenzene hybrids as multi-targeted compounds against Alzheimer's disease. Eur J Med Chem 2016;116:200–9. [DOI] [PubMed] [Google Scholar]
- 63.Bolognesi ML, Chiriano G, Bartolini M, et al. . Synthesis of monomeric derivatives to probe memoquin's bivalent interactions. J Med Chem 2011;54:8299–304. [DOI] [PubMed] [Google Scholar]
- 64.Prati F, Uliassi E, Bolognesi ML.. Two diseases, one approach: multitarget drug discovery in Alzheimer’s and neglected tropical diseases. Med Chem Comm 2014;5:853–61. [Google Scholar]
- 65.Bartolini M, Bertucci C, Bolognesi ML, et al. . Insight into the kinetic of amyloid beta (1-42) peptide self-aggregation: elucidation of inhibitors' mechanism of action. Chembiochem 2007;8:2152–61. [DOI] [PubMed] [Google Scholar]
- 66.Scherzer-Attali R, Pellarin R, Convertino M, et al. . Complete phenotypic recovery of an Alzheimer’s disease model by a quinone-tryptophan hybrid aggregation inhibitor. PLoS One 2010;5:e11101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Nepovimova E, Uliassi E, Korabecny J, et al. . Multitarget drug design strategy: quinone-tacrine hybrids designed to block amyloid-beta aggregation and to exert anticholinesterase and antioxidant effects. J Med Chem 2014;57:8576–89. [DOI] [PubMed] [Google Scholar]
- 68.Klaver DW, Wilce MC, Cui H, et al. . Is BACE1 a suitable therapeutic target for the treatment of Alzheimer's disease? Current strategies and future directions. Biol Chem 2010;391:849–59. [DOI] [PubMed] [Google Scholar]
- 69.Luo X, Yan R.. Inhibition of BACE1 for therapeutic use in Alzheimer's disease. Int J Clin Exp Pathol 2010;3:618–28. [PMC free article] [PubMed] [Google Scholar]
- 70.Bjorklund C, Oscarson S, Benkestock K, et al. . Design and synthesis of potent and selective BACE-1 inhibitors. J Med Chem 2010;53:1458–64. [DOI] [PubMed] [Google Scholar]
- 71.Venugopal C, Demos CM, Rao KS, et al. . Beta-secretase: structure, function, and evolution. CNS Neurol Disord Drug Targets 2008;7:278–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Galdeano C, Viayna E, Sola I, et al. . Huprine-tacrine heterodimers as anti-amyloidogenic compounds of potential interest against Alzheimer's and prion diseases. J Med Chem 2012;55:661–9. [DOI] [PubMed] [Google Scholar]
- 73.Spencer JP.The impact of flavonoids on memory: physiological and molecular considerations. Chem Soc Rev 2009;38:1152–61. [DOI] [PubMed] [Google Scholar]
- 74.Spencer JP.Beyond antioxidants: the cellular and molecular interactions of flavonoids and how these underpin their actions on the brain. Proc Nutr Soc 2010;69:244–60. [DOI] [PubMed] [Google Scholar]
- 75.Shimmyo Y, Kihara T, Akaike A, et al. . Flavonols and flavones as BACE-1 inhibitors: structure-activity relationship in cell-free, cell-based and in silico studies reveal novel pharmacophore features. Biochim Biophys Acta 2008;1780:819–25. [DOI] [PubMed] [Google Scholar]
- 76.Fernandez-Bachiller MI, Perez C, Monjas L, et al. . New tacrine-4-oxo-4H-chromene hybrids as multifunctional agents for the treatment of Alzheimer's disease, with cholinergic, antioxidant, and β-amyloid-reducing properties. J Med Chem 2012;55:1303–17. [DOI] [PubMed] [Google Scholar]
- 77.Baharloo F, Moslemin MH, Nadri H, et al. . Benzofuran-derived benzylpyridinium bromides as potent acetylcholinesterase inhibitors. Eur J Med Chem 2015;93:196–201. [DOI] [PubMed] [Google Scholar]
- 78.Howlett DR, Perry AE, Godfrey F, et al. . Inhibition of fibril formation in beta-amyloid peptide by a novel series of benzofurans. Biochem J 1999;340:283–9. [PMC free article] [PubMed] [Google Scholar]
- 79.Byun JH, Kim H, Kim Y, et al. . Aminostyrylbenzofuran derivatives as potent inhibitors for Abeta fibril formation. Bioorg Med Chem Lett 2008;18:5591–3. [DOI] [PubMed] [Google Scholar]
- 80.Ono M, Kung MP, Hou C, Kung HF. Benzofuran derivatives as Abeta-aggregate-specific imaging agents for Alzheimer's disease. Nucl Med Biol 2002;29:633–42. [DOI] [PubMed] [Google Scholar]
- 81.Allsop D, Gibson G, Martin IK, et al. . 3-p-Toluoyl-2-[4'-(3-diethylaminopropoxy)-phenyl]-benzofuran and 2-[4'-(3-diethylaminopropoxy)-phenyl]-benzofuran do not act as surfactants or micelles when inhibiting the aggregation of beta-amyloid peptide. Bioorg Med Chem Lett 2001;11:255–7. [DOI] [PubMed] [Google Scholar]
- 82.Zha XM, Lamba D, Zhang LL, et al. . Novel tacrine-benzofuran hybrids as potent multitarget-directed ligands for the treatment of Alzheimer’s disease: design, synthesis, biological evaluation, and X-ray crystallography. J Med Chem 2016;59:114–31. [DOI] [PubMed] [Google Scholar]
- 83.Ghosh AK, Brindisi M, Tang J.. Developing beta-secretase inhibitors for treatment of Alzheimer’s disease. J Neurochem 2012;120:71–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Padurariu M, Ciobica A, Lefter R, et al. . The oxidative stress hypothesis in Alzheimer's disease. Psychiatr Danub 2013;25:401–9. [PubMed] [Google Scholar]
- 85.Skrzydlewska E, Farbiszewski R.. Trolox-derivative antioxidant protects against methanol-induced damage. Fundam Clin Pharmacol 1997;11:460–5. [DOI] [PubMed] [Google Scholar]
- 86.Quintanilla RA, Munoz FJ, Metcalfe MJ, et al. . Trolox and 17beta-estradiol protect against amyloid beta-peptide neurotoxicity by a mechanism that involves modulation of the Wnt signaling pathway. J Biol Chem 2005;280:11615–25. [DOI] [PubMed] [Google Scholar]
- 87.Dogterom P, Nagelkerke JF, Mulder GJ.. Hepatotoxicity of tetrahydroaminoacridine in isolated rat hepatocytes: effect of glutathione and vitamin E. Biochem Pharmacol 1988;37:2311–13. [DOI] [PubMed] [Google Scholar]
- 88.Xie SS, Lan JS, Wang XB, et al. . Multifunctional tacrine-trolox hybrids for the treatment of Alzheimer's disease with cholinergic, antioxidant, neuroprotective and hepatoprotective properties. Eur J Med Chem 2015;93:42–50. [DOI] [PubMed] [Google Scholar]
- 89.Tang YZ, Liu ZQ.. Free-radical-scavenging effect of carbazole derivatives on AAPH-induced hemolysis of human erythrocytes. Bioorg Med Chem 2007;15:1903–13. [DOI] [PubMed] [Google Scholar]
- 90.Thiratmatrakul S, Yenjai C, Waiwut P, et al. . Synthesis, biological evaluation and molecular modeling study of novel tacrine-carbazole hybrids as potential multifunctional agents for the treatment of Alzheimer's disease. Eur J Med Chem 2014;75:21–30. [DOI] [PubMed] [Google Scholar]
- 91.Sharma S, Rakoczy S, Brown-Borg H.. Assessment of spatial memory in mice. Life Sci 2010;87:521–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Rosini M, Andrisano V, Bartolini M, et al. . Rational approach to discover multipotent anti-Alzheimer drugs. J Med Chem 2005;48:360–3. [DOI] [PubMed] [Google Scholar]
- 93.Fang L, Kraus B, Lehmann J, et al. . Design and synthesis of tacrine-ferulic acid hybrids as multi-potent anti-Alzheimer drug candidates. Bioorg Med Chem Lett 2008;18:2905–9. [DOI] [PubMed] [Google Scholar]
- 94.Chao X, He X, Yang Y, et al. . Design, synthesis and pharmacological evaluation of novel tacrine-caffeic acid hybrids as multi-targeted compounds against Alzheimer’s disease. Bioorg Med Chem Lett 2012;22:6498–502. [DOI] [PubMed] [Google Scholar]
- 95.Digiacomo M, Chen Z, Wang S, et al. . Synthesis and pharmacological evaluation of multifunctional tacrine derivatives against several disease pathways of AD. Bioorg Med Chem Lett 2015;25:807–10. [DOI] [PubMed] [Google Scholar]
- 96.Quintanova C, Keri RS, Marques SM, et al. . Design, synthesis and bioevaluation of tacrine hybrids with cinnamate and cinnamylideneacetate derivatives as potential anti-Alzheimer drugs. Med Chem Comm 2015;6:1969–77. [Google Scholar]
- 97.Hayashi K, Hayashi T, Otsuka H, Takeda YJ.. Antiviral activity of 5,6,7-trimethoxyflavone and its potentiation of the antiherpes activity of acyclovir. J Antimicrob Chemother 1997;39:821–4. [DOI] [PubMed] [Google Scholar]
- 98.Nakajima A, Ohizumi Y, Yamada K.. Anti-dementia activity of nobiletin, a citrus flavonoid: a review of animal studies. Clin Psychopharmacol Neurosci 2014;12:75–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Liao S, Deng H, Huang S, et al. . Design, synthesis and evaluation of novel 5, 6, 7-trimethoxyflavone-6-chlorotacrine hybrids as potential multifunctional agents for the treatment of Alzheimer’s disease. Bioorg Med Chem Lett 2015;25:1541–5. [DOI] [PubMed] [Google Scholar]
- 100.Lu C, Guo Y, Yan J, et al. . Synthesis and evaluation of multi-target-directed ligands against alzheimer's disease based on the fusion of donepezil and ebselen. J Med Chem 2013;56:9089–99. [DOI] [PubMed] [Google Scholar]
- 101.Masumoto H, Sies H.. The reaction of ebselen with peroxynitrite. Chem Res Toxicol 1996;9:262–7. [DOI] [PubMed] [Google Scholar]
- 102.Centuriao FB, Corte CL, Paixao MW, et al. . Effect of ebselen and organochalcogenides on excitotoxicity induced by glutamate in isolated chick retina. Brain Res 2005;1039:146–52. [DOI] [PubMed] [Google Scholar]
- 103.Yamagata K, Ichinose S, Miyashita A, Tagami M.. Protective effects of ebselen, a seleno-organic antioxidant on neurodegeneration induced by hypoxia and reperfusion in stroke-prone spontaneously hypertensive rat. Neuroscience 2008;153:428–35. [DOI] [PubMed] [Google Scholar]
- 104.Mao F, Chen J, Zhou Q, et al. . Novel tacrine-ebselen hybrids with improved cholinesterase inhibitory, hydrogen peroxide and peroxynitrite scavenging activity. Bioorg Med Chem Lett 2013;23:6737–42. [DOI] [PubMed] [Google Scholar]
- 105.Calabrese V, Mancuso C, Calvani M, et al. . Nitric oxide in the central nervous system: neuroprotection versus neurotoxicity . Nat Rev Neurosci 2007;8:766–75. [DOI] [PubMed] [Google Scholar]
- 106.Thatcher GR, Bennett BM, Reynolds JN. Nitric oxide mimetic molecules as therapeutic agents in Alzheimer's disease. Curr Alzheimer Res 2005;2:171–82. [DOI] [PubMed] [Google Scholar]
- 107.Chen Y, Sun J, Fang L, et al. . Tacrine-ferulic acid-nitric oxide (NO) donor trihybrids as potent, multifunctional acetyl- and butyrylcholinesterase inhibitors. J Med Chem 2012;55:4309–21. [DOI] [PubMed] [Google Scholar]
- 108.Fang L, Appenroth D, Decker M, et al. . NO-donating tacrine hybrid compounds improve scopolamine-induced cognition impairment and show less hepatotoxicity. J Med Chem 2008;51:7666–9. [DOI] [PubMed] [Google Scholar]
- 109.Chen Y, Sun J, Huang Z, et al. . Design, synthesis and evaluation of tacrine-flurbiprofen-nitrate trihybrids as novel anti-Alzheimer's disease agents. Bioorg Med Chem 2013;21:2462–70. [DOI] [PubMed] [Google Scholar]
- 110.Mohan IK, Khan M, Wisel S, et al. . Cardioprotection by HO-4038, a novel verapamil derivative, targeted against ischemia and reperfusion-mediated acute myocardial infarction. Am J Physiol Heart Circ Physiol 2009;296:140–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Kalai T, Balog M, Szabo A, et al. . New poly(ADP-ribose) polymerase-1 inhibitors with antioxidant activity based on 4-carboxamidobenzimidazole-2-ylpyrroline and -tetrahydropyridine nitroxides and their precursors. J Med Chem 2009;52:1619–29. [DOI] [PubMed] [Google Scholar]
- 112.Kalai T, Mugesh G, Roy G, et al. . Combining benzo[d] isoselenazol-3-ones with sterically hindered alicyclic amines and nitroxides: enhanced activity as glutathione peroxidase mimics. Org Biomol Chem 2005;3:3564–9. [DOI] [PubMed] [Google Scholar]
- 113.Kalai T, Altman R, Maezawa I, et al. . Synthesis and functional survey of new tacrine analogs modified with nitroxides or their precursors. Eur J Med Chem 2014;77:343–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Minarini A, Milelli A, Tumiatti V, et al. . Cystamine-tacrine dimer: a new multi-target-directed ligand as potential therapeutic agent for Alzheimer's disease treatment. Neuropharmacology 2012;62:997–1003. [DOI] [PubMed] [Google Scholar]
- 115.Yong-Seok L, Silva AJ.. The molecular and cellular biology of enhanced cognition. Nat Rev Neurosci 2009;10:126–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Cai F, Wang F, Lin FK, et al. . Redox modulation of long-term potentiation in the hippocampus via regulation of the glycogen synthase kinase-3beta pathway. Free Radic Biol Med 2008;45:964–70. [DOI] [PubMed] [Google Scholar]
- 117.Yang YJ, Wu PF, Long LH, et al. . Reversal of aging-associated hippocampal synaptic plasticity deficits by reductants via regulation of thiol redox and NMDA receptor function. Aging Cell 2010;9:709–21. [DOI] [PubMed] [Google Scholar]
- 118.Robillard JM, Gordon GR, Choi HB, et al. . Glutathione restores the mechanism of synaptic plasticity in aged mice to that of the adult. PLos One 2011;6:e20676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Wang Y, Guan XL, Wu PF, et al. . Multifunctional mercapto-tacrine derivatives for treatment of age-related neurodegenerative diseases. J Med Chem 2012;55:3588–92. [DOI] [PubMed] [Google Scholar]
- 120.Keri RS, Quintanova C, Chaves S, et al. . New tacrine hybrids with natural based cysteine derivatives as multi-targeted drugs for potential treatment of Alzheimer's disease. Chem Biol Drug Des 2016;87:101–11. [DOI] [PubMed] [Google Scholar]
- 121.Chen X, Zenger K, Lupp A, et al. . Tacrine-silibinin codrug shows neuro- and hepatoprotective effects in vitro and pro-cognitive and hepatoprotective effects in vivo. J Med Chem 2012;55:5231–42. [DOI] [PubMed] [Google Scholar]
- 122.Dong J, Atwood CS, Anderson VE, et al. . Metal binding and oxidation of amyloid-beta within isolated senile plaque cores: Raman microscopic evidence. Biochemistry 2003;42:2768–73. [DOI] [PubMed] [Google Scholar]
- 123.Zhao Y, Zhao B.. Oxidative stress and the pathogenesis of Alzheimer's disease. Oxid Med Cell Longev 2013;2013:316523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Budimir A.Metal ions, Alzheimer's disease and chelation therapy. Acta Pharm 2011;61:1–14. [DOI] [PubMed] [Google Scholar]
- 125.Li S, Jiang N, Xie S, et al. . Design, synthesis and evaluation of novel tacrine-rhein hybrids as multifunctional agents for the treatment of Alzheimer's disease. Org Biomol Chem 2014;12:801–14. [DOI] [PubMed] [Google Scholar]
- 126.Fernandezbachiller MI, Perez C, Gonzalezmunoz GC, et al. . Novel tacrine-8-hydroxyquinoline hybrids as multifunctional agents for the treatment of Alzheimer's disease, with neuroprotective, cholinergic, antioxidant, and copper-complexing properties. J Med Chem 2010;53:4927–37. [DOI] [PubMed] [Google Scholar]
- 127.Antequera D, Bolos M, Spuch C, et al. . Effects of a tacrine-8-hydroxyquinoline hybrid (IQM-622) on Aβ accumulation and cell death: Involvement in hippocampal neuronal loss in Alzheimer's disease. Neurobiol Dis 2012;46:682–91. [DOI] [PubMed] [Google Scholar]
- 128.Kozikowski AP, Campiani G, Sun LQ, et al. . Identification of a more potent analogue of the naturally occurring alkaloid huperzine A. Predictive molecular modeling of its interaction with AChE. J Am Chem Soc 1996;118:11357–62. [Google Scholar]
- 129.Sui X, Gao C.. Huperzine A ameliorates damage induced by acute myocardial infarction in rats through antioxidant, anti-apoptotic and anti-inflammatory mechanisms. Int J Mol Med 2014;33:227–33. [DOI] [PubMed] [Google Scholar]
- 130.Wall ME.Camptothecin and taxol: discovery to clinic. Med Res Rev 1998;18:299–314. [DOI] [PubMed] [Google Scholar]
- 131.Torres M, Gil S, Parra M.. New synthetic methods to 2-pyridone rings. Curr Org Chem 2005;9:1757–79. [Google Scholar]
- 132.Chand K, Alsoghier HM, Chaves S, Santos MA.. Tacrine-(hydroxybenzoyl-pyridone) hybrids as potential multifunctional anti-Alzheimer’s agents: AChE inhibition, antioxidant activity and metal chelating capacity. J Inorg Biochem 2016;163:266–77. [DOI] [PubMed] [Google Scholar]
- 133.Lin HC, Tsai SH, Chen CS, et al. . Structure-activity relationship of coumarin derivatives on xanthine oxidase-inhibiting and free radical-scavenging activities. Biochem Pharm 2008;75:1416–25. [DOI] [PubMed] [Google Scholar]
- 134.Kostova I, Bhatia S, Grigorov P, et al. . Coumarins as Antioxidants. Curr Med Chem 2011;18:3929–51. [DOI] [PubMed] [Google Scholar]
- 135.Hornick A, Lieb A, Vo NP, et al. . The coumarin scopoletin potentiates acetylcholine release from synaptosomes, amplifies hippocampal long-term potentiation and ameliorates anticholinergic- and age-impaired memory. Neuroscience 2011;197:280–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Hamulakova S, Poprac P, Jomova K, et al. . Targeting copper(II)-induced oxidative stress and the acetylcholinesterase system in Alzheimer’s disease using multifunctional tacrine-coumarin hybrid molecules. J Inorg Biochem 2016;161:52–62. [DOI] [PubMed] [Google Scholar]
- 137.Chaurasiya ND, Ganesan S, Nanayakkara NP, et al. . Inhibition of human monoamine oxidase A and B by 5-phenoxy 8-aminoquinoline analogs. Bioorg Med Chem Lett 2012;22:1701–4. [DOI] [PubMed] [Google Scholar]
- 138.Saura J, Luque JM, Cesura AM, et al. . Increased monoamine oxidase B activity in plaque-associated astrocytes of Alzheimer brains revealed by quantitative enzyme radioautography. Neuroscience 1994;62:15–30. [DOI] [PubMed] [Google Scholar]
- 139.Nebbioso M, Pascarella A, Cavallotti C, Pescosolido N.. Monoamine oxidase enzymes and oxidative stress in the rat optic nerve: age-related changes. Int J Exp Pathol 2012;93:401–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Bar-Am O, Amit T, Weinreb O, et al. . Propargylamine containing compounds as modulators of proteolytic cleavage of amyloid-beta protein precursor: involvement of MAPK and PKC activation. J Alzheimers Dis 2010;21:361–71. [DOI] [PubMed] [Google Scholar]
- 141.Lu C, Zhou Q, Yan J, et al. . A novel series of tacrine-selegiline hybrids with cholinesterase and monoamine oxidase inhibition activities for the treatment of Alzheimer's disease. Eur J Med Chem 2013;62:745–53. [DOI] [PubMed] [Google Scholar]
- 142.Sun Y, Chen J, Chen X, et al. . Inhibition of cholinesterase and monoamine oxidase-B activity by tacrine-homoisoflavonoid hybrids. Bioorg Med Chem 2013;21:7406–17. [DOI] [PubMed] [Google Scholar]
- 143.Desideri N, Bolasco A, Fioravanti R, et al. . Homoisoflavonoids: natural scaffolds with potent and selective monoamine oxidase-B inhibition properties. J Med Chem 2011;54:2155–64. [DOI] [PubMed] [Google Scholar]
- 144.Xie SS, Wang X, Jiang N, et al. . Multi-target tacrine-coumarin hybrids: Cholinesterase and monoamine oxidase B inhibition properties against Alzheimer's disease. Eur J Med Chem 2015;95:153–65. [DOI] [PubMed] [Google Scholar]
- 145.Reynolds CH, Garwood CJ, Wray S, et al. . Phosphorylation regulates tau interactions with Src homology 3 domains of phosphatidylinositol 3-kinase, phospholipase Cgamma1, Grb2, and Src family kinases. J Biol Chem 2008;283:18177–86. [DOI] [PubMed] [Google Scholar]
- 146.Spires-Jones TL, Stoothoff WH, de Calignon A, et al. . Tau pathophysiology in neurodegeneration: a tangled issue. Trends Neurosci 2009;32:150–9. [DOI] [PubMed] [Google Scholar]
- 147.Iqbal K, Liu F, Gong CX, Grundke-Iqbal I.. Tau in Alzheimer disease and related tauopathies. Curr Alzheimer Res 2010;7:656–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Pietro OD, Perezareales FJ, Juarezjimenez J, et al. . Munoztorrero D. Tetrahydrobenzo [h] [1,6] naphthyridine-6-chlorotacrine hybrids as a new family of anti-Alzheimer agents targeting β-amyloid, tau, and cholinesterase pathologies. Eur J Med Chem 2014;84:107–17. [DOI] [PubMed] [Google Scholar]
- 149.O’Leary JC, Li Q, Marinec P, et al. . Phenothiazine-mediated rescue of cognition in tau transgenic mice requires neuroprotection and reduced soluble tau burden. Mol Neurodegener 2010;1:45–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Hui AL, Chen Y, Zhu SJ, et al. . Design and synthesis of tacrine-phenothiazine hybrids as multitarget drugs for Alzheimer’s disease. Med Chem Res 2014;23:3546–57. [Google Scholar]
- 151.Cummings JL, Morstorf T, Zhong K.. Alzheimer's disease drug-development pipeline: few candidates, frequent failures. Alzheimers Res Ther 2014;6:37. [DOI] [PMC free article] [PubMed] [Google Scholar]