

Abstract
New coumaryl-carboxamide derivatives with the thiourea moiety as a linker between the alkyl chains and/or the heterocycle nucleus were synthesized and their inhibitory activity against the human carbonic anhydrase (hCA) isoforms hCA I, II, VII and IX were evaluated. While the hCA I, II and VII isoforms were not inhibited by the investigated compounds, the tumour-associated isoform hCA IX was inhibited in the high nanomolar range. 2-Oxo-N-((2-(pyrrolidin-1-yl)ethyl)carbamothioyl)-2H-chromene-3-carboxamide (e11) exhibited a selective inhibitory action against hCA IX with the Ki of 107.9 nM. In order to better understand the inhibitory profiles of studied molecules, multiscale molecular modeling approaches were used. Different molecular docking algorithms were used to investigate binding poses and predicted binding energies of studied compounds at the active sites of the CA I, II, VII and IX isoforms.
Keywords: Coumarin, carboxamid, thiourea, carbonic anhydrase, molecular docking, induced fit docking, quantum polarised ligand docking
1. Introduction
The carbonic anhydrases (CAs; EC 4.2.1.1) are a superfamily of metalloenzymes that present in all organisms and consist of metallic core of Zn2+ ion at their active center1–4. CA, explored in the beef erythrocytes for the first time, reversibly catalyses the reactions of hydration of CO2 and dehydration5 of HCO3-. Many CA isozymes involved in these processes are important therapeutic targets with the potential to be inhibited/activated for the treatment of a range of disorders such as oedema, glaucoma, obesity, cancer, epilepsy, amyloid beta, leukaemia and osteoporosis6–10. However, the physiologically relevant reaction that CAs catalyse, using as substrates CO2, COS, CS2, cyanamide, carboxylic, phosphoric and thiocarboxylicesters11–14. Sixteen different α-CA isoforms were isolated from mammals, where they play crucial physiological roles. Some of them are cytosolic (CA I, CA II, CA III, CA VII, CA XIII), others are membrane-bound (CA IV, CA IX, CA XII, CA XIV and CA XV), CA VA and CA VB are mitochondrial, and CA VI is secreted in saliva and milk11. Recent studies suggested that the necrosis formed around a tumour depends on both the excessively expressing of CA IX enzymes increased at such domain and the controlling of pH15,16. Especially, hCA IX is expressed in a restricted number of normal tissues, whereas it is over expressed in many solid tumours and considered involved in important processes connected with cancer progression. The over expression of hCA IX induces the pH imbalance of tumour tissue contributing significantly to the extracellular acidification of solid tumour; thereby hCA IX inhibitors could specifically bind hypoxic tumour cells expressing this isoform17–24. Therefore, it has been considered that the CA inhibitors are crucial molecules for the synthesis of new-generation anticancer drugs25.
Coumarin is one of the most known class of CA inhibitors (CAIs) and shows their effect as 2-hydroxycinnamic acid hydrolysis, unlike other inhibitors26,27. Coumarin derivatives show high selectivity to inhibit isoforms, especially in pharmacological applications, such as the tumour-associated ones (hCA IX and XII, which are targets for antitumour/antimetastatic drugs) or the mitochondrial ones (CA VA and VB, which are targets for antiobesity agents)28,29. Thiocoumarin, thioxocoumarin and sulphocoumarin derivatives showed high affinity for CA IX-XII even at low concentrations30–32.
Urea and thiourea compounds work as building block in the synthesis of heterocyclic compounds. These compounds, thanks to their pharmacological properties, make a significant contribution in the field of medicinal chemistry. Urea and thiourea derivatives exhibit many biological activities such as analgesic, anti-inflammatory, antimicrobial and anticancer. Thiourea derivatives are valuable building blocks for the synthesis of amides, guanidines and varieties of heterocycles33,34. It has been reported that compounds containing urea or thiourea as well as sulphonamide groups highly inhibit the enzyme carbonic anhydrase35–37.
Continuing our interest in coumarin CAIs, in this work, we report the synthesis of novel thiourea-substituted coumaryl-carboxamid derivatives and their effects on the inhibitory activity of human carbonic anhydrase hCA I, hCA II, hCA VII and hCA IX.
2. Experimental
2.1. Material and method
Melting points were taken on a Barnstead Electrothermal 9200. IR spectra were measured on a Shimadzu Prestige-21 (200 VCE) spectrometer. 1H and 13C NMR spectra were measured on a Varian Infinity Plus spectrometer at 300 and at 75 Hz, respectively. 1H and 13C chemical shifts are referenced to the internal deuterated solvent. Mass spectra were obtained using MICROMASS Quattro LC-MS-MS spectrometer. The elemental analyses were carried out with a Leco CHNS-932 instrument. Spectrophotometric analyses were performed by a BioTek Power Wave XS (Winooski, VT). The chemicals and solvents were purchased from Fluka Chemie (Taufkirchen, Germany), Merck (Taufkirchen, Germany), Alfa Aesar (Taufkirchen, Germany) and Sigma-Aldrich (Taufkirchen, Germany).
2.2. General procedures of synthesis and spectral data
2.2.1. 2-oxo-2H-chromene-3-carboxylic acid (c)
A mixture of benzaldehyde (a) (3 mmol), meldrum’s acid (b) (4.5 mmol) was stirred at reflux for 10 h. The mixture was cooled, filtered and recrystallised from methanol to get product (c). Spectral data of this compound were matched with the literature, white solid, 91% yield; mp. 145–147 °C38.
2.2.2. 2-oxo-2H-chromene-3-carbonyl chloride (d)
A 2-oxo-2H-chromene-3-carboxylic acid (c) (0.01 mol) and SOCl2 (0.05 mol) were taken in round bottom flask and it was stirred for 4 h at 80 °C temperature. After the excess SOCl2 was evaporated, the crude product was purified by ether. Spectral data of this compound was matched with the literature50.
White solid, 99% yield; IR: 3057, 1737, 1676, 1605, 1557, 1417, 1225, 1179, 1040, 761 cm−1; 1H NMR (DMSO-d6, 300 MHz) δ/ppm: 7.36–7.43 (2H, m), 7.69 (1H, td, J = 1.7, 7.3 Hz), 7.87 (1H, dd, J = 1.4, 7.6 Hz), 8.71 (1H, s); 13C NMR (DMSO-d6, 75 MHz) δ/ppm:, 116.7, 118.6, 118.9, 125.4, 130.8, 134.9, 149.1, 155.1, 157.3, 164.5.
2.2.3. N-(R-carbamothioyl)-2-oxo-2H-chromene-3-carboxamide (e1–20)
A mixture of 2-oxo-2H-chromene-3-carbonyl chloride (1 mmol) and KSCN (1,2 mmol) in CH3CN (30 ml) was heated under reflux for 30 min. Then, 1.2 mmol amine derivatives were added in the mixture and the solution was refluxed for 4 h. The solution was evaporated and the residue was extracted with water/CH2Cl2. The organic phase was washed by water for three times and dried over Na2SO4. After the organic solvent was evaporated, the crude product was recrystallised from methanol to get pure crystalline e1–20 in 25–70% yields.
2.2.4. N-(methylcarbamothioyl)-2-oxo-2H-chromene-3-carboxamide (e1)
Yellow powder, 52% yield; mp. 146–148 °C; IR: 3363, 3052, 2978, 1698, 1655, 1606, 1562, 1510, 1449, 1243, 1159, 982, 754 cm−1; 1H NMR (CDCl3, 300 MHz) δ/ppm: 3.03 (CH3NH-, 3H, d, J = 4.9 Hz), 7.36–7.43 (2H, m), 7.64–7.76 (2H, m), 8.68 (1H, s, NH), 8.93 (1H, s); 13C NMR (DMSO-d6, 75 MHz) δ/ppm: 27.0, 116.7, 119.1, 119.6, 125.7, 130.9, 134.7, 147.9, 148.5, 154.5, 160.9, 162.2. LC-MS (m/z): 300.1 [M+]. Anal. Calcd. for C12H10N2O3S; C, 54.95; H, 3.84; N, 10.68; found: C, 54.90; H, 3.86; N, 10.67.
2.2.5. 2-oxo-N-(propylcarbamothioyl)-2H-chromene-3-carboxamide (e2)
Yellow powder, 58% yield; mp. 123 °C; IR: 3339, 3053, 2964, 1703, 1655, 1607, 1518, 1450, 1362, 1158, 755 cm−1; 1H NMR (CDCl3, 300 MHz) δ/ppm: 1.00 (3H, t, J = 7.6 Hz), 1.60–1.72 (2H, m), 3.40–4.47 (2H, m), 7.35–7.42 (2H, m), 7.64–7.71 (2H, m), 8.83 (1H, s, NH), 8.92 (1H, s); 13C NMR (CDCl3, 75 MHz) δ/ppm: 11.7, 22.9, 41.8, 116.8, 118.7, 118.9, 125.5, 130.0, 134.1, 148.4, 154.6, 161.6, 161.7. LC-MS (m/z): 321.2 [M+]. Anal. Calcd. for C14H14N2O3S; C, 57.92; H, 4.86; N, 9.65; found: C, 57.90; H, 4.85; N, 9.67.
2.2.6. N-(diethylcarbamothioyl)-2-oxo-2H-chromene-3-carboxamide (e3)
Yellow powder, 40% yield; mp. 143 °C; IR: 3300, 3060, 2978, 1709, 1609, 1567, 1437, 1219, 1198, 1016, 763, 641 cm−1; 1H NMR (CDCl3, 300 MHz) δ/ppm: 1.35 (5H, s, br), 1.64 (1H, s), 3.63 (2H, s, br), 4.01 (2H, s, br), 7.27–7.46 (2H, m), 7.70–7.75 (2H, m), 8.96 (1H, s), 10.97 (N=C–SH, 1H, s, SH); 13C NMR (CDCl3, 75 MHz) δ/ppm: 11.5, 47.7, 117.0, 118.0, 118.6, 123.4, 125.9, 130.4, 135.2, 150.5, 154.8, 158.5, 161.7, 177.7. LC-MS (m/z): 321.1 [M+]. Anal. Calcd. for C15H16N2O3S; C, 59.19; H, 5.30; N, 9.20; found: C, 59.17; H, 5.31; N, 9.22.
2.2.7. N-(diisopropylcarbamothioyl)-2-oxo-2H-chromene-3-carboxamide (e4)
Yellow powder, 70% yield; mp. 150–151 °C; IR: 3231, 3047, 2978, 1713, 1672, 1608, 1499, 1333, 1200, 1107, 757, 663 cm−1; 1H NMR (CDCl3, 300 MHz) δ/ppm: 1.23–1.58 (14H, m), 7.21–7.45 (2H, m), 7.71–7.75 (2H, m), 8.98 (1H, s), 10.68 (N=C-SH, 1H, s, SH); 13C NMR (CDCl3, 75 MHz) δ/ppm:, 21.0, 46.4, 51.8, 117.0, 118.1, 118.7, 125.0, 125.9, 130.4, 135.1, 150.5, 154.8, 161.7, 164.1. LC-MS (m/z): 333.3 [M+]. Anal. Calcd. for C17H20N2O3S; C, 61.42; H, 6.06; N, 8.43; found: C, 61.40; H, 6.07; N, 8.41.
2.2.8. N-(cyclohexylcarbamothioyl)-2-oxo-2H-chromene-3-carboxamide (e5)
Yellow powder, 53% yield; mp. 164–166 °C; IR: 3321, 3051, 2925, 1704, 1665, 1609, 1523, 1452, 1366, 1163, 761, 610 cm−1; 1H NMR (CDCl3, 300 MHz) δ/ppm: 1.24–1.49 (4H, s), 1.61–1.65 (2H, m), 1.73–1.78 (2H, m), 1.97–2.00 (2H, m), 3.94–4.00 (1H, m), 7.35–7.42 (2H, m), 7.63–7.71 (2H, m), 8.76 (1H, d, J = 6.7 Hz, NH), 8.9 (1H, s); 13C NMR (CDCl3, 75 MHz) δ/ppm:, 24.9, 25.8, 32.9, 48.7, 116.8, 118.9, 125.4, 129.9, 134.1, 148.3, 150.9, 154.6, 160.6, 161.7. LC-MS (m/z): 330.2 [M+]. Anal. Calcd. for C17H18N2O3S; C, 61.80; H, 5.49; N, 8.48; found: C, 61.81; H, 5.47; N, 8.49.
2.2.9. 2-oxo-N-(pyrrolidine-1-carbonothioyl)-2H-chromene-3-carboxamide (e6)
Yellow powder, 32% yield; mp. 162–163 °C; IR: 3321, 3051, 2925, 1704, 1665, 1609, 1523, 1452, 1366, 1163, 761, 610 cm−1; 1H NMR (CDCl3, 300 MHz) δ/ppm: 1.91–2.00 (4H, m), 3.46 (2H, t, J = 6.4 Hz), 3.64 (2H, t, J = 7.0 Hz), 7.27–7.38 (2H, m), 7.53–7.62 (2H, m), 7.97 (1H, s); 13C NMR (CDCl3, 75 MHz) δ/ppm:, 24.5, 26.2, 46.4, 47.7, 116.9, 118.5, 123.4, 125.1, 126.4, 128.8, 133.0, 143.3, 154.3, 158.0, 163.4. LC-MS (m/z): 303.3 [M+]. Anal. Calcd. for C15H14N2O3S; C, 59.59; H, 4.67; N, 9.27; found: C, 59.55; H, 4.68; N, 9.29.
2.2.10. 2-oxo-N-(piperidine-1-carbonothioyl)-2H-chromene-3-carboxamide (e7)
Yellow powder, 45% yield; mp. 175–176 °C; IR: 3040, 2918, 1710, 1607, 1559, 1438, 1251, 1121, 1041, 757, 610 cm−1; 1H NMR (CDCl3, 300 MHz) δ/ppm: 1.64 (6H, s, br), 3.34 (2H, s, br), 3.71 (2H, s, br), 7.29–7.36 (2H, m), 7.52–7.61 (2H, m), 7.87 (1H, s); 13C NMR (CDCl3, 75 MHz) δ/ppm:, 24.5, 25.6, 26.4, 43.2, 48.5, 116.9, 118.6, 125.0, 126.0, 128.6, 132.8, 142.4, 154.1, 158.3, 163.5. LC-MS (m/z): 317.3 [M+]. Anal. Calcd. for C16H16N2O3S; C, 60.74; H, 5.10; N, 8.85; found: C, 60.76; H, 5.12; N, 8.81.
2.2.11. N-(4-methylpiperazine-1-carbonothioyl)-2-oxo-2H-chromene-3-carboxamide (e8)
Yellow powder, 35% yield; mp.162–164 °C; IR: 3205, 3040, 2942, 1692, 1661, 1607, 1506, 1199, 1122, 792, 551 cm−1; 1H NMR (CDCl3, 300 MHz) δ/ppm: 2.36 (3H, s), 2.59 (4H, d, J = 8.2 Hz), 3.69 (2H, s, br), 4.27 (2H, s, br), 7.40–7.46 (2H, m), 7.71–7.77 (2H, m), 8.93 (1H, s), 11.05 (N=C-SH, 1H, s, SH); 13C NMR (CDCl3, 75 MHz) δ/ppm:, 36.3, 45.9, 51.8, 117.1, 117.5, 118.5, 125.9, 130.3, 135.4, 150.6, 155.0, 157.5, 161.3, 178.1. LC-MS (m/z): 354.2 [M+]. Anal. Calcd. for C16H17N3O3S; C, 57.99; H, 5.17; N, 12.68; found: C, 57.96; H, 5.19; N, 12.65.
2.2.12. N-(morpholine-4-carbonothioyl)-2-oxo-2H-chromene-3-carboxamide (e9)
Light yellow powder, 50% yield; mp. 123–125 °C; IR: 3035, 2991, 1714, 1607, 1571, 1428, 1240, 1107, 991, 747, 564 cm−1; 1H NMR (CDCl3, 300 MHz) δ/ppm: 3.41 (2H, t, J = 4.9 Hz), 3.72 (2H, t, J = 4.3 Hz), 3.79 (4H, s), 7.28–7.88 (2H, m), 7.54–7.64 (2H, m), 7.97 (1H, s); 13C NMR (CDCl3, 75 MHz) δ/ppm:, 42.7, 47.8, 66.8, 66.9, 117.0, 118.4, 124.9, 125.2, 128.8, 133.3, 144.0, 154.3, 158.2, 163.7. LC-MS (m/z): 319.3 [M+]. Anal. Calcd. for C15H14N2O4S; C, 56.59; H, 4.43; N, 8.80; found: C, 56.55; H, 4.42; N, 8.83.
2.2.13. N-((2,3-dihydro-1H-inden-2-yl)carbamothioyl)-2-oxo-2H-chromene-3- carboxamide (e10)
Yellow powder, 61% yield; mp. 181–183 °C; IR: 3300, 3048, 2953, 1703, 1655, 1606, 1524, 1363, 1201, 797, 742, 632 cm−1; 1H NMR (CDCl3, 300 MHz) δ/ppm: 2.97 (Ar-CH2, 2H, dd, J = 6.1, 16.1 Hz), 3.43 (Ar-CH2, 2H, dd, J = 7.6, 16.1 Hz), 4.86–4.94 (1H, m), 7.17–7.38 (4H, m), 7.41–7.46 (2H, m), 7.63–7.71 (2H, m), 8.92 (1H, s), 9.05 (1H, d, J= 7.0 Hz, NH); 13C NMR (CDCl3, 75 MHz) δ/ppm:, 40.1, 51.2, 116.8, 118.6, 118.8, 124.9, 125.0, 125.5, 127.0, 127.1, 130.0, 134.3, 141.0, 148.5, 154.6, 161.5, 161.6. LC-MS (m/z): 385.1 [M+]. Anal. Calcd. for C20H16N2O3S; C, 65.92; H, 4.43; N, 7.69; found: C, 65.90; H, 4.44; N, 7.68.
2.2.14. 2-oxo-N-((2-(pyrrolidin-1-yl)ethyl)carbamothioyl)-2H-chromene-3-carboxamide (e11)
Cream powder, 42% yield; mp. 131–132 °C; IR: 3326, 3044, 2928, 1698, 1665, 1609, 1542, 1425, 1241, 1147, 995, 760, 642 cm−1; 1H NMR (CDCl3, 300 MHz) δ/ppm: 1.80 (4H, s, br), 2.59 (4H, s, br), 2.73 (-NH-CH2CH2N-, 3H, t, J = 6.4 Hz), 3.60–3.64 (-NH-CH2CH2N-, 2H, q, J = 6.4 Hz), 7.27–7.41 (2H, m), 7.63–7.70 (2H, m), 8.90 (1H, s), 9.03 (1H, s, NH); 13C NMR (CDCl3, 75 MHz) δ/ppm:, 23.8, 39.2, 54.3, 54.8, 116.8, 118.8, 125.4, 129.9, 134.1, 148.3, 154.6, 161.5, 161.7. LC-MS (m/z): 385.1 [M+]. Anal. Calcd. for C17H19N3O3S; C, 59.11; H, 5.54; N, 12.17; found: C, 59.10; H, 5.53; N, 12.19.
2.2.15. N-((2-morpholinoethyl)carbamothioyl)-2-oxo-2H-chromene-3-carboxamide (e12)
Cream powder, 35% yield; mp. 136–138 °C; IR: 3324, 3039, 2916, 1695, 1605, 1542, 1444, 1200, 1113, 765, 644 cm−1; 1H NMR (CDCl3, 300 MHz) δ/ppm: 2.52–2.69 (6H, m), 3.56–3.62 (NHCH2-, 2H, m), 3.74–3.83 (-O(CH2)2, 4H, m), 7.36–7.47 (2H, m), 7.64–7.77 (2H, m), 8.91 (1H, s), 9.17 (1H, s, NH); 13C NMR (CDCl3, 75 MHz) δ/ppm: 36.8, 53.6, 56.9, 67.2, 116.8, 118.7, 118.8, 125.4, 130.0, 134.2, 148.3, 154.6, 161.5, 161.6. LC-MS (m/z): 362.3 [M+]. Anal. Calcd. for C17H19N3O4S; C, 56.50; H, 5.30; N, 11.63; found: C, 56.52; H, 5.32; N, 11.61.
2.2.16. N-((2-(cyclohex-1-en-1-yl)ethyl)carbamothioyl)-2-oxo-2H-chromene-3-carboxamide (e13)
Yellow powder, 51% yield; mp. 142–143 °C; IR: 3349, 3049, 2926, 1703, 1650, 1610, 1527, 1452, 1243, 1160, 981, 755, 638 cm−1; 1H NMR (CDCl3, 300 MHz) δ/ppm: 1.55–1.68 (4H, m), 1.98–2.18 (4H, m), 2.25 (-NHCH2CH2-, 2H, t, J = 6.7 Hz), 3.51–3.58 (-NHCH2CH2-, 2H, m), 5.56 (-C = CH, 1H, s), 7.35–7.44 (2H, m), 7.63–7.73 (2H, m), 8.82 (1H, s, NH), 8.91 (1H, s); 13C NMR (CDCl3, 75 MHz) δ/ppm: 22.5, 23.0, 25.4, 28.0, 37.5, 38.2, 116.8, 118.7, 118.8, 124.3, 125.4, 129.9, 134.1, 134.4, 148.3, 154.5, 161.4, 161.5. LC-MS (m/z): 355.3 [M+]. Anal. Calcd. for C19H20N2O3S; C, 64.02; H, 5.66; N, 7.86; found: C, 64.05; H, 5.61; N, 7.87.
2.2.17. N-((3,4-dimethoxyphenethyl)carbamothioyl)-2-oxo-2H-chromene-3-carboxamide (e14)
Yellow powder, 60% yield; mp. 148–149 °C; IR: 3342, 3051, 2934, 1703, 1655, 1607, 1513, 1451, 1234, 1157, 1029, 747, 641 cm−1; 1H NMR (CDCl3, 300 MHz) δ/ppm: 2.89 (-NHCH2CH2-, 2H, t, J = 7.3 Hz), 3.67–3.74 (-NHCH2CH2-, 2H, m), 3.86 (-OCH3, 3H, s), 3.90 (-OCH3, 3H, s), 6.79–6.82 (3H, m), 7.36–7.42 (2H, m), 7.64–7.71 (2H, m), 8.89 (1H, s, NH), 8.91 (1H, s); 13C NMR (CDCl3, 75 MHz) δ/ppm: 35.4, 41.7, 56.0, 111.4, 112.0, 116.8, 118.5, 118.8, 120.9, 125.5, 130.0, 131.5, 134.2, 147.8, 148.4, 149.1, 154.5, 161.5, 161.6. LC-MS (m/z): 411.3 [M+]. Anal. Calcd. for C21H20N2O5S; C, 61.15; H, 4.89; N, 6.79; found: C, 61.12; H, 4.90; N, 6.76.
2.2.18. N-((benzo[d][1,3]dioxol-5-ylmethyl)carbamothioyl)-2-oxo-2H-chromene-3-carboxamide (e15)
Yellow powder, 66% yield; mp. 188–190 °C; IR: 3352, 3051, 2904, 1706, 1659, 1608, 1498, 1441, 1281, 1237, 1038, 925, 758, 645 cm−1; 1H NMR (CDCl3, 300 MHz) δ/ppm: 4.56 (-NHCH2-, 2H, d, J = 5.8 Hz), 5.95 (-OCH2O-, 2H, s), 6.76–6.85 (3H, m), 7.36–7.43 (2H, m), 7.65–7.72 (2H, m), 8.95 (1H, s), 9.12 (1H, s, NH); 13C NMR (CDCl3, 75 MHz) δ/ppm: 43.9, 101.3, 108.5, 108.6, 116.8, 118.5, 118.8, 121.3, 123.4, 125.5, 130.0, 131.9, 134.3, 147.1, 148.1, 148.8, 154.6, 161.6. LC-MS (m/z): 381.2 [M+]. Anal. Calcd. for C19H14N2O5S; C, 59.68; H, 3.69; N, 7.33; found: C, 59.65; H, 3.66; N, 7.35.
2.2.19. N-(morpholinocarbamothioyl)-2-oxo-2H-chromene-3-carboxamide (e16)
Yellow powder, 25% yield; mp. 180–182 °C; IR: 3293, 3233, 3039, 2987, 1709, 1607, 1528, 1453, 1231, 1107, 865, 761 cm−1; 1H NMR (CDCl3, 300 MHz) δ/ppm: 2.99–3.08 (-N(CH2)2, 4H, m), 3.80–3.91 (-O(CH2)2, 4H, m), 7.38–7.48 (2H, m), 7.67–7.78 (2H, m), 8.96 (1H, s), 9.66 (-N=C-SH, 1H, s, SH); 13C NMR (CDCl3, 75 MHz) δ/ppm: 55.0, 56.0, 66.3, 66.5, 116.5, 118.3, 126.0, 130.0, 131.9, 134.5, 149.3, 154.5, 159.3, 161.1, 177.1. LC-MS (m/z): 332.2 [M+]. Anal. Calcd. for C15H15N3O4S; C, 54.04; H, 4.54; N, 12.60; found: C, 54.07; H, 4.52; N, 12.62.
2.2.20. N-((4-methylpiperazin-1-yl)carbamothioyl)-2-oxo-2H-chromene-3-carboxamide (e17)
Yellow powder, 38% yield; mp. 204 °C; IR:; IR: 3205, 3040, 2942, 1692, 1661, 1607, 1506, 1199, 1122, 792, 551 cm−1; 1H NMR (CDCl3, 300 MHz) δ/ppm: 2.37 (-N-CH3, 3H, s), 2.65 (4H, s, br), 3.10 (4H, s, br), 7.43–7.48 (2H, m), 7.74–7.81 (2H, m), 8.94 (1H, s), 11.38 (1H, s, NH), 11.66 (-N=C-SH, 1H, s, SH); 13C NMR (CDCl3, 75 MHz) δ/ppm: 46.0, 53.9, 54.3, 116.5, 117.3, 118.4, 126.0, 130.5, 136.0, 151.2, 155.1, 160.5, 161.0, 177.0. LC-MS (m/z): 347.3 [M+]. Anal. Calcd. for C16H18N4O3S; C, 55.48; H, 5.24; N, 16.17; found: C, 55.45; H, 5.22; N, 16.19.
2.2.21. 2-oxo-N-(piperidin-1-ylcarbamothioyl)-2H-chromene-3-carboxamide (e18)
Orange powder, 50% yield; mp. 206 °C; IR:; IR: 3133, 3047, 2945, 1694, 1607, 1480, 1228, 1190, 1034, 758, 641 cm−1; 1H NMR (CDCl3, 300 MHz) δ/ppm: 1.50–1.52 (2H, m), 1.74–1.81 (4H, m), 2.99 (4H, s, br), 7.41–7.47 (2H, m), 7.72–7.78 (2H, m), 8.91 (1H, s), 11.32 (1H, s, NH), 11.61 (-N=C-SH, 1H, s, SH); 13C NMR (CDCl3, 75 MHz) δ/ppm: 23.4, 25.6, 55.1, 55.8, 116.8, 119.0, 119.6, 126.2, 131.5, 135.5, 150.4, 154.8, 159.8, 161.1, 176.4. LC-MS (m/z): 330.3 [M+]. Anal. Calcd. for C16H17N3O3S; C, 57.99; H, 5.17; N, 12.68; found: C, 57.97; H, 5.19; N, 12.65.
2.2.22. 2-oxo-N-((2-(piperazin-1-yl)ethyl)carbamothioyl)-2H-chromene-3-carboxamide (e19)
Yellow powder, 32% yield; mp.163–165 °C; IR:; IR: 3205, 3036, 2928, 1695, 1607, 1449, 1225, 1174, 1034, 790, 632 cm−1; 1H NMR (CDCl3, 300 MHz) δ/ppm: 2.58–2.78 (6H, m), 3.46 (2H, t, J = 4.6 Hz), 3.57–3.62 (2H, q, J = 5.8 Hz), 3.85 (2H, s, br), 7.24–7.42 (2H, m), 7.54–7.71 (2H, m), 7.91 (1H, s), 8.90 (1H, s, NH), 9.18 (1H, s, NH); 13C NMR (CDCl3, 75 MHz) δ/ppm: 37.0, 42.4, 47.5, 52.4, 53.0, 56.3, 116.8, 118.5, 125.1, 128.7, 130.0, 134.2, 143.2, 148.4, 154.3, 161.5, 163.5. LC-MS (m/z): 361.3 [M+]. Anal. Calcd. for C17H20N4O3S; C, 56.65; H, 5.59; N, 15.54; found: C, 56.63; H, 5.57; N, 15.55.
2.2.23. N-((3-(dimethylamino)propyl)carbamothioyl)-2-oxo-2H-chromene-3-carboxamide (e20)
Yellow powder, 42% yield; mp. 100 °C; IR:; IR: 3341, 3054, 2974, 1704, 1657, 1612, 1451, 1244, 1080, 966, 757, 637 cm−1; 1H NMR (CDCl3, 300 MHz) δ/ppm: 1.77–1.84 (2H, m), 2.25 (-N(CH3)2, 6H, s), 2.34–2.41 (2H, m), 3.50–3.56 (2H, m), 7.35–7.41 (2H, m), 7.63–7.71 (2H, m), 8.90 (1H, s), 9.08 (1H, s, NH); 13C NMR (CDCl3, 75 MHz) δ/ppm: 27.3, 38.7, 45.6, 57.6, 116.8, 116.8, 118.8X2, 125.4, 129.9, 134.1, 148.3, 154.6, 161.5, 161.6. LC-MS (m/z): 335.3 [M+]. Anal. Calcd. for C16H19N3O3S; C, 57.64; H, 5.74; N, 12.60; found: C, 57.61; H, 5.76; N, 12.61.
2.3. CA inhibition assays
An SX.18 MV-R Applied Photophysics (Oxford, UK) stopped-flow instrument has been used to assay the catalytic/inhibition of various CA isozymes51. Phenol Red (at a concentration of 0.2 mM) has been used as indicator, working at the absorbance maximum of 557 nm, with 10 mM Hepes (pH 7.4) as buffer, 0.1 M Na2SO4 or NaClO4 (for maintaining constant the ionic strength; these anions are not inhibitory in the used concentration)26, following the CA-catalysed CO2 hydration reaction for a period of 5–10 s. Saturated CO2 solutions in water at 25 °C were used as substrate. Stock solutions of inhibitors were prepared at a concentration of 10 mM (in DMSO-water 1:1, v/v) and dilutions up to 1 nM done with the assay buffer mentioned above. At least seven different inhibitor concentrations have been used for measuring the inhibition constant. Inhibitor and enzyme solutions were pre-incubated together for 6 h at 4 °C prior to assay, in order to allow for the formation of the E-I complex. Triplicate experiments were done for each inhibitor concentration, and the values reported throughout the paper are the mean of such results. The inhibition constants were obtained by non-linear least-squares methods using the Cheng–Prusoff equation, as reported earlier39 and represent the mean from at least three different determinations. All CA isozymes used here were recombinant proteins obtained as reported earlier by our group27,32,40,52.
2.4. Molecular modeling
Molecular modeling approaches such as molecular docking simulations were performed in terms of examination and comprehension of the details in the inhibitory profiles of these molecules. Binding poses of studied compounds at the binding pockets of the proteins were determined via molecular docking processes. The 3D crystal structures of the hCA I, II, VII and IX were obtained from Protein Data Bank with the corresponding IDs of 2FW4, 5AML, 3MDZ and 3IAI, respectively. Ligand molecules were two dimensionally sketched in Maestro package of Schrodinger Small-Molecule Drug Discovery Suite53 and were prepared via LigPrep54 module of Maestro to establish the conformations with the lowest energy in physiological pH 7.4. The three-dimensional structures of the proteins are also prepared for docking via Protein Preparation Wizard module of Maestro. Grid map generation and flexible molecular docking simulations of ligands to these four proteins were implemented using Glide module55 and Glide/HTVS (high-throughput virtual screening), Glide/SP (standard precision), Glide/XP (extra precision), QPLD (Quantum Mechanics-Polarised Ligand Docking) and IFD (Induced Fit Docking) protocols of Maestro as well as CCDC GOLD56 Docking program. As the charge polarisation that induced by the active site of the protein environment is considered, quantum mechanics (QM) modeling may give the highest level of docking accuracy. For these reasons, QPLD is also considered which uses ab initio charge calculations. Initially, Glide/SP docking was carried out to generate five poses per docked compound. These poses were submitted to QM charge calculations, which uses the 6–31 G*/LACVP* basis set, B3LYP density functional, and “Ultrafine” SCF accuracy level. In GOLD algorithm, consensus docking protocol was used to generate protein–ligand complexes with GOLD 5.3.0 software. In this respect, two docking scoring functions were combined: GoldScore and ChemScore. In this study, default genetic algorithm parameters were used and 20 poses were generated for each ligand. Search efficiency was set to its maximum value (200%) in order to increase the reliability of the docking results. Flexible amino acid side chains/rotatable groups involved in binding pocket were selected separately for all isoforms according to their protein–ligand interaction maps available in PDB. Ligand molecules were also set as flexible during all molecular docking calculations.
3. Result and discussion
3.1. Chemistry
The syntheses of the target compounds e1–e20 are depicted in Scheme 1. 3-Coumarin carboxylic acid (c) was synthesized from salicylaldehyde (a) according to literature procedures38 and it was converted to the acyl chloride by using SOCl2. To obtain thiourea-substituted coumaryl-carboxamid derivatives (e1–e20), 2-oxo-2H-chromene-3-carbonyl chloride (d) was reacted with KSCN and various amines in CH3CN, respectively.
Scheme 1.

Synthesis of new thiourea substituted coumaryl-carboxamid derivatives. Reaction conditions: (i) H2O, reflux, 10 h; (ii) SOCl2, 80 °C, 4 h; (iii) KSCN, CH3CN, 70 °C, 30 min.; (iv) RNH2, 70 °C, 4 h.
All the new compounds were characterized by 1H NMR, 13C NMR, IR, MS and elemental analysis. In the IR spectra of the synthesized compounds, it was possible to observe the absorptions about 3300 cm−1 relating to NH stretch of thiourea groups, about 1650 cm−1 relating to C=O stretch for thiourea, absorptions in about 1710 cm−1 from coumarin carbonyl moiety stretch. From the 1H NMR spectra, the signals for aromatic hydrogens were observed between 7.17 and 7.77 ppm, the signal of NH proton at thiourea was detected at about 8.90 ppm and signals observed about 11.3 ppm for SH proton at the resonance due to thiourea groups (N=C–SH). In addition, the signals of aliphatic hydrogen atoms were determined between 1.00–4.50 ppm. From the 13C NMR spectra, the signals can be seen about 177 and 163 ppm for C–SH and carbonyl of thiourea groups, respectively. The signals of the aliphatic and aromatic carbons were observed at 20–50 ppm and 110–158 ppm, respectively.
1H NMR, 13C NMR and MS spectra of the synthesized compounds are given in supplementary materials.
3.2. CA inhibition
The inhibition constants (Ki) of the synthesized compounds e1–e20 against hCA I, hCA II, hCA VII and hCA IX isoforms are given in Table 1. The hCA I, II and VII isoforms for all compounds were investigated here in the micromolar range. On the other hand, the tumour-associated isoform hCA IX was selectively inhibited by all investigated compounds with inhibition constants ranging between 107.9 and 2589.4 nM. Compound e11 showed the strongest inhibition against hCA IX with a Ki of 107.9 nM. Furthermore, the hCA IX inhibitory activity of e5, e8 and e10 are close to that of e11 (Ki=115.1 nM, 128.1 nM and 130.3 nM, respectively).
Table 1.
Carbonic anhydrase inhibitions of synthesized thiourea substituted coumaryl-carboxamid derivatives.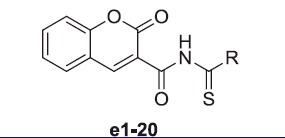
|
Ki (nM)* |
|||||
|---|---|---|---|---|---|
| Compound | R | hCA I | hCA II | hCA VII | hCA IX |
| e1 |  |
>10,000 | >10,000 | >10,000 | 322.9 |
| e2 |  |
>10,000 | >10,000 | >10,000 | 286.4 |
| e3 |  |
>10,000 | >10,000 | >10,000 | 376.2 |
| e4 |  |
>10,000 | >10,000 | >10,000 | 351.4 |
| e5 |  |
>10,000 | >10,000 | >10,000 | 115.1 |
| e6 | 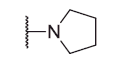 |
>10,000 | >10,000 | >10,000 | 297.5 |
| e7 |  |
>10,000 | >10,000 | >10,000 | 201.8 |
| e8 | 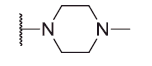 |
>10,000 | >10,000 | >10,000 | 128.1 |
| e9 | 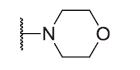 |
>10,000 | >10,000 | >10,000 | 136.5 |
| e10 | 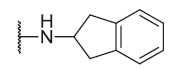 |
>10,000 | >10,000 | >10,000 | 130.3 |
| e11 | 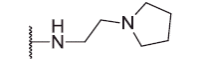 |
>10,000 | >10,000 | >10,000 | 107.9 |
| e12 |  |
>10,000 | >10,000 | >10,000 | 223.8 |
| e13 | 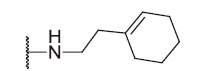 |
>10,000 | >10,000 | >10,000 | 179.8 |
| e14 |  |
>10,000 | >10,000 | >10,000 | 196.4 |
| e15 |  |
>10,000 | >10,000 | >10,000 | 184.5 |
| e16 | 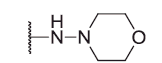 |
>10,000 | >10,000 | >10,000 | 2589.4 |
| e17 | 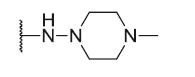 |
>10,000 | >10,000 | >10,000 | 258.9 |
| e18 |  |
>10,000 | >10,000 | >10,000 | 387.5 |
| e19 |  |
>10,000 | >10,000 | >10,000 | 249.6 |
| e20 |  |
>10,000 | >10,000 | >10,000 | 182.2 |
| AAZ | 250 | 12.1 | 6 | 25.8 | |
Mean from three different assays, by a stopped flow technique (errors were in the range of ±5–10% of the reported values).
The following structure–activity relationship (SAR) observations can be drawn from data of Table 1: (i) Replacing the methyl group on the NH of thiourea moiety (e1, Ki=322.9 nM) by a propyl (e2, Ki=286.4 nM), a cyclohexyl (e5, Ki=115.1 nM) and a 2,3-dihydro-indenyl ring (e10, Ki=130.3 nM) led to an increase in the inhibitory activity against hCA IX; on the other hand, the binding of a second alkyl group to -N atom (N,N-diethyl (e3, Ki = 376.2 nM) and N,N-diisopropyl (e4, Ki = 351.4 nM)) diminished the inhibitory activity against hCA IX. (ii) The expansion of the pyrrolidine ring of compound e6 (Ki = 297.5 nM against hCA IX) to a piperidine (compound e7, Ki = 201.8 nM against hCA IX) increased the inhibitory activity against hCA IX. Additionally, incorporated N and O atoms into the piperidine ring (R = 4-methylpiperazine (e8) and R = morpholine (e9), Ki = 128.1 nM and 136.5 nM, respectively, against hCA IX) caused a greater increase in the inhibitory activity against hCA IX. (iii) The presence of an ethyleneamine group as a spacer between the thionyl moiety and the pyrrolidine ring positively affected the inhibitory activity against hCA IX (comparing e6 (Ki = 297.5 nM) with e11 (Ki = 107.9 nM)) and the presence of a propyleneamine group between the thionyl and the N,N-dialkyl moieties did likewise (e20, Ki = 182.2 nM). On the contrary, the ethyleneamine group between the thionyl moiety and both the piperazine and morpholine rings decreased the inhibitory activity against hCA IX (comparing e8 (Ki = 128.1 nM) with e19 (Ki = 249.6 nm) and comparing e9 (Ki = 136.5 nm) with e12 (Ki = 223.8 nM)). (iv) Similarly, the presence of an amine group (-NH-) between the thionyl moiety and the piperidine, piperazine or morpholine ring led to a major decline the inhibitory activity against hCA IX (comparing e7 (Ki = 201.8 nM) with e18 (Ki = 387.5 nM), comparing e8 (Ki = 128.1 nM) with e17 (Ki = 258.9 nM) and comparing e9 (Ki = 136.5 nM) with e16 (Ki = 2589.4 nM)). (v) The replacement of the ethyleneamine group by a methyleneamine between the thionyl moiety and the aromatic ring and the cyclisation of the dimethoxy group at the phenyl ring to the dioxolane ring did not cause significant changes in the hCA IX inhibitory activity (comparing e14 (Ki = 196.4 nM) with e15 (Ki = 184.5 nM)).
According to X-ray crystallographic studies, coumarins are mechanism-based inhibitors, which undergo hydrolysis under the influence of the zinc hydroxide, nucleophilically active species of the enzyme, with the generation of substituted-2-hydroxycinnamic acids (Figure 1)26,39–41. It was reported that coumarin/sulphocoumarin inhibitors and enzyme solutions were pre-incubated together for ∼6 h prior to assay in order to allow for the formation of the E-I complex or for the eventual active site-mediated hydrolysis of the inhibitor42. Based on the above consideration, we estimate that the coumarin ring should undergo ring opening by hydrolysing coumarinic moiety to cinnamic acid derivative during pre-incubation on enzyme and inhibitor (Figure 1).
Figure 1.

Formation of 2-hydroxy-cinnamic acids A1, B1 and e11-h by the CA-mediated hydrolysis of coumarin A, B and e11.
3.3. Molecular modeling
Molecular modeling approaches, such as molecular docking calculations, are generally used techniques to qualify and quantify the important information about the ligand–receptor interaction analysis on atomistic level. There are many studies using these approaches integrated to human carbonic anhydrases (hCA) in order to clarify the molecular mechanism of action and bioactive conformation of proposed compounds at the binding site of the protein43–49. Since there is no experimental study yet on stable state (hydrolyzed/nonhydrolyzed) of studied compounds from lactone moiety at the binding pocket of the CAs, both hydrolyzed and nonhydrolyzed forms were considered at the docking. Molecular docking results were evaluated for their docking scores in the binding pocket of the hCA I, II, VII and IX isoforms. Therefore, the protein–ligand complexes with the top-docking scores were selected for further analyses. Docking results of compounds (e1–e20) at the binding pockets of hCA isoforms with Glide/HTVS (high-throughput virtual screening), Glide/SP (standard precision), Glide/XP (extra precision), IFD (induced fit docking), QPLD (quantum polarised ligand docking) and GOLD protocols were compared. Since GOLD docking program gave more successful results GOLD docking results were considered in further analysis (Tables 2 and 3). Docking scores of studied compounds (with hydrolyzed and nonhydrolyzed forms) at the hCA I, II, VII and IX isoforms were compared at the Figures 2 and 3.
Table 2.
Top-docking scores of compounds e1–e20 (non-hydrolyzed forms) at the hCAI, hCAII, hCA VII and hCA IX isoforms.
| Compounds | hCA I (2FW4) | hCA II (5AML) | hCA VII (3MDZ) | hCA IX (3IAI) |
|---|---|---|---|---|
| e1 | −3.128 | −1.163 | −5.049 | −4.605 |
| e2 | −4.747 | −2.292 | −4.400 | −8.142 |
| e3 | −2.534 | −1.996 | −5.177 | −5.259 |
| e4 | −7.622 | −2.049 | −6.114 | −4.788 |
| e5 | −5.600 | −3.613 | −4.832 | −4.903 |
| e6 | −3.339 | −5.502 | −5.150 | −5.796 |
| e7 | −5.885 | −1.811 | −5.383 | −4.591 |
| e8 | −3.745 | −4.148 | −5.370 | −6.723 |
| e9 | −3.159 | −4.798 | −6.899 | −6.678 |
| e10 | −5.625 | −5.580 | −6.330 | −5.345 |
| e11 | −4.137 | −4.298 | −4.604 | −8.429 |
| e12 | −3.805 | −4.322 | −6.770 | −4.996 |
| e13 | −4.907 | −1.443 | −5.040 | −5.491 |
| e14 | −1.843 | −2.858 | −5.397 | −5.027 |
| e15 | −3.328 | −2.898 | −4.689 | −6.798 |
| e16 | −4.407 | −3.338 | −5.441 | −6.687 |
| e17 | −4.172 | −1.909 | −4.497 | −4.167 |
| e18 | −4.258 | −2.075 | −4.446 | −8.750 |
| e19 | −2.744 | −1.885 | −4.158 | −5.103 |
| e20 | −1.473 | 0.138 | −4.866 | −4.226 |
Used protein data bank (PDB) IDs of proteins were also highlighted at the table. Docking scores are in kcal/mol.
Table 3.
Top-docking scores of compounds e1–e20 (hydrolyzed forms) at the hCAI, hCAII, hCA VII and hCA IX isoforms.
| Compounds | hCAI (2FW4) | hCAII (5AML) | hCAVII (3MDZ) | hCAIX (3IAI) |
|---|---|---|---|---|
| e1 | −4.422 | −5.837 | −5.658 | −7.025 |
| e2 | −4.980 | −6.555 | −5.468 | −6.878 |
| e3 | −4.884 | −4.292 | −6.166 | −7.064 |
| e4 | −5.270 | −7.194 | −4.855 | −1.879 |
| e5 | −5.617 | −5.991 | −6.191 | −8.287 |
| e6 | −4.802 | −6.698 | −6.690 | −7.655 |
| e7 | −4.715 | −6.375 | −5.463 | −6.833 |
| e8 | −4.441 | −3.632 | −5.703 | −4.146 |
| e9 | −5.343 | −6.699 | −6.724 | −7.310 |
| e10 | −4.726 | −7.553 | −7.104 | −8.819 |
| e11 | −4.587 | −5.816 | −5.597 | −9.006 |
| e12 | −5.377 | −5.121 | −7.164 | −8.769 |
| e13 | −5.366 | −6.667 | −5.690 | −9.481 |
| e14 | −5.554 | −5.967 | −6.518 | −9.238 |
| e15 | −4.51 | −5.879 | −6.036 | −9.445 |
| e16 | −4.929 | −6.322 | −6.652 | −4.213 |
| e17 | −4.642 | −5.634 | −6.219 | −4.123 |
| e18 | −5.149 | −6.855 | −3.487 | −2.713 |
| e19 | −5.82 | −6.421 | −7.123 | −9.301 |
| e20 | −4.507 | −5.738 | −6.672 | −9.096 |
Used protein data bank (PDB) IDs of proteins were also highlighted at the table. Docking scores are in kcal/mol.
Figure 2.

Docking scores (GOLD ChemScore dG) of studied compounds (nonhydrolyzed forms) at the hCA I, II, VII and IX isoforms.
Figure 3.

Docking scores (GOLD ChemScore dG) of studied compounds (hydrolyzed forms) at the hCA I, II, VII and IX isoforms.
The most active compound e11 at the hCA IX showed high docking scores compared to its predicted binding energies at hCA I, II and VII isoforms in both hydrolyzed and non-hydrolyzed forms. In addition, the compounds e2 and e18 in their non-hydrolyzed forms were found to have high selectivity to hCA IX. Hydrolyzed forms of compounds showed higher docking scores against hCA IX (i.e. e10–e15, e19 and e20). Figure 4 shows 2D and 3D ligand interaction diagrams of active compound e11 at the binding cavity of hCA IX.
Figure 4.

2D and 3D ligand interaction diagrams of active compound e11 at the binding cavity of hCA IX. (top) hydrolyzed form; (bottom) nonhydrolyzed form.
4. Conclusions
A series of 20 novel thiourea-substituted coumaryl-carboxamide derivatives (e1–20) were synthesized as CA inhibitors and they were evaluated for the inhibition of hCA I, II, IV and IX isoforms. All synthesized compounds exhibited selective inhibitory activity in the high nanomolar range against the tumour-associated isoform hCA IX. On the other hand, the hCA I, II and VII isoforms of were not inhibited by the investigated compounds. Multiscale molecular modeling approaches and different molecular docking algorithms were used to investigate inhibitory profiles, binding poses and predicted binding energies of studied compounds (both hydrolyzed and non-hydrolyzed forms) at the active sites of the CA I, II, VII and IX isoforms. The docking studies showed that hydrolyzed form of e11, which is the most active compound against hCA IX, interacted with His64, His94, His96 and Gln92 and non-hydrolyzed form of e11 interacted with His64, His94 and His119 in the active side of hCA IX.
1H and 13C NMR and MS spectra of the synthesized compounds are given in the Supplementary Materials.
Supplementary Material
Funding Statement
This work was supported by the Sakarya Research Fund of the Sakarya University, 10.13039/501100004473. Project Number: 2016–28-00–003.
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- 1.Pocker Y, Sarkanen S, Carbonic anhydrase: structure, catalytic versatility, and inhibition, in advances in enzymology and related areas of molecular biology. In: Meister A, ed. Hoboken, NJ: John Wiley & Sons, Inc; 1979;47:149–274. [DOI] [PubMed] [Google Scholar]
- 2.Carter MJ.Carbonic anhydrase; isoenzymes, properties, distribution, and functional significance. Biol Rev 1972;42:465–513. [DOI] [PubMed] [Google Scholar]
- 3.Supuran CT.Carbonic anhydrases: novel therapeutic applications for inhibitors and activators. Nat Rev Drug Discov 2008;7:168–81. [DOI] [PubMed] [Google Scholar]
- 4.Supuran CT.Carbonic anhydrase inhibitors and activators for novel therapeutic applications. Future Med Chem 2011;3:1165–80. [DOI] [PubMed] [Google Scholar]
- 5.Cornelio B, Laronze-Cochard M, Ceruso M, et al. Arylbenzenesulfonamides as human carbonic anhydrase inhibitors (hCAIs): synthesis by Pd nanocatalyst-mediated Suzuki − Miyaura reaction, enzyme inhibition, and X-ray crystallographic studies. J Med Chem 2016;59:721–32. [DOI] [PubMed] [Google Scholar]
- 6.Karioti A, Ceruso M, Carta F, et al. New natural product carbonic anhydrase inhibitors incorporating phenol moieties. Bioorg Med Chem 2015;23:7219–25. [DOI] [PubMed] [Google Scholar]
- 7.Özgeris B, Göksu S, Köse LP, et al. Acetylcholinesterase and carbonic anhydrase inhibitory properties of novel urea and sulfamide derivatives incorporating dopaminergic 2-aminotetralin scaffolds. Bioorg Med Chem 2016;24:2318–29. [DOI] [PubMed] [Google Scholar]
- 8.Villalba ML, Palestro P, Ceruso M, et al. Sulfamide derivatives with selective carbonic anhydrase VII inhibitory action. Bioorg Med Chem 2016;24:894–901. [DOI] [PubMed] [Google Scholar]
- 9.Fossati S, Giannoni P, Solesio ME, et al. The carbonic anhydrase inhibitor methazolamide prevents amyloid beta-induced mitochondrial dysfunction and caspase activation protecting neuronal and glial cells in vitro and in the mouse brain. Neurobiol Dis 2016;86:29–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Leppilampi M, Koistinen P, Savolainen ER, et al. The expression of carbonic anhydrase II in hematological malignancies. Clin Cancer Res 2002;8:2240–5. [PubMed] [Google Scholar]
- 11.Carta F, Osman SM, Vullo D, et al. Dendrimers incorporating benzenesulfonamide moieties strongly inhibit carbonic anhydrase isoforms I–XIV. Org Biomol Chem 2015;13:6453–7. [DOI] [PubMed] [Google Scholar]
- 12.El-Azab AS, Abdel-Aziz AAM, Ayyad RR, et al. Inhibition of carbonic anhydrase isoforms I, II, IV, VII and XII with carboxylates and sulfonamides incorporating phthalimide/phthalic anhydride scaffolds. Bioorg Med Chem 2016;24:20–5. [DOI] [PubMed] [Google Scholar]
- 13.Ombouma J, Vullo D, Dumy P, et al. Carbonic Anhydrase Glycoinhibitors belonging to the Aminoxysulfonamide Series. ACS Med Chem Lett 2015;6:819–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Barresi E, Salerno S, Marini AM, et al. Sulfonamides incorporating heteropolycyclic scaffolds show potent inhibitory action against carbonic anhydrase isoforms I, II, IX and XII. Bioorg Med Chem 2016;24:921–7. [DOI] [PubMed] [Google Scholar]
- 15.Bozdag M, Pinard M, Carta F, et al. A class of 4-sulfamoylphenyl-ω-aminoalkyl ethers with effective carbonic anhydrase inhibitory action and antiglaucoma effects. J Med Chem 2014;57:9673–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.De Simone G, Supuran CT.. Carbonic anhydrase IX: biochemical and crystallographic characterization of a novel antitumor target. Biochim Biophys Acta 2010;1804:404–9. [DOI] [PubMed] [Google Scholar]
- 17.Nishimori I, Vullo D, Minakuchi T, et al. Restoring catalytic activity to the human carbonic anhydrase (CA) related proteins VIII, X and XI affords isoforms with high catalytic efficiency and susceptibility to anion inhibition. Bioorg Med Chem Lett 2013;23:256–60. [DOI] [PubMed] [Google Scholar]
- 18.Said HM, Supuran CT, Hageman C, et al. Modulation of carbonic anhydrase 9 (CA9) in human brain cancer. Curr Pharm Des 2010;16:3288–99. [DOI] [PubMed] [Google Scholar]
- 19.Guler OO, De Simone G, Supuran CT.. Drug design studies of the novel antitumor targets carbonic anhydrase IX and XII. Curr Med Chem 2010;17:1516–26. [DOI] [PubMed] [Google Scholar]
- 20.Akurathi V, Dubois L, Lieuwes NG, et al. Synthesis and biological evaluation of a 99mTc-labelled sulfonamide conjugate for in vivo visualization of carbonic anhydrase IX expression in tumor hypoxia. Nucl Med Biol 2010;37:557–64. [DOI] [PubMed] [Google Scholar]
- 21.Genis C, Sippel KH, Case N, et al. Design of a carbonic anhydrase IX active-site mimic to screen inhibitors for possible anticancer properties. Biochemistry 2009;48:1322–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Thiry A, Supuran CT, Masereel B, Dogne JM.. Recent developments of carbonic anhydrase inhibitors as potential anticancer drugs. J Med Chem 2008;51:3051–6. [DOI] [PubMed] [Google Scholar]
- 23.Meijer TW, Bussink J, Zatovicova M, et al. Tumor microenvironmental changes induced by the sulfamate carbonic anhydrase IX inhibitor S4 in a laryngeal tumor model. PLoS One 2014;9:e108068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Krall N, Pretto F, Decurtins W, et al. A small molecule drug conjugate for the treatment of carbonic anhydrase IX expressing tumors. Angew Chem Int Ed Engl 2014;53:4231–5. [DOI] [PubMed] [Google Scholar]
- 25.Pastorek J, Pastorekova S.. Hypoxia-induced carbonic anhydrase IX as a target for cancer therapy: from biology to clinical use. Semin Cancer Biol 2015;31:52–64. [DOI] [PubMed] [Google Scholar]
- 26.Maresca A, Temperini C, Pochet L, et al. Deciphering the mechanism of carbonic anhydrase inhibition with coumarins and thiocoumarins. J Med Chem 2010;53:335–44. [DOI] [PubMed] [Google Scholar]
- 27.Touisni N, Maresca A, McDonald PC, et al. Glycosyl coumarin carbonic anhydrase IX and XII inhibitors strongly attenuate the growth of primary breast tumors. J Med Chem 2011;54:8271–7. [DOI] [PubMed] [Google Scholar]
- 28.Nocentini A, Carta F, Ceruso M, et al. Click-tailed coumarins with potent and selective inhibitory action against the tumor-associated carbonic anhydrases IX and XII. Bioorg Med Chem 2015;23:6955–66. [DOI] [PubMed] [Google Scholar]
- 29.Bozdag M, Ferraroni M, Carta F, et al. Structural insights on carbonic anhydrase inhibitory action, isoform selectivity, and potency of sulfonamides and coumarins incorporating arylsulfonylureido groups. J Med Chem 2014;57:9152–67. [DOI] [PubMed] [Google Scholar]
- 30.Grandane A, Tanc M, Di Cesare Mannelli L, et al. 6-Substituted sulfocoumarins are selective carbonic anhdydrase IX and XII inhibitors with significant cytotoxicity against colorectal cancer cells. J Med Chem 2015;58:3975–83. [DOI] [PubMed] [Google Scholar]
- 31.Tanc M, Carta F, Bozdag M, et al. 7-Substituted-sulfocoumarins are isoform-selective, potent carbonic anhydrase II inhibitors. Bioorg Med Chem 2013;21:4502–10. [DOI] [PubMed] [Google Scholar]
- 32.Ferraroni M, Carta F, Scozzafava A, Supuran CT.. Thioxocoumarins show an alternative carbonic anhydrase inhibition mechanism compared to coumarins. J Med Chem 2016;59:462–73. [DOI] [PubMed] [Google Scholar]
- 33.Imran S, Taha M, Ismail NH, et al. Synthesis, biological evaluation, and docking studies of novel thiourea derivatives of bisindolylmethane as carbonic anhydrase II inhibitor. Bioorg Chem 2015;62:83–93. [DOI] [PubMed] [Google Scholar]
- 34.Zaib S, Saeed A, Stolte K, et al. New aminobenzenesulfonamide-thiourea conjugates: synthesis and carbonic anhydrase inhibition and docking studies. Eur J Med Chem 2014;78:140–50. [DOI] [PubMed] [Google Scholar]
- 35.Supuran CT, Scozzafava A.. Carbonic anhydrase inhibitors - Part 94. 1,3,4-Thiadiazole-2-sulfonamide derivatives as antitumor agents. Eur J Med Chem 2000;35:867–74. [DOI] [PubMed] [Google Scholar]
- 36.Scozzafava A, Supuran CT.. Carbonic anhydrase inhibitors. Arylsulfonylureido and arylureido-substituted aromatic and heterocyclic sulfonamides: towards selective inhibitors of carbonic anhydrase isozyme I. J Enzyme Inhib 1999;14:343–63. [DOI] [PubMed] [Google Scholar]
- 37.Pacchiano F, Carta F, McDonald PC, et al. Ureido-substituted benzenesulfonamides potently inhibit carbonic anhydrase IX and show antimetastatic activity in a model of breast cancer metastasis. J Med Chem 2011;54:1896–902. [DOI] [PubMed] [Google Scholar]
- 38.Yang Y, Liu QW, Shi Y, et al. Design and synthesis of coumarin-3-acylamino derivatives to scavenge radicals and to protect DNA. Eur J Med Chem 2014;84:1–7. [DOI] [PubMed] [Google Scholar]
- 39.Maresca A, Temperini C, Vu H, et al. Non-zinc mediated inhibition of carbonic anhydrases: coumarins are a new class of suicide inhibitors. J Am Chem Soc 2009;131:3057–62. [DOI] [PubMed] [Google Scholar]
- 40.Maresca A, Scozzafava A, Supuran CT.. 7,8-Disubstituted- but not 6,7-disubstituted coumarins selectively inhibit the transmembrane, tumor-associated carbonic anhydrase isoforms IX and XII over the cytosolic ones I and II in the low nanomolar/subnanomolar range. Bioorg Med Chem Lett 2010;20:7255–8. [DOI] [PubMed] [Google Scholar]
- 41.Wagner J, Avvaru BS, Robbins AH, et al. Coumarinyl-substituted sulfonamides strongly inhibit several human carbonic anhydrase isoforms: solution and crystallographic investigations. Bioorg Med Chem 2010;18:4873–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tars K, Vullo D, Kazaks A, et al. Sulfocoumarins (1,2-Benzoxathiine-2,2-dioxides): A class of potent and isoform-selective inhibitors of tumor-associated carbonic anhydrases. J Med Chem 2013;56:293–300. [DOI] [PubMed] [Google Scholar]
- 43.Akincioglu A, Akincioglu H, Gulcin I, et al. Discovery of potent carbonic anhydrase and acetylcholine esterase inhibitors: novel sulfamoylcarbamates and sulfamides derived from acetophenones. Bioorg Med Chem 2015;23:3592–602. [DOI] [PubMed] [Google Scholar]
- 44.Durdagi S, Scozzafava G, Vullo D, et al. Inhibition of mammalian carbonic anhydrases I-XIV with grayanotoxin III: solution and in silico studies. J Enzyme Inhib Med Chem 2014;29:469–75. [DOI] [PubMed] [Google Scholar]
- 45.Salmas RE, Mestanoglu M, Durdagi S, et al. Kinetic and in silico studies of hydroxy-based inhibitors of carbonic anhydrase isoforms I and II. J Enzyme Inhib Med Chem 2016;31:31–7. [DOI] [PubMed] [Google Scholar]
- 46.Kocak R, Akın ET, Kalin P, et al. Synthesis of some novel norbornene‐fused pyridazines as potent inhibitors of carbonic anhydrase and acetylcholinesterase. J Heterocyclic Chem 2015;53:2049–56. [Google Scholar]
- 47.Salmas RE, Senturk M, Yurtsever M, Durdagi S.. Discovering novel carbonic anhydrase type IX (CA IX) inhibitors from seven million compounds using virtual screening and in vitro analysis. J Enzyme Inhib Med Chem 2016;31:425–33. [DOI] [PubMed] [Google Scholar]
- 48.Isik S, Vullo D, Durdagi S, et al. Interaction of carbonic anhydrase isozymes I, II, and IX with some pyridine and phenol hydrazinecarbothioamide derivatives. Bioorg Med Chem Lett 2015;25:5636–41. [DOI] [PubMed] [Google Scholar]
- 49.Fidan I, Salmas RE, Arslan M, et al. Carbonic anhydrase inhibitors: design, synthesis, kinetic, docking and molecular dynamics analysis of novel glycine and phenylalanine sulfonamide derivatives. Bioorg Med Chem 2015;23:7353–8. [DOI] [PubMed] [Google Scholar]
- 50.Ghanei-Nasab S, Khoobi M, Hadizadehd F, et al. Synthesis and anticholinesterase activity of coumarin-3-carboxamides bearing tryptamine moiety. Eur J Med Chem 2016;121:40–6. [DOI] [PubMed] [Google Scholar]
- 51.Khalifah RG.The carbon dioxide hydration activity of carbonic anhydrase I. Stop-flow kinetic studies on the native human isoenzymes B and C. J Biol Chem 1971;246:2561–73. [PubMed] [Google Scholar]
- 52.(a) Carta F, Maresca A, Scozzafava A, Supuran CT, Novel coumarins and 2-thioxo-coumarins as inhibitors of the tumor-associated carbonic anhydrases IX and XII. Bioorg Med. Chem 2012;2:2266–73. [DOI] [PubMed] [Google Scholar]; (b) Davis RA, Vullo D, Maresca A, et al. Natural product coumarins that inhibit human carbonic anhydrases. Bioorg Med Chem 2013;2:1539–43. [DOI] [PubMed] [Google Scholar]; (c) Sharma A, Tiwari M, Supuran CT.. Novel coumarins and benzocoumarins acting as isoform-selective inhibitors against the tumor-associated carbonic anhydrase IX. J Enzyme Inhib Med Chem 2014;29:292–6. [DOI] [PubMed] [Google Scholar]
- 53.Small-Molecule Drug Discovery Suite 2015-2. Schrödinger, LLC: New York, NY; 2015. [Google Scholar]
- 54.Schrödinger Release 2015-2: LigPrep. Schrödinger, LLC: New York, NY; 2015. [Google Scholar]
- 55.Halgren TA, Murphy RB, Friesner RA, et al. Glide: a new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening. J Med Chem 2004;47:1750–9. [DOI] [PubMed] [Google Scholar]
- 56.Verdonk ML, Cole JC, Hartshorn MJ, et al. Improved protein-ligand docking using GOLD. Proteins: Struct Funct Genet 2003;52:609–23. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.